

Abstract
Activated platelets stimulate thrombus formation in response to rupture of an atherosclerotic plaque or endothelial cell erosion, promoting atherothrombotic disease. Multiple pathways contribute to platelet activation. Aspirin, an irreversible inhibitor of thromboxane A2 synthesis, in combination with clopidogrel, an inhibitor of P2Y12 adenosine diphosphate platelet receptors, represent the current standard-of-care of antiplatelet therapy for patients with acute coronary syndrome and for those undergoing percutaneous coronary intervention. Although these agents have demonstrated significant clinical benefit, the increased risk of bleeding and the recurrence of thrombotic events represent substantial limitations. Thrombin is one of the most important platelet activators. The inhibition of protease-activated receptor 1 showed a good safety profile in preclinical studies. In fact, phase II studies with vorapaxar (SCH530348) and atopaxar (E5555) showed no increase of bleeding events in addition to the current standard-of-care of antiplatelet therapy. Although the results of phase III trials for both drugs are awaited, this family is a promising new addition to the current clinical practice for patients with atherothrombotic disease, not only as an alternative, but also as additional therapy.
Keywords: antiplatelet drugs, atopaxar, PAR-1, vorapaxar
Introduction
The use of an inhibitor of platelet activation via synthesis of thromboxane A2 (TXA2; aspirin) and a P2Y12 adenosine diphosphate (ADP) receptor inhibitor (clopidogrel) represents the standard-of-care for the treatment of recurrent thrombotic complications of patients with manifestations of atherosclerotic disease [1]. A large number of studies have shown the benefit of aspirin in the setting of different acute clinical scenarios, such as acute coronary syndrome (ACS) [2, 3] and percutaneous revascularization [4]. Furthermore, several studies have also shown the benefit of aspirin in the maintenance phase of these diseases as secondary prevention [5] and as primary prevention in patients with cardiovascular risk factors [2]. In contrast, the clinical efficacy of clopidogrel has been shown both as a single antiplatelet therapy in high-risk patients with various manifestations of atherosclerotic disease [6] and in combination with aspirin in patients across the ACS spectrum (unstable angina, non-ST elevation myocardial infarction and ST elevation myocardial infarction) or in those undergoing percutaneous coronary intervention (PCI) [7–12]. However, recurrent thrombotic events may continue to occur despite use of standard dual antiplatelet treatment regimens. Several reasons may explain this phenomenon, the most important of which appear to be the following: (i) there are multiple pathways contributing to platelet activation and aggregation processes, and aspirin and P2Y12 ADP receptor antagonists do not inhibit pathways other than those stimulated by TXA2 and ADP, leaving other pivotal pathways (e.g. thrombin-mediated pathways) uninhibited; and (ii) a considerable number of patients may have inadequate responsiveness to aspirin and clopidogrel therapy; this phenomenon has been shown in several studies to be associated with an increased risk of recurrent ischaemic events [13–15]. Description of the latter phenomenon goes beyond the scope of this review. The present review article provides an overview of the rationale for the use of thrombin receptor-inhibiting strategies as well as the currently available results from clinical investigations and future directions in the field. [16].
Role of platelets in thrombosis and haemostasis: basic principles and implications for thrombin-mediated signalling
The important role of platelets in normal haemostatic processes and in atherothrombotic disease are well known (Figure 1) [17]. Following plaque rupture/erosion, which may either occur spontaneously, e.g. in patients with an acute coronary event, or be iatrogenically induced, e.g. in patients undergoing percutaneous coronary intervention, platelet adhesion to the extracellular matrix occurs, which is mediated primarily by collagen receptors. After the initial adhesion of platelets to the extracellular matrix, the repair process requires a rapid response to autocrine and paracrine mediators, including ADP, thrombin, adrenaline and TXA2. These mediators amplify and sustain the initial platelet response and they recruit circulating platelets from the bloodstream to form a growing haemostatic plug [18]. Briefly, there are three main platelet membrane receptors to drive platelet activation (and maintain the response): TXA2 receptors, ADP receptors and thrombin receptors or protease-activated receptors (PARs) [18]. These are G protein-coupled receptors expressed by platelets and they mediate paracrine and autocrine platelet activation. Platelet activation by these factors leads to a change in platelet shape [19–21], expression of proinflammatory molecules, such as P-selectin [19], soluble CD40 ligand (sCD40L)[22] and other unidentified proteins [23], expression of platelet procoagulant activity [24], potentiation of aggregation by other prothrombotic factors, such as collagen [25], and, finally and most importantly, the conversion of the platelet receptor glycoprotein (GP) IIb/IIIa into an active form, which allows platelet-to-platelet adhesion, thus mediating the final aggregation process [18]. As thrombin is described as the most potent platelet activator [26, 27], inhibition of PARs provides a rational approach to define antiplatelet agents that more effectively inhibit platelet activation. The limitations of current therapies underscore the need for identifying other antiplatelet strategies to provide more comprehensive platelet inhibition.
Figure 1.
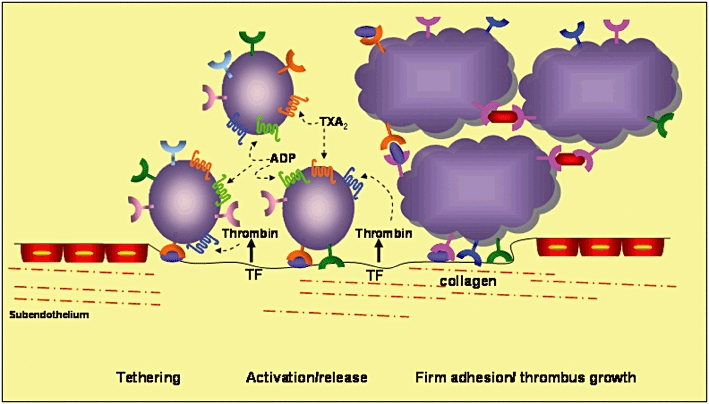
Platelet adhesion and aggregation on the extracellular matrix. The interaction between glycoprotein (GP) Ib and von Willebrand factor (vWF) mediates platelet tethering, and this enables subsequent interaction between GP VI and collagen. This triggers the shift of integrins to a high-affinity state and the release of adenosine diphosphate (ADP) and thromboxane A2 (TXA2), which bind to the P2Y12 and TP receptors, respectively. Tissue factor (TF) locally triggers thrombin formation, which also contributes to platelet activation via binding to the platelet protease-activated receptor 1 (PAR-1). Adapted from reference [29]. vWF ( ); GP Ib-V-IX (
); GP Ib-V-IX ( ); GP VI (
); GP VI ( ); Fibrinogen (
); Fibrinogen ( ); Resting GP llb/llla (
); Resting GP llb/llla ( ); Activated GP llb/llla (
); Activated GP llb/llla ( ); Resting 02β1 (
); Resting 02β1 ( ); Activated 02β1 (
); Activated 02β1 ( ); P2Y12 (
); P2Y12 ( ); TP receptor (
); TP receptor ( ); PAR-1 (
); PAR-1 ( ). TP, thromboxane receptor
). TP, thromboxane receptor
Thrombin-induced platelet activation: role of PARs
The PARs have a well-established role in vascular biology, haemostasis and atherothrombois as the receptors for thrombin. The serine protease thrombin is not only a key enzyme of the coagulation cascade but is also the strongest platelet agonist and is therefore an important contributor to atherothrombotic processes [28, 29]. The PARs are G protein-coupled receptors that mediate the cellular effects of diferent proteases, including thrombin. The proteolytic enzyme cleaves the extracellular loop of the G protein-coupled receptor, and the newly unmasked N-terminus acts as a tethered ligand that binds to the proximally located transmembrane loop of these receptors [30].
The mechanism of PAR activation and signalling is complex. Thrombin is an enzyme, and thus it can activate more than one molecule of the receptor. Another peculiarity is that the proteolytic cleavage of the receptor by thrombin is irreversible, and finally, the proteolytic cleavage of the receptor is potentially coupled to different G proteins that signal through multiple pathways (Figure 2) [16, 30].
Figure 2.
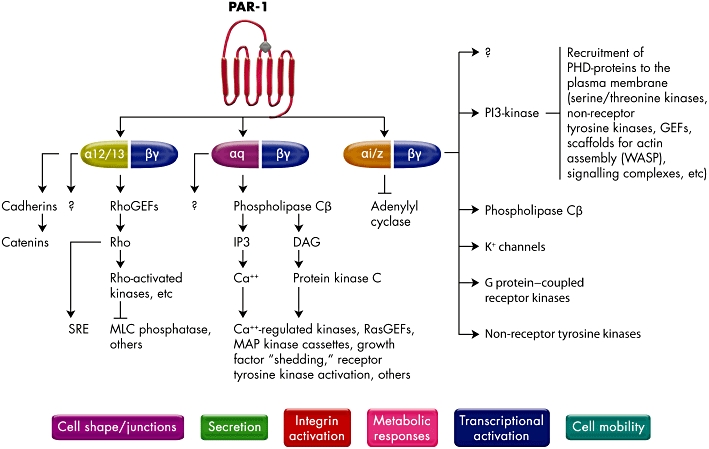
Pathways of platelet protease-activated receptor 1 (PAR-1) activation. Activated PAR-1 can signal through Gα12/13, Gαq and Gαi/z families. The α-subunits of G12 and 13 bind Rho GEFs and induce Rho-mediated cytoskeletal responses leading to changes in platelet shape. The Gαq binds phospholipase Cβ to generate IPF3, which promotes calcium mobilization and protein kinase C activation. This then activates pathways leading to granule secretion, as PAR-1-stimulated Gαq-coupled ADP release is especially important for thrombin-mediated platelet activation. The Gβγ subunits can activate PI3-kinase and other lipid-modifying enzymes, protein kinases and channels. The PI3-kinase modifies the inner leaflet of the plasma membrane to provide molecular docking sites. Activation of PAR-1 can also activate growth factor shedding and activation of receptor tyrosine kinases involved in cell growth and differentiation. GEF, guanine nucleotide exchange factors; IPF3, inositol triphosphate 3; PI3-kinase, phosphoinositide-3 kinase. Adapted from reference [16]
Currently, four PARs have been identified, and are differentially expressed by vascular cells (endothelial cells and vascular smooth muscle cells), platelets and other circulating calls. The current family of PARs in humans consists of PAR-1, -2, -3 and -4[31]. Of these, PAR-1, -3 and -4 are activated by thrombin, and only PAR-1 and -4 are expressed in human platelets. PAR-3 is unique in this family because it is not activated by synthetic peptides; its functional role has been demonstrated only in murine platelets, and its role in humans remains unclear [32, 33]. PAR-2 is activated by other proteases, such as trypsin and tryptase, and coagulation factors VIIa and Xa [30, 34].
The thrombin/PAR-mediated effects in endothelial and vascular smooth muscle cells are not fully established and sometimes controversial. In fact, in normal arteries thrombin can induce endothelium-dependent vasorelaxation [35], endothelium-dependent contraction [36] or direct smooth muscle contraction [37]. Furthermore, thrombin upregulates the expression of genes related to haemostasis (for example, tissue factor, plasminogen activator inhibitor-1) and genes related to other processes (such as cell growth, angiogenesis and cytokines). Moreover, early reports showed that thrombin acts as a potent mitogen for vascular smooth muscle cells [38].
Thrombin/PAR-mediated effects in platelets are better understood. Human platelets express PAR-1 and -4, and either of them can trigger platelet secretion and aggregation [31, 39]. PAR-1 acts as the principal thrombin receptor on human platelets and mediates platelet activation by low concentrations of thrombin (EC50 of 50 pm), whereas PAR-4 requires a higher concentration of thrombin (EC50 of 5000 pm). PAR-3 is a unique PAR that is not activated by synthetic peptides representing its tethered peptide ligand [32, 33]. PAR-1 seems to mediate platelet activation through a hirudin-like site that acts as a thrombin-binding site, and it is responsible for the cleavage of the receptor between arginine 41 and serine 42, removing the amino-terminal sequence and generating a new protonated amino group at the NH2-terminal of tethered ligand-S42FLLRN (serine-phenylalanine-leucine-leucine-argirine-asparagine) [39]. PAR-4 lacks the hirudine-like sequence and thereby requires the higher concentrations mentioned above [40, 41]. PAR-4 uses instead a negatively charged cluster of amino-acid residues to facilitate slow disassociation from the positively charged thrombin molecule [42], and could be responsible for the prolonged effects in human platelets [43].
Thrombin-mediated PAR-1 cleavage results in the activation of heteromeric G proteins of the Gα12/13 and Gαq families that interconnect several intracellular signalling pathways to various phenotypic effects of thrombin on platelets [21, 30] (Figure 2). These include TXA2 production [44], ADP release [45], serotonin and adrenaline release [46, 47], activation/mobilization of P-selectin and CD40 ligand [48, 49] and platelet aggregation [50]. Moreover, this subtype of PAR stimulates platelet procoagulant activity by leading to enhanced thrombin formation [24] and the generation of fibrin from fibrinogen [51]. Furthermore, Gi-mediated signalling is stimulated by thrombin-mediated release of platelet ADP, another key pathway in platelet-mediated processes [52] (Figure 2). PAR-4 uses the same G protein subtypes [47, 53]. There are other differences between both subtypes of PAR, such as activation of PAR-4 by cathesin G [54] and a counter-regulatory mechanism of both PAR-1 and PAR-4 in angiogenesis [55]. However, the importance of these differences is not fully understood. The current hypothesis is that PAR-4 functions as an auxiliary receptor, mediating thrombin signalling to distinct effectors or with different kinetics compared with PAR-1 [16]. Other authors have suggested that PAR-1 acts as a cofactor for thrombin activation of PAR-4 on human platelets. By inhibiting the ability of thrombin to associate with PAR-1, Leger et al. [56] showed that PAR-1 plays a critical function in assisting PAR-4 activation by thrombin, suggesting that PAR-1 and PAR-4 exist as a complex on human platelets.
Antagonism of PAR-1
In humans, PAR-1-mediated platelet activation is one the most important pathways in the development of pathological thrombosis; however, its role in protective haemostasis is not completely elucidated. The principal difference between normal and pathological haemostasis is the lack of propagation of the platelet-rich thrombus beyond the initial monolayer to become an occlusive clot [16]. PAR-4-deficient mice (PAR-4−/−) exhibit normal platelet numbers and morphology without anaemia or spontaneous bleeding, in contrast to the severe bleeding phenotype exhibited by mice deficient in fibrinogen [57, 58]. In the PAR-4−/− mice, the initial platelet accumulation at the site of injury does not differ from that in wild-type mice, and this early thrombus was proposed to support thrombin generation in sufficient amounts and allow normal fibrin deposition observed in this model [59]. In contrast, other authors have reported PAR-4−/− mice to be protected against arterial thrombosis but with markedly prolonged bleeding times, suggesting that platelet activation via thrombin is required for normal haemostasis [54].
Inhibition of PAR-1 using the PAR-1 antagonist FR171113 has been shown to reduce arterial thrombosis by reducing thrombin-induced platelet aggregation without prolonging bleeding times or influencing coagulation parameters in a guinea-pig model [60]. The use of FR171113 to inhibit PAR-1 did not inhibit ADP-induced or collagen-induced platelet aggregation, suggesting that PAR-1 antagonism does not affect other platelet signalling pathways. In the same way, the use of RWJ-58259 (PAR-1 antagonist) or antibodies to PAR-1 in primate models reduced vascular injury-induced thrombosis while sparing platelet-dependent coagulation functions, such as the activated clotting time, activated partial thromboplastin time and prothrombin times [16]. Importantly, thrombin signalling through PAR-4 remained intact in these studies, indicating a primary role of PAR-1 in mediating the prothrombotic effects of thrombin in primates. Although extrapolation of these results to humans is highly speculative, they suggest that inhibition of PAR-1 alone may be sufficient for the prevention or treatment of thrombotic events in humans. However, it cannot be excluded that preservation of thrombin signalling through platelet PAR isoforms other than PAR-1 can participate in the maintenance of normal haemostasis. These preclinical findings are also consistent with clinical observations showing substantial bleeding risk in patients treated with anticoagulants that interfere with the catalytic function of thrombin [61].
Preclinical studies
Preclinical studies have been performed with different molecules: SCH530348, SCH205831, SCH602539 and E5555. Currently, more advanced investigation is being undertaken with vorapaxar (SCH530348), currently being tested in two phase III clinical studies [62, 63], and atopaxar (E5555), which has just completed phase II testing [64–66].
Initial studies were performed in baboons and cynomolgus monkeys using the PAR-1 inhibitors SCH205831 and SCH602539, which are analogues of SCH530348. Chintala et al. [67], using a cynomolgus monkey model, showed that oral administration of SCH205831caused a complete and dose-dependent inhibition of thrombin/PAR-induced platelet aggregation until 6 h after dosing. This PAR-1 inhibitor administered orally at a dose of 10 mg kg−1 was able to inhibit platelet deposition by 50–70% onto uncoated stents in an arteriovenous shunt model of thrombosis in baboons [67]. Bleeding times, coagulation parameters and ADP-induced platelet aggregation were unchanged by the use of this drug in this model. The same group showed the efficacy of another selective and orally active PAR-1 inhibitor, SCH602539, in a dose-dependent manner using the Folts model of thrombosis in anaesthetized cynomolgus monkeys [68].
Vorapaxar (SCH530348)
Following the studies of the above-mentioned analogues, SCH530348, currently named vorapaxar, was selected for further clinical development. Vorapaxar was originally developed by Schering Plough, which has now been acquired by Merck pharmaceuticals. This PAR-1 inhibitor was shown to completely inhibit thrombin receptor agonist peptide (TRAP)-induced platelet aggregation with oral administration at doses ≥0.1 mg kg−1 for 24 h, in animal models (cynomolgus monkeys). In an animal model, vorapaxar showed a bioavailability of 86% [69]. In human platelet-rich plasma, vorapaxar inhibited thrombin and TRAP-induced platelet aggregation with an IC50 of 47 and 25 nm, respectively, without affecting aggregation induced by other reagents, such as ADP, TXA2 mimetic U46619 or collagen. However, and more importantly, vorapaxar did not affect prothrombin time or activated partial thromboplastin time, suggesting that the potential for bleeding events may not be increased [69]. These results are in agreement with the observations derived from cynomolgus monkeys that treatment with vorapaxar either alone or in combination with aspirin plus clopidogrel did not increase bleeding time or surgical blood loss compared with placebo [70]. On the basis of these promising results, vorapaxar was advanced into clinical development.
Pharmacological aspects
Vorapaxar is a synthetic tricyclic 3-phenylpyridine analogue of himbacine, a natural product that has been modified as a crystalline salt for drug development and clinical use (Figure 3) [71, 72]. Vorapaxar is a nonprotein, small-molecule, high-affinity, orally active, competitive PAR-1 inhibitor [71, 72]. It is a potent competitive antagonist of the platelet thrombin receptor PAR-1 (Ki of 2.7 nmol l−1), blocking thrombin-mediated platelet activation without interfering with cleavage of fibrinogen, the final step of coagulation, which is also provided by thrombin. In kinetic studies, this molecule showed a long receptor dissociation half-life of approximately 20 h. It is rapidly absorbed, presenting a slow elimination with a calculated half-life up to 311 h. The routes of elimination are mainly faeces and secondarily by renal clearance (less than 5%) [29]. Exposure to vorapaxar is dose related, with an interindividual variability in the range of 20–40% and without any significant difference in exposure among sex and race in fasting conditions [62]. The co-administration of food does not affect the overall drug exposure; however, time after administration when the maximum plasma concentration is reached (Tmax) and maximum plasma concentration (Cmax) are slightly delayed, and therefore, maximal platelet inhibiton is delayed by approximately 2 h. Vorapaxar is slowly but extensively metabolized by cytochrome P450 (CYP) 3A4. Therefore co-administration of drugs that modify the metabolic activity of CYP3A4 could potentially modulate vorapaxar exposure to platelets. Some evidence has been reported for ketoconazole, a potent CYP3A4 inhibitor, and rifampin, a potent CYP3A4 inducer, which may, respectively, increase or reduce, after a period of co-administration longer than 3 weeks, drug exposure for about the half of the theorical value (Table 1) [62, 63, 73].
Figure 3.
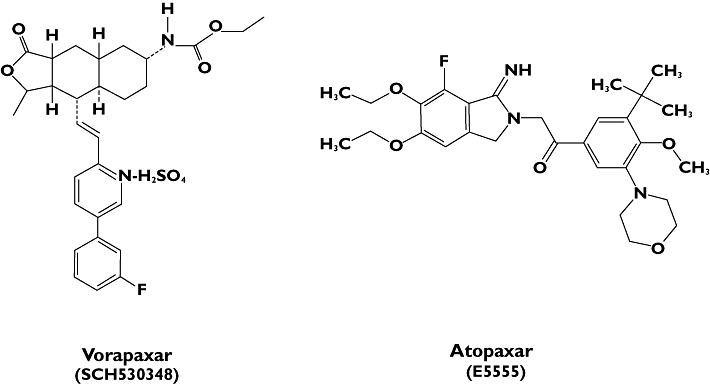
Chemical structure of vorapaxar (SCH530348) and atopaxar (E5555)
Table 1.
Characteristics of vorapaxar (SCH530348) and atopaxar (E5555)
| Vorapaxar | Atopaxar | |
|---|---|---|
| Formulation | Oral | Oral |
| Mechanism of action | Competitive and reversible | Competitive and reversible |
| Onset | ∼2 h | ∼3.5 h |
| Half-life | 311 h | 23 h |
| IC50 to inhibit thrombin-induced platelet aggregation | 47 nm | 64 nm |
| Main metabolic activation | CYP3A4 | CYP3A4 |
| Major route of elimination | Faeces | Faeces |
| Development status | Phase III | Phase II |
Adapted from reference [29].
Phase I studies
The first phase I study using vorapaxar was conducted in 50 healthy volunteers taking a single oral 5–40 mg dose [74]. This regimen was able to produce >90% mean inhibition of TRAP-induced platelet aggregation in all subjects for >72 h, with low interindividual variability in plasma concentration. In this study, vorapaxar was characterized by rapid absorption and slow elimination, with a half-life of >72 h.
Another phase I study demonstrated that a 40 mg single dose of vorapaxar was safe and well tolerated in healthy Caucasian and Japanese subjects [75, 76]. In both populations, 80% or more inhibition of TRAP-induced platelet aggregation was achieved rapidly and consistently with this drug regimen. In a separate, recently reported phase I study in healthy subjects, the dosing regimen of vorapaxar that most consistently provided the targeted level of TRAP-induced platelet inhibition throughout the entire 28 day treatment period was a 40 mg loading dose followed by a 2.5 mg once-daily maintenance dose [77].
Phase II studies
The safety profile of vorapaxar was evaluated in a multicentre, large, randomized, double-blind, placebo-controlled, dose-ranging clinical trial, the Thrombin Receptor Antagonist for Cardiovascular Event Reduction in Percutaneous Coronary Intervention (TRA-PCI) trial, which enrolled 1030 patients in 76 centres [78]. This trial evaluated the safety and efficacy of vorapaxar used in combination with standard oral antiplatelet therapy (aspirin and clopidogrel) and an antithrombotic agent (heparin or bivalirudin) over a treatment duration of 60 days in patients undergoing non-urgent PCI or coronary angiography with planned PCI. The study design was a 3:1 model randomization to receive placebo or vorapaxar on top of standard-of-care antithrombotic therapy, including aspirin, clopidogrel and an anticoagulant of choice. Patients assigned to vorapaxar received one of three single loading doses (10, 20 or 40 mg) before angiography and a maintenance therapy with one of three doses (0.5, 1.0 or 2.0 mg daily) for 2 months in the 56% of patients who underwent PCI. The primary end-point was the incidence of the composite of thrombolysis in myocardial infarction (TIMI) major or minor bleeding in the PCI cohort. No differences were observed in terms of primary end-point between vorapaxar (with all loading and maintenance doses) and placebo groups. No differences in the incidence of TIMI major and minor bleeding were also observed, although a trend towards increased bleeding rates with higher doses was observed (Tables 2 and 3). Interestingly, although the study was not powered to evaluate efficacy end-points, a strong trend was observed among patients treated with vorapaxar towards a lower rate of the composite end-point of death and major adverse cardiac events, including myocardial infarction (placebo 9% vs. vorapaxar 6%; absolute difference −2.9, 95% confidence interval −7.9 to 2.1). Vorapaxar also demonstrated a dose-dependent inhibition of TRAP-induced platelet aggregation, inhibiting almost 30% of patients as early as 0.5 h with a 40 mg loading dose. This effect was substained over 60 days with each maintenance dose [79].
Table 2.
The results of the Thrombin Receptor Antagonist for Cardiovascular Event Reduction in Percutaneous Coronary Intervention (TRA-PCI) trial by loading dose
| Safety and efficiency outcomes | Vorapaxar by loading dose | |||||
|---|---|---|---|---|---|---|
| Placebo | All doses | 10 mg | 20 mg | 40 mg | ||
| PCI | Number of patients | 151 | 422 | 129 | 120 | 173 |
| TIMI major/minor bleeding | 5 (3.3%) | 12 (2.8%) | 2 (1.6%) | 3 (2.5%) | 7 (4.0%) | |
| TIMI major bleeding | 2 (1.3%) | 3 (0.7%) | 2 (1.6%) | 0 | 1 (0.6%) | |
| TIMI minor bleeding | 3 (2.0%) | 9 (2.1%) | 0 | 3 (2.5%) | 6 (3.5%) | |
| Death/MACE | 13 (8.6%) | 24 (5.7%) | 10 (7.8%) | 6 (5.0%) | 8 (4.6%) | |
| Myocardial infarction | 11 (7.3%) | 18 (4.3%) | 7 (5.4%) | 5 (4.2%) | 6 (3.5%) | |
| CABG | Number of patients | 24 | 52 | 10 | 18 | 24 |
| TIMI major / minor bleeding | 19 (79%) | 47 (90%) | 9 (90%) | 17 (94%) | 21 (88%) | |
| TIMI major bleeding | 9 (38%) | 33 (63%) | 8 (80%) | 12 (67%) | 13 (54%) | |
| TIMI minor bleeding | 10 (42%) | 15 (29%) | 1 (10%) | 5 (28%) | 9 (38%) | |
| Chest tube drainage (ml)* | 996 (516–1724) | 988 (690–1750) | 1393 (975–1750) | 1015 (690–1670) | 870 (414–1790) | |
| PRBC transfusion (>2 units) | 5 (21%) | 9 (17%) | 2 (20%) | 2 (11%) | 5 (21%) | |
Median (interquartile). Abbreviations: CABG, coronary artery bypass graft; MACE, major adverse cardiac event; PCI, percutaneous coronary intervention; PRBC, packed red blood cells; TIMI, thrombolysis in myocardial infarction. Adapted from reference [79].
Table 3.
Rate of bleeding events in the Thrombin Receptor Antagonist for Cardiovascular Event Reduction in Percutaneous Coronary Intervention (TRA-PCI) trial by maintenance dose
| Vorapaxar by maintenance dose | ||||
|---|---|---|---|---|
| Placebo | 0.5 mg | 1.0 mg | 2.5 mg | |
| Number of patients | 63 | 136 | 139 | 138 |
| TIMI major/minor bleeding | 1 (2%) | 3 (2%) | 5 (4%) | 4 (3%) |
| TIMI major bleeding | 0 | 0 | 2 (1%) | 1 (<1%) |
| TIMI minor bleeding | 1 (2%) | 3 (2%) | 3 (2%) | 3 (2%) |
Abbreviation: TIMI, thrombolysis in myocardial infarction. Adapted from reference [79].
These results were confirmed in two small randomized trials conducted in Japanese patients: one performed in non-ST elevation acute coronary syndrome (NSTE-ACS) patients undergoing PCI (n = 117) [80], and the other among patients with previous ischaemic stroke (n = 90) [81]. Both studies showed a good safety profile, in terms of no increase in TIMI major and minor bleeding using different vorapaxar dosages (20 or 40 mg loading dose, followed by 1 or 2.5 mg daily maintenance dose in NSTE-ACS patients, and 1 or 2.5 mg daily maintenance dose for 60 days in patients with ischaemic stroke). In the first study, conducted in NSTE-ACS patients, treatment with vorapaxar was also associated with a significant reduction of periprocedural myocardial infarction [80].
Phase III studies
The favourable results derived from phase II testing led to the design of the following two large phase III clinical trials, which are currently ongoing: the Thrombin Receptor Antagonist for Clinical Event Reduction in ACS (TRACER) [62] and Thrombin Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events (TRA 2P-TIMI 50) [63] trials. These trials have been designed and powered to assess the efficacy of vorapaxar to reduce recurrent ischaemic events. However, these trials will be very important to assess the safety of vorapaxar, in particular its impact on bleeding. In fact, while phase II testing provided reassuring safety information, which allowed expansion into phase III testing, these studies are too limited in sample size to fully determine the impact of this drug on bleeding complications, which remain a major concern in the current era of antithrombotic agents. In fact, it is important to underscore the emerging data concerning the prognostic implications of bleeding, including effects on mortality, which emphasize the importance of the balance between safety and efficacy of antithrombotic agents [82].
The TRACER trial [62] is a prospective, randomized, double-blind, multicentre trial designed to assess whether the use of vorapaxar added to standard-of-care antiplatelet treatment (aspirin plus clopidogrel) will reduce the incidence of a composite end-point consisting of cardiovascular death, myocardial infarction (MI), stroke, recurrent ischaemia with rehospitalization and urgent coronary revascularization in patients admitted with non-ST elevation ACS. This study is planned to enrol approximately 10 000 patients, who will be randomized to vorapaxar (40 mg loading dose and 2.5 mg daily maintaining dose) for at least 1 year vs. placebo. This study is also designed to assess the safety of the use of vorapaxar, defined as incidence of moderate and severe bleeding events according to the Global Use of Strategies to Open Coronary Arteries (GUSTO) classification, and clinically significant bleeding, defined as TIMI major or TIMI minor bleeding, or bleeding that requires unplanned medical treatment, surgical treatment or laboratory evaluation even if not meeting the criteria for TIMI major or TIMI minor bleeding [62].
The TRA 2P-TIMI 50 trial [63] is a prospective, randomized, double-blind, multinational trial randomizing patients to vorapaxar administered only as a 2.5 mg daily maintenance dose vs. placebo in addition to current standard-of-care. This study is being conducted in patients with history/evidence of atherosclerosis involving the coronary, cerebral or peripheral vascular systems (secondary prevention). The study has completed enrollment of approximately 27 000 patients. The primary end-point is the composite of the first occurrence of any component of the composite of cardiovascular death, MI, stroke and urgent coronary revascularization, during 1 year of follow-up. Safety end-points, in particular bleeding, are also being evaluated [63].
Atopaxar (E5555)
Atopaxar, developed by Eisai pharmaceuticals, is the other PAR-1 antagonist which has completed phase II clinical development. Preclinical studies of atopaxar showed antithrombotic effects in animal models (guinea-pigs) [83]. Furthermore, atopaxar was shown to have inhibitory effects on neointimal hyperplasia in a rat balloon injury model [79] and the release of the inflammatory markers soluble CD40 ligand (sCD40L) and interleukin-6 and expression of P-selectin [83]. Moreover, this compound was also able to show inhibitory effects on thrombin-induced contractile response (spam) in a rabbit subarachnoid haemorrhage model [83, 84]. In healthy human volunteers, atopaxar was shown to inhibit thrombin- and TRAP-mediated CD40L release (IC50 47 and 38 nm, respectively) without affecting ADP-induced CD40L release [84].
Pharmacological aspects
Atopaxar is an orally active and potent PAR-1 antagonist that specifically targets the G protein-coupled receptor and binds to PAR-1 at the tethered ligand binding site on platelet membranes [29]. It is a small molecule, chemically identified as 1-(3-tert-butyl-4-methoxy-5-morpholinophenyl)-2-(5,6-diethoxy-7-fluoro-1-imino-1,3-dihydro-2H-isoindol-2yl) ethanone hydrobromide, with a molecular weight of 608.54 Da (Figure 3). The drug inhibits the binding of high-affinity [3H]-TRAP, presumably at or near the tethered ligand-binding epitope site [85]. Atopaxar has a slower onset of its effects (3.5 h) and lower half-life (23 h) in compared with vorapaxar. Like vorapaxar, atopaxar is mainly metabolized by CYP3A4, and its major route of elimination is faeces (Table 1) [29].
Phase I studies
The effects of escalating concentrations of atopaxar (20, 50 and 100 ng ml−1) on platelet function were evaluated in vitro in healthy volunteers and coronary artery disease (CAD) patients with or without clopidogrel, by Serebruany et al. [86]. Atopaxar achieves almost completeinhibition of TRAP-induced platelet aggregation, with a very minor impact on ADP- or collagen-induced aggregation (10–15% inhibition of 5 µm ml−1 ADP- or 1 µm ml−1 collagen-induced platelet aggregation in plasma). Platelet inhibition was present at a concentration of 20 ng ml−1 and was not dose dependent, and a further increase of inhibition was not observed after incubation with 50 and 100 ng ml−1. However, these effects were not shown in whole blood. Moreover, platelet biomarkers were also significantly reduced.
The pharmacodynamics and safety of oral administration were evaluated in two studies on healthy volunteers. Marion et al. [87] assessed the effects of ascending oral doses of 50, 100 and 200 mg in a randomized, double-blind and placebo-controlled manner. Doses of 100 and 200 mg achieved a similar maximum plasma concentration and area under the curve, with 91 and 95% platelet inhibition, respectively. Takeuchi et al. [88], in a similar way, found an 80% or more thrombin-induced platelet inhibition with a single dose of 50 mg orally. Repeated administration of 100 and 200 mg was associated with almost 100% platelet inhibition. There were no severe adverse events reported in either study.
Phase II studies
The results of phase II studies have been reported recently. The J-LANCELOT (Japanese-lesson from antagonizing the cellular effect of thrombin) trial [64] comprises two double-blind, placebo-controlled phase II studies conducted in ACS patients and in high-risk CAD patients. The ACS study randomized 241 patients with NSTE-ACS into four groups (placebo, 50, 100 and 200 mg atopaxar), receiving the drug daily for 12 weeks. Patients randomized to any dose of atopaxar also received a 400 mg loading dose (vs. a placebo loading dose). The CAD study randomized 263 patients into the same groups as in the ACS study, but without giving a loading dose and with a 24 week follow-up. The primary end-point was the incidence of bleeding events, classified according to the Clopidogrel in Unstable Angina to Prevent Recurrent Ischemic Events (CURE) (major and minor bleeding) and TIMI (major, minor and minimal bleeding) definitions. The secondary end-point was the incidence of major cardiovascular adverse events (MACEs), such as cardiovascular death, MI, stroke or recurrent ischaemia. In addition, TRAP-induced aggregation was performed using light transmittance aggregometry following 15 µm TRAP stimuli. Briefly, there was a low incidence of CURE bleeding in both groups of patients, without achieving any statistical significance. The entire population showed the same tendency when the events were classified according to the TIMI definition. However, there was a numerical increase in any TIMI bleeding with the dose of 200 mg (ACS study, 16.4% placebo vs. 23.0% atopaxar, P = 0.398; and CAD study, 4.5% placebo vs. 13.2% atopaxar, P = 0.081). No significant dose-dependent difference was observed in ACS patients for the incidence of TIMI bleeding (P = 0.266) and no significant difference was observed between placebo and all combined groups (P = 0.609). However, a dose-dependent trend was observed in the CAD study (P = 0.086), without significant differences between placebo and treated groups (P = 0.219). Interestingly, no patient experienced TIMI major bleeding in any group. Regarding the secondary end-point, there was a numerical reduction in MACEs among patients treated with 100 and 200 mg atopaxar. However, MACE rates in both treatment and placebo groups was not significantly different (5.0% placebo vs. 6.6% atopaxar, relative risk 0.76, 95% confidence interval 0.257–2.606, P = 0.73 in the ACS study; and 4.5% placebo vs. 1.0% atopaxar, relative risk 0.22, 95% confidence interval 0.041–1.109 in the CAD study). The most frequent MACE was recurrent ischaemia, without any cardiovascular death in both groups. The pharmacodynamic assessment of both studies (ACS, 42 patients and CAD, 80 patients) showed 20–50% platelet inhibition in the ACS study and almost 50% inhibition in the CAD study with a 50 mg daily maintenance dose of atopaxar. Using 100 or 200 mg, more than 90% inhibition was observed in both studies. In the ACS study, the mean platelet aggregation inhibition reached >80% at 3–6 h after administration of the 400 mg loading dose of atopaxar. These results are in agreement with previous phase I studies. An interesting finding of these studies was the more frequent treatment-related adverse events in patients treated with atopaxar than in patients receiving placebo (44.4 vs. 27.9%, respectively, in ACS patients, P = 0.024; 32.0 vs. 13.6%, respectively, in CAD patients, P = 0.003). In both studies, the most common adverse event related to the study drug was abnormal liver function, with a rate of around 12–15% in patients treated with 200 mg of the study drug in both studies. It has been suggested that liver function abnormalities may be attributed to concomitant thienopyridine use. There were also dose-related effects on QT corrected interval (QTc) noted with atopaxar, showing an interval prolongation with increasing doses.
Most recently, the LANCELOT ACS and CAD studies, conducted outside Japan, have been presented [65, 66]. These studies had the same design and primary end-point as J-LANCELOT. The LANCELOT ACS and CAD trials enrolled 603 and 720 patients, respectively. There was no statistical difference in the primary end-point between placebo and atopaxar in both studies. There was no difference in any TIMI bleeding event in LANCELOT ACS (placebo 10.1%, atopaxar total 9.3%, relative risk 0.91, P = 0.77). There was a trend to more TIMI bleeding (placebo 6.8%, atopaxar total 10.3%, relative risk 1.52, P = 0.17) and CURE minor bleeding events (placebo 0.6% vs. atopaxar 3.0%, relative risk 5.2, P = 0.08) in LANCELOT CAD. In LANCELOT ACS, patients receiving 100 mg of atopaxar showed a nonstatistical increase of bleeding events (both CURE and TIMI). No differences in the primary efficacy end-point were observed (7.8% in placebo vs. 8.0% in atopaxar group; P = 0.93); however, patients under atopaxar treatment showed less incidence of Holter-ECG-detected ischaemia (28.1% in placebo vs. 18.7% in atopaxar group; P = 0.02) among ACS patients. Moreover, in LANCELOT CAD, patients treated with atopaxar showed a numerically, but not statistically lower incidence of events, which did not appear to be dose related (placebo 4.6%, atopaxar total 2.6%, relative risk 0.57, P = 0.20). With regard to adverse events, the presence of liver dysfunction was observed only in 5.47% of patients on 200 mg of atopaxar among ACS patients. However, only three of these patients discontinued the drug. Overall, prolongation of QTc was seen in all study arms from randomization to end of treatment, being more exacerbated with doses of 100 and 200 mg, without any case of syncope or malignant arrhythmias. Regarding CAD patients, this transient elevation in liver transaminases and dose-dependent QTc prolongation without apparent complications were also observed in higher dose atopaxar treatment groups. Pharmacodynamic results of LANCELOT (n = 39 ACS and 80 CAD patients) corroborate the J-LANCELOT findings. Atopaxar achieved up to 92% of TRAP-induced platelet aggregation inhibition at 3–6 h after the loading dose, and 92% with a 200 mg maintenance dose at the 8th week of treatment in the ACS population, or 99% with the same dose at 12 weeks in the CAD population.
Plans for phase III development with atopaxar are not fully determined at the present time.
Perspectives and novel approaches with PAR antagonism
Although vorapaxar and atopaxar have similar pharmacological actions, there are differences between these two compounds. In particular, vorapaxar has a longer half-life than atopaxar. This property makes atopaxar more suitable for patients who may need rapid suspension of drug effects (e.g. in case of surgery). However, phase II testing suggests that atopaxar is associated with more liver dysfunction. Overall, although both drugs showed an increase in bleeding with higher doses, they demonstrated a safety bleeding risk profile when used in combination with current standard therapy (aspirin and a thienopyridine). Importantly, inhibition of the PAR-1 does not alter normal homeostasis, which is key in protecting against blood loss. This is in contrast to P2Y12 inhibition strategies, which can modulate thrombin generation profiles [89] and are associated with increased bleeding events when more potent strategies are used [11, 12, 90]. Overall, the pivotal role of thrombin-mediated platelet activation in atherothrombotic processes and the properties of PAR-1 antagonists have also led to suggestions regarding the potential for these agents to be considered not only as an ‘add-on’ therapy but also as an ‘alternative’ therapy to current standard-of-care. Indeed, future investigations will better elucidate these therapeutic possibilities.
In addition to the established antithrombotic effects of PAR-1 antagonists, other properties appear to be of potential interest. Among these, their effects on smooth muscle cell contraction are of particular interest [91]. In an animal model, Bocquet et al. [92] demonstrated that a new PAR-1 antagonist, F121958, is a potent antivasocontrictor, due to its properties in smooth muscle cells, among lesioned vessels. Moreover, this PAR-1 inhibitor did not have haemodynamic effects in normal vasculature. This shows the beneficial effect of PAR-1 inhibitors to prevent vasospasm caused by arterial injury and mediated by thrombin, in addition to their antithrombotic properties. Another interesting property is the synergistic effect on inhibition of different activation pathways. Chintala et al. [93] recently reported that the combined inhibition, in an animal model, with SCH602639 (PAR-1 antagonism) and cangrelor (P2Y12 inhibitor) was associated with synergistic antithrombotic effects in comparison with vehicle for all combinations tested in a Folts model. Other potential targets associated with PAR-1 antagonism are currently under investigation.
Conclusion
Platelets have an important role in normal haemostatic processes, but they also mediate pathological thrombosis. Multiple pathways contribute to platelet activation. Among these, thrombin-mediated platelet activation through PAR-1 signalling has a key role. Inhibition of the PAR-1 receptor is the target for novel promising antiplatelet drugs. Vorapaxar (SCH530348) and atopaxar (E5555) are PAR-1 antagonists which have completed phase II testing, showing overall favourable safety profiles and encouraging clinical outcomes (Table 4). Larger scale clinical trials will better define the efficacy and safety of PAR-1 antagonists in addition to standard-of-care in patients with atherothrombotic disease manifestations.
Table 4.
Clinical trials with vorapaxar and atopaxar
| Drug | Trial/study author | Phase | Type | Patients | n | Dose of drug | Primary end-point | Follow-up | Status | Major finding |
|---|---|---|---|---|---|---|---|---|---|---|
| Vorapaxar | TRA-PCI [75] | II | Randomized, multicentre, double-blind, placebo controlled | Elective PCI | 1030 | 10–20–40 mg (LD) 0.5–1–2.5 mg (MD) | TIMI major and minor bleeding | 60 days | Completed | No difference in primary end-point |
| Goto et al. [77] | II | Randomized, single-centre, placebo controlled | NSTEMI | 117 | 20–40 mg (LD) 1–2.5 mg (MD) | TIMI major and minor bleeding | 60 days | Completed | No difference in primary end-point; less MI in treatment arm | |
| Shinohara et al. [78] | II | Randomized, single-center placebo controlled | Previous stroke | 90 | 1–2.5 mg (MD) | TIMI major and minor bleeding | 60 days | Completed | No difference in primary end-point | |
| TRA-CER [54] | III | Randomized, multicentre, double-blind, placebo controlled | ACS | 10 000 | 40 mg (LD) 2.5 mg (MD) | Death, stroke, MI and TLR | 1 year | Recruiting | – | |
| TRA 2P-TIMI 50 [60] | III | Randomized, multicentre, double-blind, placebo controlled | History of atherosclerosis | 26 000 | 2.5 mg (MD) | Death, stroke, MI and revascularization | Anticipated average duration of 24 months (at least 1 year) | Active, not recruiting | – | |
| Atopaxar | J-LANCELOT [61] | II | Two randomized, single-centre, placebo controlled | ACS CAD | 241 263 | 400 mg (LD);50–100–200 mg (MD); 50–100–200 mg (MD) | TIMI and CURE major and minor bleeding | 12 weeks 24 weeks | Completed | No difference in primary end-point, although a higher rate of any TIMI bleeding with higher doses; most common adverse event abnormal liver function |
| LANCELOT-ACS [62] | II | Randomized, multicentre, double-blind, placebo controlled | ACS | 603 | 400 mg (LD);50–100–200 mg (MD) | TIMI major and minor bleeding | 12 weeks | Completed | No difference in primary end-point; transient elevation of liver enzymes and QTcF prolongation with higher doses | |
| LANCELOT-CAD [63] | II | Randomized, multicentre, double-blind, placebo controlled | CAD | 720 | 50–100–200 mg (MD) | TIMI major and minor bleeding | 24 weeks | Completed | No difference in primary end-point, although a higher rate of any TIMI bleeding with higher doses; transient elevation of liver enzymes and QTcF prolongation with higher doses |
Abbreviations: ACS, acute coronary syndrome; CAD, coronary artery disease; LD, loading dose; MD, maintenance dose; MI, myocardial infarction; NSTEMI, non-ST elevation myocardial infarction; PCI, percutaneous coronary intervention; TIMI, thrombolisis in myocardial infarction; TLR, target lession revascularization; QTcF, corrected QT interval (Framingham); CURE, The clopidogrel inunstable angia to prevent recurrent events.
Competing Interests
Dominick J. Angiolillo reports receiving the following: honoraria for lectures from Bristol Myers Squibb, Sanofi-Aventis, Eli Lilly Co. and Daiichi Sankyo, Inc.; consulting fees from Bristol Myers Squibb, Sanofi-Aventis, Eli Lilly Co., Daiichi Sankyo, Inc., The Medicines Company, Portola, Novartis, Medicure, Accumetrics, Arena Pharmaceuticals, Astra Zeneca and Merck; and research grants from GlaxoSmithKline, Otsuka, Eli Lilly Co., Daiichi Sankyo, Inc., The Medicines Company, Portola, Accumetrics, Schering-Plough, Astra-Zeneca and Eisai.
The other authors have no competing interests to declare.
REFERENCES
- 1.Anderson JL, Adams CD, Antman EM, Bridges CR, Califf RM, Casey DE, Jr, Chavey WE, 2nd, Fesmire FM, Hochman JS, Levin TN, Lincoff AM, Peterson ED, Theroux P, Wenger NK, Wright RS, Smith SC, Jr, Jacobs AK, Halperin JL, Hunt SA, Krumholz HM, Kushner FG, Lytle BW, Nishimura R, Ornato JP, Page RL, Riegel B, American College of Cardiology, American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non ST-Elevation Myocardial Infarction), American College of Emergency Physicians, Society for Cardiovascular Angiography and Interventions, Society of Thoracic Surgeons, American Association of Cardiovascular and Pulmonary Rehabilitation, Society for Academic Emergency Medicine ACC/AHA 2007 guidelines for the management of patients with unstable angina/non ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non ST-Elevation Myocardial Infarction): developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons: endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. Circulation. 2007;116:e148–304. doi: 10.1161/CIRCULATIONAHA.107.181940. [DOI] [PubMed] [Google Scholar]
- 2.Antithrombotic Trialists' Collaboration. Collaborative meta-analysis of randomized trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. Br Med J. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis HD, Jr, Davis JW, Archibald DG, Steinke WE, Smitherman TC, Doherty JE, 3rd, Schnaper HW, LeWinter MM, Linares E, Pouget JM, Sabharwal SC, Chesler E, DeMots H. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina. Results of a Veterans Administration Cooperative Study. N Engl J Med. 1983;309:396–403. doi: 10.1056/NEJM198308183090703. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz L, Bourassa MG, Lesperance J, Aldridge HE, Kazim F, Salvatori VA, Henderson M, Bonan R, David PR. Aspirin and dipyridamole in the prevention of restenosis after percutaneous transluminal coronary angioplasty. N Engl J Med. 1988;318:1714–19. doi: 10.1056/NEJM198806303182603. [DOI] [PubMed] [Google Scholar]
- 5.Antiplatelet Trialists' Collaboration. Collaborative overview of randomised trials of antiplatelet therapy. I: prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. Br Med J. 1994;308:81–106. [PMC free article] [PubMed] [Google Scholar]
- 6.CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE) Lancet. 1996;348:1329–39. doi: 10.1016/s0140-6736(96)09457-3. [DOI] [PubMed] [Google Scholar]
- 7.Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345:494–502. doi: 10.1056/NEJMoa010746. [DOI] [PubMed] [Google Scholar]
- 8.Steinhubl SR, Berger PB, Mann JT, 3rd, Fry ET, DeLago A, Wilmer C, Topol EJ. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. J Am Med Assoc. 2002;288:2411–20. doi: 10.1001/jama.288.19.2411. [DOI] [PubMed] [Google Scholar]
- 9.Sabatine MS, Cannon CP, Gibson CM, Lopez-Sendon JL, Montalescot G, Theroux P, Claeys MJ, Cools F, Hill KA, Skene AM, McCabe CH, Braunwald E. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med. 2005;352:1179–89. doi: 10.1056/NEJMoa050522. [DOI] [PubMed] [Google Scholar]
- 10.Chen ZM, Jiang LX, Chen YP, Xie JX, Pan HC, Peto R, Collins R, Liu LS. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomized placebo-controlled trial. Lancet. 2005;366:1607–21. doi: 10.1016/S0140-6736(05)67660-X. [DOI] [PubMed] [Google Scholar]
- 11.Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neumann FJ, Ardissino D, De Servi S, Murphy SA, Riesmeyer J, Weerakkody G, Gibson CM, Antman EM, TRITON-TIMI 38 Investigators Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–15. doi: 10.1056/NEJMoa0706482. [DOI] [PubMed] [Google Scholar]
- 12.Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, Horrow J, Husted S, James S, Katus H, Mahaffey KW, Scirica BM, Skene A, Steg PG, Storey RF, Harrington RA, PLATO Investigators. Freij A, Thorsén M. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–57. doi: 10.1056/NEJMoa0904327. [DOI] [PubMed] [Google Scholar]
- 13.Angiolillo DJ, Guzman LA, Bass TA. Current antiplatelet therapies: benefits and limitations. Am Heart J. 2008;156:S3–S9. doi: 10.1016/j.ahj.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, Alfonso F, Macaya C, Bass TA, Costa MA. Variability in individual responsiveness to clopidogrel: clinical implications, management, and future perspectives. J Am Coll Cardiol. 2007;49:1505–16. doi: 10.1016/j.jacc.2006.11.044. [DOI] [PubMed] [Google Scholar]
- 15.Krasopoulos G, Brister SJ, Beattie WS, Buchanan MR. Aspirin ‘resistance’ and risk of cardiovascular morbidity: systematic review and meta-analysis. Br Med J. 2008;336:195–8. doi: 10.1136/bmj.39430.529549.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Angiolillo DJ, Capodanno D, Goto S. Platelet thrombin receptor antagonism and atherothrombosis. Eur Heart J. 2010;31:17–28. doi: 10.1093/eurheartj/ehp504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J. 2010;74:597–607. doi: 10.1253/circj.cj-09-0982. [DOI] [PubMed] [Google Scholar]
- 18.Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;13:2482–94. doi: 10.1056/NEJMra071014. [DOI] [PubMed] [Google Scholar]
- 19.Storey RF, Judge HM, Wilcox RG, Heptinstall S. Inhibition of ADP-induced Pselectin expression and platelet-leukocyte conjugate formation by clopidogrel and the P2Y12 receptor antagonist AR-C69931MX but not aspirin. Thromb Haemost. 2002;88:488–94. [PubMed] [Google Scholar]
- 20.Foster CJ, Prosser DM, Agans JM, Zhai Y, Smith MD, Lachowicz JE, Zhang FL, Gustafson E, Monsma FJ, Jr, Wiekowski MT, Abbondanzo SJ, Cook DN, Bayne ML, Lira SA, Chintala MS. Molecular identification and characterization of the platelet ADP receptor targeted by thienopyridine antithrombotic drugs. J Clin Invest. 2001;107:1591–98. doi: 10.1172/JCI12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, Luger TA, Hollenberg MD. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev. 2005;26:1–43. doi: 10.1210/er.2003-0025. [DOI] [PubMed] [Google Scholar]
- 22.Andre P, Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, Phillips DR, Wagner DD. CD40L stabilizes arterial thrombi by a beta3 integrin-dependent mechanism. Nat Med. 2002;8:247–52. doi: 10.1038/nm0302-247. [DOI] [PubMed] [Google Scholar]
- 23.Hagihara M, Higuchi A, Tamura N, Ueda Y, Hirabayashi K, Ikeda Y, Kato S, Sakamoto S, Hotta T, Handa S, Goto S. Platelets, after exposure to a high shear stress, induce IL-10-producing, mature dendritic cells in vitro. J Immunol. 2004;172:5297–303. doi: 10.4049/jimmunol.172.9.5297. [DOI] [PubMed] [Google Scholar]
- 24.Andersen H, Greenberg DL, Fujikawa K, Xu W, Chung DW, Davie EW. Protease-activated receptor 1 is the primary mediator of thrombin-stimulated platelet procoagulant activity. Proc Natl Acad Sci USA. 1999;96:11189–93. doi: 10.1073/pnas.96.20.11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohlmann P, Eckly A, Freund M, Cazenave JP, Offermanns S, Gachet C. ADP induces partial platelet aggregation without shape change and potentiates collagen-induced aggregation in the absence of Galphaq. Blood. 2000;96:2134–39. [PubMed] [Google Scholar]
- 26.Brummel KE, Paradis SG, Butenas S, Mann KG. Thrombin functions during tissue factor-induced blood coagulation. Blood. 2002;100:148–52. doi: 10.1182/blood.v100.1.148. [DOI] [PubMed] [Google Scholar]
- 27.Davey MG, Lüscher EF. Actions of thrombin and other coagulant and proteolytic enzymes on blood platelets. Nature. 1967;216:857–8. doi: 10.1038/216857a0. [DOI] [PubMed] [Google Scholar]
- 28.Martorell L, Martínez-González J, Rodríguez C, Gentile M, Calvayrac O, Badimon L. Thrombin and protease-activated receptors (PARs) in atherothrombosis. Thromb Haemost. 2008;99:305–15. doi: 10.1160/TH07-08-0481. [DOI] [PubMed] [Google Scholar]
- 29.Ueno M, Ferreiro JL, Angiolillo DJ. Mechanism of action and clinical development of platelet thrombin receptor antagonists. Expert Rev Cardiovasc Ther. 2010;8:1191–200. doi: 10.1586/erc.10.49. [DOI] [PubMed] [Google Scholar]
- 30.Coughlin SR. Protease-activated receptors in heamostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–14. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 31.Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV, Jr, Tam C, Coughlin SR. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–4. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- 32.Chackalamannil S. Thrombin receptor (protease activated receptor-1) antagonists as potent antithrombotic agents with strong antiplatelet effects. J Med Chem. 2006;49:5389–403. doi: 10.1021/jm0603670. [DOI] [PubMed] [Google Scholar]
- 33.Shah R. Protease-activated receptors in cardiovascular health and diseases. Am Heart J. 2009;157:253–62. doi: 10.1016/j.ahj.2008.09.025. [DOI] [PubMed] [Google Scholar]
- 34.Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci USA. 1994;91:9208–12. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tesfamariam B. Thrombin receptor-mediated vascular relaxation differentiated by a receptor antagonist and desensitization. Am J Physiol. 1994;267:H1962–7. doi: 10.1152/ajpheart.1994.267.5.H1962. [DOI] [PubMed] [Google Scholar]
- 36.Derkach DN, Ihara E, Hirano K, Nishimura J, Takahashi S, Kanaide H. Thrombin causes endothelium-dependent biphasic regulation of vascular tone in porcine renal interlobar artery. Br J Pharmacol. 2000;131:1635–42. doi: 10.1038/sj.bjp.0703737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ku DD, Zaleski JK. Receptor mechanism of thrombin-induced endothelium-dependent and endothelium independent coronary vascular effects in dogs. J Cardiovasc Pharmacol. 1993;22:609–16. doi: 10.1097/00005344-199310000-00015. [DOI] [PubMed] [Google Scholar]
- 38.Chen LB, Buchanan JM. Mitogenic activity of blood components. Thrombin and prothrombin. Proc Natl Acad Sci USA. 1975;72:131–5. doi: 10.1073/pnas.72.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vu T-KH, Hung DT, Wheaton VI, Coughlin SR. Molecular of functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–68. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 40.Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103:879–87. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci USA. 1998;95:6642–46. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacques SL, Kuliopulos A. Protease-activated receptor-4 uses dual prolines and an anionic retention motif for thrombin recognition and cleavage. Biochem J. 2003;376:733–40. doi: 10.1042/BJ20030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shapiro MJ, Weiss EJ, Faruqi TR, Coughlin SR. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J Biol Chem. 2000;275:25216–21. doi: 10.1074/jbc.M004589200. [DOI] [PubMed] [Google Scholar]
- 44.Henriksen RA, Hanks VK. PAR-4 agonist AYPGKF stimulates thromboxane production by human platelets. Arterioscler Thromb Vasc Biol. 2002;22:861–6. doi: 10.1161/01.atv.0000014742.56572.25. [DOI] [PubMed] [Google Scholar]
- 45.Covic L, Singh C, Smith H, Kuliopulos A. Role of the PAR4 thrombin receptor in stabilizing platelet-platelet aggregates as revealed by a patient with Hermansky–Pudlak syndrome. Thromb Haemost. 2002;87:722–7. [PubMed] [Google Scholar]
- 46.Lova P, Campus F, Lombardi R, Cattaneo M, Sinigaglia F, Balduini C, Torti M. Contribution of protease-activated receptors 1 and 4 and glycoprotein Ib-IX-V in the G(i)-independent activation of platelet Rap1B by thrombin. J Biol Chem. 2004;279:25299–306. doi: 10.1074/jbc.M313199200. [DOI] [PubMed] [Google Scholar]
- 47.Smith CC, Wilson AP, Prichard BN, Betteridge DJ. Stimulus-induced release of endogenous catecholamines from human washed platelets. Clin Sci (Lond) 1986;70:495–500. doi: 10.1042/cs0700495. [DOI] [PubMed] [Google Scholar]
- 48.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591–4. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]
- 49.Stenberg PE, McEver RP, Shuman MA, Jacques YV, Bainton DF. A platelet alpha-granule membrane protein (GMP-140) is expressed on the plasma membrane after activation. J Cell Biol. 1985;101:880–6. doi: 10.1083/jcb.101.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997;389:183–6. doi: 10.1038/38284. [DOI] [PubMed] [Google Scholar]
- 51.Goldsack NR, Chambers RC, Dabbagh K, Laurent GJ. Thrombin. Int J Biochem Cell Biol. 1998;30:641–6. doi: 10.1016/s1357-2725(98)00011-9. [DOI] [PubMed] [Google Scholar]
- 52.Kim S, Foster C, Lecchi A, Quinton TM, Prosser DM, Jin J, Cattaneo M, Kunapuli SP. Protease-activated receptors 1 and 4 do not stimulate G(i) signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of G(i) signaling. Blood. 2002;99:3629–36. doi: 10.1182/blood.v99.10.3629. [DOI] [PubMed] [Google Scholar]
- 53.Faruqi TR, Weiss EJ, Shapiro MJ, Huang W, Coughlin SR. Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function. J Biol Chem. 2000;275:19728–34. doi: 10.1074/jbc.M909960199. [DOI] [PubMed] [Google Scholar]
- 54.Sambrano GR, Weiss EJ, Zheng YW, Huang W, Coughlin SR. Role of thrombin signalling in platelets in haemostasis and thrombosis. Nature. 2001;413:74–8. doi: 10.1038/35092573. [DOI] [PubMed] [Google Scholar]
- 55.Ma L, Perini R, McKnight W, Dicay M, Klein A, Hollenberg MD, Wallace JL. Proteinase-activated receptors 1 and 4 counter-regulate endostatin and VEGF release from human platelets. Proc Natl Acad Sci USA. 2005;102:216–20. doi: 10.1073/pnas.0406682102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, Covic L, Kuliopulos A. Blocking the protease-activated receptor 1-4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;113:1244–54. doi: 10.1161/CIRCULATIONAHA.105.587758. [DOI] [PubMed] [Google Scholar]
- 57.Hamilton JR, Cornelissen I, Coughlin SR. Impaired hemostasis and protection against thrombosis in protease-activated receptor 4-deficient mice is due to lack of thrombin signaling in platelets. J Thromb Haemost. 2004;2:1429–35. doi: 10.1111/j.1538-7836.2004.00783.x. [DOI] [PubMed] [Google Scholar]
- 58.Suh TT, Holmback K, Jensen NJ, Daugherty CC, Small K, Simon DI, Potter S, Degen JL. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–33. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]
- 59.Vandendries ER, Hamilton JR, Coughlin SR, Furie B, Furie BC. Par4 is required for platelet thrombus propagation but not fibrin generation in a mouse model of thrombosis. Proc Natl Acad Sci USA. 2007;104:288–92. doi: 10.1073/pnas.0610188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kato Y, Kita Y, Hirasawa-Taniyama Y, Nishio M, Mihara K, Ito K, Yamanaka T, Seki J, Miyata S, Mutoh S. Inhibition of arterial thrombosis by a protease-activated receptor 1 antagonist, FR171113, in the guinea pig. Eur J Pharmacol. 2003;473:163–9. doi: 10.1016/s0014-2999(03)01973-3. [DOI] [PubMed] [Google Scholar]
- 61.Schulman SP. Antiplatelet therapy in non-ST-segment elevation acute coronary syndromes. J Am Med Assoc. 2004;292:1875–82. doi: 10.1001/jama.292.15.1875. [DOI] [PubMed] [Google Scholar]
- 62.The TRA·CER Exective and Steering Committees. The Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRA·CER) trial: study design and rationale. Am Heart J. 2009;158:327–34. doi: 10.1016/j.ahj.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 63.Morrow DA, Scirica BM, Fox KA, Berman G, Strony J, Veltri E, Bonaca MP, Fish P, McCabe CH, Braunwald E, TRA 2(o)P-TIMI 50 Investigators Evaluation of a novel antiplatelet agent for secondary prevention in patients with a history of atherosclerotic disease: design and rationale for the Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events (TRA 2°P)-TIMI 50 trial. Am Heart J. 2009;158:335–41. doi: 10.1016/j.ahj.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 64.Goto S, Ogawa H, Takeuchi M, Flather MD, Bhatt DL, on behalf of the J-LANCELOT (Japanese-Lesson from Antagonizing the Cellular Effect of Thrombin) Investigators Double-blind, placebo-controlled Phase II studies of the protease-activated receptor 1 antagonist E5555 (atopaxar) in Japanese patients with acute coronary syndrome or high-risk coronary artery disease. Eur Heart J. 2010;31:2601–13. doi: 10.1093/eurheartj/ehq320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O'Donoghue M, on Behalf of the LANCELOT Investigators LANCELOT ACS: a prospective, randomized, double-blind, placebo-controlled trial of a reversible PAR-1 thrombin receptor antagonist in patients with acute coronary syndromes. Presented at Transcatheter Cardiovascular Therapeutics September 21-25, 2010, Washington DC, USA.
- 66.Wiviott SD, on Behalf of the LANCELOT Investigators LANCELOT CAD: a prospective, randomized, double-blind, placebo-controlled trial of a reversible PAR-1 thrombin receptor antagonist in patients with coronary artery disease. Presented at Transcatheter Cardiovascular Therapeutics September 21-25, 2010 Washington DC, USA.
- 67.Chintala M, Ahn H, Foster C, Xia Y, Chackalamannil S. Antithrombotic effects of SCH 205831: a potent, selective and orally active antagonist of the PAR-1 thrombin receptor. J Thromb Haemost. 2005;3 Abstract OR286. [Google Scholar]
- 68.Chintala M, Kurowski S, Vemulapalli S, Li Q, Brown A, Strony J. Efficacy of SCH 602539, a selective thrombin receptor antagonist alone and in combination with cangrelor in a Folts model of thrombosis in anesthetized monkeys. Eur Heart J. 2007;28 Abstract 188. [Google Scholar]
- 69.Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, Boykow G, Hsieh Y, Palamanda J, Agans-Fantuzzi J, Kurowski S, Graziano M, Chintala M. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J Med Chem. 2008;51:3061–4. doi: 10.1021/jm800180e. [DOI] [PubMed] [Google Scholar]
- 70.Chintala M, Vemulapalli S, Kurowski S, Sabin C, Reynolds D, Prevete K, Friedrich G. SCH 530348, a novel oral antiplatelet agent, demonstrated no bleeding risk alone or in combination with aspirin and clopidogrel in cynomolgus monkeys. Arterioscler Thromb Vasc Biol. 2008;28:e138–e139. [Google Scholar]
- 71.Chackalamannil S, Davies RJ, Asberom T, Doller D, Leone D. A highly effi cient total synthesis of (+)-himbacine. J Am Chem Soc. 1996;118:9812–13. [Google Scholar]
- 72.Doller D, Chackalamannil S, Czarniecki M, McQuade R, Ruperto V. Design, synthesis, and structure-activity relationship studies of himbacine derived muscarinic receptor antagonists. Bioorg Med Chem Lett. 1999;9:901–06. doi: 10.1016/s0960-894x(99)00101-8. [DOI] [PubMed] [Google Scholar]
- 73.Oestreich J. SCH-530348, a thrombin receptor (PAR-1) antagonist for the prevention and treatment of atherothrombosis. Curr Opin Investig Drugs. 2009;10:988–96. [PubMed] [Google Scholar]
- 74.Kosoglou T, Reyderman L, Tiessen R, Fales RR, Cutler DL, Keller R, Yang B, Van Lier JJ, van Vliet AA. TRAP-induced platelet aggregation following single and multiple rising oral doses of SCH 530348, a novel thrombin receptor antagonist, in healthy volunteers. Pharmacol Ther. 2009;85(Suppl 1) PI–40,–21. [Google Scholar]
- 75.Kosoglou T, Tiessen R, Van Vliet AA, Fales RR, Keller R, Cutler DL. Safety and tolerability of SCH 530348, a novel antiplatelet agent, after single and multiple oral dosing in healthy subjects. Eur Heart J. 2008;29:201. P1340. [Google Scholar]
- 76.Reyderman L, Kosoglou T, Kasserra C, Young S, Pei J, Schiller J, Lin H, Cutler DL. Lack of ethnic differences in the pharmacodynamics (PD) and pharmacokinetics (PK) of SCH 530348, a novel oral antiplatelet agent, in Japanese and Caucasian subjects. Clin Pharmacol Ther. 2009;85:S21. PI-41. [Google Scholar]
- 77.Kosoglou T, Reyderman L, Kasserra C, Young S, Pei J, Maxwell SE, Schiller J, Cutler DL. Optimizing dose of the novel thrombin receptor antagonist SCH 530348 based on pharmacodynamics and pharmacokinetics in healthy subjects. Clin Pharmacol Ther. 2008;83:55. [Google Scholar]
- 78.Becker RC, Moliterno DJ, Jennings LK, Pieper KS, Pei J, Niederman A, Ziada KM, Berman G, Strony J, Joseph D, Mahaffey KW, Van de Werf F, Veltri E, Harrington RA, TRA-PCI Investigators Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomised, double-blind, placebo-controlled phase II study. Lancet. 2009;373:919–28. doi: 10.1016/S0140-6736(09)60230-0. [DOI] [PubMed] [Google Scholar]
- 79.Jennings LK, Earhart A, Becker RC, Reyderman L, Veltri E, Harrington RA. Thrombin receptor antagonist (TRA;SCH 530348) is a selective, potent inhibitor of PAR1 activity with predictable pharmacokinetics. Orlando, FL: Presented at American Heart Association Scientific Sessions November 4–7; 2007.
- 80.Goto S, Yamaguchi T, Ikeda Y, Kato K, Yamaguchi H, Jensen P. Safety and exploratory efficacy of the novel thrombin receptor (PAR-1) antagonist SCH530348 for non-ST-segment elevation acute coronary syndrome) J Atheroscler Thromb. 2010;17:156–64. doi: 10.5551/jat.3038. [DOI] [PubMed] [Google Scholar]
- 81.Shinohara Y, Goto S, Shimizu K, Jensen P. A phase II safety study of novel antiplatelet agent, SCH 530348, in Japanese patients with prior ischemic stroke. Int J Stroke. 2008;3:139. PO101-193. [Google Scholar]
- 82.Ferreiro JL, Sibbing D, Angiolillo DJ. Platelet function testing and risk of bleeding complications. Thromb Haemost. 2010;103:1128–35. doi: 10.1160/TH09-11-0799. [DOI] [PubMed] [Google Scholar]
- 83.Kogushi M, Yokohama H, Kitamura S, Hishinuma I. Effects of E5555, a protease-activated receptor-1 antagonist, on the inflammatory markers in vitro. J Thromb Haemost. 2007;5 P-M-059. [Google Scholar]
- 84.Kai Y, Hirano K, Maeda Y, Nishimura J, Sasaki T, Kanaide H. Prevention of the hypercontractile response to thrombin by proteinase-activated receptor-1 antagonist in subarachnoid hemorrhage. Stroke. 2007;38:3259–65. doi: 10.1161/STROKEAHA.107.487769. [DOI] [PubMed] [Google Scholar]
- 85.Matsuoka T, Kogushi M, Kawata T, Kimura A, Chiba K, Musha T, Kobayashi H, Kawahara T, Suzuki S, Kajiwara A, Hishinuma L. Inhibitory effect of E5555, an orally active thrombin receptor antagonist, onintimal hyperplasia following balloon injury. J Am Coll Cardiol. 2004;43:68A–A. [Google Scholar]
- 86.Serebruany VL, Kogushi M, Dastros-Pitei D, Flather M, Bhatt DL. The in-vitro effects of E5555, a protease-activated receptor (PAR)-1 antagonist, on platelet biomarkers in healthy volunteers and patients with coronary artery disease. Thromb Haemost. 2009;102:111–9. doi: 10.1160/TH08-12-0805. [DOI] [PubMed] [Google Scholar]
- 87.Marion A, Reyes J, Chen H, Wittek A. Safety, Pharmacokinetics, and Pharmacodynamics of ascending multiple oral doses of a new protease activated receptor-1 inhibitor in healthy volunteers. American College of Clinical Pharmacology. 35th Annual meeting September 17-19, 2006 in Cambridge, MA.
- 88.Takeuchi M, Kageyama M, Kitamura S, Hagino R, Ozawa H, Tanaka H. Pharmacodinamics and safety of a novel protease activated receptor-1 antagonist E5555 in healthy volunteers. Eur Heart J. 2007;28:14. [Google Scholar]
- 89.Angiolillo DJ, Capranzano P, Desai B, Shoemaker SB, Charlton R, Zenni MM, Guzman LA, Bass TA. Impact of P2Y(12) inhibitory effects induced by clopidogrel on platelet procoagulant activity in type 2 diabetes mellitus patients. Thromb Res. 2009;124:318–22. doi: 10.1016/j.thromres.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 90.CURRENT-OASIS 7 Investigators. Mehta SR, Bassand JP, Chrolavicius S, Diaz R, Eikelboom JW, Fox KA, Granger CB, Jolly S, Joyner CD, Rupprecht HJ, Widimsky P, Afzal R, Pogue J, Yusuf S. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med. 2010;363:930–42. doi: 10.1056/NEJMoa0909475. [DOI] [PubMed] [Google Scholar]
- 91.Létienne R, Leparq-Panissié A, Calmettes Y, Nadal-Wollbold F, Perez M, Le Grand B. Antithrombotic activity of F 16618, a new PAR1 antagonist evaluated in extracorporeal arterio-venous shunt in the rat. Biochem Pharmacol. 2010;79:1616–21. doi: 10.1016/j.bcp.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 92.Bocquet A, Létienne R, Sablayrolles S, De Vries L, Perez M, Le Grand B. Effects of a new PAR1 antagonist, F 16618, on smooth muscle cell contraction. Eur J Pharmacol. 2009;611:60–3. doi: 10.1016/j.ejphar.2009.03.056. [DOI] [PubMed] [Google Scholar]
- 93.Chintala M, Strony J, Yang B, Kurowski S, Li Q. SCH 602539, a protease-activated receptor-1 antagonist, inhibits thrombosis alone and in combination with cangrelor in a folts model of arterial thrombosis in cynomolgus monkeys. Arterioscler Thromb Vasc Biol. 2010;30:2143–9. doi: 10.1161/ATVBAHA.110.203414. [DOI] [PubMed] [Google Scholar]