

Background: PS is an allosteric regulator lipid that appears on activated platelets along with PE.
Results: We show that PE 1) increases the kcat and decreases Km of thrombin formation; 2) reduces membrane surface packing to promote Va binding; and 3) binds to Va and Xa to trigger their tight association.
Conclusion: This suggests a role in initiating blood coagulation.
Significance: PE may also be a regulator lipid.
Keywords: Blood Coagulation Factors, Enzyme Kinetics, Lipids, Phosphatidylethanolamine, Phosphatidylserine, Prothrombin, Serine Protease, Phosphatidylethanolamine, Factor Va, Factor Xa
Abstract
Constituents of platelet membranes regulate the activity of the prothrombinase complex. We demonstrate that membranes containing phosphatidylcholine and phosphatidylethanolamine (PE) bind factor Va with high affinity (Kd = ∼10 nm) in the absence of phosphatidylserine (PS). These membranes support formation of a 60–70% functional prothrombinase complex at saturating factor Va concentrations. Although reduced interfacial packing does contribute to factor Va binding in the absence of PS, it does not correlate with the enhanced activity of the Xa-Va complex assembled on PE-containing membranes. Instead, specific protein-PE interactions appear to contribute to the effects of PE. In support of this, soluble C6PE binds to recombinant factor Va2 (Kd = ∼6.5 μm) and to factor Xa (Kd = ∼91 μm). C6PE and C6PS binding sites of factor Xa are specific, distinct, and linked, because binding of one lipid enhances the binding and activity effects of the other. C6PE triggers assembly (Kdapp = ∼40 nm) of a partially active prothrombinase complex between factor Xa and factor Va2, compared with Kdapp for C6PS ∼2 nm. These findings provide new insights into the possible synergistic roles of platelet PE and PS in regulating thrombin formation, particularly when exposed membrane PS may be limiting.
Introduction
Localization of blood coagulation enzymes and cofactors to the surface of procoagulant anionic phospholipid membranes is a central paradigm in hemostasis and thrombosis (1, 2). The prototype for these complexes is the prothrombinase complex that catalyzes the conversion of prothrombin to thrombin. This enzyme complex consists of the enzyme factor Xa, the cofactor factor Va, and calcium bound to a phospholipid surface. Formation of the prothrombinase complex results in a greater than 105-fold rate enhancement in the conversion of prothrombin to thrombin (3, 4). Abnormal regulation of the prothrombinase complex has been demonstrated to be critical in the pathogenesis of thrombotic disorders (5).
PS has long been considered the most important phospholipid contributing to the activity of coagulation factors on phospholipid membranes (6). Near optimal binding of Xa and Va to model membranes occurs at ∼10–12% PS,4 which supports optimal prothrombinase activity (7, 8). Membrane PE has been shown to enhance prothrombin activation particularly in the presence of low concentrations of PS (1–5%) (9). PE does not affect prothrombin activation rates at optimal PS concentrations but reduces the requirement for PS ∼10-fold (10). The presence of PE in a membrane decreases the Kmapp for prothrombin, as well as the Kdapp for formation of the factor Va-factor Xa complex (10). These authors hypothesized that the observed effects on Km and Kdeff might result from reduced interfacial packing generally associated with the presence of PE in a membrane. In the current paper, we test this hypothesis and ask whether there are additional effects of PE on membrane binding of factors Xa or Va or on the activity of factors Xa or Va that might also contribute to the effects of PE on prothrombinase activity and prothrombinase assembly.
EXPERIMENTAL PROCEDURES
Proteins and Reagents
Bovine and human coagulation proteins, as well as the thrombin inhibitor dansylarginine-N-(3-ethyl-1,5-pentanediyl) amide (DAPA), were obtained from Hematologic Technologies, Inc. (Essex Junction, VT). Human dansyl-Glu-Gly-Arg-chloromethylketone (DEGR-Xa) was purchased from Hematologic Technologies Inc. All of the phospholipids were obtained from Avanti Polar Lipids (Alabaster, AL). 1,2-Dihexanoyl-sn-glycero-3-phospho-d-serine was custom synthesized by Avanti Polar Lipids (Alabaster, AL). The fluorescent probe 1-[4 (trimethylammonium)-6-phenyl]-1,3,5-hexatriene (TMA-DPH) was obtained from Molecular Probes (Eugene, OR). Chromogenic substrates were from Chromogenix (Molndal, Sweden). Cell culture media were from Invitrogen (Carlsbad, CA).
Recombinant Proteins
Native recombinant factor Va2 (rHFVa) was expressed in COS-7 and purified using established methods (11).
Phospholipid Preparations
Phospholipid (both synthetic and natural) vesicles were prepared by sonication as previously described (11). Measured suspensions of C6PS and C6PE were prepared as previously described (12).
Prothrombinase Assay
Rates of thrombin generation were determined in reaction mixtures containing factor Va, factor Xa, 1.4 μm prothrombin, and phospholipid vesicles in 25 mm Tris, pH 7.4, 150 mm NaCl, 2.7 mm KCl, 10 mg/ml BSA, and 3 mm CaCl2. Experiments were performed using either all bovine coagulation proteins or all human coagulation proteins. Rates of thrombin generation were measured using the substrate S2238 as described previously (11). Thrombin generation was also assayed in some cases using DAPA exactly as described (12) in the presence of 3 mm CaCl2.
Membrane Binding of Factors Xa and Va
Fluorescence energy transfer from Trp to a membrane-located dansyl probe was measured using either an SLM-8100 or a SPEX® FluoroLog-3 fluorescence spectrophotometer, as described by Gilbert et al. (13) and modified by Peng et al. (11).
Binding of Factors Xa and Va to Soluble Lipids
A SPEX® FluoroLog-3 spectrofluorometer (Horiba Jobin Yvon Inc., Edison, NJ) was used to measure intrinsic fluorescence intensity, using an excitation wavelength of 280 nm (4-nm band pass) and an emission wavelength of 340 nm (4-nm band pass) as described previously (14). In dual-ligand experiments, human factor Xa and rHFVa were preincubated with C6PS at 23 °C and then titrated with C6PE and vice versa. Fluorescence data were fit to a single-site binding model using a Marquardt-Levenberg algorithm (14).
Interfacial Packing: TMA-DPH Fluorescence Anisotropy
TMA-DPH was incorporated into membranes by adding a small volume (0.04–0.08% of vesicle sample volumes) of TMA-DPH in methanol to vesicle suspensions to a final lipid:probe ratio of 200:1. Fluorescence anisotropy was recorded after the fluorescence signal stabilized (excitation wavelength, 360 nm; emission wavelength, 450 nm) (15).
RESULTS
Prothrombinase Activity on PE Membranes
Previous studies reported that PE:PC membranes have either limited or no capacity to promote formation of an active prothrombinase complex (16). We recorded the activity of the bovine prothrombinase complex on DOPE:DOPC membranes in the presence of varying factor Va concentrations, 0.2 nm factor Xa, 1.4 μm prothrombin, 3 mm CaCl2, and 5 μm phospholipid vesicles. Fig. 1A shows that membranes containing 10–30% PE, 90–70% PC, and no PS supported prothrombinase complex activity. Activity increased with increasing DOPE membrane content and saturated at high factor Va (Vsat), indicating that activity reflects formation of a Xa-Va complex on PE:PC membranes, with a Kdapp of 0.8 ± 0.1 nm on 30% PE membranes. The effect of DOPE on Vsat was optimal by 20% DOPE and, Kdapp for prothrombinase formation at 30% DOPE was comparable with values reported for prothrombinase formation on PS-containing membranes (17). For this reason and because the PE content of platelet microparticles or platelet membranes is roughly 30% (18, 19), our focus will be on membranes containing 30% PE.
FIGURE 1.

Prothrombinase activity on DOPC:DOPE:DOPS membranes. A, rates of thrombin generation were determined at 37 °C in reaction mixtures containing 25 mm Tris, pH 7.4, 150 mm NaCl, 2.7 mm KCl, 0.6% PEG 8000, 3 mm CaCl2, 0.2 nm bovine factor Xa, a fixed concentration of prothrombin (1.4 μm), varying factor Va concentrations, and 5 μm of DOPC:DOPE vesicles of varying compositions: 70:30 (○), 80:20 (♢), and 90:10 (▿). Three independent experiments are shown for each phospholipid vesicle composition. The line represents global fitting (GraphPad Prism) of the data while varying parameters Kdapp and Vsat, as described under “Experimental Procedures.” The inset shows the rates of thrombin formation in the presence of 30% egg PE:70% egg PC membranes. B, binding of DEGR-Xa to factor Va2 in the presence of 30:70 DOPE:DOPC membranes was determined in the presence of increasing concentration of factor Va2 and at a fixed concentration of DEGR-Xa (10 nm). The buffer was same as in A. The line represents the best fit of the data to a simple single-site binding model as obtained using Sigma Plot. C, rates of thrombin generation were determined at 37 °C in reaction mixtures containing 3 mm CaCl2, 0.2 nm bovine factor Xa, 5 nm bovine factor Va, 5 μm of vesicles of varying compositions: 100% DOPC (●), 80:20 DOPC:DOPE (▴), 69:30:1, and DOPC:DOPE:DOPS (■). The lines show the fit of these data using Sigma Plot to a standard Michaelis-Menten model to determine the kcat and Km of prothrombin activation with membranes of different lipid compositions, as described in the text.
Membrane mimetic phospholipid vesicles have been used to model the assembly and activity of the prothrombinase complex on platelet membranes. Researchers have used synthetic lipids (16, 20), as well as lipids derived from natural sources (21, 22). In our work, we use synthetic lipids because they are chemically more stable. In control experiments, we investigated the activity of the prothrombinase complex assembled on phospholipid membranes composed of 70 or 80% egg PC and 20 or 30% egg PE to ensure that our results are not artifacts associated with a particular synthetic lipid species. Nearly identical results were obtained with membranes containing egg yolk-derived lipid (Vsat = 245 ± 25 IIa min−1 and Kdapp = 0.7 ± 0.2 nm for 30% egg PE, as shown in Fig. 1A, inset) and with DOPE:DOPC-containing membranes (Table 1 and Fig. 1A).
TABLE 1.
Contribution of PS, PE, and DPhPC to prothrombinase activity
Rates of prothrombin activation were determined on phospholipid vesicles with the compositions specified using bovine coagulation proteins as described under “Experimental Procedures.” The parameters Kdapp and Vsat for prothrombinase complex formation (Fig. 1) are expressed as the means ± S.E. of the parameter, where standard errors are derived from fitting a single experiment or globally fitting N independent experiments to a hyperbola (see “Experimental Procedures”). ND, not detected.
| Membrane | Kdapp | Vsat | N |
|---|---|---|---|
| nm | nm·min−1 | ||
| DOPC:DOPE | |||
| 90:10 | 12 ± 4 | 127 ± 22 | 3 |
| 80:20 | 9.8 ± 1.5 | 246 ± 18 | 3 |
| 70:30 | 0.8 ± 0.1 | 234 ± 5 | 3 |
| DOPC:DOPS | |||
| 99:1 | 17 ± 8 | 8 ± 3 | 3 |
| 95:5 | 1.1 ± 0.2 | 629 ± 26 | 3 |
| 90:10 | 0.2 ± 0.0 | 675 ± 10 | 3 |
| 75:25 | 0.3 ± 0.1 | 800 ± 30 | 1 |
| DOPC:DOPE:DOPS | |||
| 69:30:1 | 1.3 ± 0.2 | 459 ± 15 | 4 |
| 65:30:5 | 0.3 ± 0.0 | 754 ± 15 | 6 |
| 60:30:10 | 0.3 ± 0.1 | 762 ± 21 | 1 |
| DOPC:DPhPC:DOPS | |||
| 70:30:0 | 30 ± 4 | 0.8 ± 0.1 | 2 |
| 69:30:1 | 1.3 ± 0.4 | 7.9 ± 0.6 | 3 |
| 65:30:5 | 0.3 ± 0.0 | 643 ± 14 | 1 |
| 60:30:10 | 0.3 ± 0.0 | 685 ± 14 | 1 |
Our small Kdapp is consistent with that reported by Smirnov et al. (10), who interpreted this as the Kd for assembly of the prothrombinase complex. To confirm this interpretation, the fluorescence of DEGR-FXa in the presence of 30% DOPE:DOPC membranes was recorded (23) (Fig. 1B) to obtain the Kd for association of factors Va and Xa (2 ± 0.4 nm). This was comparable with the Kdapp recorded in Table 1, confirming that the Kdapp values do reflect assembly of the Xa-Va complex on DOPC:DOPE membranes.
Because previous studies (10) focused only on PE effects on Km and not on kcat, we determined the initial rates of thrombin generation in the presence of pure DOPC or DOPC:DOPE (70:30) membranes, 0.2 nm Xa, 5 nm Va, and increasing concentrations of prothrombin (0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, and 2 μm) (Fig. 1C). Fitting these data resulted in a Km of 0.43 ± 0.02 μm for 70:30 DOPC:DOPE membranes, which is ∼15-fold lower than for DOPC membranes (Kmapp = ∼6 ± 1 μm; data not shown). We note that prothrombinase activity did not saturate with respect to Va addition for DOPC membranes because Va binds very weakly to such membranes (24). The kcat for thrombin generation obtained with DOPC membrane was only 0.08 ± 0.01 s−1, compared with 21.5 ± 2 s−1 for 70:30 DOPC:DOPE membranes, nearly a 28-fold increase caused by the presence of DOPE. Overall, the kcat/Km for 70:30 DOPC:DOPE membranes (∼5 × 107 m−1 s−1) was approximately that reported for bovine Va-Xa complex on 25:75 PS:PC membranes (17, 25). The rate of thrombin generation in the absence of any lipid was not measurable in our reaction condition (0.2 nm FXa, 5 nm FVa2). Thus, the effect of DOPE on kcat was comparable with, if not larger than, the effect on Km. This is significant because a kcat effect implies a change in enzyme conformation, whereas a Km effect could reflect substrate binding to the membrane-bound complex, which may or may not reflect changes in enzyme structure. Including 1% DOPS in DOPC:DOPE membranes (69:30:1) (Fig. 2A) produced an identical Km (0.40 ± 0.03 μm) and a 2-fold increase in kcat (43.5 ± 3 s−1). This effect is seen in the Vsat values collected in Table 1, where we see that additional membrane DOPS content led to only a small further increased activity (∼2-fold). This small amount of PS also reduced Kdapp (Table 1 and Fig. 2A). Additional PS (5–10%) roughly doubled Vsat (Table 1 and Fig. 2, B and C), meaning that only 1% PS is needed to form a nearly optimal prothrombinase in the presence of 30% PE. The experiments summarized in Table 1 were all carried out with 1.4 μm prothrombin, which is well above the Km for prothrombinase on 30% DOPE vesicles with or without 1% PS. Because PS had no measurable effect on Km for membranes containing 30% DOPE, we conclude that the increase in saturating activity (Vmax) with increasing PS is due to increases in kcat.
FIGURE 2.
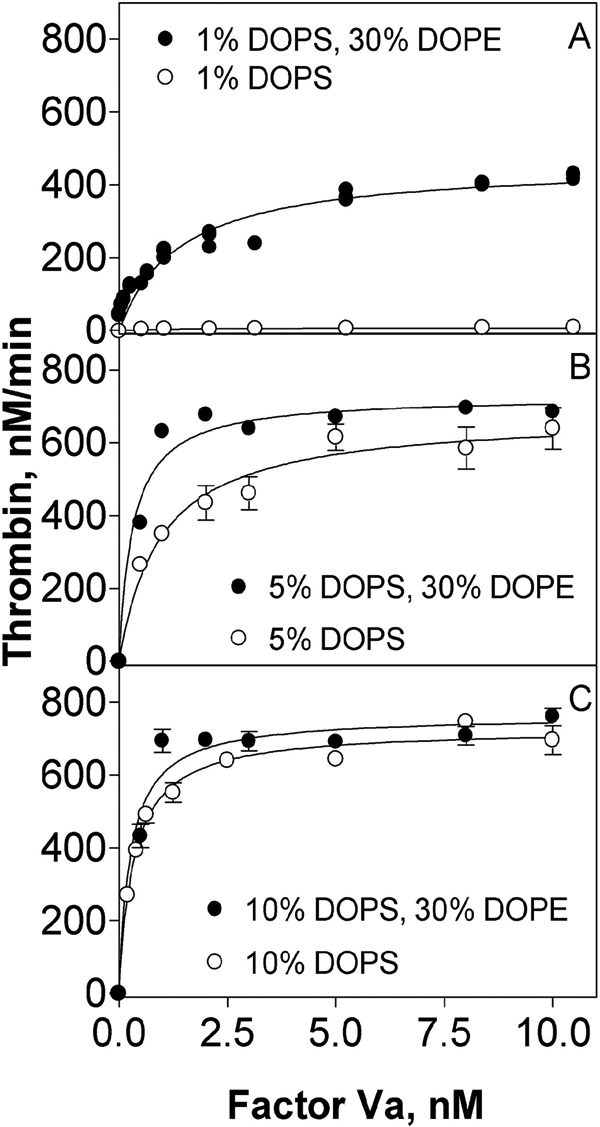
DOPE enhances prothrombinase activity on membranes containing low concentrations of DOPS. Rates of thrombin generation were determined at 37 °C in buffer containing 25 mm Tris, pH 7.4, 150 mm NaCl, 2.7 mm KCl, 10 mg/ml BSA, 3 mm CaCl2, 0.2 nm bovine factor Xa, 1.4 μm bovine prothrombin, bovine factor Va, and 5 μm DOPC:DOPS:DOPE vesicles of varying compositions: 69:1:30 and 99:1:0 (A), 65:5:30 and 95:5:0 (B), and 60:10:30 and 90:10:0 (C). In A, four independent experiments are shown. Global fitting of the data for Vsat and Kdapp was performed using GraphPad Prism version 4.00. In B and C, the error bars represent one standard error of the parameter for triplicate measurements in a representative experiment.
From the results summarized in Table 1, it is clear that DOPE significantly promoted prothrombinase complex assembly and activity only for membranes containing little or no DOPS, whereas DOPE had little effect when incorporated into membranes containing ≥5% DOPS, in agreement with Smirnov et al. (10). However, the current results establish that PE also increases kcat and that PS further increases both kcat and Kdapp but not Km.
Because of the expense of isolating human factor Va and because many earlier experiments used bovine factor Va (26), bovine coagulation proteins were used in most of these experiments. However, control experiments using human coagulation proteins confirmed that similar kinetics are seen for human prothrombinase assembled on 70:30 DOPC:DOPE membranes (kcat = 12 ± 2 s−1, Km = 0.6 ± 0.04 μm, and kcat/Km = ∼2 × 107 m−1 s−1) as we found using bovine proteins.
Binding of Factor Va to PE Membranes
We used intrinsic fluorescence to follow the binding of bovine factor Va to membranes containing variable concentrations of DOPE and DOPS (Fig. 3 and Table 2). Surprisingly, factor Va bound fairly tightly to membranes containing only 30% (Kd = 10 ± 3 nm; Fig. 3A) or 20% DOPE (Kd = 12 ± 8 nm) but more weakly (Kd = 51 ± 8 nm) to membranes containing only 10% DOPE (Table 2). Bovine factor Va interacts extremely weakly (Kd = ∼3 μm) with membranes composed of only egg PC (24), demonstrating that even a small fraction of PE is very important for factor Va membrane binding. Factor Va bound similarly but somewhat more tightly to 30% PE membranes composed of highly unsaturated natural egg yolk-derived lipids (Fig. 3A, inset; Kd = ∼3 ± 0.5 nm) compared with the synthetic lipids used in this study. This is consistent with the observation that factor Va binding free energy derives principally from insertion of Trp residues into the membrane (27), an event that should be promoted by the reduced interfacial packing associated with highly unsaturated lipids. Adding 1% PS to 30% DOPE membranes had little effect on factor Va binding, although increasing PS content to 5–10% did improve binding somewhat but not beyond what was observed in the absence of DOPE (Table 2). We conclude that although PE does promote Va binding to membranes, PS promotes tighter binding in a PE-independent fashion. As a control, we showed that the Kd for bovine Va binding to 75:25 DOPC:DOPS membranes (0.35 nm) (Table 2) was comparable with that reported for human recombinant factor Va to synthetic vesicles (Kd = 0.24 nm) (28), indicating that our results are not species-specific.
FIGURE 3.
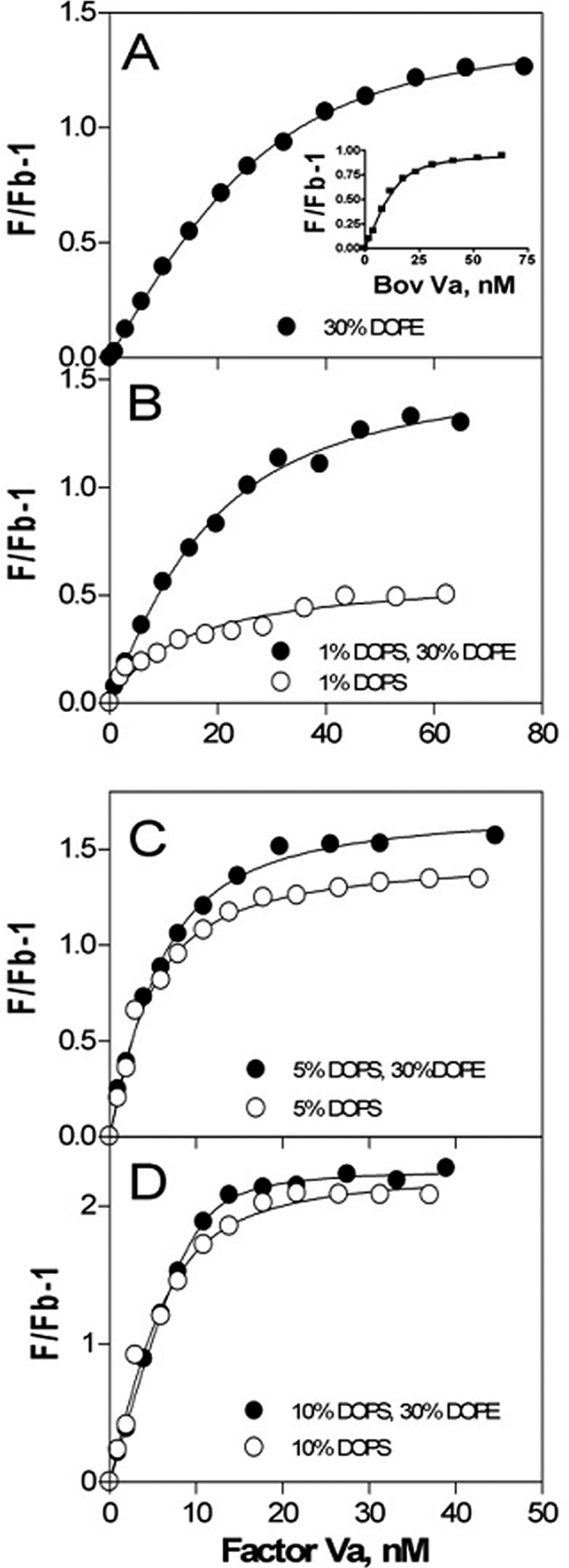
Binding of factor Va to phospholipid vesicles. Samples contained 150 mm NaCl, 20 mm Tris, pH 7.4, and 0.6 μm labeled DOPC:DOPS:DOPE:dansyl-PE vesicles of varying compositions: 70:0:27.5:2.5 (A), 69:1:27.5:2.5 and 96.5:1:0:2.5 (B), 65:5:27.5:2.5 and 92.5:5:0:2.5 (C), and 60:10:27.5:2.5 and 87.5:10:0:2.5 (D). Fluorescence measurements (F) were made as described under “Experimental Procedures” after incremental addition of bovine factor Va at 25 °C. The lines represent the best fit of data in one representative experiment. The inset in A shows the binding of bovine Va to egg PE:egg PC (20:80) membranes.
TABLE 2.
Contribution of PS, PE, and DPhPC to binding of bovine factor Va to membranes
The binding of bovine factor Va to phospholipid vesicles with the compositions specified was analyzed as described under “Experimental Procedures.” The parameters Kd, n, and (F∞/Fb) − 1 were calculated by fitting the data as shown in Fig. 3. Kd, dissociation constant for factor Va binding to phospholipid vesicles; n, number of phospholipid monomers composing a single binding site; N, number of independent experiments; ND, no measurable binding detected.
| Membrane | Kd | n | N |
|---|---|---|---|
| nm | |||
| DOPC:DOPE | |||
| 90:10 | 51 ± 8 | 21 ± 27 | 2 |
| 80:20 | 12 ± 8 | 20 ± 4 | 3 |
| 70:30 | 10 ± 3 | 22 ± 3 | 2 |
| DOPC:DOPS | |||
| 99:1 | 17 ± 8 | a | 2 |
| 95:5 | 2.3 ± 0.3 | 136 ± 9 | 2 |
| 90:10 | 1.4 ± 0.2 | 74 ± 3 | 4 |
| 75:25 | 0.35 ± 0.17 | 51 ± 2 | 2 |
| DOPC:DOPE:DOPS | |||
| 69:30:1 | 9 ± 5 | 36 ± 12 | 2 |
| 65:30:5 | 1.9 ± 0.3 | 93 ± 7 | 4 |
| 60:30:10 | 1.3 ± 0.3 | 69 ± 5 | 2 |
| DOPC:DPhPC:DOPS | |||
| 70:30:0 | ND | ND | 2 |
| 69:30:1 | 7 ± 3 | 85 ± 34 | 2 |
| 65:30:5 | 3.4 ± 0.8 | 101 ± 14 | 2 |
| 60:30:10 | 1.6 ± 0.2 | 63 ± 2 | 3 |
a The value for n did not converge.
Although addition of >1% DOPS to membranes containing 30% DOPE decreased Kd, it simultaneously increased the stoichiometry of binding (number of lipids per bound protein at saturation), such that the combined effect of 30% DOPE and DOPS was roughly that of DOPS alone. Even at 1% DOPS, the addition of 30% DOPE had little effect on Va binding (Kd for binding to 1% DOPS decreased only from 17 ± 8 to 9 ± 5 nm with inclusion of 30% DOPE; Fig. 3B and Table 2). We conclude that there is minimal synergistic effect of DOPE and DOPS on factor Va membrane binding, even at very low DOPS content.
Effects of Interfacial Packing on Prothrombinase Activity and Factor Va Binding
PE can adopt a nonlamellar, reverse hexagonal (HII) phase because it has a small head group and two acyl chains (29, 30). Because the HII phase has negative curvature with respect to its symmetry axis, PE is said to have a negative “intrinsic membrane curvature” and can induce curvature elastic stress in a monolayer, such that packing in the interface of that monolayer is reduced, especially for unsaturated acyl chains and/or high temperature (31). Although this effect actually depends on the other lipids present in the monolayer, it is commonly assumed for convenience that monolayer curvature is an additive property of the intrinsic curvatures of all lipids in the monolayer (31). Based on the assumption of intrinsic curvature additivity, it was proposed that the stimulatory effect of PE on prothrombinase caused by its molecular shape should reduce membrane surface packing (10).
To test this proposal, we constructed model membranes having experimentally defined relative interfacial packing and containing defined amounts of DOPC, DOPE, and DOPS. To adjust measured interfacial packing, we used another synthetic phospholipid, 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC), also shown to induce HII phase and having a cross-sectional area ratio of lipid head groups to acyl chains similar to DOPE (32). Unlike DOPE, which has a head group with reduced cross-sectional area, DPhPC derives its thermal shape from very bulky acyl chains. We use changes in TMA-DPH fluorescence anisotropy, previously shown to report changes in interfacial order in outer monolayers of membranes to which it absorbs (33, 34), to reflect differences in interfacial order between membranes.
Our results demonstrate that both PS and PE promote prothrombinase activity but that there is no correlation between membrane interfacial order, as detected using an established probe of interfacial order, and increased activity. We designed a PS-free composition by a combination of rational design and trial and error (DOPC:DPhPC:DOPE, 65:5:30) having the same interfacial order as pure DOPC membranes. Membranes with this composition supported significantly more activity than pure DOPC membranes (Table 3), demonstrating a PE-induced activity increase without a decrease in surface packing. Adding just 1% DOPS to these membranes reduced interfacial order substantially (Table 3), demonstrating that in this model membrane system, monolayer intrinsic curvature indeed cannot be approximated as an additive function of composition. The addition of this small amount of DOPS also increased activity roughly 9-fold. Although DPhPC increased interfacial order in PS-free vesicles, the addition of 1% DOPS produced a dramatic decrease in order, but still only a 10-fold increase in activity (Table 3). These activity increases are comparable with that seen for adding 1% DOPS to pure DOPC membranes (Table 3) and thus are likely effects of DOPS, not of bilayer packing changes. By contrast, the addition of 30% DOPE to DOPC membranes produced only a small decrease in packing (Table 3) but a 600-fold increase in activity.
TABLE 3.
Interfacial packing of factor Xa activity on and factor Va binding to vesicles containing different concentration of DOPC and DOPE and in the absence and presence of 1% DOPS
| Lipids | 0% DOPS |
1% DOPC replaced by 1% DOPS |
||||
|---|---|---|---|---|---|---|
| TMA-DPH Anisotropy | Activity Vsat | Va Binding | TMA-DPH Anisotropy | Activity Vsat | Va Binding | |
| nm/min | nm | nm/min | nm | |||
| 100% PC | 0.2271 | 0.6 ± 0.1 | ND | 0.2231 | 8 ± 3 | 17 ± 8 |
| 70% PC + 30% PE | 0.2119 | 234 ± 5 | 10 ± 3 | 0.2007 | 459 ± 15 | 9 ± 5 |
| 65% PC + 5% DPhPC + 30% PE | 0.2242 | 52 ± 4 | 6350 ± 700 | 0.2036 | 486 ± 12 | 10 ± 2 |
| 70% PC + 30% DPhPC | 0.2416 | 0.8 ± 0.2 | ND | 0.1948 | 7.9 ± 0.6 | 7 ± 3 |
In contrast to activity, Table 3 demonstrates that a reduction in membrane packing did enhance factor Va binding. If reduced membrane packing can promote factor Va binding, could it be that PE also promotes factor Xa binding, thereby helping to bring both to the surface of a 30% PE membrane to promote prothrombinase assembly?
Factor Xa Binding to PE Membranes
Using concentrations of bovine factor Xa up to 1 μm and our FRET binding assay, we were unable to obtain reliable Xa binding data with membranes containing 30% DOPE or <25% DOPS. The latter result is consistent with previous reports (Kd of ∼1.5 μm for 10% bovine brain PS membranes) (8). However, the presence of 30% DOPE increased the affinity of factor Xa for membranes containing 10% PS (Kd = ∼187 nm), similar to the value for binding to 75:25 DOPC:DOPS membranes (Kd = ∼219 nm), which is consistent with previous Kd estimates for 25% PS membranes (8, 25). These observations suggest that 30% DOPE in membranes containing less than optimal DOPS might enhance factor Xa binding slightly but likely not sufficiently to explain the demonstrated prothrombinase supporting ability of 30% DOPE membranes (Fig. 2). Next, we consider another possible explanation, specific binding of PE to either factor Xa or Va to trigger changes in these proteins that might increase factor Xa activity or enhance formation of a prothrombinase complex.
Soluble C6PE Binds to rHFVa and Factor Xa
Because of the large effect of PE on kcat, we hypothesized that PE might bind to regulatory sights on either factor Xa or factor Va, as we have documented for PS binding to these proteins (12, 35, 36). To investigate this, direct binding of C6PE to rHFVa2 was examined by measuring changes in intrinsic fluorescence as reported previously (36). We utilized only the factor Va2 glycoform because we showed that soluble PS does not stimulate formation of an active prothrombinase complex from the Va1 glycoform (23). We determined at each experimental condition the critical micelle concentration of C6PE (12) to confirm the absence of C6PE micelles. The critical micelle concentrations of C6PE in the presence of 100 nm human rHFVa2 and factor Xa were 800 and 850 μm, respectively. C6PE bound to rHFVa2 with a Kd of 6.5 ± 0.5 μm based on a single-site model for binding (Fig. 4A), comparable with values previously reported for C6PS binding to rHFVa2 (∼20 μm) (14). Interspecies control experiments confirmed that C6PE bound with affinity comparable with that of bovine Va2 (shown in the inset to Fig. 4A; Kd = ∼8 μm) and also comparable with C6PS binding to bovine factor Va (9 μm) (36).
FIGURE 4.

Binding of rHFVa and Factor Xa to soluble C6PE. The intrinsic fluorescence intensity of 0.1 μm rHFVa (A) or 0.1 μm human factor Xa (B) in 20 mm Tris, 150 mm NaCl, and 5 mm CaCl2, pH 7.5, was measured as a function of C6PE concentration at 23 °C. The data were analyzed according to a simple single-binding site model to yield a Kd of 6.5 ± 0.5 μm for rHFVa and 91 ± 1.4 μm for factor Xa. The insets to A and B show the binding of bovine factors Va/Xa with C6PE (Kd values are 8 ± 1.2 and 90 ± 14 μm for factors Va and Xa, respectively). C presents fluorescence of DEGR-Xa with the addition of rHVa2 in the presence of 400 μm C6PE. The fit of a single-binding site model to the data provided the Kd (40 ± 3 nm) for Xa-DEGR-Va2 complex formation.
We also used intrinsic fluorescence to detect binding of C6PE to human factor Xa (Fig. 4B) and bovine factor Xa (Fig. 4B, inset). When analyzed using a single-site model, these data yielded Kd values of 91 ± 13 and 90 ± 10 μm, respectively, also comparable with previously reported values for C6PS binding (73–90 μm) (35, 37).
To test directly whether PE binding promotes assembly of the Xa-rHVa2 complex, binding of DEGR-Xa to rHVa2 in the presence of 400 μm C6PE was monitored by following DEGR-Xa fluorescence (23), as shown in Fig. 4C. The titration curve was well described by a single-binding site model with a dissociation constant KdVa-Xa, = 40 ± 3 nm for assembly of the Xa-Va2 complex. This is 20-fold weaker than for assembly in the presence of C6PS (2 nm at 22 °C) (12) but >25-fold tighter than estimated for assembly in the absence of lipid (>1 μm) (12). A factor of 25 in binding affinity corresponds to a free energy of interaction that is more favorable by >3 kBT. This is a very significant free energy change for a two-dimensional binding reaction at low membrane concentration. Thus, the Kdapp for prothrombinase assembly for 5 μm 30:70 DOPE:DOPC membranes (∼ 0.8 nm; Table 1) is much smaller than for DOPC membranes, for which prothrombinase assembly was undetectable (data not shown).
Synergy between Binding of C6PS and C6PE to Human Factors Xa and rHFVa
Our results and those of others (38) demonstrate synergy between PE and PS in membranes. In experiments analogous to those reported in Fig. 4, we recorded at increasing C6PS concentrations (0–200 μm) the intrinsic fluorescence intensity of 0.1 μm human factor Xa preincubated for 15 min with a saturating concentration of C6PE. The data were again appropriately analyzed according to a single-binding site model to obtain an apparent Kd of 40 ± 5 μm for C6PS binding in the presence of 200 μm C6PE (Table 4). Similarly, we obtained the apparent Kd of C6PE binding to human factor Xa preincubated with a saturating concentration of C6PS (400 μm) (Kd = 7 ± 0.7 μm; Table 4). The results summarized in Table 4 clearly indicate synergy between C6PS and C6PE binding to factor Xa. To determine whether this effect was specific for the PE species, we tested whether C6(D)PS or C6PC binding was similarly linked to C6PS binding. The results in Table 4 show clearly that preincubating with these lipids did not alter the affinity of C6PS for factor Xa, nor did preincubating with C6PS alter the affinity for C6(D)PS or C6PC (Table 4). We conclude that the synergy between C6PS and C6PE binding to factor Xa is specific for these lipids. Synergy between C6PS and C6PE binding to 0.1 μm rHFVa was not observed (Table 4).
TABLE 4.
Linkage between sites for C6PS and C6PE on human factors Xa and rHFVa2
Binding of soluble phospholipids to 0.1 μm rHFVa or 0.1 μm human factor Xa was monitored using intrinsic fluorescence intensity in 20 mm Tris, 150 mm NaCl, and 5 mm CaCl2, pH 7.5, as a function of C6PS, C6PE, C6PC, or C6(D)PS and the concentration at 23 °C. The data were analyzed according to a single binding site model to reveal the effects of preincubation with one lipid C6PE or C6PS on the binding of the other.
| Protein | Lipid titrant | Conditions | Kd |
|---|---|---|---|
| μm | |||
| Xa | C6PS | No other lipid | 86 ± 10 (35) |
| C6PS | Preincubated with 200 μm C6PE | 40 ± 5 | |
| C6PS | Preincubated with 400 μm C6(D)PS | 95 ± 12 | |
| C6PS | Preincubated with 400 μm C6PC | 90 ± 8 | |
| Xa | C6PE | No other lipid | 91 ± 13 |
| C6PE | Preincubated with 400 μm C6PS | 7 ± 0.7 | |
| Xa | C6(D)PS | No other lipid | 71 (35) |
| C6(D)PS | Preincubated with 400 μm C6PS | 67 ± 24 | |
| Xa | C6PC | No other lipid | 203 ± 27 (35) |
| C6PC | Preincubated with 400 μm C6PS | 198 ± 68 | |
| Va | C6PS | No other lipid | 20 ± 1 (14) |
| C6PS | Preincubated with 200 μm C6PE | 17 ± 3 | |
| Va | C6PE | No other lipid | 6.5 ± 0.5 |
| Preincubated with 400 μm C6PS | 5.6 ± 0.5 |
DISCUSSION
Earlier studies (9, 10, 16) found that PE was synergistic with PS in enhancing prothrombinase activity on membranes containing low concentrations of PS. Smirnov et al. (10) focused on the effect of PS on Km for prothrombin activation and the Kdapp for Va-Xa assembly at high (50%) surface concentrations of PE. They had reported that 50% PE promoted prothrombin activation only on membranes having 0 or 1% PS and that the effect of 50% PE alone was only ∼15% of that for PE with any amount of PS, i.e. that the effect of PE on prothrombinase activity was much less than the effect of PS. We report that 30% DOPE membranes are surprisingly ∼35% as active as membranes containing 30% DOPE and 10% DOPS, roughly the proportion of these species exposed on activated platelet membranes (18, 19), making it important to understand the mechanism for PE and the ability of PE both to promote prothrombinase assembly and activity, both synergistically with PS and in the absence of PS. In this regard, Smirnov et al. (10) hypothesized that the effects of PE on prothrombinase assembly and activity might be due to reduced interfacial packing expected for membranes containing PE. The present study was designed to test this hypothesis and thereby define the mechanism by which the presence of PE in a membrane enhanced prothrombin activation by membrane-bound factors Va and Xa.
We report seven significant new observations: 1) PE effects kcat more than Km (Fig. 1C), and a kcat effect strongly implies an enzyme or cofactor conformational change, whereas a Km effect could be due to conformational changes or to altered binding of enzyme cofactor and substrate to the membrane; 2) PE promotes binding of factor Va to membranes (Fig. 3 and Table 2), and because we showed that reduced membrane packing enhanced factor Va binding, the effect of PE on Va binding is probably due to its ability to reduce membrane packing; 3) PE promotes assembly of a prothrombinase complex (Fig. 1, A and B) because of its effects on factors Va and Xa independent of the presence of a membrane (Fig. 4C) and thus not because of its effects on membrane packing; 4) 30% PE does not produce a measurable effect on factor Xa membrane binding; 5) C6PE and C6PS bind synergistically to sites on factor Xa to alter its activity (Table 4); 6) C6PE binds to factor Va with greater affinity than does C6PS, but binding of these lipids to sites on this protein is not synergistic (Table 4); and 7) both PS and PE promote prothrombinase activity, but the magnitudes of these effects do not correlate with their effects on membrane interfacial packing (Table 3).
All of these observations are consistent with a new hypothesis for how PE promotes prothrombin activation. First, factor Va binds with significant affinity to 30% PE membranes (Table 2) because its membrane-binding free energy derives almost exclusively from penetration of the membrane interface by two Trp residues (39). Even though factor Xa binds poorly to 30% DOPE membranes, once a small amount of Xa binds to the membrane, PE will occupy regulatory sites on it and factor Va, resulting in formation of an active prothrombinase complex. Because C6PS is more effective than C6PE in increasing factor Xa activity and in promoting Xa-Va interaction (Fig. 4), even a small amount of PS in the 30% DOPE membrane will occupy the regulatory sites on Xa and Va to increase in Vsat and decrease in Kdapp (Table 1). Even though prothrombin does not bind tightly to a 30% PE membrane, the well known reduction of Km upon binding to PS-containing membranes is due to binding of lipids to the regulatory site on Xa and not to the availability of membrane-associated prothrombin (40). In this picture, the effect of PE on membrane interfacial packing contributes only to bringing factor Va to the membrane, but not for the reduction in Km, the increase in kcat, and the decrease in Kdapp, which result from PE regulation of both factors Xa and Va.
The lipid-binding regions through which C6PS influences factors Va and Xa have been identified (14, 27, 41). Are these the same sites occupied by PE? Although we cannot say with certainty, our data suggest that this is the case for factor Xa. The initial rate of factor Xa-mediated prothrombin activation increased by 4-fold in the presence of saturating (400 μm) C6PE (data not shown). A subsaturating concentration of C6PS (50 μm) produced a 15-fold activity increase, whereas subsequent addition of a saturating concentration (400 μm) of C6PE increased factor Xa activity by an additional 2.7-fold (total 40-fold increase, as compared with no lipid). This effect is less than the 60-fold increase seen with C6PS alone (37). This is consistent with C6PE and C6PS competing for common sites, because the addition of C6PE would displace C6PS, resulting in decreased activity. If these lipids bound two different sites, we would expect a 4 × 15 = 60-fold) change, which was not observed. However, why do we observe synergy in Kd values if the two lipids have common sites? This could be explained by C6PE binding to two linked sites, as has been reported for C6PS (41). Our current hypothesis is that C6PS and C6PE share the regulatory site on Xa (non-additive effects on activity) but also bind to the anomalous site in the catalytic domain to which the regulatory site is linked (41). More extensive experiments are needed to prove this hypothesis. We expect that this is not the case for factor Va2, because only the site in the C1 domain regulates the interaction between factors Xa and Va (27). We will need additional studies to confirm that this site also binds C6PE to regulate assembly.
The effects of PE in model membrane studies are insignificant for membranes containing >1% PS (our results and Ref. 10). Platelet activation is generally recognized to expose plasma to membranes containing upwards of 11–13% PS and 25–35% PE (18). If we view coagulation in terms of the four stages (initiation, amplification, propagation, and shutdown) proposed in the emerging cell based model (42), our results make it unlikely that PE plays a significant role during the propagation stage, at which point platelet activation has already occurred. However, during initiation and perhaps the early part of amplification, our results show that the small amount (∼10 mol%) of PE exposed on resting platelets along with the even smaller amount of PS (∼1–2 mol%) could promote formation of prothrombinase if factors Xa and Va were available. Because small amounts of these proteins are produced via the TF-VIIa reactions at the earliest stages of platelet plug formation, they could assemble an active prothrombinase on unactivated platelets and trigger formation of sufficient thrombin to activate more platelets and initiate the amplification stage.
Even in the absence of PS, we show that membranes containing 30% PE are remarkably effective in assembling an active prothrombinase reaction. This is particularly interesting in light of the report that very LDLs promote prothrombin activation even though they contain essentially no PS (43). Even more interesting is the report that oxidized LDL particles, even though they contain no PS, elicit a strong prothrombin activating activity, likely because of small amounts of PE oxidation products that resemble PS (44). Thus, our results are consistent with the possibility that PE plays an important role in the etiology of atherosclerotic lesion-induced clot formation.
This work was supported, in whole or in part, by National Institutes of Health Grants GM32707 and HL 072827 (to B. R. L.) and HL43106 (to W. H. K.).
- PS
- phosphatidylserine
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- DAPA
- dansylarginine-N-(3-ethyl-1,5-pentanediyl) amide
- DEGR
- dansyl-Glu-Gly-Arg-chloromethylketone
- TMA-DPH
- [4 (trimethylammonium)-6-phenyl]-1,3,5-hexatriene
- DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DPhPC
- 1,2-diphytanoyl-sn-glycero-3-phosphocholine.
REFERENCES
- 1. Kane W. H., Davie E. W. (1988) Blood 71, 539–555 [PubMed] [Google Scholar]
- 2. Mann K. G., Nesheim M. E., Church W. R., Haley P., Krishnaswamy S. (1990) Blood 76, 1–16 [PubMed] [Google Scholar]
- 3. Nesheim M. E., Taswell J. B., Mann K. G. (1979) J. Biol. Chem. 254, 10952–10962 [PubMed] [Google Scholar]
- 4. Weinreb G. E., Mukhopadhyay K., Majumder R., Lentz B. R. (2003) J. Biol. Chem. 278, 5679–5684 [DOI] [PubMed] [Google Scholar]
- 5. Bertina R. M., Koeleman B. P., Koster T., Rosendaal F. R., Dirven R. J., de Ronde H., van der Velden P. A., Reitsma P. H. (1994) Nature 369, 64–67 [DOI] [PubMed] [Google Scholar]
- 6. Jones M. E., Lentz B. R., Dombrose F. A., Sandberg H. (1985) Thromb. Res. 39, 711–724 [DOI] [PubMed] [Google Scholar]
- 7. Cutsforth G. A., Koppaka V., Krishnaswamy S., Wu J. R., Mann K. G., Lentz B. R. (1996) Biophys. J. 70, 2938–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cutsforth G. A., Whitaker R. N., Hermans J., Lentz B. R. (1989) Biochemistry 28, 7453–7461 [DOI] [PubMed] [Google Scholar]
- 9. Smeets E. F., Comfurius P., Bevers E. M., Zwaal R. F. (1996) Thromb. Res. 81, 419–426 [DOI] [PubMed] [Google Scholar]
- 10. Smirnov M. D., Ford D. A., Esmon C. T., Esmon N. L. (1999) Biochemistry 38, 3591–3598 [DOI] [PubMed] [Google Scholar]
- 11. Peng W., Quinn-Allen M. A., Kim S. W., Alexander K. A., Kane W. H. (2004) Biochemistry 43, 4385–4393 [DOI] [PubMed] [Google Scholar]
- 12. Majumder R., Weinreb G., Lentz B. R. (2005) Biochemistry 44, 16998–17006 [DOI] [PubMed] [Google Scholar]
- 13. Gilbert G. E., Furie B. C., Furie B. (1990) J. Biol. Chem. 265, 815–822 [PubMed] [Google Scholar]
- 14. Majumder R., Quinn-Allen M. A., Kane W. H., Lentz B. R. (2005) Biochemistry 44, 711–718 [DOI] [PubMed] [Google Scholar]
- 15. Haque M. E., Koppaka V., Axelsen P. H., Lentz B. R. (2005) Biophys. J. 89, 3183–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Billy D., Willems G. M., Hemker H. C., Lindhout T. (1995) J. Biol. Chem. 270, 26883–26889 [DOI] [PubMed] [Google Scholar]
- 17. Krishnaswamy S., Nesheim M. E., Pryzdial E. L., Mann K. G. (1993) Methods Enzymol. 222, 260–280 [DOI] [PubMed] [Google Scholar]
- 18. Bevers E. M., Comfurius P., Zwaal R. F. (1983) Biochim. Biophys. Acta 736, 57–66 [DOI] [PubMed] [Google Scholar]
- 19. Zwaal R. F., Bevers E. M. (1983) Subcell. Biochem. 9, 299–334 [DOI] [PubMed] [Google Scholar]
- 20. Kung C., Hayes E., Mann K. G. (1994) J. Biol. Chem. 269, 25838–25848 [PubMed] [Google Scholar]
- 21. Boskovic D. S., Troxler T., Krishnaswamy S. (2004) J. Biol. Chem. 279, 20786–20793 [DOI] [PubMed] [Google Scholar]
- 22. Kamath P., Krishnaswamy S. (2008) J. Biol. Chem. 283, 30164–30173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Majumder R., Weinreb G., Zhai X., Lentz B. R. (2002) J. Biol. Chem. 277, 29765–29773 [DOI] [PubMed] [Google Scholar]
- 24. Koppaka V., Lentz B. R. (1996) Biophys. J. 70, 2930–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krishnaswamy S., Jones K. C., Mann K. G. (1988) J. Biol. Chem. 263, 3823–3834 [PubMed] [Google Scholar]
- 26. Smirnov M. D., Esmon C. T. (1994) J. Biol. Chem. 269, 816–819 [PubMed] [Google Scholar]
- 27. Majumder R., Quinn-Allen M. A., Kane W. H., Lentz B. R. (2008) Blood 112, 2795–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saleh M., Peng W., Quinn-Allen M. A., Macedo-Ribeiro S., Fuentes-Prior P., Bode W., Kane W. H. (2004) Thromb. Haemost. 91, 16–27 [DOI] [PubMed] [Google Scholar]
- 29. Gruner S. M. (1985) Proc. Natl. Acad. Sci. U.S.A. 82, 3665–3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cullis P. R., Hope M. J., Tilcock C. P. (1986) Chem. Phys. Lipids 40, 127–144 [DOI] [PubMed] [Google Scholar]
- 31. Rand R. P., Fuller N. L., Gruner S. M., Parsegian V. A. (1990) Biochemistry 29, 76–87 [DOI] [PubMed] [Google Scholar]
- 32. Haque M. E., Lentz B. R. (2004) Biochemistry 43, 3507–3517 [DOI] [PubMed] [Google Scholar]
- 33. Ho C., Slater S. J., Stubbs C. D. (1995) Biochemistry 34, 6188–6195 [DOI] [PubMed] [Google Scholar]
- 34. Wu H., Zheng L., Lentz B. R. (1996) Biochemistry 35, 12602–12611 [DOI] [PubMed] [Google Scholar]
- 35. Banerjee M., Drummond D. C., Srivastava A., Daleke D., Lentz B. R. (2002) Biochemistry 41, 7751–7762 [DOI] [PubMed] [Google Scholar]
- 36. Zhai X., Srivastava A., Drummond D. C., Daleke D., Lentz B. R. (2002) Biochemistry 41, 5675–5684 [DOI] [PubMed] [Google Scholar]
- 37. Koppaka V., Wang J., Banerjee M., Lentz B. R. (1996) Biochemistry 35, 7482–7491 [DOI] [PubMed] [Google Scholar]
- 38. Smirnov M. D., Safa O., Esmon N. L., Esmon C. T. (1999) Blood 94, 3839–3846 [PubMed] [Google Scholar]
- 39. Majumder R., Allen M. A., Kane W. H., Lentz B. R. (2005) Biophys. J. 86, 92A–93A [Google Scholar]
- 40. Banerjee M., Majumder R., Weinreb G., Wang J., Lentz B. R. (2002) Biochemistry 41, 950–957 [DOI] [PubMed] [Google Scholar]
- 41. Srivastava A., Wang J., Majumder R., Rezaie A. R., Stenflo J., Esmon C. T., Lentz B. R. (2002) J. Biol. Chem. 277, 1855–1863 [DOI] [PubMed] [Google Scholar]
- 42. Hoffman M., Monroe D. M., 3rd. (2001) Thromb. Haemost. 85, 958–965 [PubMed] [Google Scholar]
- 43. Klein S., Spannagl M., Engelmann B. (2001) Arterioscler. Thromb. Vasc. Biol. 21, 1695–1700 [PubMed] [Google Scholar]
- 44. Zieseniss S., Zahler S., Muller I., Hermetter A., Engelmann B. (2001) J. Biol. Chem. 276, 19828–19835 [DOI] [PubMed] [Google Scholar]