

Abstract
The human papillomavirus (HPV) directly infects cervical keratinocytes and interferes with TLR signalling. To shed light on the effect of HPV on upstream receptors, we evaluated TLRs 1–9 gene expression in HPV-negative normal and HPV-positive pre-malignant and malignant ex vivo cervical tissue. Quantitative real-time polymerase chain reaction was performed separately for epithelial and stromal tissue compartments. Differences in gene expression were analyzed by the Jonckheere-Terpstra trend test or the Student's t-test for pairwise comparison. Laser capture microdissection revealed an increase in TLR3 and a decrease in TLR1 mRNA levels in dysplastic and carcinoma epithelium, respectively. In the stroma, a trend of increasing TLR 1, 2, 5, 6, and 9 mRNA levels with disease severity was found. These findings implicate the involvement of TLR3 and TLR1 in early and late cervical carcinogenesis, respectively, suggesting that stromal upregulation of TLRs may play a role in cervical disease progression.
1. Introduction
The most important role of toll-like receptors (TLRs) in host defence is the regulation of innate and adaptive immune responses by epithelial cells, the first line of protection at, for example, the respiratory, gastrointestinal tract, skin and genitourinary mucosal sites. Present on a variety of cell types, TLRs play an essential role in innate immune system function by recognizing discrete exogenous pathogen-associated molecular patterns (PAMPs) and endogenous damage-associated molecular pattern (DAMP) ligands to induce an innate immune response [1]. Ten functional TLRs have been identified and characterized in humans. Among these, TLRs 3, 4, 7, 8, and 9 have been found to play a critical role in antiviral immunity by triggering the downstream production of interferons (IFNs) including IFN-α, IFN-β and IFN-γ [1, 2]. IFNs are important components of the innate immune response against invading pathogens. They are synthesized during initial infection and evoke antiviral, antitumour, and immune-regulatory activity which gives protection to surrounding cells [3–5]. The IFN group is split into two classes; Type I IFNs encompass IFN-α, -β, -κ, -δ, -ε, -ω, and -τ [6], and generate antiviral activity [7], while IFN-γ is the only type II IFN [7] and is involved in the regulation of immune and inflammatory responses [8]. IFN-κ is a newly identified type I IFN and is known to have similar antiviral properties. However, it is distinct from other type I IFNs as it signals in a discrete autocrine, rather than paracrine manner [9].
TLRs 3, 7, 8, and 9 can be activated by viral products such as double stranded RNA [10], single stranded RNA [11], and double stranded CpG-rich DNA [12], respectively, whereas cell-surface TLR4 is stimulated by lipopolysaccharide (LPS) and some viral proteins [13]. TLRs 1 and 6 recognize bacterial and mycoplasma lipoproteins, respectively [14–16] while acting in cooperation with TLR2, whereas TLR5 is specifically stimulated by bacterial flagellin [17, 18]. In addition, damage-associated molecular patterns (DAMPS) (which include heat shock proteins, high-mobility group box 1 (HMGB1), uric acid crystals, hyaluronan, heparin sulfate, messenger RNA, surfactant protein A, and various products of the extracellular matrix such as fibronectin and fibrinogen) have been suggested to activate TLRs [1].
The critical function of TLRs in innate immune functioning is well studied, but, in recent years, their role in tumour genesis and cancer progression has become an active field of research. Because of their innate immunity signalling, TLRs play an important gate keeper role in controlling the production of proinflammatory cytokines and chemokines as well as IFNs. It has recently been speculated that aberrant TLR expression and signalling in cancer cells and the resulting downstream cascades can directly promote tumour progression [19]. Indeed, aberrant TLR1, 2, 3, 4, 5, 6, and 9 signalling has been implicated in a variety of human cancers [1].
Cervical cancer is the second most common malignancy in women worldwide with human papillomavirus (HPV) recognized as its causative agent [20]. HPV is a double stranded DNA virus which infects the squamous epithelium of the uterine cervix. Most infections are cleared by the host, but a proportion of women are unable to clear the infection, resulting in cervical lesions. Although persistent HPV infection is necessary, it is insufficient for the development of cervical cancer. HPV can directly inhibit the function of TLR downstream molecules involved in the IFN pathway [21, 22] and accumulating evidence supports an interaction between TLRs and HPV [23–25]. TLR expression throughout the course of cervical carcinogenesis has not been addressed previously and requires further exploration taking the entire tumour microenvironment into consideration. In this regard, we have recently characterized cytokine and IFN gene expression levels in the normal, premalignant, and malignant epithelium and surrounding stroma of ex vivo cervical tissue by laser capture microscopy and quantitative real-time polymerase chain reaction [26]. Compared to normal tissue, these molecules were lacking or showed low expression in the diseased epithelium while all of them were aberrantly increased in the tumour stroma. To provide a complete picture of innate immune responses in the same tissue samples, here we report a comprehensive transcriptome analysis of all upstream TLRs involved in these downstream pathway disturbances.
2. Materials and Methods
2.1. Sample Preparation
Cervical biopsies were obtained with written consent from women attending the Colposcopy Clinic at the Thunder Bay Regional Health Sciences Centre (TBRHSC) between November 2005 and November 2006. The study has been approved by the local Research Ethics Team at the TBRHSC (no. 21.05). Biopsies were snap frozen in liquid nitrogen and immediately transferred to −80°C. Tissue was sectioned on a cryostat (Leica CM1850, Leica Microsystems, Richmond Hill, ON, Canada), maintaining tissue temperature at −20°C using Tissue Tek embedding medium (O.C.T. Compound, Sakura Finetek, Torrance, Calif, USA) and sectioned. For laser capture microdissection (LCM) analysis, 8 μm-thick tissue sections were cut, adhered to uncharged slides, and kept in −20°C prior to LCM preparation. All tweezers, brushes, and surfaces were cleaned with DEPC-treated 70% (v/v) ethanol between specimens to reduce RNAse activity and RNA cross-contamination.
All samples collected in this study were thoroughly tested for RNA integrity, as previously described [26, 27]. Of 110 samples, 25 had enough remaining tissue suitable for LCM analysis. The 25 samples utilized in this study along with their HPV infection status are listed in Table 1. In total, 11 of 25 samples were negative for HPV and diagnosed as morphologically normal. Of the 14 diseased samples, 4 were diagnosed as low-grade lesions (LSIL), 6 were high-grade lesions (HSIL), and 4 were invasive carcinomas. Importantly, all normal tissue was negative for HPV infection, and all diseased tissue was positive for HPV infection. Histological diagnosis of the snap-frozen biopsy was verified before and after LCM and correlated with the diagnosis of the biopsy that was taken for clinical purposes.
Table 1.
Sample size. Twenty-five frozen cervical biopsies were categorized into 3 groups; normal tissue, case IDs N1–N11; dysplastic tissue, case IDs D1–D10; invasive carcinoma tissue, case IDs C1–C4. Laser capture microdissection was used to separate each specimen into epithelium and stroma compartments. The HPV infection status is shown, and the specific HPV type is noted when known.
| Case ID | Morphological diagnosis | HPV infection status | Case ID | Morphological diagnosis | HPV infection status |
|---|---|---|---|---|---|
| N1 | Normal | Negative | D3 | LSIL | Types 66, 82 |
| N2 | Normal | Negative | D4 | LSIL | Types 6, 66 |
| N3 | Normal | Negative | D5 | HSIL | Type 16 |
| N4 | Normal | Negative | D6 | HSIL | Type 16 |
| N5 | Normal | Negative | D7 | HSIL | Type 33 |
| N6 | Normal | Negative | D8 | HSIL | Type 16 |
| N7 | Normal | Negative | D9 | HSII | Type 16 |
| N8 | Normal | Negative | D10 | HSII | TYPe 39 |
| N9 | Normal | Negative | C1 | Carcinoma | Type 16 |
| N10 | Normal | Negative | C2 | Carcinoma | HR HPV+ |
| N11 | Normal | Negative | C3 | Carcinoma | HR HPV+ |
| D1 | LSIL | Type 31 | C4 | Carcinoma | HR HPV+ |
| D2 | LSIL | Type 51 |
LSIL: low-grade squamous intraepithelial lesion; HR: high risk; HSIL: high-grade squamous intraepithelial lesion.
2.2. HPV Typing and Specimen Classification
Ten tissue sections were cut and then taken immediately for LCM analysis, and placed into 1.5 mL microcentrifuge tubes for DNA extraction. DNA extraction was performed using the Qiagen QIAamp DNA micro kit (Qiagen, Mississauga, ON, Canada) following the manufacturer's protocol for tissue samples. Samples were HPV genotyped using Luminex hybridization at the National Microbiology Laboratory (Winnipeg, B, Canada), as previously described [26]. In addition, PCR analysis [38] and p16-based testing using the CINtec p16 immunocytochemistry staining kit (CINtec p16INK4a Cytology Kit; mtm laboratories, Westborough, Mass, USA) were performed to visualize HPV infection. For histopathological diagnosis, sections were cut and processed for hematoxylin and eosin staining [39] and diagnosed by the same pathologist (N.E.). Tissue samples were diagnosed as morphologically normal, low/high-grade lesion, and cervical carcinoma tissue. For this study, samples were categorized as HPV-negative and morphologically normal (Normal; n = 11), HPV-positive and morphologically dysplastic (Dysplasia; n = 10) and HPV-positive and morphologically malignant (Carcinoma; n = 4).
2.3. RNA Extraction and Integrity Assessment
RNA was extracted from 10 × 10 μm thick tissue sections of microdissected cervical cells or keratinocytes as previously described [27]. The quality and quantity of RNA extracted from cervical tissue was assessed using the Bio-Rad Experion automated electrophoresis system. RNA integrity from samples was assessed as previously described [27].
2.4. Laser Capture Microdissection
Tissue specimens were prepared for laser capture microdissection (LCM) using the Arcturus Histogene Frozen Section Staining Kit, as previously described [27]. Up to five thousand captures (~25,000 cells) of both epithelium and stroma for each tissue specimen were taken. Macrocaps were cleaned of unwanted debris using CapSure pads (Arcturus) prior to being deposited into 0.5 mL microfuge tubes for RNA extraction. The laser spot size was consistently 15 μm in diameter whereas the laser power and duration ranged from 80 to 95 mW and 0.65 to 0.8 millisecond duration between specimens. Images of the excision process have been previously reported [27].
2.5. Quantitative Real-Time Polymerase Chain Reaction
RNA isolated from samples was reverse transcribed to complementary DNA (cDNA) using the High Capacity cDNA Archive Kit (Applied Biosystems) according to manufacturer's directions with random hexamer primers. RNA isolated from microdissected samples was reverse transcribed at a minimum of 0.6 ng/μL (ranging from 0.6–2.8 ng/μL), depending on acquired LCM sample RNA concentration. Complementary DNA (cDNA) was amplified using the TaqMan PreAmp Master Mix Kit (Applied Biosystems). Gene amplification uniformity was assessed as previously described [27]. Reactions were carried out according to the quantitative real-time polymerase chain reaction (qRT-PCR) protocol specified in the TaqMan PreAmp Master Mix Kit. Triplicate reactions of 25 μL volume were added to a 96-optical well plate (Applied Biosystems) and incubated at standard qRT-PCR conditions (50°C for 5 minutes 95°C for 10 minutes and then cycled at 95°C for 15 seconds and 60°C for 1 minute for 40 cycles (detection limit)). qRT-PCR was repeated for 10% of ex vivo human samples to ensure result reliability. Target genes were normalized to the housekeeping gene hypoxanthine phosphoribosyltransferase1 (HPRT1), as previous results indicate its expression is unaffected by HPV infection [27]. TaqMan gene expression on demand assays for HPRT1 and TLRs 1 through 9 were used (Applied Biosystems). Negative controls where cDNA was omitted or the enzyme was missing in the RT reaction were run to monitor for contamination or nonspecific primer binding. A positive control was included on every plate to control for variation between runs. Relative quantification of target genes was performed using auto Ct and baseline settings and a threshold of 0.20 (Applied Biosystems 7300/7500/7500 Fast Real-Time PCR System Software).
2.6. Statistical Analysis
The difference in cycle thresholds (ΔCT) between HPRT1 and the TLRs was calculated for each gene in each biological sample. These values were converted to relative expression values using 2−ΔCT [40]. Statistical differences in gene expression levels were assessed using the Jonckheere-Terpstra [41] test for trends when analyzing the three tissue groups together and using the Student's t-test for pairwise comparison when comparing diseased versus normal tissue. P-values for all statistical comparisons are reported with at most two significant digits. Statistical significance is stated when P ≤ 0.05. All tests are two sided if not otherwise indicated and based on the “exact” version. Calculations were performed using Microsoft Excel version 12 (2008) and Cytel Studio StatXact7 version 7.0.0 (2005), Cytel Software Corporation (http://www.cytel.com/) and the SAS statistical analysis package (Version 9.1.2, http://support.sas.com/) for statistical tests.
3. Results
3.1. Baseline TLR mRNA Levels in Normal, HPV-Negative Ex Vivo Cervical Tissue
To define baseline mRNA levels of TLRs in healthy cervical tissue, 11 cervical biopsies from histologically normal, HPV-negative women were analyzed, as shown in Table 1. Gene expression levels in the epithelium and stroma of healthy samples are shown in Figures 1(a) and 1(b), respectively. High levels of TLRs 1, 2, 3, and 5 mRNA were found in relation to lower levels of TLRs 4, 6, 7, and 9, with TLR 8 found at the lowest levels in normal epithelium. Alternatively, high levels of TLRs 1, 3, 4 and 5 mRNA were found in relation to lower levels of TLRs 2, 6, 7 and 9, with TLR 8 again found at the lowest levels in normal stroma. Importantly, mRNA was found for all TLRs in healthy cervical epithelium and stroma.
Figure 1.
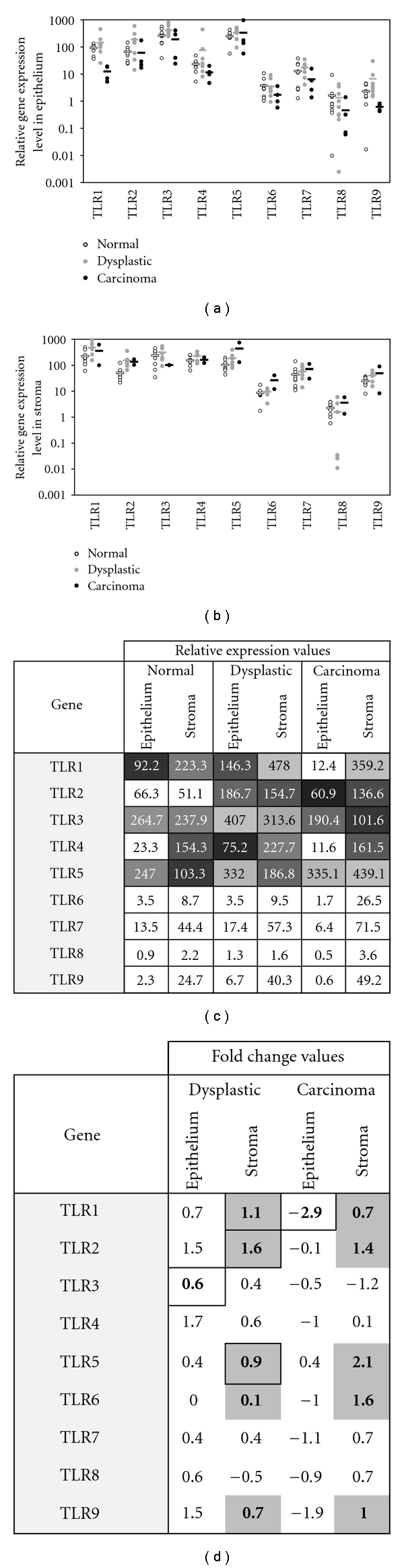
TLR gene expression in normal, dysplastic and carcinoma ex vivo cervical tissue. Scatterplot indicating relative TLR gene expression levels (logged) in normal, dysplastic and carcinoma samples in the epithelium (a) and stroma (b) compartment. Horizontal bars show the average expression levels per group for each gene. Asterisks show significant expression differences from normal samples according to the Student's t-test for pairwise comparisons. Reported average relative expression values for each gene in both tissue compartments (c), values calculated according to 2−ΔCT ∗ 1,000. Fold change values for TLRs in dysplastic and carcinoma samples (d). Significant fold change values according to the Student's t-test for pairwise comparison of two types are indicated with a thick border, and genes with significant upregulation trends according to the Jonckheere-Terpstra test are shaded gray.
3.2. TLR3 Increases in Dysplasia Epithelium While TLR1 Decreases in Carcinoma Epithelium
To characterize TLR mRNA levels in dysplastic and carcinoma epithelium, 14 HPV-positive, diseased cervical epithelial samples were analyzed, as shown in Table 1. Previous studies have demonstrated an interaction between TLR signalling and HPV [25] upstream of the IFN pathway. HPV infections disrupt cytokine expression, and the E6 and E7 oncoproteins particularly target the type I IFN pathway [42]. We previously reported IFN-κ and IL-10 downregulation in dysplastic and carcinoma epithelium [26]. We therefore hypothesized that TLR mRNA expression levels would be altered due to the presence of HPV in the epithelium, to possibly reflect these downstream findings in the stroma. Using LCM to isolate the epithelium in whole cervical biopsy sections, the current analysis revealed several significant differences in TLR mRNA levels in HPV-positive, diseased compared to healthy, HPV-negative epithelium (Figure 1(a)). Importantly, all epithelial samples were positive for TLR expression (Figure 1(c)). While TLR3 levels were found to be significantly higher in dysplastic epithelial samples (P = 0.05, Student's t-test; Figure 1(d)), TLR1 levels were found to be significantly lower in carcinoma epithelium (P = 0.01, Student's t-test; Figure 1(d)). A trend analysis did not reveal any further differences in TLR expression levels in the epithelium. Therefore, the most striking differences found in the epithelium were the increased TLR3 gene expression levels in dysplastic epithelium and the decreased TLR1 gene expression levels in carcinoma epithelium; the latter of which corresponds to decreased IFN-κ levels previously reported in the same samples [26].
3.3. TLRs 1, 2, 6 as well as 5 and 9 Increase with Disease Severity in Cervical Stroma
To characterize TLR mRNA levels in dysplastic and carcinoma stroma, 14 HPV-positive, diseased cervical stromal samples were analyzed, as shown in Table 1. Previous LCM analysis of the same tissue samples revealed increased downstream IFN mRNA levels in dysplastic and carcinoma stroma [26]. TLRs are upstream elicitors of the IFN pathways, and we therefore expected to see TLR alterations in the stroma with cervical disease progression. Using LCM to isolate the stroma in whole cervical biopsy sections, the current analysis revealed several significant differences in TLR mRNA levels in HPV-positive, diseased compared to healthy, HPV-negative stroma (Figure 1(b)). Importantly, all stromal samples were positive for TLR expression (Figure 1(c)). A trend analysis revealed increasing expression trends with disease severity in diseased stroma for TLRs 1, 2, 6 as well as 5 and 9 (P = 0.05, 0.001, 0.02, 0.01, and 0.04 resp., Jonckheere-Terpstra test; Figure 1(d)). Although a trend exists, strong increases in TLR 1, 2, and 5 mRNA levels were found predominately in dysplastic stroma (P = 0.02, 0.001 and 0.05 resp., Student's t-test). Thus, the most striking results found in the stroma compartment were the increased mRNA levels for five TLRs, which coincides with our previous reported increase in downstream IL-10, IFN-κ, IFN-β, and IFN-γ in the stroma of the same samples [26].
4. Discussion
Through the ex vivo analysis of gene expression in HPV-negative, healthy as well as HPV-positive premalignant and malignant cervical tissue, we report a transcriptome model with an emphasis on tissue compartmentalization for innate immune signalling pathways in cervical carcinogenesis. By conducting a systematic literature survey (Table 2), we did not find a comparable study which elucidated the transcriptome of TLRs (current study) and downstream effectors [26] in cervical carcinogenesis using LCM and qPCR. Only one transcriptome study of cervical cancer development utilizing LCM to separate tissue compartments was found [43]. However, differential expression of TLR or IFN expression was not discovered in that investigation [43], likely due to low transcription levels undetectable using microarray analysis. Other transcriptome studies (Table 2) used primary human keratinocytes and/or cervical cancer-derived cell lines [33, 35], whole biopsies [24, 28–31, 34, 36] or both [25, 32, 37] or cytobrush samples [23, 44], but to date none have analyzed the transcriptome in isolated tissue components. Here, we found all TLRs to be expressed ex vivo by healthy, HPV-negative cervical keratinocytes and stroma, albeit at different levels: mRNA for TLRs 1, 3 and 5 tended to be most abundant compared to TLRs 6 to 9, especially TLR8, which were expressed at consistently lower levels in either tissue compartment (Figures 1(a) and 1(b)), while TLR 2 was expressed highest in epithelium, and TLR4 was expressed highest in stroma. These findings are partly concordant with a previous investigation [35], where TLRs 1, 2, 3, 5, and 6 were found most abundant and TLRs 4, 7, 8, s and 9 were expressed at lower levels in normal ectocervical keratinocytes. Discordant TLR 6 findings may be a result of keratinocytes being analyzed within their natural tissue context, complete with signals from the underlying dermal stroma, in the current analysis.
Table 2.
Summary of literature of dysregulated innate immunity genes in cervical tissue. A systematic literature search for investigations into TLR and IFN expression in HPV-infected cervical tissue or cell lines was performed using the following search terms in the PubMed database: “Cervical” and “interferon gamma” (489 hits) or “interferon beta” (202 hits) or “interferon kappa” (3 hits) or “toll-like receptor” (59 hits). Acronyms IFN-γ, -β and -κ as well as TLR in combination with “cervical” were also tried. The listed citations are articles published up to 5 January 2011 which describe TLR and IFN gene regulation and dysregulation. Only articles published in English were considered, and only articles relevant for our study were included in the table.
| Gene | Material | Sample type | Method | Group | ||
|---|---|---|---|---|---|---|
| Normal | CIN | Carcinoma | ||||
| 15 | 11 | 13 | Full biopsy | qPCR | Pao et al. 1995 [28] | |
| 4 | 12 | N/S | Full biopsy, HeLa | qPCR, IHC | de Gruijl et al. 1999 [29] | |
| N/A | N/A | 52 | Full biopsy | qPCR, IHC | Gey et al. 2003 [30] | |
| IFN-γ | 10 | N/A | 29 | Full biopsy | qPCR, IHC | Alcocer-González et al. 2006 [31] |
| 6 | N/A | 6 | Cytobrush | cDNA array | Manavi et al. 2007 [31] | |
| 150 | 198 | N/A | Full biopsy | qPCR | Scott et al. 2009 [32] | |
| N/S | N/S | N/S | Full biopsy, LCM | qPCR | DeCarlo et al. 2008 [27] | |
| 11 | 10 | 4 | Full biopsy, LCM | qPCR | DeCarlo et al. 2010 [19] | |
|
| ||||||
| N/A | N/A | N/A | CK | cDNA array, qPCR | Nees et al. 2001 [33] | |
| IFN-β | N/S | N/S | N/S | Full biopsy, LCM | qPCR | DeCarlo et al. 2008 [27] |
| 11 | 10 | 4 | Full biopsy, LCM | qPCR | DeCarlo et al. 2010 [19] | |
|
| ||||||
| N/S | N/S | N/S | Full biopsy, LCM | qPCR | DeCarlo et al. 2008 [27] | |
| IFN-κ | N/S | 10 | N/A | Full biopsy, PHFK | qPCR, IHC | Rincon-Orozco et al. 2009 [34] |
| 11 | 10 | 4 | Full biopsy, LCM | qPCR | DeCarlo et al. 2010 [19] | |
|
| ||||||
| 3 | 4 | N/A | CK, PHK, SiHa, CaSki, HeLa, C33A | qPCR | Hasan et al. 2007 [25] | |
| TLR1 | 12 | N/A | N/A | CK, VK, ME180, HeLa | qPCR | Herbst-Kralovetz et al. 2008 [35] |
| 65 | 65 | N/A | Cytobrush | qPCR | Daud et al. 2010 [24] | |
|
| ||||||
| 3 | 4 | N/A | CK, PHK, SiHa, CaSki, HeLa, C33A | qPCR | Hasan et al. 2007 [26] | |
| TLR2 | 12 | N/A | N/A | CK, VK, ME180, HeLa | qPCR | Herbst-Kralovetz et al. 2008 [35] |
| 65 | 65 | N/A | Cytobrush | qPCR | Daud et al. 2010 [24] | |
|
| ||||||
| 3 | 4 | N/A | CK, PHK, SiHa, CaSki, HeLa, C33A | qPCR, IF | Hasan et al. 2007 [26] | |
| TLR3 | 12 | N/A | N/A | CK, VK, ME180, HeLa | qPCR | Herbst-Kralovetz et al. 2008 [35] |
| 65 | 65 | N/A | Cytobrush | qPCR | Daud et al. 2010 [24] | |
|
| ||||||
| 3 | 4 | N/A | CK, PHK, SiHa, CaSki, HeLa, C33A | qPCR | Hasan et al. 2007 [26] | |
| TLR4 | 12 | N/A | N/A | CK, VK, ME180, HeLa | qPCR | Herbst-Kralovetz et al. 2008 [35] |
| 20 | 125 | 49 | Full biopsy | qPCR, IHC | Yu et al. 2010 [25] | |
|
| ||||||
| 3 | 4 | N/A | CK, PHK, SiHa, CaSki, HeLa, C33A | qPCR | Hasan et al. 2007 [19] | |
| TLR5 | 12 | N/A | N/A | CK, VK, ME180, HeLa | qPCR | Herbst-Kralovetz et al. 2008 [35] |
| 9 | 22 | 24 | Full biopsy | qPCR | Kim et al. 2008 [36] | |
|
| ||||||
| 3 | 4 | N/A | CK, PHK, SiHa, CaSki, HeLa, C33A | qPCR | Hasan et al. 2007 [26] | |
| TLR6 | 12 | N/A | N/A | CK, VK, ME180, HeLa | qPCR | Herbst-Kralovetz et al. 2008 [35] |
| 65 | 65 | N/A | Cytobrush | qPCR | Daud et al. 2010 [24] | |
|
| ||||||
| 3 | 4 | N/A | CK, PHK, SiHa, CaSki, HeLa, C33A | qPCR | Hasan et al. 2007 [26] | |
| TLR7 | 12 | N/A | N/A | CK, VK, ME180, HeLa | qPCR | Herbst-Kralovetz et al. 2008 [35] |
| 65 | 65 | N/A | Cytobrush | qPCR | Daud et al. 2010 [24] | |
|
| ||||||
| 3 | 4 | N/A | CK, PHK, SiHa, CaSki, HeLa, C33A | qPCR | Hasan et al. 2007 [19] | |
| TLR8 | 12 | N/A | N/A | CK, VK, ME180, HeLa | qPCR | Herbst-Kralovetz et al. 2008 [35] |
| 65 | 65 | N/A | Cytobrush | qPCR | Daud et al. 2010 [24] | |
|
| ||||||
| 3 | 4 | N/A | Full biopsy, CK, PHK, SiHa, CaSki, HeLa, C33A | qPCR, IF, IHC | Hasan et al. 2007 [26] | |
| 9 | 22 | 24 | Full biopsy | qPCR | Lee et al. 2007 [37] | |
| TLR9 | 12 | N/A | N/A | CK, VK, ME180, HeLa | qPCR | Herbst-Kralovetz et al. 2008 [35] |
| 65 | 65 | N/A | Cytobrush | qPCR | Daud et al. 2010 [24] | |
| 20 | 125 | 49 | Full biopsy | qPCR, IHC | Yu et al. 2010 [25] | |
CIN, cervical intraepithelial neoplasia; CK, in vitro cervical keratinocytes; IF, immunofluorescence; IHC, immunohistochemistry; LCM, laser capture microdissection; N/A, not applicable; N/S, not stated; PHFK, primary human foreskin keratinocytes; PHK, primary human keratinocytes, anatomical origin not specified; qPCR, quantitative real-time polymerase chain reaction; VK, vaginal keratinocytes.
Compared to normal tissue, TLR3 was found to be higher expressed in premalignant epithelium and TLR1 to be expressed lower in malignant epithelium. Similar to previous findings, [24, 25] we found a trend for TLRs 4 and 9 to be decreased in carcinoma epithelium, although not significantly. Likewise, other TLR receptors were also decreased, with the exception of TLR8, in the carcinoma epithelium, but statistical significance was not reached, possibly due to individual differences in expression levels in ex vivo samples. This decreasing trend in TLR expression coincides with our previous finding that IFN-κ was abolished in diseased epithelium, [26] and other studies have similarly revealed lower levels of type I and II IFNs in cervical malignancy, [30, 36, 37] although the isolation of cervical epithelium was not performed in these studies. In contrast to low TLR levels in carcinoma epithelium, TLRs 1, 2, 6, as well as 5 and 9 showed higher expression in premalignant and malignant stroma compared to normal tissue. These results coincide with previously reported increases in IL-10, IFN-κ, IFN-β, and IFN-γ mRNA levels as well as infiltrating monocytes and dendritic cells in the cervical stroma in these samples [26]. Previous investigations have also revealed elevated mRNA level of TLRs 5 and 9 in cervical carcinoma tissue, [29, 34] and in contrast, higher IFN-γ and IL-10 have been associated with lower incidences of cervical dysplasia, [28] however neither investigation entailed tissue microdissection to reveal the cell types responsible.
Based on our combined gene expression findings, we propose a disease scenario in the cervical microenvironment (Figure 2). During early cervical carcinogenesis, when the cervical epithelium is still undergoing differentiation, which typically takes place parallel to the viral life cycle of HPV, TLR3, a classical receptor eliciting antiviral responses via IFN-regulatory factor (IRF) 3, expression increases in dysplastic epithelium. This upregulation may be an attempt by the infected cells to initiate immune responses against the HPV infection [25]. The concomitant downregulation of downstream IFN-κ in premalignant epithelium samples could likely be attributed to interference of HPV E6 with the IFN pathway by blocking IRF 3 [45]. We also show that TLR1 was significantly decreased in carcinoma epithelium and may represent the sole upstream TLR significantly affected in keratinocytes in late carcinogenesis. With disease severity, the presence of HPV in the epithelium may lead to higher expression of TLRs in diseased stroma, which can account for the increases in IFNs previously found [26]. Altogether, these data demonstrate that the TLR pathways are not being induced in late carcinogenesis in the cervical epithelium, yet the presence of HPV indirectly triggers TLR pathway activation in the cervical stroma, eliciting downstream innate immune responses. However, IFNs may play a direct role in the expression levels of TLRs, as recent evidence points towards an intriguing mechanism by which type I IFNs can directly induce TLRs 1, 3, 5 and 7, [46] which are some of the TLRs found to be up-regulated in diseased cervical stroma in the present study. The presence of up-regulated responses in the stroma and not in the epithelium may contribute to the persistence of HPV and ultimately cultivate the progression of the lesion.
Figure 2.
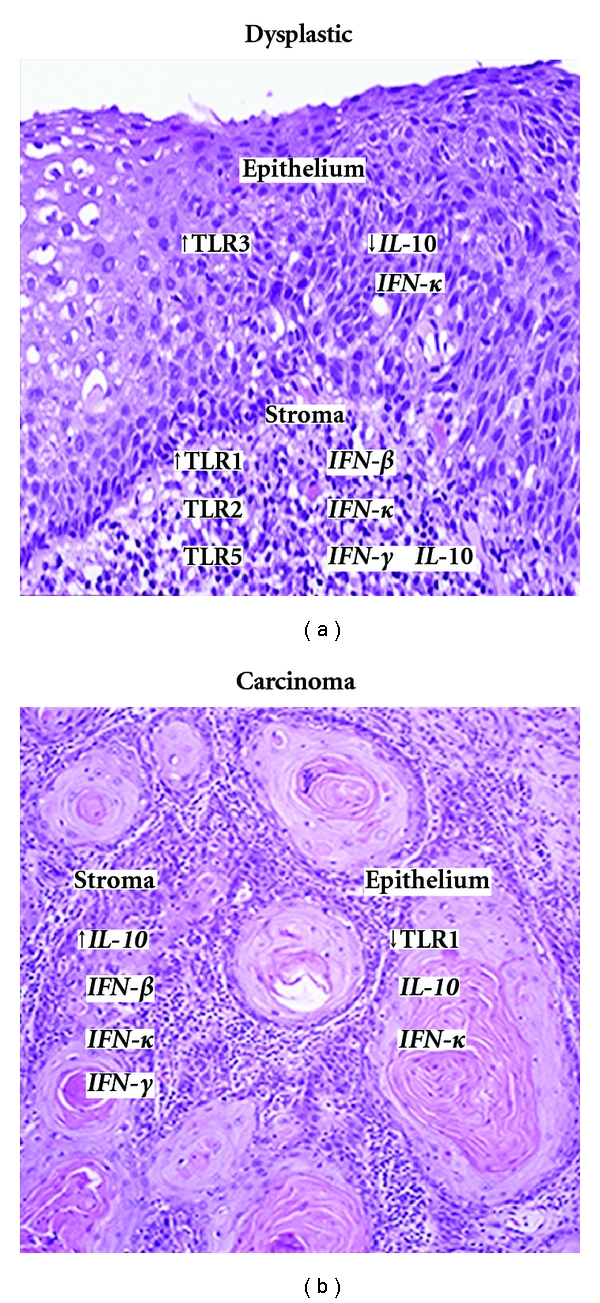
Overall gene expression models for ex vivo dysplastic (a) and carcinoma (b) cervical epithelium and stroma. Genes presented from the current analysis as either up or down-regulated compared to HPV-negative, normal cervical tissue. Italicized genes are based on data we reported previously [19]. In addition to the data presented, trend analyses also revealed a significant up-regulation of TLRs 1, 2, 5, 6, and 9 in the stroma with disease severity.
Several mechanisms have been suggested for IFN-κ downregulation in malignant epithelium: (i) downregulation of TLR9 by HPV16 E6, [25] (ii) methylation of the IFN-κ promoter [32] and (iii) downregulation of IFN-κ by the HPV16 E6 oncoprotein [47]. The present findings suggest yet another potential underlying mechanism which is linked to the three-fold and statistically significant decreased expression level of TLR1 in malignant epithelium. In contrast, the increased TLR1 expression in malignant stroma is likely due to increased monocyte infiltration, implicating DAMPS from dying cancer cells as potential ligands. This notion requires further investigation because TLR1 is usually involved in the activation of downstream proinflammatory cytokines rather than IFNs. However, recent research suggests that in addition to the canonical TLRs 3, 4, 7, 8, and 9, the TLR2 subfamily (TLR1, 2 and 6) is associated with IFN responses [48].
HPV-associated cancers are a worldwide health concern, and since the recently approved prophylactic vaccine against common high-srisk HPV types is not a treatment option, there is a great need for studies to characterize the effect of HPV on innate immune responses to advance treatment options. Because of their wide-ranging impact upon both innate and adaptive immunity in several disease settings, TLRs and their signalling pathways emerge as attractive therapeutic targets [49]. Based on our present findings using a highly defined sample size, TLRs 3 and 1 could be further investigated for diagnostic and eventually prognostic marker use. Although TLRs can mediate host innate immune system signalling, overexpression of TLRs has been paradoxically found in many tumour cases [50]. In line with our findings in diseased cervical stroma, TLR antagonists, for example, which act to block MyD-88, could confer therapeutic benefit in cervical cancer patients.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Acknowledgments
This work was supported by the Northern Health Fund (to I.Z.), Grant no. GA1-2005-009, a Discovery Grant (to I.Z.) from the Natural Sciences and Engineering Research Council of Canada (NSERC), Grant no. 355858-2008 and by an Ontario Graduate Scholarship (to C.D.). R. Jackson and S. Niccoli contributed equally. C. D. is now a graduate student in the Department of Psychology, University of Victoria.
References
- 1.Sato Y, Goto Y, Narita N, Hoon DSB. Cancer cells expressing toll-like receptors and the tumor microenvironment. Cancer Microenvironment. 2009;2(1):S205–S214. doi: 10.1007/s12307-009-0022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K. Toll-like receptor signalling. Nature Reviews Immunology. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 3.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proceedings of the Royal Society of London. Series B. 1957;147(927):258–267. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- 4.Takaoka A, Hayakawa S, Yanai H, et al. Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424(6948):516–523. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- 5.Stetson DB, Medzhitov R. Type I Interferons in Host Defense. Immunity. 2006;25(3):373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and their actions. Annual Review of Biochemistry. 1987;56:727–777. doi: 10.1146/annurev.bi.56.070187.003455. [DOI] [PubMed] [Google Scholar]
- 7.Bach EA, Aguet M, Schreiber RD. The IFNγ receptor: a paradigm for cytokine receptor signaling. Annual Review of Immunology. 1997;15:563–591. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- 8.LaFleur DW, Nardelli B, Tsareva T, et al. Interferon-κ, a novel type I interferon expressed in human keratinocytes. Journal of Biological Chemistry. 2001;276(43):39765–39771. doi: 10.1074/jbc.M102502200. [DOI] [PubMed] [Google Scholar]
- 9.Buontempo PJ, Jubin RG, Buontempo CA, Wagner NE, Reyes GR, Baroudy BM. Antiviral activity of transiently expressed IFN-κ is cell-associated. Journal of Interferon and Cytokine Research. 2006;26(1):40–52. doi: 10.1089/jir.2006.26.40. [DOI] [PubMed] [Google Scholar]
- 10.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413(6857):732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 11.Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 12.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 13.Janssens S, Beyaert R. Role of Toll-like receptors in pathogen recognition. Clinical Microbiology Reviews. 2003;16(4):637–646. doi: 10.1128/CMR.16.4.637-646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeuchi O, Kawai T, Mühlradt PF, et al. Discrimination of bacterial lipoproteins by Toll-like recepttor 6. International Immunology. 2001;13(7):933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 15.Takeuchi O, Sato S, Horiuchi T, et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. Journal of Immunology. 2002;169(1):10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 16.Farhat K, Riekenberg S, Heine H, et al. Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. Journal of Leukocyte Biology. 2008;83(3):692–701. doi: 10.1189/jlb.0807586. [DOI] [PubMed] [Google Scholar]
- 17.Hayashi F, Smith KD, Ozinsky A, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410(6832):1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 18.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. Journal of Immunology. 2001;167(4):1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 19.Chen R, Alvero AB, Silasi DA, Steffensen KD, Mor G. Cancers take their Toll—the function and regulation of Toll-like receptors in cancer cells. Oncogene. 2008;27(2):225–233. doi: 10.1038/sj.onc.1210907. [DOI] [PubMed] [Google Scholar]
- 20.Zur Hausen H. Papillomaviruses in human cancer. Applied Pathology. 1987;5(1):19–24. [PubMed] [Google Scholar]
- 21.Li S, Labrecque S, Gauzzi MC, et al. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-α . Oncogene. 1999;18(42):5727–5737. doi: 10.1038/sj.onc.1202960. [DOI] [PubMed] [Google Scholar]
- 22.Chang YE, Laimins LA. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. Journal of Virology. 2000;74(9):4174–4182. doi: 10.1128/jvi.74.9.4174-4182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daud II, Scott ME, Ma Y, Shiboski S, Farhat S, Moscicki AB. Association between toll-like receptor expression and human papillomavirus type 16 persistence. International Journal of Cancer. 2011;128(4):879–886. doi: 10.1002/ijc.25400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu L, Wang L, Li M, Zhong J, Wang Z, Chen S. Expression of toll-like receptor 4 is down-regulated during progression of cervical neoplasia. Cancer Immunology, Immunotherapy. 2010;59(7):1021–1028. doi: 10.1007/s00262-010-0825-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasan UA, Bates E, Takeshita F, et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. Journal of Immunology. 2007;178(5):3186–3197. doi: 10.4049/jimmunol.178.5.3186. [DOI] [PubMed] [Google Scholar]
- 26.Decarlo CA, Severini A, Edler L, et al. IFN-κ, a novel type i IFN, is undetectable in HPV-positive human cervical keratinocytes. Laboratory Investigation. 2010;90(10):1482–1491. doi: 10.1038/labinvest.2010.95. [DOI] [PubMed] [Google Scholar]
- 27.DeCarlo CA, Escott NG, Werner J, et al. Gene expression analysis of interferon κ in laser capture microdissected cervical epithelium. Analytical Biochemistry. 2008;381(1):59–66. doi: 10.1016/j.ab.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott ME, Ma Y, Kuzmich L, Mescicki AB. Diminished IFN-γ and IL-10 and elevated Foxp3 mRNA expression in the cervix are associated with CIN 2 or 3. International Journal of Cancer. 2009;124(6):1379–1383. doi: 10.1002/ijc.24117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim WY, Lee JW, Choi JJ, et al. Increased expression of Toll-like receptor 5 during progression of cervical neoplasia. International Journal of Gynecological Cancer. 2008;18(2):300–305. doi: 10.1111/j.1525-1438.2007.01008.x. [DOI] [PubMed] [Google Scholar]
- 30.Gey A, Kumari P, Sambandam A, et al. Identification and characterisation of a group of cervical carcinoma patients with profound downregulation of intratumoral Type 1 (IFNγ) and Type 2 (IL-4) cytokine mRNA expression. European Journal of Cancer. 2003;39(5):595–603. doi: 10.1016/s0959-8049(02)00839-0. [DOI] [PubMed] [Google Scholar]
- 31.Alcocer-González JM, Berumen J, Taméz-Guerra R, et al. In vivo expression of immunosuppressive cytokines in human papillomavirus-transfonned cervical cancer cells. Viral Immunology. 2006;19(3):481–491. doi: 10.1089/vim.2006.19.481. [DOI] [PubMed] [Google Scholar]
- 32.Rincon-Orozco B, Halec G, Rosenberger S, et al. Epigenetic silencing of interferon-κ in human papillomavirus type 16-positive cells. Cancer Research. 2009;69(22):8718–8725. doi: 10.1158/0008-5472.CAN-09-0550. [DOI] [PubMed] [Google Scholar]
- 33.Nees M, Geoghegan JM, Hyman T, Frank S, Miller L, Woodworth CD. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-κB-responsive genes in cervical keratinocytes. Journal of Virology. 2001;75(9):4283–4296. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JW, Choi JJ, Eun SS, et al. Increased toll-like receptor 9 expression in cervical neoplasia. Molecular Carcinogenesis. 2007;46(11):941–947. doi: 10.1002/mc.20325. [DOI] [PubMed] [Google Scholar]
- 35.Herbst-Kralovetz MM, Quayle AJ, Ficarra M, et al. Quantification and comparison of toll-like receptor expression and responsiveness in primary and immortalized human female lower genital tract epithelia. American Journal of Reproductive Immunology. 2008;59(3):212–224. doi: 10.1111/j.1600-0897.2007.00566.x. [DOI] [PubMed] [Google Scholar]
- 36.Pao CC, Lin CY, Yao DS, Tseng CJ. Differential expression of cytokine genes in cervical cancer tissues. Biochemical and Biophysical Research Communications. 1995;214(3):1146–1151. doi: 10.1006/bbrc.1995.2405. [DOI] [PubMed] [Google Scholar]
- 37.De Gruijl TD, Bontkes HJ, Van den Muysenberg AJC, et al. Differences in cytokine mRNA profiles between premalignant and malignant lesions of the uterine cervix. European Journal of Cancer. 1999;35(3):490–497. doi: 10.1016/s0959-8049(98)00371-2. [DOI] [PubMed] [Google Scholar]
- 38.Zehbe I, Rylander E, Edlund K, Wadell G, Wilander E. Detection of human papillomavirus in cervical intra-epithelial neoplasia, using in situ hybridization and various polymerase chain reaction techniques. Virchows Archiv. 1996;428(3):151–157. doi: 10.1007/BF00200657. [DOI] [PubMed] [Google Scholar]
- 39.Culling CFA, Allison RT, Barr WT. Microorganisms. 4th edition. London, UK: Butterworths & Co.; 1985. Cellular pathology techniques; pp. 55–59. [Google Scholar]
- 40.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 41.Hollander M, Wolfe DA. Nonparametric Statistical Methods. 2nd edition. Hoboken, NJ, USA: Wiley-Interscience; 1999. [Google Scholar]
- 42.Stanley MA, Pett MR, Coleman N. HPV: from infection to cancer. Biochemical Society Transactions. 2007;35(6):1456–1460. doi: 10.1042/BST0351456. [DOI] [PubMed] [Google Scholar]
- 43.Gius D, Funk MC, Chuang EY, et al. Profiling microdissected epithelium and stroma to model genomic signatures for cervical carcinogenesis accommodating for covariates. Cancer Research. 2007;67(15):7113–7123. doi: 10.1158/0008-5472.CAN-07-0260. [DOI] [PubMed] [Google Scholar]
- 44.Manavi M, Hudelist G, Fink-Retter A, Gschwandtler-Kaulich D, Pischinger K, Czerwenka K. Gene profiling in Pap-cell smears of high-risk human papillomavirus-positive squamous cervical carcinoma. Gynecologic Oncology. 2007;105(2):418–426. doi: 10.1016/j.ygyno.2006.12.032. [DOI] [PubMed] [Google Scholar]
- 45.Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes and Development. 1998;12(13):2061–2072. doi: 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khoo JJ, Forster S, Mansell A. Toll-like receptors as interferon-regulated genes and their role in disease. Journal of Interferon and Cytokine Research. 2011;31(1):13–25. doi: 10.1089/jir.2010.0095. [DOI] [PubMed] [Google Scholar]
- 47.Dell’Oste V, Azzimonti B, Mondini M, et al. Altered expression of UVB-induced cytokines in human papillomavirus-immortalized epithelial cells. Journal of General Virology. 2008;89(10):2461–2466. doi: 10.1099/vir.0.83586-0. [DOI] [PubMed] [Google Scholar]
- 48.Dietrich N, Lienenklaus S, Weiss S, Gekara NO. Murine Toll-like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PLoS One. 2010;5(4) doi: 10.1371/journal.pone.0010250. Article ID e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mohan C, Zhu J. Toll-like receptor signaling pathways—therapeutic opportunities. Mediators of Inflammation. 2010;2010:7 pages. doi: 10.1155/2010/781235. Article ID 781235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.So EY, Ouchi T. The application of toll like receptors for cancer therapy. International Journal of Biological Sciences. 2010;6(7):675–681. doi: 10.7150/ijbs.6.675. [DOI] [PMC free article] [PubMed] [Google Scholar]