

Abstract
Aspirin and P2Y12 antagonists are commonly used anti-platelet agents. Aspirin produces its effects through inhibition of thromboxane A2 (TXA2) production, while P2Y12 antagonists attenuate the secondary responses to ADP released by activated platelets. The anti-platelet effects of aspirin and a P2Y12 antagonist are often considered to be separately additive. However, there is evidence of an overlap in effects, in that a high level of P2Y12 receptor inhibition can blunt TXA2 receptor signalling in platelets and reduce platelet production of TXA2. Against this background, the addition of aspirin, particularly at higher doses, could cause significant reductions in the production of prostanoids in other tissues, e.g. prostaglandin I2 from the blood vessel wall. This review summarizes the data from clinical studies in which dose-dependent effects of aspirin on prostanoid production have been evaluated by both plasma and urinary measures. It also addresses the biology underlying the cardiovascular effects of aspirin and its influences upon prostanoid production throughout the body. The review then considers whether, in the presence of newer, more refined P2Y12 receptor antagonists, aspirin may offer less benefit than might have been predicted from earlier clinical trials using more variable P2Y12 antagonists. The possibility is reflected upon, that when combined with a high level of P2Y12 blockade the net effect of higher doses of aspirin could be removal of anti-thrombotic and vasodilating prostanoids and so a lessening of the anti-thrombotic effectiveness of the treatment.
Keywords: prostaglandins, thienopyridines, thrombosis, thromboxane A2
Introduction
Aspirin and P2Y12-receptor antagonists are commonly used anti-platelet agents. The anti-platelet effect of aspirin is mediated by reduction of the production of thromboxane A2 (TXA2) while P2Y12 antagonists reduce the secondary responses to ADP released by activated platelets. In principle, the anti-platelet effects of aspirin and a P2Y12 antagonist could be considered additive because they act at different steps. However, there is accumulating evidence that there may, in fact, be an overlap in effects inasmuch that a high level of P2Y12-receptor inhibition can reduce both platelet responses to TXA2 and platelet production of TXA2. This could have important consequences for combination therapy, in particular when higher doses of aspirin are used, which cause significant reductions in the production of other prostanoids, notably the anti-thrombotic and vasodilating prostanoid, prostaglandin I2 (PGI2). This review explores the biology underlying the cardiovascular effects of aspirin and P2Y12 antagonists with particular regard to the balance of prostanoids produced by the cyclo-oxygenase system and addresses the potential impact of combining a high dose of aspirin with a high level of P2Y12-receptor inhibition.
Aspirin
Aspirin mediates its cardioprotective effect through irreversible inhibition of platelet COX-1 and blockade of the production of TXA2. However, the effects of aspirin are not platelet-specific and the inhibition of COX-1 and, to some extent COX-2, in other cell types can reduce the production of other prostanoids. The consequences of this can include inhibition of PGI2 production [1], a prostanoid whose biological actions oppose those of TXA2. The extent to which aspirin can have effects beyond the platelet is influenced by a number of factors, including individual differences in aspirin response, aspirin pharmacokinetics, the turnover of COX enzymes in different cell types and the selectivity of aspirin for COX-1 over COX-2 [2]. Another crucial factor could be the dose of aspirin given, which can vary from 75 mg day−1 up to 1500 mg day−1 depending on clinical decision making and also geographical area. These arguments are best pursued with an understanding of the mechanism of action of aspirin.
How the prostanoids are formed
The pathways leading to the production of prostanoids have been reviewed many times [3–13] (Figure 1). In general terms, arachidonic acid released from membrane phospholipids by the action of phospholipase A2 is converted by prostaglandin H synthase, better known as cyclo-oxygenase or COX, in two independent enzymatic reactions producing first prostaglandin (PG) G2, via a cyclo-oxygenase function, and then PGH2 via a peroxidase function.
Figure 1.
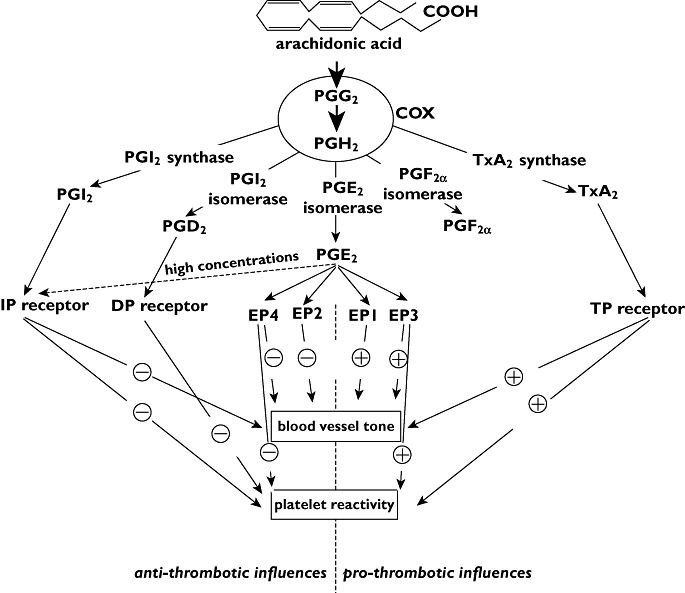
Schematic representation of predominant pathways of prostanoid formation and effect with regard to platelets and blood vessels
Prostaglandin G2 and PGH2 are also known as the prostaglandin endoperoxides and have some direct biological actions of their own, generally through actions on receptors for TXA2. However, the prostaglandin endoperoxides are unstable molecules whose main purpose is to serve as substrates for secondary enzyme systems to produce end product prostanoids. In the cardiovascular system, the three most important synthases are PGI synthase, TX synthase and PGE synthase, responsible for the conversion of PGH2 to PGI2, TXA2 and PGE2, respectively (Figure 1) [3, 6, 9, 10, 13–25].
Important to our understanding of the roles of prostanoids within the cardiovascular system is the existence of different isoforms of COX. The first evidence that two molecularly distinct forms of COX exist came from studies using lung epithelia [26] from which the conclusion was drawn that separate genes encoding different COX isoforms exist. Soon after, two isoforms of COX were clearly identified [27–31], and a consensus developed that in general terms, COX-1 represented the constitutive form of the enzyme and COX-2 the inducible form associated with inflammation. It is now well understood that COX-2 also has constitutive functions at discrete sites of the body, such as within the kidney and central nervous system.
Prostanoid formation within the cardiovascular system
The two prostanoids that have attracted the most attention with regard to the cardiovascular system are PGI2 and TXA2, principally because of their opposing effects upon platelet function and thrombosis [4–6, 8–10, 13–15, 18–21, 32]. In physiological conditions, most systemic blood vessels produce PGI2, and, to a lesser extent, PGE2 from the endothelium and vascular smooth muscle cells. The principal source of TXA2 in the circulation is platelets, although low concentrations may also be produced by vascular smooth muscle cells. In addition to TXA2, platelets can produce PGE2 to some extent, but not PGI2. It has also been reported that there may be an exchange of endoperoxides between platelets and endothelial cells such that one cell type could provide endoperoxides to support prostanoid production by the other cell type [33].
The vast majority of studies indicate that platelets contain only COX-1, as would be expected of an anucleated cell type in which the COX-2 gene could neither be induced nor COX-2 protein expressed. However, as platelets are derived from nucleated megakaryocytes, there is the possibility that COX-2 induced in these precursor cells could carry over to the mature platelet [34, 35], or even that residual mRNA within formed platelets could be transcribed into protein [36]. This phenomenon has been demonstrated following coronary artery bypass surgery, presumably as a consequence of megakaryocytes being exposed to the systemic rise in cytokines that follows such an invasive procedure [37].
In contrast to the platelet, the isoform of COX normally present within endothelial cells is a point of some controversy. By measuring the concentrations of urinary metabolites of prostanoids, in particular metabolites of PGI2 and TXA2, it has been shown that while dosing with NSAIDs reduces the concentrations of both PGI2 and TXA2 metabolites, dosing with selective inhibitors of COX-2 reduces only the concentrations of PGI2 metabolites [38, 39]. As it has been assumed that urinary PGI2 metabolites come from endothelial cells and urinary TXA2 metabolites from platelets this has been taken as indicating that COX-2 underlies the normal production of PGI2 by endothelial cells within the circulation [9, 14–17, 20]. However, COX-2 is not usually expressed by endothelial cells in culture under normal conditions. Similarly, immunohistochemistry provides evidence for COX-1, but not COX-2 expression, within human blood vessels in the absence of vascular disease [40–44]. The same has been widely reported for large vessels taken from laboratory animals [45–48] and even for vessels taken from animal models of experimental atherosclerosis [49]. Interestingly, while COX-2 expression is upregulated in transplant atherosclerosis or flow-induced vascular remodelling [50], this induction follows from a local reduction, rather than an increase, in flow and is associated with local inflammatory responses rather than shear.
Effects of the prostanoids within the cardiovascular system
The three most abundant prostanoids within the cardiovascular system, PGI2, TXA2 and PGE2, have differential, and sometimes opposing effects (Table 1). PGI2 promotes vasodilatation, inhibits vascular smooth muscle cell proliferation and reduces platelet activation, TXA2 promotes vasoconstriction, vascular smooth muscle cell proliferation and platelet activation and PGE2 tends to promote vasodilatation, while at low concentrations it increases platelet reactivity and at high concentrations inhibits platelet reactivity. These effects are mediated by binding to the cognate GPCR receptors, IP and TP for PGI2 and TXA2, respectively, and EP1 to 4 for PGE2[51]. Differential coupling of the four EP receptors explains the varied effects of PGE2 in the cardiovascular system [52]. For example, activation of EP1 and EP3 receptors is associated with vasoconstriction whereas activation of EP2 and EP4 is associated with vasodilatation. On platelets, activation of EP3 receptors inhibits adenylyl cyclase, reduces intraplatelet cAMP concentrations and increases platelet excitability [10, 53–55], whereas activation of EP4 and EP2 has been proposed to increase intraplatelet cAMP levels and thus counteract the platelet activation by EP3 [54–57]. Possibly at high concentrations, PGE2 may also activate platelet IP receptors and inhibit platelet reactivity through stimulation of adenylyl cyclase activity.
Table 1.
Summary of prostanoid receptors and associated intracellular signalling pathways with respect to platelets and vascular smooth muscle
| Prostanoid | Receptor | Signalling pathway | Vascular smooth muscle tone | Platelet reactivity |
|---|---|---|---|---|
| PGD2 | DP | ↑ Adenylyl cyclase | ↓ | |
| PGE2 | EP1 | ↑ IP3/DAG/Ca2+ | ↑ | |
| EP2 | ↑ Adenylyl cyclase | ↓ | ↓ | |
| EP3 | ↓ Adenylyl cyclase | ↑ | ↑ | |
| EP4 | ↑ Adenylyl cyclase | ↓ | ↓ | |
| IP | ↑ Adenylyl cyclase | ↓ | ↓ | |
| PGF2α | FP | ↑ IP3/DAG/Ca2+ | ↑ | |
| PGH2 | TP | ↑ IP3/DAG/Ca2+ | ↑ | ↑ |
| PGI2 | IP | ↑ Adenylyl cyclase | ↓ | ↓ |
| TXA2 | TP | ↑ IP3/DAG/Ca2+ | ↑ | ↑ |
Aspirin and the platelet
Aspirin mediates its cardio-protective effects through irreversible inhibition of platelet COX-1 and subsequent blockade of the production of TXA2, reducing thrombus formation [58]. It is widely held that there is a non-linear relationship between potency in vitro and efficacy in vivo with the consequence that a high level of inhibition of platelet COX-1 is required in vivo to obtain anti-platelet efficacy [2, 58, 59]. To achieve this effect rapidly in an acute setting, a dose of 300 mg may be required. However, complete inhibition of platelet COX-1 can be achieved in long-term therapy using doses between 50 and 100 mg day−1[2, 58, 60]. When taken p.o., aspirin is rapidly absorbed and has a systemic bioavailability of approximately 50% at doses between 20 and 1300 mg, reaching peak plasma exposure within 30 to 40 min. However, the half-life of aspirin in the circulation is only about 15 to 20 min. Despite the high clearance, the effects of aspirin on platelet TXA2 production exist for the lifetime of the platelet, a consequence of aspirin's ability to acetylate irreversibly COX-1 and the inability of anucleated platelets to synthesize new COX-1 protein. Furthermore, as circulating cells, platelets are exposed to peak concentrations of aspirin in the portal circulation before metabolism of aspirin by the liver. The effects of aspirin at any given time are therefore influenced by the plasma exposure and rate of platelet renewal; human platelets have a mean lifetime of 8 to 10 days and approximately 10 to 12% of platelets are replaced each day [2].
In the clinic, aspirin is used at doses from 75 mg to 1500 mg day–1. Daily doses at or below 162 mg are generally referred to as ‘low-dose’ aspirin. The optimal dose for cardio-protection has not been empirically measured as there has never been a large randomized study that has directly compared different doses of aspirin on cardiovascular outcomes, except, as discussed below, in the recently reported CURRENT-OASIS 7 trial, which compared aspirin doses in the presence of clopidogrel [61, 62]. However, randomized trials have proven that aspirin is an effective anti-thrombotic agent when used long term at doses between 50 and 100 mg day−1[2, 58, 60]. When different doses of aspirin have been compared in randomized trials, there has been no evidence for increased efficacy at higher doses [2, 58, 61–63]. In 2007 Campbell et al. conducted a comprehensive review of published literature to evaluate the most clinically relevant aspirin dose [64]. They concluded that the available evidence, although predominantly from secondary prevention observational studies, indicated that doses of aspirin greater than 75–81 mg day−1 produced no enhancement in anti-thrombotic efficacy but were associated with increased bleeding events. This is in accord with the conclusion of Patrono and colleagues who recommended that the lowest effective dose of aspirin (50 to 100 mg day−1) is the most appropriate strategy to maximize anti-platelet efficacy and minimize toxicity [63]. With regard to higher doses of aspirin, there is also limited evidence for a blunting of the anti-thrombotic effects and a dose-related increased risk of unwanted bleeding [65–68].
Aspirin's effects on the cardiovascular system other than the platelet
Because of its primary effect in platelets, it is often forgotten that aspirin is also inhibitory at other sites within the cardiovascular system. Recent concerns about the potential pro-thrombotic effects of the COX-2-selective drugs were initially prompted by studies showing that consumption of either celecoxib [39] or rofecoxib [38] reduced urinary PGI2 metabolites, an effect that was interpreted as being consistent with an increased risk of thrombosis because of a loss in anti-thrombotic PGI2. However, aspirin was shown more than 10 years earlier to reduce urinary PGI2 metabolites, although the reduction was less marked than the reduction in TXA2 metabolites [1]. These investigators used a wide dose range of aspirin, 20–2600 mg day−1, and found that lower doses of aspirin had greater inhibitory effects upon TXA2 than PGI2 metabolites. However, they found that inhibition of platelet function was not maximal at the lower aspirin dosage and notably that aspirin at doses greater than 80 mg day−1 caused substantial inhibition of endogenous PGI2 production. They concluded that it was ‘unlikely that any dose of aspirin can maximally inhibit thromboxane generation without also reducing endogenous prostacyclin biosynthesis’. In support of this idea, local infusion of aspirin to the human coronary bed has been shown to increase coronary vascular resistance and reduce coronary blood flow [69], as has i.v. infusion of indomethacin [70]. So if it is able to reduce the intravascular production of PGI2, aspirin could also release a brake upon atherosclerotic disease progression and platelet activation, as well as promoting vasoconstriction. Taken together these could increase the risk of adverse cardiovascular events, an idea supported by the report that individuals with dysfunctional IP receptors have accelerated cardiovascular disease [71]. Further support for this concept can be derived from studies using mouse models. For example, in a mouse model of atherosclerosis, deletion of the IP receptor was found to enhance disease progression, whereas deletion of the TP receptor or treatment with a TP receptor antagonist reduced atherogenesis [72, 73]. Similarly, platelet and vascular responses following experimental injury are enhanced in knockout mice lacking the IP receptor, and may be depressed in mice lacking the TP receptor [74, 75]. Finally, a gene/dose dependent relationship between blood pressure, platelet aggregation and thrombogenesis has been demonstrated using heterozygote and homozygote knock out mice for the IP receptor [76].
The effects of aspirin at sites other than the platelet are informed by the recent understanding that inhibition of COX-2 isoforms by NSAIDs is associated with an increased risk of adverse cardiovascular events [4, 5, 8, 11, 18, 77]. While on the one hand these effects could be associated with inhibition of the vascular production of PGI2 leading directly to local increases in platelet reactivity, on the other hand it is also important to realize that aspirin and NSAIDs can increase blood pressure in normotensive subjects and in those with existing hypertension [4, 5, 9, 11, 14, 15, 18, 20, 23, 77] thereby increasing the risk of thrombotic events through exacerbation of the development of atherosclerotic disease [78]. Indeed, the use of these drugs is weakly associated with an increased risk of congestive heart failure [79] and an increased risk of hypertension [80]. As these effects are COX mechanism-driven and dose-related, it is clear that higher doses of aspirin and longer exposures have greater effects than lower doses and shorter exposures [80].
In addition to the PGI2 and TXA2 pathways, aspirin also affects the production of other prostanoids within the circulation, most notably PGE2. PGE2 has been identified as both an inhibitor and a potentiator of platelet aggregation via interaction with different isoforms of the EP receptor (see above). Interestingly, deletion of the F prostanoid receptor (FP) in mice reduces blood pressure and atherogenesis associated with disruption of renin release in the kidney [81], so it is important not to become too narrowly focused on aspirin and platelet endothelial cell interactions although this is very much where the weight of evidence lies.
Aspirin dose and cardiovascular effects
From the above it is clear that aspirin has mixed effects upon the cardiovascular system; while attention is generally paid to the platelet, aspirin affects other sites notably the blood vessel wall and the kidney and these effects are dose-related. At a low dose (75–81 mg) aspirin has proportionally greater effects upon the platelet than other sites, but there is still notable inhibition of urinary PGI2 metabolites consistent with around a 50% inhibition of systemic COX. As the dose of aspirin is increased so is the inhibition of systemic COX and so one way to explore the dose dependent effects of aspirin is to measure the impact of different doses on different prostanoids. In the large prospective trials that have assessed the effect of aspirin on clinical outcomes, concentrations of prostanoids have not been measured [64]. However, prostanoid concentrations have been analysed in some smaller mechanistic studies using either healthy controls or patients (Table 2). For the reasons discussed above it is the impact of aspirin dose on the concentrations of TXA2 and PGI2 that has drawn most attention. It is important to note that although such studies provide a source of data to assess the dose dependent effects of aspirin, it is not straightforward to compare directly the results between studies due to differences such as study design, subjects and exposure times. In particular, the methodology used to analyse different prostanoids, the number of doses and the timing of measurements after the last dose can have an impact on the interpretation of the results. In patients, there is also evidence for different levels of prostanoid production in response to disease leading to the involvement of different cell types and COX isoforms which can confound comparisons.
Table 2.
Summary of clinical studies in which dose-dependent effects of aspirin on prostanoid production were evaluated
| Study design | Aspirin dose(s) | Treatment time | Prostanoid measurements | Effect | Reference |
|---|---|---|---|---|---|
| Healthy volunteers (n = 5) | 20, 40, 80, 160, 325 and 650 mg day−1, and 650 mg twice and four times daily | Each dose taken for 7 days in ascending order in consecutive weeks | Urine 2,3-dinor-TXB2 | Dose-dependent reduction in 2,3-dinor-TXB2 and PGI-M concentrations using aspirin between 20 and 325 mg day−1 | [1] |
| Urine PGI-M | |||||
| Healthy volunteers (n = 8) | 20 and 100 mg given to the same volunteers | Each dose given once with a 2-week washout period between doses | TXB2 and 6-keto- PGF1α formation during blood clotting | Dose-dependent inhibition of PGI2 production | [93] |
| Healthy volunteers (n = 5) | 150 and 300 mg | Once | Platelet TXB2 production PGI2 production in vein segments | Inhibition of TXB2 and PGI2 production by both doses | [89] |
| Randomized parallel study in healthy volunteers (n = 52) | 80 and 325 mg day−1 or 325 mg enteric coated aspirin | Daily or every other day dosing for 14 days | TXB2 and 6-keto-PGF1α in blood | PGI2 production inhibited by 325 mg only | [95] |
| Placebo-controlled trial in healthy volunteers (n = 45) | 75 and 162.5 mg day−1; 325 mg every second day; 75 mg controlled release aspirin day−1 | 4 days | PGI2 synthesis following i.v. infusion of bradykinin | All doses of standard aspirin suppressed bradykinin-stimulated PGI2 | [87] |
| Double-blind trial using healthy volunteers (n = 45) | 162.5 mg day−1 or 325 mg every second day | 28 days | Serum TXB2 | Dose-dependent inhibition of PGI-M | [87] |
| Urine 2,3-dinor-TXB2 | |||||
| Urine PGI-M | |||||
| Single-blind randomized prospective study in healthy volunteers (n = 10) | 50 mg p.o. aspirin or 500 mg i.v. infusion over 60 min | Once | Platelet TXB2 | Dose-dependent inhibition of urinary metabolites | [96] |
| Urine 2,3-dinor-TXB2 | |||||
| Urine PGI-M | |||||
| Urine PGE2 | |||||
| Post-stroke patients (n = 19) | 40, 320 and 1280 mg day−1 given in ascending doses | 7 weeks; 2-week washout periods between doses | Serum TXB2 | Dose-dependent inhibition of urinary metabolites | [94] |
| Urine 11-dehydro-TXB2 | |||||
| Urine PGI-M | |||||
| Placebo-controlled study in patients with cardiovascular metabolic syndrome (n = 121) | 100 and 300 mg day−1 | 2 weeks | Serum TXB2 | Only TXB2 production inhibited | [97] |
| Serum 6-keto-PGF1α | |||||
| Crossover study including patients with type I diabetes (n = 8) and healthy controls (n = 7) | 80 mg every second day or 325 mg day−1 | 14 days with 2-week washout period between treatments | Urine TXB2 | 80 mg every second day suppressed PGI2 production to a greater extent than 325 mg day−1 | [103] |
| Urine 6-keto-PGF1α | |||||
| Patients undergoing aortocoronary bypass surgery (n = 70) | 40, 80 or 325 mg | Aspirin administered once 12 to 16 h before surgery | Serum TXB2 | Dose-dependent inhibition of TXB2 and PGI2 production | [90] |
| PGI2 production by aortic and saphenous vein tissue measured ex vivo | |||||
| Patients undergoing surgery for varicose veins (n = 47) | 40, 81 and 300 mg | 14 h pre-operation | PGI2 synthesis from vein sections ex vivo measured by bioassay | 81 and 300 mg aspirin significantly inhibited PGI2 synthesis whereas 40 mg had no effect | [88] |
| Patients undergoing bowel resection (n = 62) | 40, 75 or 300 mg | Once, 24 h before operation | 6-keto-PGF1α synthesis by mesenteric arteries or veins segments obtained during surgery | Arterial and venous 6-keto-PGF1α synthesis significantly inhibited by all doses | [91] |
| Placebo-controlled study in subjects with prior sporadic colorectal adenoma(s) (n = 60) | 81, 325 and 650 mg day−1 | 4 weeks | Rectal mucosal PGE2 concentrations at baseline and after 4-week treatment | All doses significantly suppressed PGE2 | [137] |
| Randomized double-blind placebo-controlled crossover study in healthy volunteers (n = 12) | 75 and 300 mg day−1 or 300 mg day−1 enteric coated aspirin | Each treatment period for 5 days with 2-week washout period between treatments | TXB2 and PGE2 in rectal dialysates at baseline and after 5 days | TXB2 but not PGE2 concentrations inhibited by all doses | [138] |
Prostanoids are generally too unstable to measure in the circulation and they are therefore usually quantified by measuring metabolites in plasma or in urine [1, 38, 39, 82–84]. TXA2 released by platelets in the circulation is rapidly hydrolyzed non-enzymically to TXB2, the concentrations of which can be quantified in plasma and used as an index of TXA2 production. However, TXB2 is itself metabolized (t1/2 = 5 to 7 min) to the urinary metabolites 11-dehydro TXB2 and 2,3-dinor TXB2 for clearance by the kidneys. Both of these metabolites can be measured in plasma or urine and can provide an estimate of in vivo TXA2 production [85, 86]. PGI2 is hydrolyzed to the by-product 6-keto PGF1α which can be metabolized further in the liver to 2,3-dinor-6-keto PGF1α (PGI-M). PGI2 production can also be measured following i.v. infusion of bradykinin. This is believed to promote PGI2 production and has been used as an index of aspirin-sensitive PGI2 production in some studies [87]. In addition to direct measurement in plasma, PGE2 production can be followed by measuring the metabolite 11-hydroxy-9,15-dioxo-2,3,4,5-tetranorprostane-1,20-dioic acid (PGE-M).
An issue with measuring prostanoid metabolites in urine is that this does not provide accurate information on the cellular source of the production, and is not sensitive enough to address local effects. This can be particularly important when attempting to assess the local impact of aspirin on the production of PGI2 by the vascular endothelium. Urinary concentrations of 6-keto PGF1α are generally used to estimate the local generation of PGI2 in the kidney, whereas concentrations of PGI-M are used to estimate the systemic, non-renal production of PGI2, including production by the vascular endothelium. However, PGI-M can also be produced in the kidneys, because of constitutive expression of COX-2 and PGI synthase in the kidneys, and this could make a significant contribution to the overall concentrations of PGI-M measured in urine. Indeed, the case has been made that urinary PGI-M may bear only a weak association with the whole body production of PGI2[18]. In several studies, PGI2 production has been evaluated in artery and vein sections removed during surgery or biopsy [88–91]. These studies can provide relevant information on the local effect of aspirin in the vascular wall, but are compromised by the need to make measurements ex vivo.
Bearing in mind the qualifications outlined above, it is evident from the studies summarized in Table 2 that aspirin can inhibit TXA2 production completely at low and high doses, consistent with its irreversible inhibition of platelet COX-1. In contrast, the majority of studies in both patients and healthy volunteers provide evidence for a dose-dependent relationship between aspirin and PGI2 production. With the exception of one study [92], aspirin doses <81 mg day−1, when used acutely or over a prolonged period, have been reported to inhibit selectively TXA2 production over PGI2 production. In studies in which different aspirin doses have been directly compared [90, 91, 93–97], the lowest aspirin dose used has been found to provide the best PGI2-sparing effect. Moreover, even where PGI2 production was inhibited, the extent of inhibition was generally lower than that observed for TXA2.
Cells producing PGI2 have nuclei and can synthesize new COX enzymes. Consequently, the recovery time for PGI2 production following aspirin treatment can differ from the recovery time for platelet TXA2 production. The recovery time for PGI2 production following treatment with aspirin has been assessed in cultured endothelial and vascular smooth muscle cells [98–102]. In both cell types, synthesis of new COX enzyme can overcome the effect of aspirin, but the recovery time may be shorter for vascular smooth muscle cells (within 3 h) than for endothelial cells (within 24 h). In several of the studies the effect of aspirin on prostanoid production has been measured at multiple time points allowing some assessment of recovery time [89, 93, 103–107]. Using i.v. infusion of bradykinin to monitor vascular production of PGI2, Heavey et al. [105] and Ritter et al. [107] observed that PGI2 production recovered within 6 h following 600-mg aspirin given p.o. or i.v. Similarly, urine PGI-M concentrations returned to baseline within 3 h following 500-mg aspirin twice daily in a separate study [106]. In contrast, using measurement of 6-keto-PGF1α concentrations in blood, Gerrard et al. [103] observed that PGI2 production did not return to baseline for up to 72 h following 600-mg aspirin. Similarly, Preston et al. [104] observed that 6-keto-PGF1α production by vein segments ex vivo was inhibited >70% for up to 72 h following a 500-mg aspirin dose. Unfortunately, different aspirin doses were not used in any of these studies so it is not possible to assess whether aspirin dose can affect the time to recovery.
Aspirin and P2Y12 receptor antagonist interactions
In addition to aspirin treatment, antagonism of platelet P2Y12 receptors has become established as a clinically relevant anti-platelet approach. Very briefly, as a secondary agonist involved in platelet aggregation, ADP released from platelet α granules has a central role in haemostatic plug and pathological thrombus formation, effects mediated by the functionally distinct purine receptors P2Y1 and P2Y12[108–112]. Activation of P2Y1 receptors induces a phospholipase C-mediated increase of intracellular calcium leading to platelet-shape change and initial reversible aggregation via Gαq. P2Y12 receptors in their turn complete the aggregation response initiated by P2Y1 receptors via Gαi-mediated inhibition of adenylyl cyclase and through a less well-defined activation of phosphoinositol-3-kinase (PI3-K) [113]. By blocking the response to ADP, P2Y12-receptor antagonists can effectively attenuate the primary agonist response and reduce platelet aggregation. P2Y12-receptor antagonists in clinical use include the thienopyridines, clopidogrel and prasugrel [111, 112, 114, 115]. These are both pro-drugs that require hepatic activation to generate an active metabolite that binds irreversibly to the receptor and thereby like aspirin they inhibit the platelet for its remaining lifetime. In addition to the thienopyridines, a number of direct acting reversibly binding P2Y12-receptor antagonists are currently under development. Most advanced is ticagrelor (also known as AZD6140) that belongs to a new class, the cyclopentyl-triazolo-pyridines [114, 116]. The standard dosing of clopidogrel results in a partial and very variable inhibition [117, 118] whereas both prasugrel and ticagrelor provide a more complete and consistent platelet inhibition [115].
When considering the potential combined effects of treatment with aspirin and a P2Y12-receptor antagonist, it appears an attractive idea to use the two together as dual therapy, particularly if the platelet is mechanistically considered in isolation. However, several lines of evidence suggest that there might not be a simple additive effect of the two treatments. In particular, antagonists of P2Y12 receptors reduce the aggregatory responses of platelets to TXA2, reduce the production of TXA2 by platelets and sensitize platelets to the anti-aggregatory effects of PGI2 by blocking P2Y12 receptor-mediated inhibition of adenyl cyclase [119–125]. Together, this evidence indicates that administration of a P2Y12-receptor antagonist can achieve three related pharmacological effects on the platelet: (i) blockade of P2Y12 receptors; (ii) inhibition of TXA2 pathways of platelet activation; and (iii) sensitization of platelets to endogenous anti-aggregatory PGI2. This then leads us to the question of what might be the systemic results of providing aspirin to an individual already receiving a P2Y12-receptor antagonist. In doing this we must, of course, consider that the magnitude of any effects will be dependent upon the level of P2Y12-receptor inhibition. One could hypothesize that the net effects will be different when combining aspirin with different P2Y12 antagonists that achieve different levels of P2Y12-receptor inhibition in clinical use, e.g. when platelets are weakly P2Y12-receptor inhibited, aspirin may produce a substantial additional anti-platelet effect, whereas when platelets are strongly P2Y12-receptor inhibited, aspirin may add relatively little. So addition of aspirin, particularly at high doses, may actually provide no further anti-thrombotic benefit in the presence of high level P2Y12-receptor blockade, as TXA2-dependent pathways of platelet aggregation will already be largely blocked (these ideas are captured in Figure 2). Despite this, addition of aspirin will cause dose-dependent increases in the risk of major and minor gastrointestinal bleeds [65, 67, 68, 126], which have recently been judged to be as potentially frequent in individuals receiving dual anti-platelet therapy as in individuals receiving warfarin [127]. As well as being immediately dangerous, bleeds are associated with a reduction in patient compliance to anti-thrombotic therapy and a consequent increase in patient risk of thrombotic events [128, 129]. As detailed above, aspirin will also produce dose-dependent reductions in the production of PGI2 within the circulation (Table 2) and promote increases in fluid retention and blood pressure [79, 80].
Figure 2.
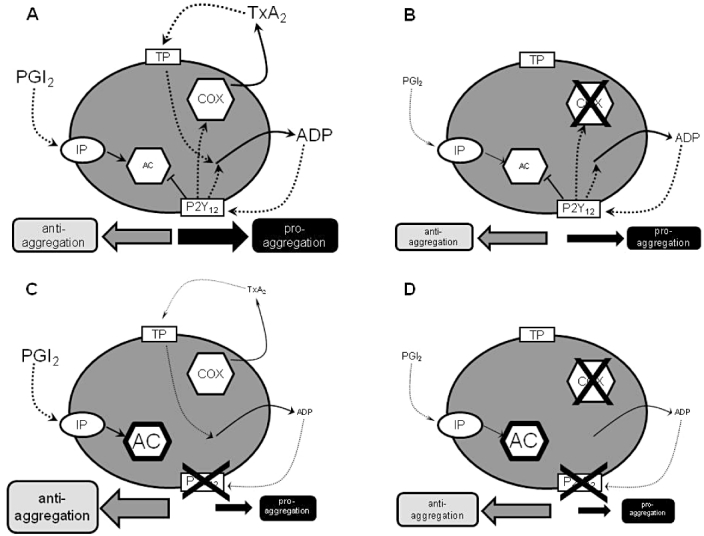
Schematic representation of effects of aspirin and P2Y12-receptor blockers on platelet pathways. (A) In physiological conditions there is balance between anti-aggregatory and pro-aggregatory influences on the platelet from the prostanoid and P2Y12 pathways. PGI2 formed by the blood vessel wall is anti-aggregatory through the stimulation of adenylyl cyclase (AC) secondary to activation of platelet PGI2 receptors (IP receptors). Thromboxane A2 (TXA2) produced by the activity of cyclooxygenase (COX) within the platelet is pro-aggregatory, acting through platelet TXA2 receptors (TP receptors). Activation of TP receptors also promotes the release of ADP from platelets which further drives aggregation. ADP produces its pro-aggregatory effects through activation of platelet ADP receptors (P2Y12 receptors) and inhibition of platelet AC. Activation of P2Y12 receptors drives more production of TXA2 and more release of ADP. (B) In the presence of aspirin, platelet COX is blocked and the TXA2 pathway of aggregation abolished. The P2Y12 pathway of platelet activation is unaffected. Overall, there is a lessening of pro-aggregatory drive. There is an accompanying lessening in anti-aggregatory influence, as aspirin will also reduce the production of PGI2 by the blood vessel wall. (C) In the presence of a blocker of P2Y12 receptors, AC inhibition is lessened and so the anti-aggregatory effects of PGI2 are enhanced. As the effects of the TXA2 pathway are amplified through the P2Y12 pathway, the pro-aggregatory effects of TXA2 are also lessened and TXA2 production is reduced. (D) When aspirin is added to a P2Y12-receptor blocker, there may be some small additional reduction in pro-aggregatory influences by the complete removal of TXA2 production; however, there is also a loss of PGI2 which might lead to an even greater reduction in anti-aggregatory influence
Overall, therefore, if one ignores its direct effects upon platelet COX, aspirin has a rather negative influence upon the cardiovascular system. When used as a single therapy, however, these deleterious cardiovascular effects are more than outweighed by aspirin's direct effects upon the platelet. However, if P2Y12-receptor antagonists strongly reduce platelet pathways of TXA2 aggregation and enhance the effects of endogenous PGI2, one is forced to ask the question, what now is the net effect of aspirin? Unfortunately, we have relatively little clinical data to explore this possibility as most large-scale outcome studies of P2Y12-receptor antagonists have been conducted in the presence of aspirin. Results from the TRITON-TIMI 38 trial of prasugrel vs. clopidogrel are of little use in exploring this point as there have been no data shown of outcomes in relation to aspirin dose and most outcomes occurred very early in the trial before the impact of changes in vascular resistance could occur. While the trial enrolment was approximately one-third in North America, one quarter in Western Europe and one quarter in Eastern Europe, and aspirin was taken by 99% of participants, the dose of aspirin was recommended to be between 75 and 162 mg day–1 to reduce aspirin variability [130–132]. Analysis of data from the CURE study of aspirin plus clopidogrel vs. aspirin alone demonstrated that there was no increased anti-thrombotic benefit with the addition of doses of aspirin above 100 mg [133]. In this trial there were clear geographical differences with low-dose aspirin being primarily used in Europe and high-dose aspirin primarily used in North America. There was, however, an increase in bleeding risk with increased dosing of aspirin, such that taking account of the net adverse clinical events (composite of death, myocardial infarction, stroke or major bleeding); there was a benefit to using lower doses of aspirin. Interestingly, it is possible that results from the PLATO study may indicate an interaction between strong P2Y12-receptor blockade and aspirin. Overall, PLATO showed ticagrelor to be superior to clopidogrel in reducing the rate of the composite efficacy end point of cardiovascular death, myocardial infarction or stroke after acute coronary syndrome events [116]. However, in a pre-specified subgroup analysis of multiple patient characteristics and baseline variables, a weak interaction between randomized treatment and region was observed for the primary end point, in that while the hazard ratio favoured ticagrelor in the three non-North American regions it numerically favoured clopidogrel in the North American region (interaction P = 0.045) [116]. Further evaluation indicated that the observation was driven primarily by results in the USA compared with the non-US countries. In analysis of patient characteristics and treatment patterns it was noted that there was a confounding factor regarding aspirin use in the different regions, such that in North America maintenance doses above 300 mg were predominantly used whereas in other regions less than 100-mg maintenance was predominantly used. Extensive evaluation of the data revealed that 80 to 100% of the observed interaction was explained by the maintenance aspirin dose depending on its definition. High maintenance doses of aspirin were associated with decrease in relative efficacy of ticagrelor, while those who received low maintenance aspirin doses, the vast majority (92%) in PLATO, had significant reductions in cardiovascular events compared with clopidogrel [134].
In helping us understand results from studies using more potent and less variable P2Y12 antagonists, data from studies using clopidogrel may be of relatively limited use. In particular, the variability in response to clopidogrel in study populations [117, 118] means that it is difficult to draw conclusions regarding the mechanistic effects following addition of aspirin; possibly in poor clopidogrel responders addition of aspirin would be beneficial, whereas in patients who are good responders it is less certain. Such a consideration may well explain why in the CURRENT OASIS 7 study relatively little difference was seen in short-term outcomes (30 days) between patients receiving either low- (75 mg) or high-dose (150 mg) clopidogrel plus low- (75–100 mg) or high-dose aspirin (300–325 mg) [61, 62]. In line with these thoughts about clopidogrel, it is interesting to reflect that there have been some suggestions that in at-risk populations higher doses of aspirin may be associated with a greater thrombotic risk than lower doses [135], and as pointed out by these authors much of our information about the use of aspirin has come from trials conducted some 20 years or so ago, since which time blood pressure and lipid control have greatly improved. Notably, against this background of improved and stable therapy, recommendations for aspirin use have changed, it now being apparent that aspirin has little benefit for primary prevention [60], whereas analyses some 15 years or so ago suggested it was of use [136]. These trends appear consistent; i.e. as control of other risk factors is improved, the benefit of aspirin becomes less clear. So it might well be as P2Y12-receptor antagonists become more refined, we see that aspirin offers less benefit in dual anti-platelet therapy for secondary prevention than might have been predicted from earlier clinical trials using more variable P2Y12 antagonists, particularly clopidogrel. If correct then this would suggest a class effect of P2Y12-receptor antagonists interacting with aspirin, with the effect being dependent upon the level of P2Y12-receptor inhibition.
Conclusion
In summary, in vitro, ex vivo and in vivo mechanistic studies link the anti-thrombotic effects of aspirin to irreversible inhibition of platelet COX-1 and formation of TXA2. However, aspirin also produces dose-dependent inhibition of COX at other sites within the body and some of these inhibitory effects, notably reduction in endothelial cell production of PGI2 and increase in blood pressure, are associated with an increase in overall cardiovascular risk. P2Y12-receptor antagonists have also been shown to be anti-thrombotic because of their blockade of ADP-dependent pathways of platelet activation, and dual therapy with aspirin has now become standard care for many patients at risk of thrombosis. While clinical trials using clopidogrel have investigated interactions with aspirin, large outcome studies of newer and more potent P2Y12-receptor antagonists, notably prasugrel and ticagrelor, have not randomized the dose of aspirin. As potent P2Y12-receptor antagonists can strongly inhibit TXA2-dependent pathways of platelet activation, i.e. those targeted by aspirin, and sensitize platelets to the anti-thrombotic effects of endogenously produced PGI2, there is the possibility that additional dosing with aspirin, in particular high-dose aspirin, will not confer any additional cardioprotective effect. On the contrary, there is a possibility that combining a high level of P2Y12 antagonism with high doses of aspirin could unmask an effect of aspirin on the production of anti-thrombotic prostanoids, notably PGI2, increase the risk of fluid retention and hypertension, and increase the risk of bleeds, particularly gastrointestinal bleeds. Clearly this currently is only a hypothesis. However, as these effects could impair the overall therapeutic benefit of the treatment, it will be important to evaluate further this concept in pre-clinical and clinical studies.
Competing Interests
T. D. W. has received research funding and consultancy fees from AstraZeneca. S. N. and C. A. W. are employees of and hold stocks and shares in AstraZeneca.
REFERENCES
- 1.FitzGerald GA, Oates JA, Hawiger J, Maas RL, Roberts LJ, 2nd, Lawson JA, Brash AR. Endogenous biosynthesis of prostacyclin and thromboxane and platelet function during chronic administration of aspirin in man. J Clin Invest. 1983;71:676–88. doi: 10.1172/JCI110814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patrono C, Baigent C. Low-dose aspirin, coxibs, and other NSAIDS: a clinical mosaic emerges. Mol Interv. 2009;9:31–9. doi: 10.1124/mi.9.1.8. [DOI] [PubMed] [Google Scholar]
- 3.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 4.Mitchell JA, Warner TD. COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat Rev Drug Discov. 2006;5:75–86. doi: 10.1038/nrd1929. [DOI] [PubMed] [Google Scholar]
- 5.Warner TD, Mitchell JA. Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB J. 2004;18:790–804. doi: 10.1096/fj.03-0645rev. [DOI] [PubMed] [Google Scholar]
- 6.Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–73. [PubMed] [Google Scholar]
- 7.Flower RJ, Vane JR. Inhibition of prostaglandin biosynthesis. Biochem Pharmacol. 1974;23:1439–50. doi: 10.1016/0006-2952(74)90381-5. [DOI] [PubMed] [Google Scholar]
- 8.Warner TD, Mitchell JA. COX-2 selectivity alone does not define the cardiovascular risks associated with non-steroidal anti-inflammatory drugs. Lancet. 2008;371:270–3. doi: 10.1016/S0140-6736(08)60137-3. [DOI] [PubMed] [Google Scholar]
- 9.Funk CD, FitzGerald GA. COX-2 inhibitors and cardiovascular risk. J Cardiovasc Pharmacol. 2007;50:470–9. doi: 10.1097/FJC.0b013e318157f72d. [DOI] [PubMed] [Google Scholar]
- 10.Deeb RS, Upmacis RK, Lamon BD, Gross SS, Hajjar DP. Maintaining equilibrium by selective targeting of cyclooxygenase pathways: promising offensives against vascular injury. Hypertension. 2008;51:1–7. doi: 10.1161/HYPERTENSIONAHA.107.092866. [DOI] [PubMed] [Google Scholar]
- 11.Hennekens CH, Borzak S. Cyclooxygenase-2 inhibitors and most traditional nonsteroidal anti-inflammatory drugs cause similar moderately increased risks of cardiovascular disease. J Cardiovasc Pharmacol Ther. 2008;13:41–50. doi: 10.1177/1074248407312990. [DOI] [PubMed] [Google Scholar]
- 12.Smith WL, Song I. The enzymology of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediat. 2002;68–69:115–28. doi: 10.1016/s0090-6980(02)00025-4. [DOI] [PubMed] [Google Scholar]
- 13.Hinz B, Brune K. Antipyretic analgesics: nonsteroidal antiinflammatory drugs, selective COX-2 inhibitors, paracetamol and pyrazolinones. Handb Exp Pharmacol. 2007;177:65–93. doi: 10.1007/978-3-540-33823-9_3. [DOI] [PubMed] [Google Scholar]
- 14.Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634–42. doi: 10.1161/CIRCULATIONAHA.106.181424. [DOI] [PubMed] [Google Scholar]
- 15.Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351:1709–11. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- 16.Fitzgerald GA. COX-2 in play at the AHA and the FDA. Trends Pharmacol Sci. 2007;28:303–7. doi: 10.1016/j.tips.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 17.FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001;345:433–42. doi: 10.1056/NEJM200108093450607. [DOI] [PubMed] [Google Scholar]
- 18.Flavahan NA. Balancing prostanoid activity in the human vascular system. Trends Pharmacol Sci. 2007;28:106–10. doi: 10.1016/j.tips.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Flower RJ. The development of COX2 inhibitors. Nat Rev. 2003;2:179–91. doi: 10.1038/nrd1034. [DOI] [PubMed] [Google Scholar]
- 20.Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patrono C, Patrignani P, Garcia Rodriguez LA. Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J Clin Invest. 2001;108:7–13. doi: 10.1172/JCI13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whelton A. COX-2-specific inhibitors and the kidney: effect on hypertension and oedema. J Hypertens Suppl. 2002;20:S31–5. [PubMed] [Google Scholar]
- 23.White WB. Cardiovascular effects of the cyclooxygenase inhibitors. Hypertension. 2007;49:408–18. doi: 10.1161/01.HYP.0000258106.74139.25. [DOI] [PubMed] [Google Scholar]
- 24.Guan Y, Zhang Y, Wu J, Qi Z, Yang G, Dou D, Gao Y, Chen L, Zhang X, Davis LS, Wei M, Fan X, Carmosino M, Hao C, Imig JD, Breyer RM, Breyer MD. Antihypertensive effects of selective prostaglandin E2 receptor subtype 1 targeting. J Clin Invest. 2007;117:2496–505. doi: 10.1172/JCI29838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sellers MM, Stallone JN. Sympathy for the devil: the role of thromboxane in the regulation of vascular tone and blood pressure. Am J Physiol. 2008;294:H1978–86. doi: 10.1152/ajpheart.01318.2007. [DOI] [PubMed] [Google Scholar]
- 26.Rosen GD, Birkenmeier TM, Raz A, Holtzman MJ. Identification of a cyclooxygenase-related gene and its potential role in prostaglandin formation. Biochem Biophys Res Commun. 1989;164:1358–65. doi: 10.1016/0006-291x(89)91819-6. [DOI] [PubMed] [Google Scholar]
- 27.Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci U S A. 1992;89:7384–8. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem. 1991;266:12866–72. [PubMed] [Google Scholar]
- 29.Masferrer JL, Seibert K, Zweifel B, Needleman P. Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proc Natl Acad Sci U S A. 1992;89:3917–21. doi: 10.1073/pnas.89.9.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Banion MK, Winn VD, Young DA. cDNA cloning and functional activity of a glucocorticoid-regulated inflammatory cyclooxygenase. Proc Natl Acad Sci U S A. 1992;89:4888–92. doi: 10.1073/pnas.89.11.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci U S A. 1991;88:2692–6. doi: 10.1073/pnas.88.7.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vane JR, Botting RM. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104:2S–8S. doi: 10.1016/s0002-9343(97)00203-9. discussion 21S-22S. [DOI] [PubMed] [Google Scholar]
- 33.Marcus AJ, Weksler BB, Jaffe EA, Broekman MJ. Synthesis of prostacyclin from platelet-derived endoperoxides by cultured human endothelial cells. J Clin Invest. 1980;66:979–86. doi: 10.1172/JCI109967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rocca B, Secchiero P, Ciabattoni G, Ranelletti FO, Catani L, Guidotti L, Melloni E, Maggiano N, Zauli G, Patrono C. Cyclooxygenase-2 expression is induced during human megakaryopoiesis and characterizes newly formed platelets. Proc Natl Acad Sci U S A. 2002;99:7634–9. doi: 10.1073/pnas.112202999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guthikonda S, Lev EI, Patel R, DeLao T, Bergeron AL, Dong JF, Kleiman NS. Reticulated platelets and uninhibited COX-1 and COX-2 decrease the antiplatelet effects of aspirin. J Thromb Haemost. 2007;5:490–6. doi: 10.1111/j.1538-7836.2007.02387.x. [DOI] [PubMed] [Google Scholar]
- 36.Evangelista V, Manarini S, Di Santo A, Capone ML, Ricciotti E, Di Francesco L, Tacconelli S, Sacchetti A, D'Angelo S, Scilimati A, Sciulli MG, Patrignani P. De novo synthesis of cyclooxygenase-1 counteracts the suppression of platelet thromboxane biosynthesis by aspirin. Circ Res. 2006;98:593–5. doi: 10.1161/01.RES.0000214553.37930.3e. [DOI] [PubMed] [Google Scholar]
- 37.Weber AA, Przytulski B, Schumacher M, Zimmermann N, Gams E, Hohlfeld T, Schror K. Flow cytometry analysis of platelet cyclooxygenase-2 expression: induction of platelet cyclooxygenase-2 in patients undergoing coronary artery bypass grafting. Br J Haematol. 2002;117:424–6. doi: 10.1046/j.1365-2141.2002.03423.x. [DOI] [PubMed] [Google Scholar]
- 38.Catella-Lawson F, McAdam B, Morrison BW, Kapoor S, Kujubu D, Antes L, Lasseter KC, Quan H, Gertz BJ, FitzGerald GA. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J Pharmacol Exp Ther. 1999;289:735–41. [PubMed] [Google Scholar]
- 39.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999;96:272–7. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker CS, Hall RJ, Evans TJ, Pomerance A, Maclouf J, Creminon C, Yacoub MH, Polak JM. Cyclooxygenase-2 is widely expressed in atherosclerotic lesions affecting native and transplanted human coronary arteries and colocalizes with inducible nitric oxide synthase and nitrotyrosine particularly in macrophages. Arterioscler Thromb Vasc Biol. 1999;19:646–55. doi: 10.1161/01.atv.19.3.646. [DOI] [PubMed] [Google Scholar]
- 41.Belton O, Byrne D, Kearney D, Leahy A, Fitzgerald DJ. Cyclooxygenase-1 and -2-dependent prostacyclin formation in patients with atherosclerosis. Circulation. 2000;102:840–5. doi: 10.1161/01.cir.102.8.840. [DOI] [PubMed] [Google Scholar]
- 42.Schonbeck U, Sukhova GK, Graber P, Coulter S, Libby P. Augmented expression of cyclooxygenase-2 in human atherosclerotic lesions. Am J Pathol. 1999;155:1281–91. doi: 10.1016/S0002-9440(10)65230-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stemme V, Swedenborg J, Claesson H, Hansson GK. Expression of cyclo-oxygenase-2 in human atherosclerotic carotid arteries. Eur J Vasc Endovasc Surg. 2000;20:146–52. doi: 10.1053/ejvs.2000.1145. [DOI] [PubMed] [Google Scholar]
- 44.Zidar N, Odar K, Glavac D, Jerse M, Zupanc T, Stajer D. Cyclooxygenase in normal human tissues – is COX-1 really a constitutive isoform, and COX-2 an inducible isoform? J Cell Mol Med. 2009;13:3753–63. doi: 10.1111/j.1582-4934.2008.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warner TD, Vojnovic I, Giuliano F, Jimenez R, Bishop-Bailey D, Mitchell JA. Cyclooxygenases 1, 2, and 3 and the production of prostaglandin I2: investigating the activities of acetaminophen and cyclooxygenase-2-selective inhibitors in rat tissues. J Pharmacol Exp Ther. 2004;310:642–7. doi: 10.1124/jpet.103.063875. [DOI] [PubMed] [Google Scholar]
- 46.Hamilton LC, Mitchell JA, Tomlinson AM, Warner TD. Synergy between cyclo-oxygenase-2 induction and arachidonic acid supply in vivo: consequences for nonsteroidal antiinflammatory drug efficacy. FASEB J. 1999;13:245–51. doi: 10.1096/fasebj.13.2.245. [DOI] [PubMed] [Google Scholar]
- 47.Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, Kaley G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90:1159–66. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- 48.Pratico D, Tillmann C, Zhang ZB, Li H, FitzGerald GA. Acceleration of atherogenesis by COX-1-dependent prostanoid formation in low density lipoprotein receptor knockout mice. Proc Natl Acad Sci U S A. 2001;98:3358–63. doi: 10.1073/pnas.061607398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong E, Huang JQ, Tagari P, Riendeau D. Effects of COX-2 inhibitors on aortic prostacyclin production in cholesterol-fed rabbits. Atherosclerosis. 2001;157:393–402. doi: 10.1016/s0021-9150(00)00756-5. [DOI] [PubMed] [Google Scholar]
- 50.Rudic RD, Brinster D, Cheng Y, Fries S, Song WL, Austin S, Coffman TM, FitzGerald GA. COX-2-derived prostacyclin modulates vascular remodeling. Circ Res. 2005;96:1240–7. doi: 10.1161/01.RES.0000170888.11669.28. [DOI] [PubMed] [Google Scholar]
- 51.Kobayashi T, Narumiya S. Function of prostanoid receptors: studies on knockout mice. Prostaglandins Other Lipid Mediat. 2002;68–69:557–73. doi: 10.1016/s0090-6980(02)00055-2. [DOI] [PubMed] [Google Scholar]
- 52.Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–7. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 53.Gross S, Tilly P, Hentsch D, Vonesch JL, Fabre JE. Vascular wall-produced prostaglandin E2 exacerbates arterial thrombosis and atherothrombosis through platelet EP3 receptors. J Exp Med. 2007;204:311–20. doi: 10.1084/jem.20061617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heptinstall S, Espinosa DI, Manolopoulos P, Glenn JR, White AE, Johnson A, Dovlatova N, Fox SC, May JA, Hermann D, Magnusson O, Stefansson K, Hartman D, Gurney M. DG-041 inhibits the EP3 prostanoid receptor – a new target for inhibition of platelet function in atherothrombotic disease. Platelets. 2008;19:605–13. doi: 10.1080/09537100802351073. [DOI] [PubMed] [Google Scholar]
- 55.Smith JP, Haddad EV, Downey JD, Breyer RM, Boutaud O. PGE2 decreases reactivity of human platelets by activating EP2 and EP4. Thromb Res. 2010;126:e23–9. doi: 10.1016/j.thromres.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paul BZ, Ashby B, Sheth SB. Distribution of prostaglandin IP and EP receptor subtypes and isoforms in platelets and human umbilical artery smooth muscle cells. Br J Haematol. 1998;102:1204–11. doi: 10.1046/j.1365-2141.1998.00910.x. [DOI] [PubMed] [Google Scholar]
- 57.Mao GF, Jin JG, Bastepe M, Ortiz-Vega S, Ashby B. Prostaglandin E2 both stimulates and inhibits adenyl cyclase on platelets: comparison of effects on cloned EP4 and EP3 prostaglandin receptor subtypes. Prostaglandins. 1996;52:175–85. doi: 10.1016/s0090-6980(96)00095-0. [DOI] [PubMed] [Google Scholar]
- 58.Patrono C, Garcia Rodriguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353:2373–83. doi: 10.1056/NEJMra052717. [DOI] [PubMed] [Google Scholar]
- 59.Reilly IA, FitzGerald GA. Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood. 1987;69:180–6. [PubMed] [Google Scholar]
- 60.Baigent C, Blackwell L, Collins R, Emberson J, Godwin J, Peto R, Buring J, Hennekens C, Kearney P, Meade T, Patrono C, Roncaglioni MC, Zanchetti A. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849–60. doi: 10.1016/S0140-6736(09)60503-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mehta SR, Bassand JP, Chrolavicius S, Diaz R, Eikelboom JW, Fox KA, Granger CB, Jolly S, Joyner CD, Rupprecht HJ, Widimsky P, Afzal R, Pogue J, Yusuf S. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med. 2010;363:930–42. doi: 10.1056/NEJMoa0909475. [DOI] [PubMed] [Google Scholar]
- 62.Mehta SR, Tanguay JF, Eikelboom JW, Jolly SS, Joyner CD, Granger CB, Faxon DP, Rupprecht HJ, Budaj A, Avezum A, Widimsky P, Steg PG, Bassand JP, Montalescot G, Macaya C, Di Pasquale G, Niemela K, Ajani AE, White HD, Chrolavicius S, Gao P, Fox KA, Yusuf S. Double-dose versus standard-dose clopidogrel and high-dose versus low-dose aspirin in individuals undergoing percutaneous coronary intervention for acute coronary syndromes (CURRENT-OASIS 7): a randomised factorial trial. Lancet. 2010;376:1233–43. doi: 10.1016/S0140-6736(10)61088-4. [DOI] [PubMed] [Google Scholar]
- 63.Patrono C, Baigent C, Hirsh J, Roth G. Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition) Chest. 2008;133:199S–233S. doi: 10.1378/chest.08-0672. [DOI] [PubMed] [Google Scholar]
- 64.Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA. 2007;297:2018–24. doi: 10.1001/jama.297.18.2018. [DOI] [PubMed] [Google Scholar]
- 65.Hernandez-Diaz S, Garcia Rodriguez LA. Cardioprotective aspirin users and their excess risk of upper gastrointestinal complications. BMC Med. 2006;4:22. doi: 10.1186/1741-7015-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.The Dutch TIA Trial Study Group. A comparison of two doses of aspirin (30 mg vs. 283 mg a day) in patients after a transient ischemic attack or minor ischemic stroke. N Engl J Med. 1991;325:1261–6. doi: 10.1056/NEJM199110313251801. [DOI] [PubMed] [Google Scholar]
- 67.Lanas A, Garcia-Rodriguez LA, Arroyo MT, Gomollon F, Feu F, Gonzalez-Perez A, Zapata E, Bastida G, Rodrigo L, Santolaria S, Guell M, de Argila CM, Quintero E, Borda F, Pique JM. Risk of upper gastrointestinal ulcer bleeding associated with selective cyclo-oxygenase-2 inhibitors, traditional non-aspirin non-steroidal anti-inflammatory drugs, aspirin and combinations. Gut. 2006;55:1731–8. doi: 10.1136/gut.2005.080754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340:1888–99. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
- 69.Duffy SJ, Castle SF, Harper RW, Meredith IT. Contribution of vasodilator prostanoids and nitric oxide to resting flow, metabolic vasodilation, and flow-mediated dilation in human coronary circulation. Circulation. 1999;100:1951–7. doi: 10.1161/01.cir.100.19.1951. [DOI] [PubMed] [Google Scholar]
- 70.Friedman PL, Brown EJ, Jr, Gunther S, Alexander RW, Barry WH, Mudge GH, Jr, Grossman W. Coronary vasoconstrictor effect of indomethacin in patients with coronary-artery disease. N Engl J Med. 1981;305:1171–5. doi: 10.1056/NEJM198111123052002. [DOI] [PubMed] [Google Scholar]
- 71.Arehart E, Stitham J, Asselbergs FW, Douville K, Mackenzie T, Fetalvero KM, Gleim S, Kasza Z, Rao Y, Martel L, Segel S, Robb J, Kaplan A, Simons M, Powell RJ, Moore JH, Rimm EB, Martin KA, Hwa J. Acceleration of cardiovascular disease by a dysfunctional prostacyclin receptor mutation. Potential implications for cyclooxygenase-2 inhibition. Circ Res. 2008;102:986–93. doi: 10.1161/CIRCRESAHA.107.165936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kobayashi T, Tahara Y, Matsumoto M, Iguchi M, Sano H, Murayama T, Arai H, Oida H, Yurugi-Kobayashi T, Yamashita JK, Katagiri H, Majima M, Yokode M, Kita T, Narumiya S. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J Clin Invest. 2004;114:784–94. doi: 10.1172/JCI21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Egan KM, Wang M, Fries S, Lucitt MB, Zukas AM, Pure E, Lawson JA, FitzGerald GA. Cyclooxygenases, thromboxane, and atherosclerosis: plaque destabilization by cyclooxygenase-2 inhibition combined with thromboxane receptor antagonism. Circulation. 2005;111:334–42. doi: 10.1161/01.CIR.0000153386.95356.78. [DOI] [PubMed] [Google Scholar]
- 74.Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, Lawson JA, FitzGerald GA. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science (New York, NY) 2002;296:539–41. doi: 10.1126/science.1068711. [DOI] [PubMed] [Google Scholar]
- 75.Xiao CY, Hara A, Yuhki K, Fujino T, Ma H, Okada Y, Takahata O, Yamada T, Murata T, Narumiya S, Ushikubi F. Roles of prostaglandin I(2) and thromboxane A(2) in cardiac ischemia-reperfusion injury: a study using mice lacking their respective receptors. Circulation. 2001;104:2210–5. doi: 10.1161/hc4301.098058. [DOI] [PubMed] [Google Scholar]
- 76.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006;116:1391–9. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332:1302–8. doi: 10.1136/bmj.332.7553.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cooney MT, Dudina AL, Graham IM. Value and limitations of existing scores for the assessment of cardiovascular risk: a review for clinicians. J Am Coll Cardiol. 2009;54:1209–27. doi: 10.1016/j.jacc.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 79.McGettigan P, Han P, Jones L, Whitaker D, Henry D. Selective COX-2 inhibitors, NSAIDs and congestive heart failure: differences between new and recurrent cases. Br J Clin Pharmacol. 2008;65:927–34. doi: 10.1111/j.1365-2125.2008.03121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Forman JP, Rimm EB, Curhan GC. Frequency of analgesic use and risk of hypertension among men. Arch Intern Med. 2007;167:394–9. doi: 10.1001/archinte.167.4.394. [DOI] [PubMed] [Google Scholar]
- 81.Yu Y, Lucitt MB, Stubbe J, Cheng Y, Friis UG, Hansen PB, Jensen BL, Smyth EM, FitzGerald GA. Prostaglandin F2alpha elevates blood pressure and promotes atherosclerosis. Proc Natl Acad Sci U S A. 2009;106:7985–90. doi: 10.1073/pnas.0811834106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.FitzGerald GA, Brash AR, Falardeau P, Oates JA. Estimated rate of prostacyclin secretion into the circulation of normal man. J Clin Invest. 1981;68:1272–6. doi: 10.1172/JCI110373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Patrono C, Ciabattoni G, Pugliese F, Pierucci A, Blair IA, FitzGerald GA. Estimated rate of thromboxane secretion into the circulation of normal humans. J Clin Invest. 1986;77:590–4. doi: 10.1172/JCI112341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McAdam BF, Byrne D, Morrow JD, Oates JA. Contribution of cyclooxygenase-2 to elevated biosynthesis of thromboxane A2 and prostacyclin in cigarette smokers. Circulation. 2005;112:1024–9. doi: 10.1161/CIRCULATIONAHA.105.542696. [DOI] [PubMed] [Google Scholar]
- 85.Catella F, Healy D, Lawson JA, FitzGerald GA. 11-Dehydrothromboxane B2: a quantitative index of thromboxane A2 formation in the human circulation. Proc Natl Acad Sci U S A. 1986;83:5861–5. doi: 10.1073/pnas.83.16.5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Murphy RC, FitzGerald GA. Current approaches to estimation of eicosanoid formation in vivo. Adv Prostaglandin Thromboxane Leukot Res. 1994;22:341–8. [PubMed] [Google Scholar]
- 87.Clarke RJ, Mayo G, Price P, FitzGerald GA. Suppression of thromboxane A2 but not of systemic prostacyclin by controlled-release aspirin. N Engl J Med. 1991;325:1137–41. doi: 10.1056/NEJM199110173251605. [DOI] [PubMed] [Google Scholar]
- 88.Hanley SP, Bevan J, Cockbill SR, Heptinstall S. Differential inhibition by low-dose aspirin of human venous prostacyclin synthesis and platelet thromboxane synthesis. Lancet. 1981;1:969–71. doi: 10.1016/s0140-6736(81)91733-5. [DOI] [PubMed] [Google Scholar]
- 89.Preston FE, Whipps S, Jackson CA, French AJ, Wyld PJ, Stoddard CJ. Inhibition of prostacyclin and platelet thromboxane A2 after low-dose aspirin. N Engl J Med. 1981;304:76–9. doi: 10.1056/NEJM198101083040203. [DOI] [PubMed] [Google Scholar]
- 90.Weksler BB, Pett SB, Alonso D, Richter RC, Stelzer P, Subramanian V, Tack-Goldman K, Gay WA., Jr Differential inhibition by aspirin of vascular and platelet prostaglandin synthesis in atherosclerotic patients. N Engl J Med. 1983;308:800–5. doi: 10.1056/NEJM198304073081402. [DOI] [PubMed] [Google Scholar]
- 91.Hanley SP, Bevan J. Inhibition by aspirin of human arterial and venous prostacyclin synthesis. Prostaglandins Leukot Med. 1985;20:141–9. doi: 10.1016/0262-1746(85)90005-8. [DOI] [PubMed] [Google Scholar]
- 92.Kyrle PA, Eichler HG, Jager U, Lechner K. Inhibition of prostacyclin and thromboxane A2 generation by low-dose aspirin at the site of plug formation in man in vivo. Circulation. 1987;75:1025–9. doi: 10.1161/01.cir.75.5.1025. [DOI] [PubMed] [Google Scholar]
- 93.Davi G, Custro N, Novo S, Mattina A, Strano A. The effect of two low doses of aspirin on whole blood thromboxane and prostacyclin generation in healthy subjects. Thromb Haemost. 1983;50:669–70. [PubMed] [Google Scholar]
- 94.Tohgi H, Konno S, Tamura K, Kimura B, Kawano K. Effects of low-to-high doses of aspirin on platelet aggregability and metabolites of thromboxane A2 and prostacyclin. Stroke. 1992;23:1400–3. doi: 10.1161/01.str.23.10.1400. [DOI] [PubMed] [Google Scholar]
- 95.Gow JA, Ebbeling L, Gerrard JM. The effect of regular and enteric-coated aspirin on bleeding time, thromboxane, and prostacyclin. Prostaglandins Leukot Essent Fatty Acids. 1993;49:515–20. doi: 10.1016/0952-3278(93)90040-4. [DOI] [PubMed] [Google Scholar]
- 96.Boger RH, Bode-Boger SM, Gutzki FM, Tsikas D, Weskott HP, Frolich JC. Rapid and selective inhibition of platelet aggregation and thromboxane formation by intravenous low dose aspirin in man. Clin Sci (Lond) 1993;84:517–24. doi: 10.1042/cs0840517. [DOI] [PubMed] [Google Scholar]
- 97.Gao XR, Adhikari CM, Peng LY, Guo XG, Zhai YS, He XY, Zhang LY, Lin J, Zuo ZY. Efficacy of different doses of aspirin in decreasing blood levels of inflammatory markers in patients with cardiovascular metabolic syndrome. J Pharm Pharmacol. 2009;61:1505–10. doi: 10.1211/jpp/61.11.0010. [DOI] [PubMed] [Google Scholar]
- 98.Jaffe EA, Weksler BB. Recovery of endothelial cell prostacyclin production after inhibition by low doses of aspirin. J Clin Invest. 1979;63:532–5. doi: 10.1172/JCI109332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Whiting J, Salata K, Bailey JM. Aspirin: an unexpected side effect on prostacyclin synthesis in cultured vascular smooth muscle cells. Science. 1980;210:663–5. doi: 10.1126/science.6776627. [DOI] [PubMed] [Google Scholar]
- 100.Salata KF, Whiting JD, Bailey JM. Regeneration of prostacyclin synthetase activity in cultured vascular cells following aspirin treatment. Adv Prostaglandin Thromboxane Res. 1980;6:537–40. [PubMed] [Google Scholar]
- 101.Bailey JM, Muza B, Hla T, Salata K. Restoration of prostacyclin synthase in vascular smooth muscle cells after aspirin treatment: regulation by epidermal growth factor. J Lipid Res. 1985;26:54–61. [PubMed] [Google Scholar]
- 102.Hla TT, Bailey JM. Differential recovery of prostacyclin synthesis in cultured vascular endothelial vs. smooth muscle cells after inactivation of cyclooxygenase with aspirin. Prostaglandins Leukot Essent Fatty Acids. 1989;36:175–84. doi: 10.1016/0952-3278(89)90059-8. [DOI] [PubMed] [Google Scholar]
- 103.Gerrard JM, Taback S, Singhroy S, Docherty JC, Kostolansky I, McNicol A, Kobrinsky NL, McKenzie JK, Rowe R. In vivo measurement of thromboxane B2 and 6-keto-prostaglandin F1 alpha in humans in response to a standardized vascular injury and the influence of aspirin. Circulation. 1989;79:29–38. doi: 10.1161/01.cir.79.1.29. [DOI] [PubMed] [Google Scholar]
- 104.Preston FE, Greaves M, Jackson CA, Stoddard CJ. Low-dose aspirin inhibits platelet and venous cyclo-oxygenase in man. Thromb Res. 1982;27:477–84. doi: 10.1016/0049-3848(82)90065-2. [DOI] [PubMed] [Google Scholar]
- 105.Heavey DJ, Barrow SE, Hickling NE, Ritter JM. Aspirin causes short-lived inhibition of bradykinin-stimulated prostacyclin production in man. Nature. 1985;318:186–8. doi: 10.1038/318186a0. [DOI] [PubMed] [Google Scholar]
- 106.Vesterqvist O. Rapid recovery of in vivo prostacyclin formation after inhibition by aspirin. Evidence from measurements of the major urinary metabolite of prostacyclin by GC-MS. Eur J Clin Pharmacol. 1986;30:69–73. doi: 10.1007/BF00614198. [DOI] [PubMed] [Google Scholar]
- 107.Ritter JM, Cockcroft JR, Doktor HS, Beacham J, Barrow SE. Differential effect of aspirin on thromboxane and prostaglandin biosynthesis in man. Br J Clin Pharmacol. 1989;28:573–9. doi: 10.1111/j.1365-2125.1989.tb03544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gachet C. Regulation of platelet functions by P2 receptors. Annu Rev Pharmacol Toxicol. 2006;46:277–300. doi: 10.1146/annurev.pharmtox.46.120604.141207. [DOI] [PubMed] [Google Scholar]
- 109.Cattaneo M. ADP receptors: inhibitory strategies for antiplatelet therapy. Drug News Perspect. 2006;19:253–9. doi: 10.1358/dnp.2006.19.5.985936. [DOI] [PubMed] [Google Scholar]
- 110.Gachet C. The platelet P2 receptors as molecular targets for old and new antiplatelet drugs. Pharmacol Ther. 2005;108:180–92. doi: 10.1016/j.pharmthera.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 111.Gachet C. P2 receptors, platelet function and pharmacological implications. Thromb Haemost. 2008;99:466–72. doi: 10.1160/TH07-11-0673. [DOI] [PubMed] [Google Scholar]
- 112.Cattaneo M. Platelet P2 receptors: old and new targets for antithrombotic drugs. Expert Rev Cardiovasc Ther. 2007;5:45–55. doi: 10.1586/14779072.5.1.45. [DOI] [PubMed] [Google Scholar]
- 113.Kahner BN, Shankar H, Murugappan S, Prasad GL, Kunapuli SP. Nucleotide receptor signaling in platelets. J Thromb Haemost. 2006;4:2317–26. doi: 10.1111/j.1538-7836.2006.02192.x. [DOI] [PubMed] [Google Scholar]
- 114.Cattaneo M. New P2Y12 blockers. J Thromb Haemost. 2009;7(Suppl. 1):262–5. doi: 10.1111/j.1538-7836.2009.03382.x. [DOI] [PubMed] [Google Scholar]
- 115.Jakubowski JA, Winters KJ, Naganuma H, Wallentin L. Prasugrel: a novel thienopyridine antiplatelet agent. A review of preclinical and clinical studies and the mechanistic basis for its distinct antiplatelet profile. Cardiovasc Drug Rev. 2007;25:357–74. doi: 10.1111/j.1527-3466.2007.00027.x. [DOI] [PubMed] [Google Scholar]
- 116.Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, Horrow J, Husted S, James S, Katus H, Mahaffey KW, Scirica BM, Skene A, Steg PG, Storey RF, Harrington RA, Freij A, Thorsen M. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–57. doi: 10.1056/NEJMoa0904327. [DOI] [PubMed] [Google Scholar]
- 117.Cattaneo M. Aspirin and clopidogrel: Efficacy, safety, and the issue of drug resistance. Arterioscler Thromb Vasc Biol. 2004;24:1980–87. doi: 10.1161/01.ATV.0000145980.39477.a9. [DOI] [PubMed] [Google Scholar]
- 118.Fitzgerald DJ, Maree A. Aspirin and clopidogrel resistance. Hematology Am Soc Hematol Educ Program. 2007;2007:114–20. doi: 10.1182/asheducation-2007.1.114. [DOI] [PubMed] [Google Scholar]
- 119.Li Z, Zhang G, Le Breton GC, Gao X, Malik AB, Du X. Two waves of platelet secretion induced by thromboxane A2 receptor and a critical role for phosphoinositide 3-kinases. J Biol Chem. 2003;278:30725–31. doi: 10.1074/jbc.M301838200. [DOI] [PubMed] [Google Scholar]
- 120.Hobson AR, Qureshi Z, Banks P, Curzen NP. Effects of clopidogrel on ‘aspirin specific’ pathways of platelet inhibition. Platelets. 2009;20:386–90. doi: 10.1080/09537100903003227. [DOI] [PubMed] [Google Scholar]
- 121.Armstrong PC, Dhanji AR, Truss NJ, Zain ZN, Tucker AT, Mitchell JA, Warner TD. Utility of 96-well plate aggregometry and measurement of thrombi adhesion to determine aspirin and clopidogrel effectiveness. Thromb Haemost. 2009;102:772–8. doi: 10.1160/TH09-04-0215. [DOI] [PubMed] [Google Scholar]
- 122.Guidetti GF, Lova P, Bernardi B, Campus F, Baldanzi G, Graziani A, Balduini C, Torti M. The Gi-coupled P2Y12 receptor regulates diacylglycerol-mediated signaling in human platelets. J Biol Chem. 2008;283:28795–805. doi: 10.1074/jbc.M801588200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cattaneo M, Lecchi A. Inhibition of the platelet P2Y12 receptor for adenosine diphosphate potentiates the antiplatelet effect of prostacyclin. J Thromb Haemost. 2007;5:577–82. doi: 10.1111/j.1538-7836.2007.02356.x. [DOI] [PubMed] [Google Scholar]
- 124.Armstrong PC, Dhanji AR, Tucker AT, Mitchell JA, Warner TD. Reduction of platelet thromboxane A2 production ex vivo and in vivo by clopidogrel therapy. J Thromb Haemost. 2010;8:613–5. doi: 10.1111/j.1538-7836.2009.03714.x. [DOI] [PubMed] [Google Scholar]
- 125.Armstrong PC, Leadbeater PD, Chan MV, Kirkby NS, Jakubowski JA, Mitchell JA, Warner TD. In the presence of strong P2Y(12) receptor blockade, aspirin provides little additional inhibition of platelet aggregation. J Thromb Haemost. 2011;9:552–61. doi: 10.1111/j.1538-7836.2010.04160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hallas J, Dall M, Andries A, Andersen BS, Aalykke C, Hansen JM, Andersen M, Lassen AT. Use of single and combined antithrombotic therapy and risk of serious upper gastrointestinal bleeding: population based case-control study. BMJ. 2006;333:726. doi: 10.1136/bmj.38947.697558.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shehab N, Sperling LS, Kegler SR, Budnitz DS. National estimates of emergency department visits for hemorrhage-related adverse events from clopidogrel plus aspirin and from warfarin. Arch Intern Med. 2010;170:1926–33. doi: 10.1001/archinternmed.2010.407. [DOI] [PubMed] [Google Scholar]
- 128.Wang TY, Xiao L, Alexander KP, Rao SV, Kosiborod MN, Rumsfeld JS, Spertus JA, Peterson ED. Antiplatelet therapy use after discharge among acute myocardial infarction patients with in-hospital bleeding. Circulation. 2008;118:2139–45. doi: 10.1161/CIRCULATIONAHA.108.787143. [DOI] [PubMed] [Google Scholar]
- 129.Roy P, Bonello L, Torguson R, de Labriolle A, Lemesle G, Slottow TL, Steinberg DH, Kaneshige K, Xue Z, Satler LF, Kent KM, Suddath WO, Pichard AD, Lindsay J, Waksman R. Impact of ‘nuisance’ bleeding on clopidogrel compliance in patients undergoing intracoronary drug-eluting stent implantation. Am J Cardiol. 2008;102:1614–7. doi: 10.1016/j.amjcard.2008.07.063. [DOI] [PubMed] [Google Scholar]
- 130.Wiviott SD, Antman EM, Gibson CM, Montalescot G, Riesmeyer J, Weerakkody G, Winters KJ, Warmke JW, McCabe CH, Braunwald E. Evaluation of prasugrel compared with clopidogrel in patients with acute coronary syndromes: design and rationale for the TRial to assess Improvement in Therapeutic Outcomes by optimizing platelet InhibitioN with prasugrel Thrombolysis In Myocardial Infarction 38 (TRITON-TIMI 38) Am Heart J. 2006;152:627–35. doi: 10.1016/j.ahj.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 131.Wiviott SD, Braunwald E, McCabe CH, Horvath I, Keltai M, Herrman JP, Van de Werf F, Downey WE, Scirica BM, Murphy SA, Antman EM. Intensive oral antiplatelet therapy for reduction of ischaemic events including stent thrombosis in patients with acute coronary syndromes treated with percutaneous coronary intervention and stenting in the TRITON-TIMI 38 trial: a subanalysis of a randomised trial. Lancet. 2008;371:1353–63. doi: 10.1016/S0140-6736(08)60422-5. [DOI] [PubMed] [Google Scholar]
- 132.Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neumann FJ, Ardissino D, De Servi S, Murphy SA, Riesmeyer J, Weerakkody G, Gibson CM, Antman EM. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–15. doi: 10.1056/NEJMoa0706482. [DOI] [PubMed] [Google Scholar]
- 133.Peters RJ, Mehta SR, Fox KA, Zhao F, Lewis BS, Kopecky SL, Diaz R, Commerford PJ, Valentin V, Yusuf S. Effects of aspirin dose when used alone or in combination with clopidogrel in patients with acute coronary syndromes: observations from the Clopidogrel in Unstable angina to prevent Recurrent Events (CURE) study. Circulation. 2003;108:1682–7. doi: 10.1161/01.CIR.0000091201.39590.CB. [DOI] [PubMed] [Google Scholar]
- 134.AstraZeneca. Ticagrelor NDA 22-433 Briefing Document for Cardiovascular and Renal Drugs Advisory Committee Meeting. Available at http://www.fda.gov/downloads/AdvisoryCommitteesMeetingMaterials/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/UCM220197.pdf (last accessed 30 July 2010)
- 135.Kong DF, Hasselblad V, Kandzari DE, Newby LK, Califf RM. Seeking the optimal aspirin dose in acute coronary syndromes. Am J Cardiol. 2002;90:622–5. doi: 10.1016/s0002-9149(02)02566-3. [DOI] [PubMed] [Google Scholar]
- 136.Antiplatelet Trialists' Collaboration. Collaborative overview of randomised trials of antiplatelet therapy – I: prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. BMJ. 1994;308:81–106. [PMC free article] [PubMed] [Google Scholar]
- 137.Sample D, Wargovich M, Fischer SM, Inamdar N, Schwartz P, Wang X, Do KA, Sinicrope FA. A dose-finding study of aspirin for chemoprevention utilizing rectal mucosal prostaglandin E(2) levels as a biomarker. Cancer Epidemiol Biomarkers Prev. 2002;11:275–9. [PubMed] [Google Scholar]
- 138.Cole AT, Filipowicz B, Hyman-Taylor P, Hawkey CJ. Low dose aspirin: selective inhibition of rectal dialysis thromboxane B2 in healthy volunteers. Aliment Pharmacol Ther. 1994;8:521–6. doi: 10.1111/j.1365-2036.1994.tb00325.x. [DOI] [PubMed] [Google Scholar]