

Abstract
Glycoprotein (GP) IIb-IIIa antagonists inhibit the aggregation of activated platelets. Three agents are approved for clinical use. In this review, the characteristics of each agent, their pharmacodynamic profile, results in pivotal clinical trials and the associated clinical implications are discussed. GP IIb-IIIa antagonists have greatest benefit when used as adjunctive therapy during percutaneous coronary intervention (PCI) when the patient has intra-coronary thrombosis. These agents appear to provide greatest benefit when used in combination with heparin. The clinical niche for parenteral GP IIb-IIIa antagonists is evolving. The rapid onset and offset of GP IIb-IIIa antagonists plus dosing designed to inhibit extensively platelet aggregation differentiates them from oral agents. The contemporary niche appears to include patients in transition, such as individuals requiring emergent PCI before oral agents are fully active and for unstable patients requiring transport to PCI centres, particularly in patients likely to have intracoronary thrombus. Subsequent studies should evaluate the optimal duration of therapy with GP IIb-IIIa antagonists.
Keywords: acute coronary syndromes, antiplatelet, coronary intervention, glycoprotein IIb-IIIa, pharmacodynamics, platelet
Identification of glycoprotein IIb-IIIa as a therapeutic target
Glanzmann thrombasthenia is a hereditary disorder associated with mucocutaneous bleeding in which platelets do not aggregate [1, 2]. Two glycoproteins, designated IIb (αIIb) and IIIa (β3), were found to be missing from the surface of platelets taken from patients with Glanzmann thrombasthenia. These glycoproteins form a calcium-dependent complex on the platelet surface [3]. Subsequent studies demonstrated that binding of fibrinogen to the platelet surface mediates platelet aggregation and that GP IIb-IIIa is an activation dependent receptor for fibrinogen and to a lesser extent, fibronectin, von Willebrand factor and vitronectin [4–6]. Each molecule of fibrinogen can bind to two GP IIb-IIIa complexes thereby cross-linking or aggregating platelets [7, 8]. GP IIb-IIIa is the most abundant integrin on the platelet surface with approximately 50 000 to 80 000 complexes on the surface of each platelet. Activation of platelets that occurs after endothelial injury or rupture of an atherosclerotic plaque leads to activation of GP IIb-IIIa and the aggregation of platelets (Figure 1).
Figure 1.
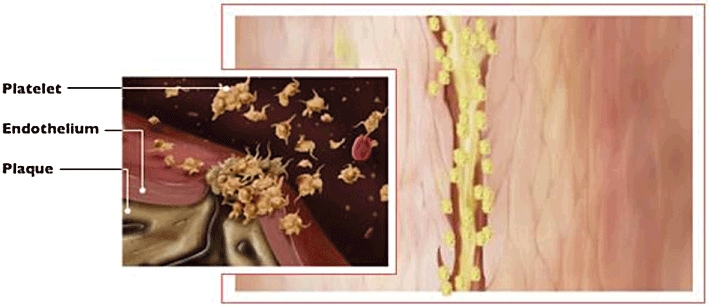
Rupture of an atherosclerotic plaque leads to the activation of platelets, a conformational change in GP IIb-IIIa and the aggregation of platelets
Binding of adhesive proteins to GP IIb-IIIa is mediated by the sequence arginine-glycine-aspartic acid (RGD) and the gamma chain dodecapeptide (Figure 2). This sequence was identified initially in fibronectin, but subsequently demonstrated in fibrinogen, von Willebrand factor and vitronectin [9]. Peptides that contain the RGD sequence are capable of inhibiting interaction between GP IIb-IIIa and fibrinogen [10]. Another peptide sequence capable of mediating binding of fibrinogen to GP IIb-IIIa is lysine-glutamine-alanine-glycine-aspartic acid-valine [11]. Unlike RGD, this sequence is found only in fibrinogen [12]. Although various adhesive proteins can bind to GP IIb-IIIa under appropriate conditions, fibrinogen appears to be the predominant protein that mediates aggregation [13], while von Willebrand factor appears to play a more prominent role in the setting of increased shear stress [14].
Figure 2.
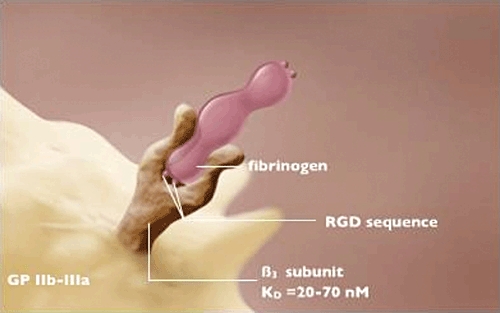
Each molecule of fibrinogen can bind to two GP IIb-IIIa molecules. This leads to cross-linking/aggregation of platelets. The RGD sequence is pivotal to binding of fibrinogen that forms an initial more reversible complex (KD 150–180 nm) and a subsequent more stable complex with a KD of 20–70 nm
Development of glycoprotein IIb-IIIa antagonists
A number of antibodies directed against platelet GP IIb-IIIa were developed to inhibit binding of fibrinogen to platelets [15, 16]. One such monoclonal antibody (7E3), was chosen, in part, because of its cross-reactivity in dogs, a well characterized and preferred animal in which to induce arterial thrombosis [17]. To prevent clearance of platelets to which the antibody had bound, the Fc component was cleaved. The cleaved product, 7E3-F(ab′)2, was shown to inhibit aggregation of platelets in dogs without the development of spontaneous haemorrhage [18]. To limit immunologic response to the Fab fragment in humans, a mouse/ human chimera was developed (c7E3 Fab) which is known as abciximab [19].
Disintegrins are a class of agents identified in snake venom. One of these peptides, barbourin, contains a KGD (lysine-glycine-aspartic acid) sequence that has unique specificity for GP IIb-IIIa [20]. Although naturally occurring disintegrins are too immunogenic for human use, their structure provided a template for the development of synthetic peptide antagonists. The cyclic heptapeptide, epifibatide, contains the KGD sequence identified in barbourin. Specifically engineered synthetic compounds are the third method developed to inhibit binding of fibrinogen to GP IIb-IIIa and their development entailed imitation of the charge and spatial conformation of the RGD sequence. The parenteral peptidomimetic antagonist, lamifiban (not available for clinical use), and the nonpeptide antagonist, tirofiban, are examples of synthetic compounds.
Characteristics of abciximab
The characteristics of abciximab (molecular weight of approximately 50 000 Daltons) are consistent with its antibody derivation [21]. Abciximab has a short half-life in the fluid phase of blood. The majority of the bolus is bound rapidly to platelets that serve as the circulating pool of abciximab. The bolus dose was designed to achieve greater than 80% blockade of GP IIb-IIIa on platelets in blood. The remaining unbound fraction of abciximab is cleared rapidly [22]. Abciximab displays a rapid on-rate, binding to platelets in less than 1 min. The dissociation (off-rate) of abciximab is measured in hours [21]. The prolonged platelet-bound half-life of abciximab accounts for both its prolonged effect on platelet function after termination of infusion and the gradual return of platelet function thereafter [23]. Abciximab has the greatest affinity for GP IIb-IIIa (reflected by its dissociation constant [KD] of 5 nmol l−1, Figure 3) among the GP IIb-IIIa antagonists that are available for clinical use [21].
Figure 3.
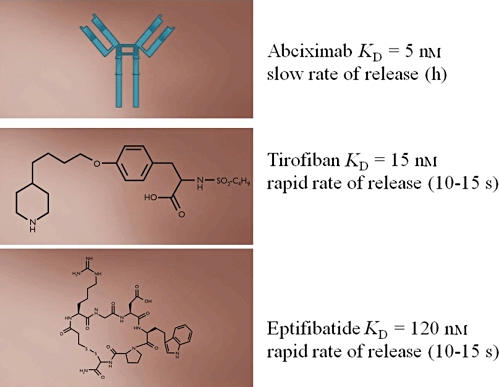
The structure and kinetics of the available GP IIb-IIIa antagonists
Clinical implications of the characteristics of abciximab include the following. It has a longer duration of effect than the other agents developed. Because the circulating pool of abciximab is bound to platelets, the effects of the agent can be reversed with the transfusion of platelets. One mechanism by which platelet transfusion decreases the antiplatelet effect is through the binding of abciximab to GP IIb-IIIa on the transfused platelets that decreases the occupancy of GP IIb-IIIa by abciximab on all platelets. The prolonged half-life led to initial testing of a bolus of abciximab without subsequent infusion. Subsequent pharmacodynamic studies demonstrated that recruitment of both platelets and surface expression of IIb-IIIa can occur after PCI [22]. Thus, to inhibit this recruitable reservoir of platelets and GP IIb-IIIa a regimen that included an initial bolus plus subsequent infusion was tested and was adopted. Because testing with abciximab occurred before thienpyridines were commonly used, the necessity of the subsequent infusion has not been assessed in patients treated with the combination of aspirin plus a second antiplatelet agent (e.g. clopidogrel).
Abciximab, unlike tirofiban and eptifibatide, binds with equal affinity to GP IIb-IIIa and αvβ3 (vitronectin receptor) [24]. In addition, abciximab binds, albeit with lower affinity, to the leukocyte Mac-1 receptor [25]. Abciximab inhibits αvβ3 receptor mediated smooth muscle migration and proliferation in vitro[26]. The influence in vivo of short term (1–2 days) inhibition of the αvβ3 receptor on the migration and proliferation of vascular smooth muscle cells that occurs over weeks to months has not been determined.
Characteristics of synthetic antagonists to GP IIb-IIIa, tirofiban and eptifibatide
Both eptifibatide and tirofiban (molecular weight of less than 1000 Daltons for each) exhibit a longer half-life in the fluid phase of blood compared with abciximab. The half-life of eptifibatide is 2.5 h and that of tirofiban is 2 h [27, 28]. Administration of eptifibatide and tirofiban is associated with a considerable fraction of these drugs that are in the plasma component of blood until cleared by hepatic and renal mechanisms.
A key difference between abciximab and the small molecules eptifibatide and tirofiban is the rate at which these agents dissociate from GP IIb-IIIa (Figure 3). The off-rate of eptifibatide and tirofiban is 10–15 s compared with hours for abciximab [29, 30]. Because of the rapid binding and release of the small molecules, the concentration of these agents in the fluid phase of blood is a critical determinant of receptor occupancy and hence inhibitory effects.
Although tirofiban and eptifibatide are similar with respect to their off-rate, they differ markedly with respect to their affinity for GP IIb-IIIa. Of the three agents available for clinical use, the affinity of abciximab is greatest (KD = 5 nmol l−1) followed by that of tirofiban (KD = 15 nmol l−1) and eptifibatide (KD = 120 nmol l−1). Because of the rapid off-rate, tirofiban and eptifibatide are in competition with fibrinogen for binding to GP IIb-IIIa. The lower affinity of eptifibatide necessitates a greater molar concentration of eptifibatide compared with tirofiban to achieve a similar extent of inhibition. The implications of the differences in the affinity with respect to inhibition of platelet involvement in thrombosis in vivo have not been defined.
Pharmacodynamic assessment of GP IIb-IIIa antagonists
The development of this class of agents highlighted limitations in the means by which platelet function can be assessed as well as the importance of the methods used in sample preparation, assay conditions and the timing of pharmacodynamic assessment. Development of optimal dosages would have been facilitated by the availability of a clinically validated established method to assess platelet function [31]. The development of GP IIb-IIIa antagonists relied heavily on turbidometric platelet aggregation for pharmacodynamic assessment.
Aggregometry was developed in the 1960s [32]. Turbidometric platelet aggregation is performed in platelet rich plasma that is prepared from anticoagulated blood. The platelet suspension limits transmission of light through the sample and is defined as 0% aggregation. Maximal (100%) aggregation is defined as the transmission of light through platelet poor plasma. Although platelets are activated in vivo by multiple agonists simultaneously, the agonist or combination of agonists and their concentration that simulates thrombosis in vivo has not been defined. ADP has been used most commonly to assess pharmacodynamic effects of GP IIb-IIIa antagonists.
Different concentrations of ADP were used to characterize inhibitory effects of GP IIb-IIIa antagonists. Pharmacodynamic studies performed during the development of tirofiban used 5 µm ADP [28]. By contrast, pharmacodynamic studies during the development of abciximab and eptifibatide used 20 µm ADP [8, 28]. The inhibitory effects of any antiplatelet agent will be less when a more powerful stimulus (i.e. greater concentration or more potent agonist) is used to induce aggregation. Accordingly, even though a similar extent of inhibition was apparent during the development of tirofiban and abciximab, the inhibitory effect of abciximab was likely to be greater because inhibitory effects were assessed in the presence of a greater concentration of agonist.
Platelet aggregometry has been performed traditionally with platelet rich plasma prepared from blood treated with trisodium citrate. Chelation of calcium limits enzyme activity and prevents activity of the coagulation cascade. In addition, the anticoagulation of blood with the use of a calcium chelator alters platelet function [33]. Although activation of platelets in the absence of agonist is not seen, increased reactivity (i.e. increased activation in response to an agonist) is apparent [33]. A potential for interaction between calcium chelators and GP IIb-IIIa is suggested by the critical role of calcium in maintaining the structure and function of GP IIb-IIIa [3]. Inhibitory effects of GP IIb-IIIa antagonists are enhanced when assessed in blood or plasma that has been anticoagulated with a calcium chelator [34]. The influence of calcium chelation on the inhibitory effects is greatest with eptifibatide but evident also with tirofiban and abciximab [35]. This observation has led the use of non-calcium chelating anticoagulants such as the serine protease inhibitor D-phenylalanyl-L-arginine chloromethyl ketone (PPACK) to inhibit the activity of thrombin and prevent clotting in vitro.
The importance of the anticoagulant used in pharmacodynamic evaluation is underscored by the following observation. The concentration of eptifibatide necessary to inhibit ADP-induced platelet aggregation by 50% (i.e. IC50) is four-fold greater when blood has been anticoagulated with PPACK compared with trisodium citrate [36]. Inhibition of the binding of fibrinogen to GP IIb-IIIa by eptifibatide exhibited an IC50 of 63 ± 34 nmol l−1 in 1 mmol l−1 calcium and an IC50 of 8.7 ± 1.2 nmol l−1 in 50 µmol l−1 calcium [36].
The timing of assessment of pharmacodynamic effects is critical and must be influenced by the characteristics of the agent. Because the off-rate of abciximab is hours, the pharmacodynamic assessment characterized inhibitory effects shortly after the bolus and then 2 h and greater after onset of treatment. Despite marked differences in the off-rate, early pharmacodynamic assessment of the inhibitory effects of tirofiban and eptifibatide focused also on the effects immediately after the bolus and the inhibitory effects observed 2 h and greater after onset of treatment [27, 28]. One implication of the rapid off-rate is that a decrease in the concentration of drug in blood, even if transient, will be associated with decreased inhibitory effects. Accordingly, the dosing regimen for tirofiban and eptifibatide must ensure that the loading dose (bolus) is sufficient to maintain an effective concentration of drug until steady state concentrations are achieved.
The dosing regimen of tirofiban that was used in PCI trials did not consistently inhibit aggregation during the first 2 h of treatment. The extent of inhibition of maximal platelet aggregation induced by 20 µm ADP in PPACK anticoagulated blood was consistently greater after treatment with abciximab than tirofiban between 15 min and 1 h after onset of treatment in patients with acute coronary syndromes [37–39]. The extent of inhibition of aggregation ranged from 60% to 66% between 15 and 60 min after onset of treatment with tirofiban. By contrast, the extent of inhibition of aggregation ranged from 90% to 94% from 15 to 60 min after onset of treatment with abciximab. An increase in the bolus of tirofiban from 10 µg kg−1 to 25 µg kg−1 without change in the rate of infusion has been associated with an increase in the extent of inhibition of aggregation to 92–95% [40, 41].
Pharmacodynamic assessment of the effect of GP IIb-IIIa antagonists played a pivotal role in the development of these agents. Unfortunately, as frequently occurs with the development of a new class of drugs, limitations of the methods available to assess the effect of novel drugs are demonstrated by the development process. Although the optimal method to assess platelet function has not been defined, critical lessons learned include: 1) the anticoagulant used to prevent clotting in vitro can influence the results obtained. In the case of GP IIb-IIIa antagonists, chelation of calcium augments their ability to inhibit binding of fibrinogen to GP IIb-IIIa, 2) the agonist used to induce activation of platelets influences the observed efficacy of the agent. Although it could be argued that platelets will not be exposed to 20 µm ADP in vivo, the culmination of agonist effect in response to diverse agonists in vivo is likely to manifest a powerful stimulus for activation. The agonist or combination and their concentration that best reflects thrombosis in vivo have not been defined and 3) the measurement of antiplatelet effects of agents with a rapid off-rate must prevent a transient reduction in the concentration of drug and hence the inhibitory effect.
Clinical trials of GP IIb-IIIa inhibitors in PCI
Randomized clinical trials have demonstrated that adjunctive therapy with GP IIb-IIIa antagonists decrease the combined endpoint of death, myocardial infarction and target vessel revascularization after PCI, particularly in patients with ACS [42, 43]. Of the three agents available for clinical use, abciximab, eptifibatide and tirofiban, abciximab has been most studied. Because of the common mechanism of action, results with these agents are frequently combined and several of the more recent trials allowed use of any agent in this class. For clarity, results of pivotal trials with each agent will be discussed separately with a focus on clinical insight provided.
Abciximab
The EPIC study was the first large clinical trial demonstrating that blockade of GP IIb-IIIa with abciximab decreased the risk of cardiac events after PCI [44]. Administration of a 0.25 mg kg−1 intravenous loading dose followed by a 12 h infusion of 0.125 µg kg−1 min−1 reduced the 30 days incidence of cardiac events from 12.8% to 8.3% whereas the bolus alone had a modest effect (30 days event rate of 11.5%). As mentioned previously, pharmacodynamic characterization demonstrated that both platelets and an internal pool of GP IIb-IIIa can be recruited after PCI [8, 22]. Although the bolus plus infusion of abciximab led to a greater reduction in the subsequent incidence of cardiac events, the necessity of the infusion has not been assessed when a 600 mg loading dose of clopidogrel or newer and more powerful antagonists of P2Y12 have been used.
The EPILOG study demonstrated that adjuvant treatment with abciximab plus a lower dose of heparin (target activated clotting time of 250 s) decreased the risk of bleeding without increasing the risk of ischaemic cardiac events after PCI [45]. The EPISTENT study extended the beneficial effects of abciximab to PCI performed with intracoronary stents [46].
The adjunctive value of abciximab during PCI in patients who have been treated before PCI with a more effective loading dose of clopidogrel (600 mg at least 2 h before the procedure) was investigated by the ISAR group. In lower risk patients undergoing predominantly elective PCI, treatment with abciximab offered no benefit [47]. By contrast, ACS patients with elevated markers of cardiac injury exhibited a lower incidence of peri-procedural myocardial infarction when treated with abciximab [48].
The GUSTO IV study assessed the potential benefit of abciximab in the treatment of ACS without PCI [49]. The dosage of abciximab developed for PCI or the same bolus plus a longer infusion did not decrease the subsequent incidence of cardiac events. The mechanism underlying this lack of benefit has not been defined. The results are consistent, however, with a limited benefit of short-term intense antiplatelet therapy in the absence of mechanical therapy (PCI) or transition to longer-term more powerful antiplatelet therapy (i.e. thienopyridine plus aspirin). Accordingly, it supported use of abciximab primarily in association with PCI.
In the GUSTO V study, the combination of the PCI dosage of abciximab plus half dose thrombolytic agent for the treatment of ST elevation myocardial infarction appears to promote earlier vessel patency and decreases recurrent ischaemic events but does not decrease the 30 days or 1 year mortality [50, 51]. Adjunctive treatment with abciximab during primary PCI treatment of ST elevation myocardial infarction decreased subsequent cardiac events. However, the beneficial effects appeared to be greater when PCI was performed without intracoronary stents [52, 53].
More recently, intra-coronary injection of abciximab during primary PCI for patients with ST elevation myocardial infarction has been investigated. The characteristics of abciximab (rapid on-rate and slow off-rate) support a potential benefit associated with this approach that should lead to transient very high local concentrations of abciximab in the vicinity of a thrombus. In one study, intra-coronary abciximab decreased microvascular obstruction, improved perfusion and decreased infarct size [54].
Although treatment with abciximab reduces the incidence of peri-procedural myocardial infarction in patients undergoing PCI, the ISAR studies demonstrated that adjunctive therapy influenced that benefit. For elective PCI, adequate pretreatment with clopidogrel (600 mg at least 2 h before the procedure) obviated the beneficial effects of abciximab. Similarly, the use of bivalirudin rather than the combination of unfractionated heparin plus a GP IIb-IIIa inhibitor was associated with a statistically non-inferior incidence of major adverse cardiac events (driven predominantly by peri-procedural MI) in patients undergoing predominantly elective PCI [55]. These results were extended to ACS patients in the ACUITY trial [56]. In ACUITY, the addition of a GP IIb-IIIa inhibitor to treatment with bivalirudin did not reduce the incidence of major adverse cardiac events after PCI. Further, results with bivalirudin alone were statistically non-inferior to those with heparin plus a GP IIb-IIIa inhibitor. Treatment with bivalirudin alone has been consistently associated with a lower risk of bleeding compared with an anticoagulant plus a GP IIb-IIIa inhibitor.
In summary, randomized clinical trials testing the efficacy of abciximab have provided important clinical insight. Beneficial effects of abciximab are likely to be greatest during PCI, particularly when PCI is performed in patients most likely to have intracoronary thrombosis – ST-elevation and non-ST-elevation myocardial infarction. The beneficial effects were seen predominantly when treatment with abcixmab was combined with heparin (or low molecular weight fraction of heparin) but not with the direct thrombin inhibitor, bivalirudin. Thienopyridines (e.g. clopidogrel) appear to obviate the need for abciximab in elective PCI for relief of stable angina.
Eptifibatide
Treatment with eptifibatide during PCI was associated with limited benefit in the IMPACT II study [57]. Subsequent pharmacodynamic studies demonstrated the pivotal role of calcium when inhibitory effects of eptifibatide were characterized in vitro[36]. A regimen that included a double bolus (two 180 µg kg−1 doses 10 min apart), plus a 2µg kg−1 min−1 infusion for 18–24 h was developed to prevent a transient decrease in the concentration and to maintain consistent inhibitory effects during the first hour after onset of treatment [58]. In the ESPRIT trial, adjunctive treatment with this regimen of eptifibatide during PCI decreased the 30 days incidence of death, myocardial infarction and target vessel revascularization from 10.5% to 6.6% [59]. Treatment with eptifibatide of patients with ACS was assessed in the PURSUIT trial [60]. Those treated with eptifibatide had a 1.5% absolute reduction in the 30 days incidence of death and myocardial infarction. Consistent with the results seen with abciximab, patients in whom PCI was performed exhibited the greatest benefit [61].
More recently, the potential value of initiation of therapy with eptifibatide at least 12 h before PCI (upstream treatment) was tested in patients with non-ST-elevation ACS in the EARLY ACS trial [62]. Treatment with eptifibatide for at least 12 h before angiography was not superior to the provisional use of eptifibatide at the time of PCI and was associated with a greater risk of bleeding. The results in EARLY ACS were consistent with those reported in ACUITY trial [63]. Upstream treatment with a GP IIb-IIIa inhibitor was not superior to provisional use during PCI and upstream use was associated with a greater risk of bleeding. Based on the results of these two trials, the regular use of an upstream GP IIb-IIIa inhibitor for ACS patients cannot be recommended.
Tirofiban
The clinical impact of tirofiban was assessed in both PCI and the treatment of ACS [64, 65]. Different dosages were used in each setting. The ACS dosage of tirofiban included a 30 min loading with 0.4 µg kg−1 min−1 followed by an infusion of 0.1 µg kg−1 min−1 whereas the rate of infusion when used as an adjunct to PCI was 0.15 µg kg−1 min−1. The PRISM-PLUS study demonstrated that treatment of patients with ACS with the combination of aspirin, heparin and tirofiban reduced the 30 days incidence of death, myocardial infarction or refractory ischaemia from 22.3% to 18.5% [66]. The TACTICS-TIMI 18 study extended these observations by demonstrating that patients with ACS who were treated with tirofiban derive the greatest benefit from an invasive strategy (coronary angiography in all subjects) rather than a non-invasive strategy of coronary angiography selectively in those with provocable ischaemia on stress testing [66]. Analysis of patients treated with upstream tirofiban identified patients who were being transferred to a tertiary (PCI) centre who derived particular benefit from treatment with tirofiban [67]. Thus, although regular use of an upstream GP IIb-IIIa inhibitor cannot be recommended based on the results of EARLY ACS, unstable patients who must be transferred to a PCI centre are a group of patients in whom upstream therapy should still be considered.
The use of tirofiban as an adjunct to PCI did not reduce significantly the 30 days incidence of death, myocardial infarction and urgent target vessel revascularization in the RESTORE study. A subsequent study was performed to establish the utility of tirofiban in PCI. That trial, TARGET, remains as the only large scale direct comparison of outcomes in patients randomized to either abciximab or the same dosage of tirofiban that was used in the RESTORE study [68]. Those treated with tirofiban had a 7.6% incidence of cardiac events after 30 days and those treated with abciximab had a 6.0% incidence. Thus, adjunctive treatment with abcximab was superior to adjunctive treatment with tirofiban (with the RESTORE dose).
Subsequent pharmacodynamic studies demonstrated limited inhibition 15–60 min after onset of treatment with the PCI dosage of tirofiban [37–39] that can be overcome by increasing the bolus to 25 µg kg−1[40, 41]. A large scale trial, TENACITY, was begun but ended after limited enrolment because of financial limitations of the sponsor. A series of moderate sized studies demonstrated that the high bolus dose of tirofiban appeared to be safe and effective compared with placebo and abciximab. A recent meta-analysis summarized the results of those trials [69].
The use of tirofiban (high bolus dose) early in the treatment of patients with ST elevation myocardial infarction (STEMI) improved resolution of ST segment elevation [70]. Resolution of ST segment elevation was chosen as the primary endpoint because it has been associated with a greater risk of death, an observation that was confirmed in the ON-TIME 2 trial [70]. Consistent with recent results with intracoronary abciximab, GP IIb-IIIa inhibition at the time of PCI in patients with ST elevation (and intra-coronary thrombus) appeared to improve reperfusion.
Additional analysis from the ON-TIME 2 trial identified another potential use of GP IIb-IIIa inhibition in patients with STEMI, reduction in the early incidence of stent thrombosis [71]. Early stent thrombosis was more common among patients with STEMI [72]. Moreover, many of these patients have not been treated before their acute event with anti-thrombotic therapy. The importance of very early intervention poses a risk of very early stent thrombosis that can be attenuated by more than 50% with the use of a GP IIb-IIIa inhibitor as demonstrated in the ON-TIME 2 trial [71].
Trials with oral GP IIb-IIIa antagonists
Oral agents with specificity for the RGD sequence that were evaluated in clinical trials were all prodrugs and included xemilofiban, orbofiban, sibrafiban and lotrafiban. These drugs were tested in five randomized clinical trials in patients with acute coronary syndromes or undergoing PCI. Because of the risk of bleeding associated with long-term treatment, dosages associated with a more modest extent of inhibition of platelet aggregation were used in these trials. Nevertheless, the risk of bleeding was increased in patients treated with oral GP IIb-IIIa antagonists [73–77]. In each trial an increased risk of mortality was associated with treatment with an oral GP IIb-IIIa antagonist and a meta-analysis confirmed an increased risk of death (odds ratio of 1.37, P = 0.001) in those treated with an oral GP IIb-IIIa antagonist [78]. Accordingly, further development of these agents was abandoned.
Subsequent studies have focused on the mechanisms responsible for the increased mortality. Examination of platelet function in those treated with orbofiban in the OPUS study demonstrated increased platelet reactivity [79, 80] as well as increased activation of neutrophils [81]. Accordingly, one mechanism potentially contributing to an increased risk of mortality is a paradoxical increase in platelet reactivity. Biophysical studies have demonstrated that antagonists which bind to the RGD sequence of GP IIb-IIIa induce a conformational change of the integrin from its resting state [82, 83]. A model has been proposed whereby binding of antagonists to GP IIb-IIIa caused weak outside-in signaling insufficient to activate platelets and that pre-activation with ADP was required for strong platelet activation [84]. A recent study demonstrated that GP IIb-IIIa antagonists potentiate activation of glycoprotein VI-associated FcR gamma-chain phosphorylation [85]. Phosphorylation of downstream signaling molecules ensued and correlated with increased dense granule secretion, cytosolic calcium release and exposure of phosphatidylserine on the platelet surface. Antagonism of P2Y12 abolished the potentiated phosphatidylserine exposure and dense granule secretion but not the cytosolic calcium release. Accordingly, these results support the hypothesis that binding of antagonists to GP IIb-IIIa initiates outside-in signaling that can potentiate activation of platelets. This effect can be attenuated by agents such as clopidogrel which blocks P2Y12.
Genetic polymorphisms and GP IIb-IIIa antagonists
Genetic polymorphisms have been shown to influence platelet function. The glycoprotein IIIa Leu33Pro polymorphism has been associated with increased platelet microaggregation but does not affect the inhibitory effect of GP IIb-IIIa antagonists [86]. The PlA2 allele of GP IIIa appears to confer an additional risk for development of atherosclerosis and arterial thrombosis when combined with other pro-atherogenic/pro-thrombotic factors such as type 2 diabetes [87]. This polymorphism has not been associated with alteration in the effect of GP IIb-IIIa antagonists. Genotyping of known genetic variants implicated in primary coronary disease in 924 Caucasians with acute coronary syndromes participating in the OPUS-TIMI16 trial of the GPIIb/IIIa antagonist orbofiban demonstrated no significant impact on the composite endpoint (P = 0.88), which included myocardial infarction, death, recurrent ischaemia, urgent revascularization and stroke, but a significant impact on recurrent myocardial infarction alone (P = 0.04). There was a significant interaction of the polymorphisms with orbofiban treatment influencing bleeding outcomes (P = 0.004) [88]. Thus, although genetic polymorphisms do not appear to influence the effect of the agents, they may influence outcomes.
Summary, implications and future directions
In summary, intravenous GP IIb-IIIa antagonists reduce the subsequent incidence of cardiac events when used in the setting of PCI. The clinical benefit of these agents is greatest when used in patients with intra-coronary thrombus who are undergoing PCI. Despite the availability of more powerful oral anti-platelet agents, the use of GP IIb-IIIa antagonists will be associated with greatest clinical benefit in the following clinical scenarios:
Conditions associated with a high likelihood of intracoronary thrombosis, such as ST-elevation and non-ST-elevation myocardial infarction where an invasive/interventional strategy is planned.
When the anticoagulant is heparin rather than bivalirudin.
When the patient has not been treated before PCI with a P2Y12 antagonist.
In unstable ACS patients who require transfer to a PCI centre.
To reduce the risk of stent thrombosis in a patient with ST-elevation myocardial infarction (particularly when there is not adequate pretreatment with a P2Y12 antagonist).
The development of newer antiplatelet agents that have a more rapid onset of action and are more powerful antagonists of platelet function demands a reassessment of the role of GP IIb-IIIa antagonists. GP IIb-IIIa antagonists are likely to be of greatest value when short term intense antiplatelet therapy is indicated, such as during PCI. The optimal duration of treatment with GP IIb-IIIa antagonists should be re-evaluated.
Competing Interests
During the past five years DS was served a consultant, accepted research grants, and had been reimbursed for speaking from the following pharmaceutical companies: Merck, Guilford Pharmaceuticals, the Medicine Company, BMS, and Sanofi-Aventis.
REFERENCES
- 1.Nurden AT, Caen JP. An abnormal platelet glycoprotein pattern in threes cases of Glanzmann thrombasthenia. Br J Haematol. 1974;28:253–60. doi: 10.1111/j.1365-2141.1974.tb06660.x. [DOI] [PubMed] [Google Scholar]
- 2.Phillips DR, Jenkins CSP, Luscher EF, Larrieu MJ. Molecular differences of exposed surface proteins on thrombasthenic platelet plasma membranes. Nature. 1975;257:599–600. doi: 10.1038/257599a0. [DOI] [PubMed] [Google Scholar]
- 3.Kunicki TJ, Pidard D, Rosa JP, Nurden AT. The formation of calcium-dependent complexes of platelet membrane glycoproteins IIb and IIIa in solution as determined by crossed immunoelectrophoresis. Blood. 1981;58:268–78. [PubMed] [Google Scholar]
- 4.Coller BS. Interaction of normal, thrombasthenic, and Bernard-Soulier platelets with immobilized fibrinogen: defective platelet-fibrinogen interaction in thrombasthenia. Blood. 1980;55:169–78. [PubMed] [Google Scholar]
- 5.Bennett JS, Vilaire G. Exposure of platelet fibrinogen receptors by ADP and epinephrine. J Clin Invest. 1979;64:1393–401. doi: 10.1172/JCI109597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peerschke EI. The platelet fibrinogen receptor. Semin Hematol. 1985;22:241–59. [PubMed] [Google Scholar]
- 7.Gogstad GO, Brosstad F, Krutnes MB, Hagen I, Solum NO. Fibrinogen-binding properties of the human platelet glycoprotein IIb-IIIa complex: a study using crossed-radioimmunoelectrophoresis. Blood. 1982;60:663–71. [PubMed] [Google Scholar]
- 8.Parise LV, Phillips DR. Reconstitution of the purified platelet fibrinogen receptor: fibrinogen binding properties of the glycoprotein IIb-IIIa complex. J Biol Chem. 1985;260:10698–707. [PubMed] [Google Scholar]
- 9.Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 1984;309:30–3. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- 10.Plow EF, Marguerie G, Ginsberg M. Fibrinogen, fibrinogen receptors, and the peptides that inhibit these interactions. Biochem Pharmacol. 1987;36:4035–40. doi: 10.1016/0006-2952(87)90558-2. [DOI] [PubMed] [Google Scholar]
- 11.Kloczewiak M, Timmons S, Hawiger J. Recognition site for the platelet receptor is present on the 15-residue carboxy-terminal fragment of the gamma chain of human fibrinogen and is not involved in the fibrin polymerization reaction. Thromb Res. 1983;29:249–55. doi: 10.1016/0049-3848(83)90147-0. [DOI] [PubMed] [Google Scholar]
- 12.Weisel JW, Nagaswami C, Vilaire G, Bennett JS. Examination of the platelet membrane glycoprotein IIb-IIIa complex and its interaction with fibrinogen and other ligands by electron microscopy. J Biol Chem. 1992;267:16637–43. [PubMed] [Google Scholar]
- 13.Gralnick HR, Williams SB, Coller BS. Fibrinogen competes with von Willebrand factor for binding to the glycoprotein IIb/IIIa complex when platelets are stimulated with thrombin. Blood. 1984;64:797–800. [PubMed] [Google Scholar]
- 14.Weiss HJ, Hawiger J, Ruggeri ZM, Turritto VT, Thiagarajan P, Hoffman T. Fibrinogen-independent platelet adhesion and thrombus formation on subendothelium mediated by glycoprotein IIb-IIIa complex at high shear rate. J Clin Invest. 1989;83:288–97. doi: 10.1172/JCI113871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett JS, Hoxie JA, Leitman SF, Vilaire G, Cines DB. Inhibition of fibrinogen binding to stimulated human platelets by a monoclonal antibody. Proc Natl Acad Sci USA. 1983;80:2417–21. doi: 10.1073/pnas.80.9.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coller BS, Peerschke EI, Scudder LE, Sullivan CA. A murine monoclonal antibody that completely blocks the binding of fibrinogen to platelets produces a thrombasthenic-like state in normal platelets and binds to glycoproteins IIb and/or IIIa. J Clin Invest. 1983;72:325–38. doi: 10.1172/JCI110973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coller BS, Scudder LE. Inhibition of dog platelet function by in vivo infusion of F(ab′)2 fragments of a monoclonal antibody. Blood. 1985;66:1456–9. [PubMed] [Google Scholar]
- 18.Tcheng JE, Ellis SG, George BS, Kereiakes DJ, Kleiman NS, Talley JD, Wang AL, Weisman HF, Califf RM, Topol EJ. Pharmacodynamics of chimeric glycoprotein IIb/IIIa integrin antiplatelet antibody Fab 7E3 in high-risk coronary angioplasty. Circulation. 1994;90:1757–64. doi: 10.1161/01.cir.90.4.1757. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki K, Sakai Y, Hisamichi N, Taniuchi Y, Sato K, Terazaki C, Kaku S, Kawasaki T, Yano S, Inagaki O, Masuho Y. Comparison of the antiplatelet effect of YM 337 and abciximab in rhesus monkeys. Eur J Pharmacol. 1997;336:169–76. doi: 10.1016/s0014-2999(97)01241-7. [DOI] [PubMed] [Google Scholar]
- 20.Philip DR, Charo IF, Scarborough RM. GP IIb/IIIa; the responsive integrin. Cell. 1991;65:359–62. doi: 10.1016/0092-8674(91)90451-4. [DOI] [PubMed] [Google Scholar]
- 21.Jordan RE, Wagner CL, Mascelli MA, Treacy G, Nedelman MA, Weissman HF, Coller BS. Preclinical development of c7E3: a mouse/human chimeric monoclonal antibody fragment that inhibits platelet function by blockade of GP IIb/IIIa receptors with observations on the immunogenicity of c7E3 in humans. In: Horton MA, editor. Adhesion Receptors As Therapeutic Targets. Boca Raton, FL: CRC Press; 1996. pp. 281–305. [Google Scholar]
- 22.Kleiman NS, Raizner AE, Jordan R, Wang AL, Norton D, Mace KF, Joshi A, Coller BS, Weisman HF. Differential inhibition of platelet aggregation induced by adenosine triphosphate or a thrombin receptor-activating peptide in patients treated with bolus chimeric 7E3 Fab: implications for inhibition of the internal pool of GP IIb/IIIa inhibitors. J Am Coll Cardiol. 1995;26:1665–71. doi: 10.1016/0735-1097(95)00391-6. [DOI] [PubMed] [Google Scholar]
- 23.Mascelli MA, Lance ET, Damaraju L, Wagner CL, Weisman HF, Jordan RE. Pharmacodynamic profile of short-term abciximab treatment demonstrates prolonged platelet inhibition with gradual recovery from GP IIb/IIIa receptor blockade. Circulation. 1998;97:1680–8. doi: 10.1161/01.cir.97.17.1680. [DOI] [PubMed] [Google Scholar]
- 24.Tam SH, Sassoli PM, Jordan RE, Nakada MT. Abciximab (Reopro, chimeric 7E3 Fab) demonstrates equivalent affinity and functional blockade of glycoprotein IIb/IIIa and αvβ3 integrins. Circulation. 1998;98:1085–91. doi: 10.1161/01.cir.98.11.1085. [DOI] [PubMed] [Google Scholar]
- 25.Simon DI, Xu H, Ortlepp S, Rogers C, Rao NK. 7E3 monoclonal antibody directed against the platelet glycoprotein IIb/IIIa cross-reacts with the leukocyte integrin Mac-1 and blocks adhesion to fibrinogen and ICAM-1. Arterioscler Thromb Vasc Biol. 1997;17:528–35. doi: 10.1161/01.atv.17.3.528. [DOI] [PubMed] [Google Scholar]
- 26.Stouffer GA, Hu Z, Sajid M, Li H, Jin G, Nakada MT, Hanson SR, Runge MS. Beta 3 integrins are upregulated after vascular injury and modulate thrombospondin- and thrombin-induced proliferation of cultured smooth muscle cells. Circulation. 1998;97:907–15. doi: 10.1161/01.cir.97.9.907. [DOI] [PubMed] [Google Scholar]
- 27.Tcheng JE, Harrington RA, Kottke-Marchant K, Kleiman NS, Ellis SG, Kereiakes DJ, Mick MJ, Navetta FI, Smith JE, Worley SJ, Miller JA, Joseph DM, Sigmon KN, Kitt MM, DuMee CM, Califf RM, Topor EJ. Multicenter, randomized, double-blind, placebo-controlled trial of the platelet integrin glycoprotein IIb/IIIa blocker integrelin in elective coronary intervention. Circulation. 1995;91:2151–7. doi: 10.1161/01.cir.91.8.2151. [DOI] [PubMed] [Google Scholar]
- 28.Kereiakes DJ, Kleiman NS, Ambrose J, Cohen M, Rodriguez S, Palabrica T, Herrmann HC, Sutton JM, Weaver WD, McKee DB, Fitzpatrick V, Sax FL. Randomized, double-blind study of tirofiban (MK-383) platelet IIb-IIIa blockade in high risk patients undergoing coronary angioplasty. J Am Coll Cardiol. 1996;27:536–42. doi: 10.1016/0735-1097(95)00500-5. [DOI] [PubMed] [Google Scholar]
- 29.Phillips DR, Scarborough RM. Clinical pharmacology of eptifibatide. Am J Cardiol. 1997;80:11B–20B. doi: 10.1016/s0002-9149(97)00572-9. [DOI] [PubMed] [Google Scholar]
- 30.Moussa SA, Bennet JS. Platelets in health and disease: platelet GP IIb/IIIa structure and function: recent advances in antiplatelet therapy. Drugs Future. 1996;21:1141–54. [Google Scholar]
- 31.Coller BS. Monitoring platelet GP IIb/IIIa antagonist therapy. Circulation. 1998;97:5–9. doi: 10.1161/01.cir.97.1.5. [DOI] [PubMed] [Google Scholar]
- 32.Born GV. Platelets in thrombogenesis: mechanism and inhibition of platelet aggregation. Ann R Coll Surg Engl. 1965;36:200–6. [PMC free article] [PubMed] [Google Scholar]
- 33.Schneider DJ, Tracy PB, Mann KG, Sobel BE. Differential effects of anticoagulants on the activation of platelets ex vivo. Circulation. 1997;96:2877–83. doi: 10.1161/01.cir.96.9.2877. *In vitro study demonstrating that chelation of calcium alters platelet function. [DOI] [PubMed] [Google Scholar]
- 34.Rebello SS, Huang J, Faul JD, Lucchesi BR. Role of extracellular ionized calcium in the in vitro assessment of GPIIb/IIIa receptor antagonists. J Thromb Thrombolysis. 2000;9:23–8. doi: 10.1023/a:1018679708251. [DOI] [PubMed] [Google Scholar]
- 35.Kereiakes DJ, Lorenz T, Young JJ, Kukielka G, Mueller MN, Nanniazzi-Alaimo L, Phillips DR. Differential effects of citrate versus PPACK anticoagulation on measured platelet inhibition by abciximab, eptifibatide and tirofiban. J Thromb Thrombolysis. 2001;12:123–7. doi: 10.1023/a:1012991303381. [DOI] [PubMed] [Google Scholar]
- 36.Phillips DR, Teng W, Arfsten A, Nannizzi-Alaimo L, White MM, Longhurst C, Shattil SJ, Randolph A, Jakubowski JA, Jennings LK, Scarborough RM. Effect of Ca2+ on GP IIb-IIIa interactions with integrelin: enhanced GP IIb-IIIa binding and inhibition of platelet aggregation by reductions in concentration of ionized calcium in plasma anticoagulated with citrate. Circulation. 1997;96:1488–94. doi: 10.1161/01.cir.96.5.1488. [DOI] [PubMed] [Google Scholar]
- 37.Kabbani SS, Aggarwal A, Terrien ET, DiBattiste PM, Sobel BE, Schneider DJ. Suboptimal early inhibition of platelets by treatment with tirofiban and implications for coronary interventions. Am J Cardiol. 2002;8:647–50. doi: 10.1016/s0002-9149(01)02319-0. [DOI] [PubMed] [Google Scholar]
- 38.Herrmann HC, Swierkosz TA, Kapoor S, Tardiff DC, DiBattiste PM, Hirshfeld JW, Klugherz BD, Kolansky DM, Magness K, Valettas N, Wilensky RL. Comparison of degree of platelet inhibition by abciximab-vs-tirofiban in patients with unstable angina and non-q-wave myocardial infarction undergoing percutaneous coronary intervention. Am J Cardiol. 2002;89:1293–6. doi: 10.1016/s0002-9149(02)02329-9. [DOI] [PubMed] [Google Scholar]
- 39.Lakkis N, Lakiss N, Bobek J, Farmer J. Platelet inhibition with tirofiban early during PCI: dosing revisited. Catheter Cardiovasc Interv. 2002;56:474–7. doi: 10.1002/ccd.10238. [DOI] [PubMed] [Google Scholar]
- 40.Schneider DJ, Herrmann HC, Lakkis N, Aguirre F, Wan Y, Aggarwal A, Kabbani SS, DiBattiste PM. Enhanced early inhibition of platelet aggregation with an increased bolus of tirofiban. Am J Cardiol. 2002;90:1421–3. doi: 10.1016/s0002-9149(02)02892-8. [DOI] [PubMed] [Google Scholar]
- 41.Schneider DJ, Herrmann HC, Lakkis N, Aguirre F, Lo MW, Yin KC, Aggarwal A, Kabbani SS, DiBattiste PM. Increased concentrations of tirofiban in blood and their correlation with inhibition of platelet aggregation after greater bolus doses of tirofiban. Am J Cardiol. 2003;91:334–6. doi: 10.1016/s0002-9149(02)03163-6. [DOI] [PubMed] [Google Scholar]
- 42.Lincoff AM, Califf RM, Topol EJ. Platelet glycoprotein IIb/IIIa receptor blockade in coronary artery disease. J Am Coll Cardiol. 2000;35:1103–15. doi: 10.1016/s0735-1097(00)00554-4. [DOI] [PubMed] [Google Scholar]
- 43.Boersma E, Akkerhuis KM, Theroux P, Califf RM, Topol EJ, Simoons ML. Platelet glycoprotein IIb/IIIa receptor inhibition in non-ST-elevation acute coronary syndromes: early benefit during medical treatment only, with additional protection during percutaneous coronary intervention. Circulation. 1999;100:2045–8. doi: 10.1161/01.cir.100.20.2045. [DOI] [PubMed] [Google Scholar]
- 44.EPIC Investigators. Use of a monoclonal antibody directed against the platelet glycoprotein IIb/IIIa receptor in high-risk coronary angioplasty. N Engl J Med. 1994;330:956–61. doi: 10.1056/NEJM199404073301402. [DOI] [PubMed] [Google Scholar]
- 45.EPILOG Investigators. Platelet glycoprotein IIb/IIIa blockade with abciximab with low-dose heparin during percutaneous coronary revascularization. N Engl J Med. 1997;336:1689–96. doi: 10.1056/NEJM199706123362401. [DOI] [PubMed] [Google Scholar]
- 46.EPISTENT Investigators. Randomized placebo-controlled and balloon-angioplasty controlled trial to assess safety of coronary stenting with use of platelet glycoprotein IIb/IIIa blockade. Lancet. 1998;352:87–92. doi: 10.1016/s0140-6736(98)06113-3. [DOI] [PubMed] [Google Scholar]
- 47.Kastrati A, Mehilli J, Schühlen H, Dirschinger J, Dotzer F, ten Berg JM, Neumann FJ, Bollwein H, Volmer C, Gawaz M, Berger PB, Schömig A, Intracoronary Stenting and Antithrombotic Regimen-Rapid Early Action for Coronary Treatment Study Investigators A clinical trial of abciximab in elective percutaneous coronary intervention after pretreatment with clopidogrel. N Engl J Med. 2004;350:232–8. doi: 10.1056/NEJMoa031859. [DOI] [PubMed] [Google Scholar]
- 48.Kastrati A, Mehilli J, Neumann FJ, Dotzer F, ten Berg J, Bollwein H, Graf I, Ibrahim M, Pache J, Seyfarth M, Schühlen H, Dirschinger J, Berger PB, Schömig A, Intracoronary Stenting and Antithrombotic Regimen-Rapid Early Action for Coronary Treatment Study Investigators Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA. 2006;295:1531–8. doi: 10.1001/jama.295.13.joc60034. [DOI] [PubMed] [Google Scholar]
- 49.Simoons ML, GUSTO IV-ACS Investigators Effect of glycoprotein IIb/IIIa receptor blocker abciximab on outcome in patients with acute coronary syndromes without early coronary revascularisation: the GUSTO IV-ACS randomised trial. Lancet. 2001;357:1915–24. doi: 10.1016/s0140-6736(00)05060-1. [DOI] [PubMed] [Google Scholar]
- 50.Topol EJ, GUSTO V Investigators Reperfusion therapy for acute myocardial infarction with fibrinolytic therapy or combination reduced fibrinolytic therapy and platelet glycoprotein IIb/IIIa inhibition: the GUSTO V randomised trial. Lancet. 2001;357:1905–14. doi: 10.1016/s0140-6736(00)05059-5. [DOI] [PubMed] [Google Scholar]
- 51.Lincoff AM, Califf RM, Van de Werf F, Willerson JT, White HD, Armstrong PW, Guetta V, Gibler WB, Hochman JS, Bode C, Vahanian A, Steg PG, Ardissino D, Savonitto S, Bar F, Sadowski Z, Betriu A, Booth JE, Wolski K, Waller M, Topol EJ, Global Use of Strategies to Open Coronary Arteries Investigators (GUSTO) Mortality at 1 year with combination platelet glycoprotein IIb/IIIa inhibition and reduced-dose fibrinolytic therapy vs conventional fibrinolytic therapy for acute myocardial infarction: GUSTO V randomized trial. JAMA. 2002;288:2130–5. doi: 10.1001/jama.288.17.2130. [DOI] [PubMed] [Google Scholar]
- 52.Montalescot G, Barragan P, Wittenberg O, Ecollan P, Elhadad S, Villain P, Boulenc JM, Morice MC, Maillard L, Pansieri M, Choussat R, Pinton P, ADMIRAL Investigators Platelet glycoprotein IIb/IIIa inhibition with coronary stenting for acute myocardial infarction. N Engl J Med. 2001;344:1895–903. doi: 10.1056/NEJM200106213442503. [DOI] [PubMed] [Google Scholar]
- 53.Stone GW, Grines CL, Cox DA, Garcia E, Tcheng JE, Griffin JJ, Guagliumi G, Stuckey T, Turco M, Carroll JD, Rutherford BD, Lansky AJ, Controlled Abciximab and Device Investigation to Lower Late Angioplasty Complications (CADILLAC) Investigators Comparison of angioplasty with stenting, with or without abciximab, in acute myocardial infarction. N Engl J Med. 2002;346:957–66. doi: 10.1056/NEJMoa013404. [DOI] [PubMed] [Google Scholar]
- 54.Thiele H, Schindler K, Friedenberger J, Eitel I, Fürnau G, Grebe E, Erbs S, Linke A, Möbius-Winkler S, Kivelitz D, Schuler G. Intracoronary compared with intravenous bolus abciximab application in patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention: the randomized Leipzig immediate percutaneous coronary intervention abciximab IV versus IC in ST-elevation myocardial infarction trial. Circulation. 2008;118:49–57. doi: 10.1161/CIRCULATIONAHA.107.747642. [DOI] [PubMed] [Google Scholar]
- 55.Lincoff AM, Bittl JA, Harrington RA, Feit F, Kleiman NS, Jackman JD, Sarembock IJ, Cohen DJ, Spriggs D, Ebrahimi R, Keren G, Carr J, Cohen EA, Betriu A, Desmet W, Kereiakes DJ, Rutsch W, Wilcox RG, de Feyter PJ, Vahanian A, Topol EJ, REPLACE-2 Investigators Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA. 2003;289:853–63. doi: 10.1001/jama.289.7.853. [DOI] [PubMed] [Google Scholar]
- 56.Stone GW, McLaurin BT, Cox DA, Bertrand ME, Lincoff AM, Moses JW, White HD, Pocock SJ, Ware JH, Feit F, Colombo A, Aylward PE, Cequier AR, Darius H, Desmet W, Ebrahimi R, Hamon M, Rasmussen LH, Rupprecht HJ, Hoekstra J, Mehran R, Ohman EM, ACUITY Investigators Bivalirudin for patients with acute coronary syndromes. N Engl J Med. 2006;355:2203–16. doi: 10.1056/NEJMoa062437. [DOI] [PubMed] [Google Scholar]
- 57.IMPACT II investigators. Randomized placebo-controlled trial of effect of eptifibatide on complications of percutaneous coronary intervention. IMPACT II. Lancet. 1997;349:1422–8. [PubMed] [Google Scholar]
- 58.Gilchrist IC, O'Shea JC, Kosoglou T, Jennings LK, Lorenz TJ, Kitt MM, Kleiman NS, Talley D, Aguirre F, Davidson C, Runyon J, Tcheng JE. Pharmacodynamics and pharmacokinetics of higher-dose, double-bolus eptifibatide in percutaneous coronary intervention. Circulation. 2001;104:406–11. doi: 10.1161/hc2901.093504. [DOI] [PubMed] [Google Scholar]
- 59.The ESPRIT Investigators. Novel dosing regimen of eptifibatide in planned coronary stent implantation (ESPRIT): a randomized, placebo-controlled trial. Lancet. 2000;356:2037–44. doi: 10.1016/S0140-6736(00)03400-0. [DOI] [PubMed] [Google Scholar]
- 60.The PURSUIT Trial Investigators. Inhibition of platelet glycoprotein IIb/IIIa with eptifibatide in patients with acute coronary syndromes. N Engl J Med. 1998;339:436–43. doi: 10.1056/NEJM199808133390704. [DOI] [PubMed] [Google Scholar]
- 61.Kleiman NS, Lincoff AM, Flaker GC, Pieper KS, Wilcox RG, Berdan LG, Lorenz TJ, Cokkinos DV, Simoons ML, Boersma E, Topol EJ, Califf RM, Harrington RA. Early percutaneous coronary intervention, platelet inhibition with eptifibatide, and clinical outcomes in patients with acute coronary syndromes. Circulation. 2000;101:751–7. doi: 10.1161/01.cir.101.7.751. [DOI] [PubMed] [Google Scholar]
- 62.Giugliano RP, White JA, Bode C, Armstrong PW, Montalescot G, Lewis BS, van't Hof A, Berdan LG, Lee KL, Strony JT, Hildemann S, Veltri E, Van de Werf F, Braunwald E, Harrington RA, Califf RM, Newby LK, EARLY ACS Investigators Early versus delayed, provisional eptifibatide in acute coronary syndromes. N Engl J Med. 2009;360:2176–90. doi: 10.1056/NEJMoa0901316. [DOI] [PubMed] [Google Scholar]
- 63.Stone GW, Bertrand ME, Moses JW, Ohman EM, Lincoff AM, Ware JH, Pocock SJ, McLaurin BT, Cox DA, Jafar MZ, Chandna H, Hartmann F, Leisch F, Strasser RH, Desaga M, Stuckey TD, Zelman RB, Lieber IH, Cohen DJ, Mehran R, White HD, ACUITY Investigators Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA. 2007;297:591–602. doi: 10.1001/jama.297.6.591. [DOI] [PubMed] [Google Scholar]
- 64.The RESTORE Investigators. Effects of platelet glycoprotein IIb/IIIa blockade with tirofiban on adverse cardiac events in patients with unstable angina or acute myocardial infarction undergoing coronary angioplasty. Circulation. 1997;96:1445–53. doi: 10.1161/01.cir.96.5.1445. [DOI] [PubMed] [Google Scholar]
- 65.The PRISM-PLUS Investigators. Inhibition of the platelet glycoprotein IIb/IIIa receptor with tirofiban in unstable angina and non-Q-wave myocardial infarction. N Engl J Med. 1998;338:1488–97. doi: 10.1056/NEJM199805213382102. [DOI] [PubMed] [Google Scholar]
- 66.Cannon CP, Weintraub WS, Demopoulos LA, Vicari R, Frey MJ, Lakkis N, Neumann FJ, Robertson DH, DeLucca PT, DiBattiste PM, Gibson CM, Braunwald E. Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med. 2001;344:1879–87. doi: 10.1056/NEJM200106213442501. [DOI] [PubMed] [Google Scholar]
- 67.Théroux P, Alexander J, Jr, Dupuis J, Pesant Y, Gervais P, Grandmont D, Kouz S, Laramée P, Huynh T, Barr E, Sax FL, PRISM-PLUS Investigators Upstream use of tirofiban in patients admitted for an acute coronary syndrome in hospitals with or without facilities for invasive management. PRISM-PLUS Investigators. Am J Cardiol. 2001;87:375–80. doi: 10.1016/s0002-9149(00)01386-2. [DOI] [PubMed] [Google Scholar]
- 68.Topol EJ, Moliterno DJ, Herrmann HC, Powers ER, Grines CL, Cohen DJ, Cohen EA, Bertrand M, Neumann FJ, Stone GW, DiBattiste PM, Demopoulos L. Comparison of two platelet glycoprotein IIb/IIIa inhibitors, tirofiban and abciximab, for the prevention of ischemic events with percutaneous coronary revascularization. N Engl J Med. 2001;344:1888–94. doi: 10.1056/NEJM200106213442502. [DOI] [PubMed] [Google Scholar]
- 69.Valgimigli M, Biondi-Zoccai G, Tebaldi M, van't Hof AW, Campo G, Hamm C, ten Berg J, Bolognese L, Saia F, Danzi GB, Briguori C, Okmen E, King SB, Moliterno DJ, Topol EJ. Tirofiban as adjunctive therapy for acute coronary syndromes and percutaneous coronary intervention: a meta-analysis of randomized trials. Eur Heart J. 2010;31:35–49. doi: 10.1093/eurheartj/ehp376. [DOI] [PubMed] [Google Scholar]
- 70.Van't Hof AW, ten Berg J, Heestermans T, Dill T, Funck RC, van Werkum W, Dambrink JH, Suryapranata H, van Houwelingen G, Ottervanger JP, Stella P, Giannitsis E, Hamm C. Prehospital initiation of tirofiban in patients with ST-elevation myocardial infarction undergoing primary angioplasty (On-TIME 2): a multicentre, double-blind, randomised controlled trial. Lancet. 2008;372:537–46. doi: 10.1016/S0140-6736(08)61235-0. [DOI] [PubMed] [Google Scholar]
- 71.Heestermans AA, Van Werkum JW, Hamm C, Dill T, Gosselink AT, De Boer MJ, Van Houwelingen G, Hoorntje JC, Koopmans PC, Ten Berg JM, Van't Hof AW. Marked reduction of early stent thrombosis with pre-hospital initiation of high-dose tirofiban in ST-segment elevation myocardial infarction. J Thromb Haemost. 2009;7:1612–8. doi: 10.1111/j.1538-7836.2009.03573.x. [DOI] [PubMed] [Google Scholar]
- 72.Beinart R, Abu Sham'a R, Segev A, Hod H, Guetta V, Shechter M, Boyko V, Behar S, Matetzky S. The incidence and clinical predictors of early stent thrombosis in patients with acute coronary syndrome. Am Heart J. 2010;159:118–24. doi: 10.1016/j.ahj.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 73.O'Neil WW, Serruys P, Knudtson M, van Es GA, Timmis GC, van der Zwaan C, Kleiman J, Gong J, Roecker EB, Dreiling R, Alexander J, Anders R. Long-term treatment with a platelet glycoprotein-receptor antagonist after percutaneous coronary revascularization: EXCITE Trial Investigators: Evaluation of Oral Xemilofiban in Controlling Thrombotic Events. N Engl J Med. 2000;342:1316–24. doi: 10.1056/NEJM200005043421803. [DOI] [PubMed] [Google Scholar]
- 74.Cannon CP, McCabe CH, Wilcox RG, Langer A, Caspi A, Berink P, Lopez-Sendon J, Toman J, Charlesworth A, Anders RJ, Alexander JC, Skene A, Braunwald E. Oral glycoprotein IIb/IIIa inhibition with orofiban in patients with unstable coronary syndromes (OPUS-TIMI 16) trial. Circulation. 2000;102:149–56. doi: 10.1161/01.cir.102.2.149. [DOI] [PubMed] [Google Scholar]
- 75.SYMPHONY Investigators. Comparison of sibrafiban with aspirin for prevention of cardiovascular events after acute coronary syndromes: a randomized trial: Sibrafiban Versus Aspirin to Yield Maximum Protection From Ischemic Heart Events Post-Acute Coronary Syndromes. Lancet. 2000;355:337–45. [PubMed] [Google Scholar]
- 76.Second SYMPHONY Investigators. Randomized trial of aspirin, sibrafiban, or both for secondary prevention after acute coronary syndromes. Circulation. 2001;103:1727–33. doi: 10.1161/01.cir.103.13.1727. [DOI] [PubMed] [Google Scholar]
- 77.Topol EJ, Easton JD, Amarenco P, Califf R, Harrington R, Graffagnino C, Davis S, Diener HC, Ferguson J, Fitzgerald D, Shuaib A, Koudstaal PJ, Theroux P, Van de Werf F, Willerson JT, Chan R, Samuels R, Ilson B, Granett J. Design of the blockade of the glycoprotein IIb/IIIa receptor to avoid vascular occlusion (BRAVO) trial. Am Heart J. 2000;139:927–33. doi: 10.1067/mhj.2000.105107. [DOI] [PubMed] [Google Scholar]
- 78.Chew DP, Bhatt DL, Sapp S, Topol EJ. Increased mortality with oral platelet glycoprotein IIb/IIIa antagonists. A meta-analysis of phase III multicenter randomized trials. Circulation. 2001;103:201–6. doi: 10.1161/01.cir.103.2.201. [DOI] [PubMed] [Google Scholar]
- 79.Holmes MB, Sobel BE, Cannon CP, Schneider DJ. Increased platelet reactivity in patients given orbofiban after an acute coronary syndrome: an OPUS-TIMI 16 substudy. Orbofiban in Patients with Unstable coronary syndromes. Thrombolysis in Myocardial Infarction. Am J Cardiol. 2000;85:491–3. doi: 10.1016/s0002-9149(99)00778-x. [DOI] [PubMed] [Google Scholar]
- 80.Cox D, Smith R, Quinn M, Theroux P, Crean P, Fitzgerald DJ. Evidence of platelet activation during treatment with a GPIIb/IIIa antagonist in patients presenting with acute coronary syndromes. J Am Coll Cardiol. 2000;36:1514–9. doi: 10.1016/s0735-1097(00)00919-0. [DOI] [PubMed] [Google Scholar]
- 81.Serrano CV, Jr, Nicolau JC, Venturinelli M, Baracioli LM, Anders RJ, Cannon CP, Ramires JA. Role of oral blockade of platelet glycoprotein IIb/IIIa on neutrophil activation in patients with acute coronary syndromes. Cardiovasc Drugs Ther. 2003;17:129–32. doi: 10.1023/a:1025335718142. [DOI] [PubMed] [Google Scholar]
- 82.Hantgan RR, Stahle MC. Integrin priming dynamics: mechanisms of integrin antagonist-promoted alphaIIbbeta3:PAC-1 molecular recognition. Biochemistry. 2009;48:8355–65. doi: 10.1021/bi900475k. [DOI] [PubMed] [Google Scholar]
- 83.Hantgan RR, Stahle MC, Connor JH, Connor RF, Mousa SA. alphaIIbbeta3 priming and clustering by orally active and intravenous integrin antagonists. J Thromb Haemost. 2007;5:542–50. doi: 10.1111/j.1538-7836.2007.02351.x. [DOI] [PubMed] [Google Scholar]
- 84.Bassler N, Loeffler C, Mangin P, Yuan Y, Schwarz M, Hagemeyer CE, Eisenhardt SU, Ahrens I, Bode C, Jackson SP, Peter K. A mechanistic model for paradoxical platelet activation by ligand-mimetic alphaIIb beta3 (GPIIb/IIIa) antagonists. Arterioscler Thromb Vasc Biol. 2007:27. doi: 10.1161/01.ATV.0000255307.65939.59. [DOI] [PubMed] [Google Scholar]
- 85.Jones ML, Harper MT, Aitken EW, Williams CM, Poole AW. RGD-ligand mimetic antagonists of integrin alphaIIbbeta3 paradoxically enhance GPVI-induced human platelet activation. J Thromb Haemost. 2010;8:567–76. doi: 10.1111/j.1538-7836.2009.03719.x. [DOI] [PubMed] [Google Scholar]
- 86.Sirotkina OV, Khaspekova SG, Zabotina AM, Shimanova YV, Mazurov AV. Effects of platelet glycoprotein IIb-IIIa number and glycoprotein IIIa Leu33Pro polymorphism on platelet aggregation and sensitivity to glycoprotein IIb-IIIa antagonists. Platelets. 2007;18:506–14. doi: 10.1080/09537100701326739. [DOI] [PubMed] [Google Scholar]
- 87.Pellitero S, Reverter JL, Tàssies D, Pizarro E, Monteagudo J, Salinas I, Aguilera E, Sanmartí A, Reverter JC. Polymorphisms in platelet glycoproteins Ia and IIIa are associated with arterial thrombosis and carotid atherosclerosis in type 2 diabetes. Thromb Haemost. 2010;103:630–7. doi: 10.1160/TH09-07-0453. [DOI] [PubMed] [Google Scholar]
- 88.Shields DC, Fitzgerald AP, O'Neill PA, Muckian C, Kenny D, Moran B, Cannon CP, Byrne CE, Fitzgerald DJ. The contribution of genetic factors to thrombotic and bleeding outcomes in coronary patients randomised to IIb/IIIa antagonists. Pharmacogenomics J. 2002;2:182–90. doi: 10.1038/sj.tpj.6500100. [DOI] [PubMed] [Google Scholar]