

Abstract
Currently available medications have significant limitations, most notably low response rate and time lag for treatment response. Recent clinical studies have demonstrated that ketamine, an NMDA receptor antagonist produces a rapid antidepressant response (within hours) and is effective in treatment resistant depressed patients. Molecular and cellular studies in rodent models demonstrate that ketamine rapidly increases synaptogenesis, including increased density and function of spine synapses, in the prefrontal cortex (PFC). Ketamine also produces rapid antidepressant actions in behavioral models of depression, and reverses the deficits in synapse number and behavior resulting from chronic stress exposure. These effects of ketamine are accompanied by stimulation of the mammalian target of rapamycin (mTOR), and increased levels of synaptic proteins. Together these studies indicate that ketamine rapidly reverses the atrophy of spines in the PFC and thereby causes a functional reconnection of neurons that underlies the rapid behavioral responses. These studies identify new targets for rapid acting antidepressants that are safer than ketamine.
Keywords: Rapamycin, glutamate, depression, synaptogenesis
1. Introduction
Depression is a widespread and heterogeneous disorder with severe health and socioeconomic consequences (Kessler et al, 2003). Despite years of research and efforts to develop effective treatments, available antidepressant medications have serious limitations. This includes low rates of treatment response (~one in three respond to the first medication prescribed, and up to two in three after testing multiple medications) (Trivedi et al, 2006). Moreover, there is a time lag of several weeks to months before a therapeutic effect is observed, a serious problem given the high rate of suicide in depressed patients.
However, recent clinical studies provide evidence of novel experimental medications that address the limitations of current antidepressant drugs. In particular these studies demonstrate that a low dose of NMDA receptor antagonist, ketamine, produces a rapid antidepressant response within hours (Berman et al, 2000; Zarate et al, 2006). Moreover, these rapid actions of ketamine are observed in patients who are resistant to two or more typical antidepressants (i.e, considered treatment resistant). Identification of a rapid acting, efficacious agent with a completely different mechanism of action represents a significant advance for the treatment of depression.
The neurobiological mechanisms underlying the antidepressant actions of ketamine are more complex than simple blockade of NMDA receptors. This hypothesis is based on the time course of the therapeutic response and sustained actions of ketamine. The very low dose of ketamine used for these studies first produces mild psychotomimetic and dissociative effects 30–40 minutes after administration, effects that are transient and completely dissipate by 80 minutes (Zarate et al., 2006). This is presumably because of the rapid metabolism of ketamine (half-life is 180 min in humans; Clements et al., 1982). After this initial psychotomimetic phase, the antidepressant effects are observed at 110 min and are sustained for approximately 7 days after a single dose of ketamine (Zarate et al., 2006). These findings indicate that ketamine initiates a cascade of events that results in a rapid response that is sustained even after the drug has been metabolized.
We have investigated the possibility that ketamine activates a change in synaptogenesis that is delayed but long-lasting. The results demonstrate that ketamine stimulates a signaling cascade that leads to increased dendritic protein synthesis and increased density and function of spine synapses. These findings are discussed in the context of the deleterious effects of stress and depression on synaptogenesis, and the implications for designing novel, rapid acting antidepressants.
2. Rapid Antidepressant Actions of Ketamine
The regulation of synapse formation or synaptogenesis is a subcellular neuronal alteration that contributes to synaptic plasticity, a fundamental function of the brain. Synaptic plasticity is the ability to process information from other neuronal inputs, store that information, and make the appropriate future adaptive responses. Synaptic plasticity and synaptogenesis have been studied primarily in models of learning and memory in the hippocampus, but this important process also plays an important role in multiple functions in other brain regions (Figure 1). An increase in functional synaptogenesis is typically accompanied by an increase in the number of dendritic spines, the physical site of synaptic connections (Holtmaat et al, 2009; Kessels and Malinow, 2009; Yoshihara et al, 2009). Dendritic spines can be visualized by Golgi staining or by filling individual neurons with a dye that diffuses throughout the dendritic arbor, allowing for analysis of the density of spines. The shape of spines also provides information about synaptic function: shapes range from narrow and spindly to round or mushroom shaped, correlating with low to high levels of synaptic maturity, stability, and functional neurotransmitter activity (Holtmaat et al, 2009; Kessels and Malinow, 2009; Yoshihara et al, 2009).
Figure 1.
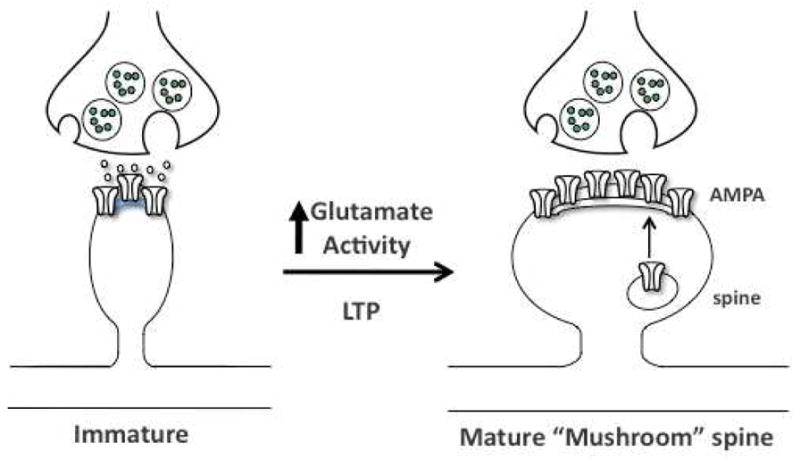
Model for activity-dependent induction of mature spines. Diagram shows a presynaptic terminal with synaptic vesicle adjacent to an immature spine. Synaptic activity leads to spine maturation and insertion of glutamate AMPA receptors into the postsynaptic membrane. The signaling mechanisms underlying synaptogenesis are studied in a cellular model of learning and memory, long- term potentiation (LTP). See text for details.
2.1. Ketamine increases synaptogenesis
The potential role of dendrites and spines in stress-related illnesses such as depression is supported by basic studies demonstrating that exposure to stress causes atrophy of neurons in limbic brain regions implicated in depression, including the prefrontal cortex (PFC) and hippocampus (McEwen 2008; Shansky and Morrison, 2009). This includes a decrease in the density of spines, as well as a decrease in the number and length of dendrite branches. These effects could contribute to the reduction in volume of PFC and hippocampus determined by imaging the brains of depressed patients (Drevets and Furey, 2010; Macqueen et al, 2008).
In contrast to the effects of stress, we have recently reported that administration of a low dose of ketamine results in the rapid induction of spine number in layer V pyramidal neurons of the PFC (Li et al, 2010). Spine analysis was conducted 24 hr after ketamine administration, and it is possible that the induction of spine formation occurs sooner, given the rapid induction of synaptic proteins (i.e., as early as 2 hrs after ketamine administration, see below). Additional studies will be required to determine the time course for induction, as well as maintenance of spine formation.
At the 24 hr time point, there is an increase in the number of mushroom or mature spines, indicating that ketamine increases spine stability and function. This possibility was directly tested by analysis of neurotransmitter-induced excitatory postsynaptic currents (EPSCs) in the same PFC layer V pyramidal neurons that were analyzed for spine density. The results demonstrate that ketamine administration significantly increases the frequency and amplitude of both 5-HT- and hypocretin-induced EPSCs. The increase in EPSP amplitude is consistent with the increase in the density of mushroom spines by ketamine. The increase in 5-HT- and hypocretin-induced EPSCs indicates that there is an increase in corticocortical and thalamocortical connections, respectively. It will be important in future studies to determine the full time course for the induction of spine density by ketamine, including how long these new spines last.
Consistent with the rapid induction of synaptogenesis, we also found that ketamine produced rapid antidepressant effects in several different behavioral models. This included a significant decrease in immobility in the forced swim test (FST), decreased escape failure and latency to escape in the learned helplessness (LH) paradigm, and decreased latency to feed in the novelty suppressed feeding test (NSFT) (Li et al, 2010). Although the FST is responsive to acute administration of typical antidepressant agents, LH is only responsive to subchronic (7 days) treatment, and the NSFT, although a model of anxiety, is responsive to chronic (21 days) administration of a typical antidepressant. Together, these findings provide evidence of the fast antidepressant behavioral actions of ketamine in these rodent models.
2.2. Ketamine rapidly reverses the effects of chronic stress
In addition to studies in normal animals, we have also examined the influence of ketamine in animals exposed to chronic unpredictable stress (CUS), considered one of the better rodent models of depression. In the CUS model, repeated exposure to stress over the course of several weeks results in the development of anhedonia, a core symptom of depression, and this effect is reversed by chronic administration of a typical antidepressant (Willner, 2005; Banasr et al., 2007). In addition, chronic stress exposure decreases the density of spines in the PFC (Liu et al, 2008; Shansky and Morrison, 2009), providing a morphological endpoint that can be measured and that is relevant to the atrophy of PFC in depression (Drevets and Furey, 2010).
We found that exposure to CUS for 3 weeks decreased the density of spines in layer V pyramidal neurons, as expected, and decreased 5-HT and hypocretin-induced EPSCs in the same neurons (Li et al, 2011). A single dose of ketamine completely reversed the deficit in spine density, and this was accompanied by a reversal of the deficit in 5-HT and hypocretin-induced EPSC. Together these studies demonstrate that ketamine reverses the spine loss caused by chronic stress exposure and normalizes PFC connectivity.
To examine the possibility that reversal of the synapse loss caused by CUS influences behavior, we also examined sucrose preference, which provides a measure of anhedonia in rodents. CUS exposure for 3 weeks significantly decreased the preference for a sweetened solution, and this effect was completely reversed by a single dose of ketamine (Li et al, 2011). In addition, CUS increased the latency to feed in the NSFT, and this effect was also reversed by a single dose of ketamine. These effects of ketamine were also sustained for approximately 7 days, similar to the time course for the antidepressant actions of ketamine in treatment resistant depressed patients (Zarate et al, 2006).
3. Signaling pathways underlying the rapid antidepressant actions of ketamine
The signaling pathways and mechanisms that control synapse formation and neuroplasticity have been studied primarily in models of learning and memory (Figure 1). In particular, cellular and behavioral forms of long-term memory are dependent on new protein synthesis and are accompanied by an increase in the number of mature synapses (Holtmaat et al, 2009; Kessels and Malinow, 2009; Yoshihara et al, 2009). These studies demonstrate that protein synthesis dependent long-term memory is dependent on stimulation of the mammalian target of rapamycin (mTOR) (Hoeffer et al, 2010; Livingston et al, 2010).
mTOR is a large serine/threonine kinase that regulates the initiation of protein translation. It is ubiquitously expressed, including localization in dendritic processes where it can control new protein synthesis when required for synaptogenesis (Figure 2). Stimulation of mTOR, or more precisely the mTORC1 complex occurs via phosphorylation, particularly of the mTOR kinase domain by Akt or protein kinase B. Akt also contributes to activation of mTORC1 via repression of the tuberous sclerosis complex (TSC) 1 and 2. Akt can be activated by neurotrophic factor signaling cascades, including the phosphoinositide-3 kinase (PI3K)-phosphoinositide-dependent kinase 1 (PDK1) and by extracellular signal regulated protein kinase (ERK) pathways (Hoeffer and Klann, 2010). Another important regulatory domain is the binding site for FK506 binding protein 12 (FKBP12), which is involved in activation of mTOR signaling (Hoeffer and Klann 2010).
Figure 2.
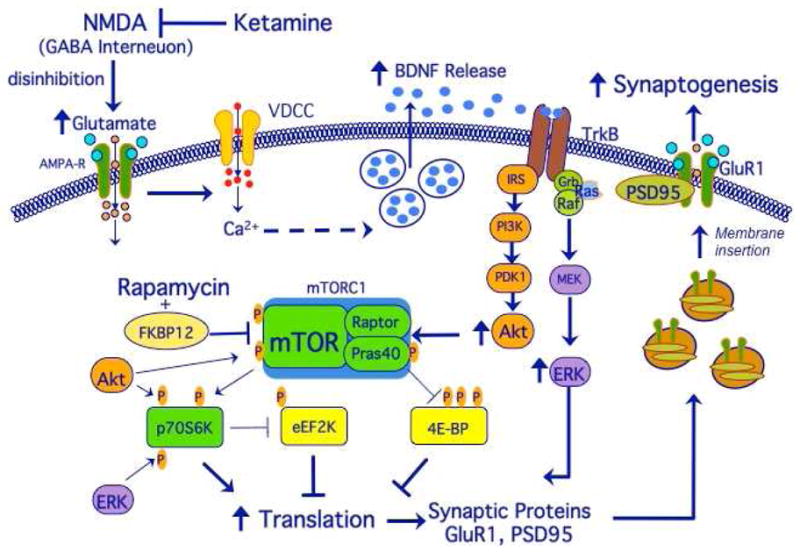
Ketamine stimulates mTOR and synaptogenesis: neurotransmitter and intracellular signaling mechanisms. Ketamine stimulates glutamate transmission, resulting in BDNF release and activation of Akt and ERK signaling, which in turn stimulate mTOR and synaptic protein synthesis. This leads to insertion of GluR1 and increased synaptogenesis, which contributes to the rapid antidepressant effects of ketamine. See text for details.
Activation of mTORC1 leads to phosphorylation and activation of p70S6 kinase and repression of the inhibitory 4E binding proteins (4E-BPs). Stimulation of p70S6 kinase, in turn controls translation at a number of levels, including the synthesis of S6 ribosomal subunit, phosphorylation of the RNA helicase cofactor eIF4A, and activation of eukaryotic elongation factor 2 (eEF2) kinase (Hoeffer and Klann, 2010; Livingston et al, 2010). The activity of p70S6 kinase is also stimulated by ERK. Phosphorylation of 4E-BPs by mTOR results in derepression of the cap-binding activity of eIF4E thereby enhancing translation initiation.
3.1. Ketamine stimulates mTOR signaling and increases synaptic protein synthesis
Based on these studies, we tested the hypothesis that the synaptogenic actions of ketamine occur via stimulation of mTOR signaling and synaptic protein synthesis (Figure 2). Levels of phosphorylated and activated forms of mTOR and p70S6 kinase were analyzed in synaptosome enriched fractions of the PFC. The results demonstrate that ketamine administration causes a very rapid induction of phospho-mTOR and phospho-p70S6 kinase, as well as increased levels of phospho-4E-BP1, which together would lead to activation of mTOR dependent translation and protein synthesis (Li et al, 2010). The induction of mTOR signaling occurs within 30 min of ketamine administration and is transient, with the mTOR phospho-proteins returning to basal, non-stimulated levels by 2 hr. Further studies demonstrate an inverted U dose response for ketamine, with low doses (5 and 10 mg/kg) stimulating and a higher, anesthetic dose (80 mg/kg) having no effect on mTOR signaling.
We also found that levels of phospho-Akt, as well as phospho-ERK, possible upstream activators of mTOR, were rapidly increased by ketamine (Figure 2). The possibility that ketamine-induction of mTOR signaling is mediated by Akt and ERK signaling was directly tested by pretreatment with inhibitors of each of these two kinases. Infusions (intracerebroventricular, ICV) of selective inhibitors of either PI3K-Akt (LY294002) or of MEK-ERK (U0126) completely blocked the ability of ketamine to stimulate the phosphorylation of mTOR, p70S6 kinase and 4E-PB1 (Li et al, 2010).
A recent paper has demonstrated that ketamine increases the phosphorylation of glycogen synthase kinase-3 (GSK-3), and that mice with a knock in mutation that blocks the phosphorylation of GSK-3 do not respond to ketamine in a behavioral model of depression (Beurel et al, 2011). Phosphorylation of GSK-3, a target of mood stabilizing agents, inhibits the activity of this kinase (Li and Jope, 2010). Further studies are required to determine the mechanisms underlying the induction of GSK-3 phosphorylation by ketamine, but it is interesting to speculate that this occurs via Akt, which is activated by ketamine and is known be a major regulator of GSK-3 (Lie et al., 2010).
To determine if there was an increase in synaptic protein synthesis, levels of activity regulated cytoskeletal protein (Arc), glutamate AMPA receptor-1 (GluR1), postsynaptic density protein-95 (PSD95), and synapsin I were examined in the synaptosome enriched PFC preparations. A single dose of ketamine significantly increased levels of these synaptic proteins 1 hr (Arc) to 2 hrs (GluR1, PSD95, and synapsin I) after administration (Li et al, 2010). Surprisingly, the induction of GluR1, PSD95, and synapsin I was relatively stable, as levels remained elevated 72 hr after ketamine treatment (Li et al, 2010), consistent with the formation and sustained induction of mature spine synapses (Holtmaat and Svoboda, 2009; Kessels and Malinow, 2009; Yoshihara et al, 2009). In contrast, the induction of Arc was transient, with levels returning to vehicle controls by 6 hr after ketamine. Arc has been implicated in both early and late phase LTP and maturation of synapses, and the functional consequences of its transient induction by ketamine are unknown (Bramham et al, 2008).
3.2. Ketamine-induction of synaptogenesis is dependent on mTOR
To directly examine the role of mTOR signaling in the actions of ketamine, animals were pretreated with rapamycin, a selective mTOR inhibitor (Figure 2). Rapamycin binds to FKBP12, and disrupts its interaction with mTOR, which is necessary for activation of mTOR (Hoeffer and Klann, 2010). For these studies, rats were infused with rapamycin (ICV) 30 min before administration of ketamine, and spine density and EPSCs were determined 24 hr later. The results demonstrate that rapamycin pretreatment completely blocks the induction of spine formation by ketamine, as well as the induction of 5-HT and hypocretin-induced EPSCs (Li et al, 2010). Rapamycin pretreatment also completely blocked the induction of synaptic proteins (GluR1, PSD95, and synapsin I) by ketamine. Similarly, the ability of ketamine to reverse the deficit in synaptic proteins resulting from exposure to CUS was blocked by rapamycin infusion (Li et al, 2010).
We also examined the requirement for mTOR signaling in the behavioral actions of ketamine. Infusion of rapamycin (ICV) completely blocked the antidepressant actions of ketamine in the FST, LH, and NSFT (Li et al, 2010). Moreover, the ability of ketamine to reverse the behavioral deficits in the CUS model were also blocked by rapamycin, including the decrease in sucrose preference and increase in latency in NSFT caused by CUS exposure (Li et al, 2010).
A recent study has also reported that the behavioral actions of ketamine are dependent on protein synthesis (Autry et al., 2011). However, this paper reports that protein synthesis is dependent on activation of eukaryotic elongation factor 2 (eEF2) and was not able to detect ketamine-induction of mTOR signaling or rapamycin blockade of the behavioral actions of ketamine (Autry et al., 2011). There are several possible technical reasons that could explain these differences. First, in the study by Autry and colleagues mTOR signaling was measured in crude homogenates of hippocampus, not synaptosome enriched fractions of PFC as reported by Li et al (2010). Since mTOR is expressed throughout neuronal and glial cell bodies, analysis of crude homogenates could mask changes in the smaller dendritic cellular compartment. Second, analysis of behavior was conducted at an early time point (30 min) after ketamine administration, a time point when patients experience mild psychotomimetic and dissociative effects (Berman et al., 2000; Zarate et al., 2006). This also corresponds to the time when levels of extracellular glutamate are increased by ketamine (Moghaddam et al., 1997). Increased glutamate transmission could underlie the increased activity observed in the FST, and as pointed out by the authors (Autrie et al., 2011), rapamycin would not be expected to block the effects of ketamine at this time point since the induction of synaptic proteins and synaptogenesis is delayed by approximately 2 hrs. Extracellular glutamate returns to basal levels after 2 hrs (Moghaddam et al., 1997), and this point corresponds very closely to the earliest time point at which an antidepressant response is observed in depressed patients (Berman et al., 2000; Zarate et al., 2006). Thus, the increase in glutamate transmission and induction of mTOR precede and are required for the induction of synaptic protein synthesis, synaptogenesis, and antidepressant behavioral responses, which are then sustained. Another difference is that the Autry study (2011) administered rapamycin systemically, which could result in peripheral side effects, whereas it was infused ICV in the Li et al study (2010).
The Autry study (2011) reports that a key action of ketamine is the rapid (30 min) induction of BDNF, demonstrating that blockade of protein synthesis by pretreatment with anisomycin blocks both the induction of BDNF and the behavioral actions of ketamine in the FST (Autry et al., 2011). It is also possible that anisomycin pretreatment decreases basal levels of BDNF protein synthesis in dendrites and spines, which would reduce activity dependent release of BDNF.
3.3. Ro 25-6981, a selective NR2B antagonist, stimulates mTOR and synaptic protein synthesis
Ketamine is a nonselective NMDA receptor antagonist, and there have been reports that selective antagonists of the NR2B subtype produce antidepressant actions in rodent models (Maeng et al, 2008a) and in humans (Preskorn et al, 2007). Therefore, we examined the influence of a selective NR2B antagonist, Ro 25-6981 on mTOR signaling and synaptic protein synthesis. The results demonstrate that the selective NR2B antagonist produces effects similar to ketamine. Ro 25-6981 administration rapidly (1 hr) stimulated mTOR signaling (increased levels of phospho-mTOR, phospho-p70S6 kinase, and phospho-4E-BP1), and increased levels of GluR1, PSD95, and synapsin I, determined at a 6 hr time point (Li et al, 2010).
The behavioral actions of the NR2B selective antagonist were also similar to ketamine. Ro 25-6981 administration produced an antidepressant response in the FST and NSFT, and these effects were blocked by infusion of rapamycin. In the CUS paradigm, the NR2B selective antagonist completely blocked the deficits in sucrose consumption and novelty suppressed feeding resulting from CUS exposure (Li et al., 2011). These findings suggest that the actions of ketamine are mediated by blockade of NR2B receptors, and provide further evidence of a functional connection between induction of synaptogenesis and antidepressant behavioral responses.
4. Role of Glutamate in the rapid antidepressant actions of NMDA Receptor antagonists
The neurotransmitter mechanisms underlying the molecular and cellular actions of ketamine have not been fully elucidated, although there is evidence that the effects are mediated by changes in glutamate transmission. There is a previous report that the behavioral actions of ketamine are blocked by pretreatment with a glutamate receptor antagonist, 2,3-dihydroxy-6-nitro-7- sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) (Maeng et al, 2008). NBQX is a selective antagonist of a glutamate ionotropic receptor subtype, a-amino-3- hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA). In addition, ketamine is reported to increase extracellular glutamate in the PFC, determined by extracellular dialysis studies (Moghaddam et al, 1997). The latter study also demonstrated that the time course for ketamine-induction of extracellular glutamate is similar to the rapid induction of mTOR signaling (i.e., increased at 30–60 min, return to baseline by 2 hrs). Moreover, the dose response for the induction of extracellular glutamate is similar to that for mTOR (i.e., inverted U, with low doses increasing but no effect or slight decrease with higher anesthetic doses). In addition, cellular and behavioral studies of synaptic plasticity demonstrate that stimulation of mTOR signaling and synaptic protein synthesis is dependent on activation of AMPA receptors (Hoeffer and Klann, 2010; Livingston et al, 2010).
Based on these reports, the requirement for glutamate-AMPA receptor activation in the ketamine-induction of mTOR signaling was determined. Pretreatment with NBQX completely blocked ketamine stimulation of phospho-mTOR, phospho-p70S6 kinase, and phospho-4E-BP1 (Li et al, 2010). NBQX pretreatment also blocked ketamine-induction of phospho-Akt and phospho-ERK, the putative upstream activators of mTOR signaling. Together, these studies are consistent with the hypothesis that ketamine-induction of mTOR signaling and synaptogenesis occurs via stimulation of glutamate transmission and AMPA receptor activation.
The cellular mechanisms underlying the ability of an NMDA receptor antagonist to increase glutamate-AMPA receptor activation has been examined using electrophysiological approaches. Administration of ketamine decreases the spontaneous activity of GABAergic interneurons in the PFC of awake rats (Homayoun and Moghaddam, 2007). In the same study, but with a delayed onset, ketamine administration was found to increase the firing rate of glutamatergic pyramidal neurons. These findings indicate that NMDA receptor antagonists block spontaneous GABAergic activity, and thereby result in disinhibition of glutamate transmission, a hypothesis that requires further testing. In addition, it is possible that ketamine also has direct effects on pyramidal neurons that further enhance synaptogenesis and the maturation of spines.
4.1. Potential role of BDNF in the actions of NMDA receptor antagonists
The mechanisms by which stimulation of glutamate-AMPA receptor activation could lead to induction of mTOR signaling have been examined in models of learning and memory. These studies demonstrate that AMPA receptor regulation of synaptic function involves the activation of L-type voltage-dependent calcium channels (VDCCs) and activity-dependent release of BDNF (Hoeffer and Klann, 2010; Jourdi et al, 2009) (Figure 2). BDNF is a key regulator of mTOR signaling and synaptic plasticity, stimulating dendritic protein synthesis, spine maturation, and synaptic transmission (Jourdi et al, 2009; Takei et al, 2004). Stimulation of AMPA receptors leads to activity-dependent release of BDNF, which in turn leads to activation of Akt and ERK, and stimulation of mTOR and synaptic protein synthesis (Hoeffer and Klann, 2010; Jourdi et al, 2009; Slipczuk et al, 2009; Takei et al, 2004).
Together, these reports are consistent with the hypothesis that the actions of ketamine are dependent on glutamate-AMPA receptor stimulation of BDNF release in the PFC, which then stimulates mTOR and spine formation. This hypothesis is supported by the report demonstrating that the behavioral actions of ketamine are blocked in BDNF conditional deletion mutants Autry et al., 2011). Together these studies suggest that increased synthesis and activity-dependent release could contribute to the actions of ketamine. Release of BDNF could stimulate TrkB receptors and PI3K-Akt and ERK signaling, which is required for ketamine activation of mTOR (Li et al, 2010). Previous studies demonstrate that typical antidepressant treatments increase the expression of BDNF in limbic brain regions (Duman and Monteggia, 2006; Krishnan et al, 2008). However, there is no evidence that typical antidepressants increase BDNF release, as demonstrated for activity dependent release of BDNF via activation of AMPA receptors and VDCC (Jourdi et al., 2009). Activity dependent release of BDNF may be a key step in the rapid actions of ketamine. Studies are currently being conducted in BDNF mutant mice to examine this possibility.
There are several BDNF mutant lines, including constitutive (heterozygous) and conditional deletion mutants. Another line of interest is a knock-in of a single nucleotide polymorphism (SNP), Val66Met, found in humans. The Val66Met SNP is located in the BDNF prodomain and blocks trafficking into the regulated secretion pathway and activity-dependent release of BDNF (Casey et al, 2009). The Met allele is found in ~20-30% of humans, and is associated with several functional deficits, including impairments in episodic memory and executive function (Egan et al, 2003; Frodl et al, 2006), decreased hippocampal volume, both in normal and depressed patients (Bueller et al, 2006; Frodl et al, 2007; Pezawas et al, 2004), and increased susceptibility to depression in patients previously experiencing trauma or stress (Gatt et al, 2009; Kaufman et al, 2006; Kim et al, 2007). The BDNF Met knock-in mice mimic the human polymorphism, with reduced hippocampal volume, atrophy of hippocampal neurons, and increased anxiety (Chen et al, 2006). These mice will be particularly interesting for studies to determine if the actions of ketamine on mTOR signaling and spine formation require activity dependent release of BDNF.
4.2. MKP-1, a negative regulator of BDNF-ERK-mTOR signaling
In our whole genome microarray studies to elucidate the pathophysiology of depression, we have identified a negative regulator of the BDNF-ERK cascade, mitogen activated protein (MAP) kinase phosphatase-1 (MKP-1) (Duric et al, 2010). MKP-1 is a dual specificity phosphatase that dephosphorylates both threonine and tyrosine residues and is a key negative regulator of the ERK signaling cascade (Jeffrey et al, 2007). In our microarray studies of postmortem tissue, we found that levels of MKP-1 were significantly increased in the hippocampus of depressed subjects relative to matched controls (Duric et al, 2010). There was also a significant decrease in the levels of ERK, as previously reported (Dwivedi et al, 2006a). In rodent studies, we found that levels of MKP-1 were increased in the hippocampus by exposure to CUS, and this effect was reversed by chronic antidepressant treatment. Increased expression of MKP-1 in depressed subjects and in response to CUS is consistent with reports that MKP-1 is an immediate early gene that is induced by cellular stress and adrenal glucocorticoids (Keyse and Emslie, 1992; Seta et al, 2001).
The functional consequences of altered MKP-1 levels were examined using viral vector and mutant mouse approaches. Over expression of MKP-1 in the hippocampus using a recombinant adeno-associated virus was sufficient to produce a depressive phenotype in rats, including a decrease in sucrose preference, an increase in escape failures in the LH model, and an increase in latency to feed in the NSFT (Duric et al, 2010). Conversely, MKP-1 deletion mutant mice displayed a resilient behavioral phenotype upon exposure to CUS (i.e., no decrease in sucrose consumption). MKP-1 null mice exposed to CUS also displayed higher levels of phospho-ERK compared to wild type controls exposed to the same CUS paradigm.
Abnormal and sustained elevation of MKP-1, combined with decreased expression of ERK, could lead to decreased BDNF-ERK signaling, and thereby contribute to decreased basal, as well as activity dependent activation of mTOR signaling. Both the PFC and hippocampus and related circuitry have been implicated in depression and antidepressant response (Drevets and Furey, 2010; Macqueen et al, 2008), and in the actions of ketamine (Li et al., 2010; Autry et al., 2011). Additional studies are required to determine if mTOR signaling and synaptogenesis are regulated by MKP-1 in both regions, as well as other limbic structures.
5. Rapid acting antidepressant targets
The discovery of ketamine as a rapid acting antidepressant that is effective in treatment resistant depressed patients is a major breakthrough for mood disorders therapeutics. Ketamine is also effective for bipolar depression (DiazGranados et al, 2010) and suicide ideation (DiazGranados et al., 2010b; Larkin and Beautrais, 2011). However, there are limitations: ketamine is a street drug with abuse potential, and is reported to cause neurotoxicity with repeated use (Behrens et al, 2007).
Characterization of the cellular signaling mechanisms underlying the effects of ketamine has provided potential targets for new medications that might share the therapeutic actions of ketamine, but without the side effects. Another possibility would be to identify agents that sustain the actions of ketamine. We have discussed evidence that a selective NR2B receptor antagonist produces ketamine like effects in rodent models (Li et al, 2010) and in human subjects (Preskorn et al, 2007). However, the NR2B agent used in the latter study, CP-101,606, did cause some dissociative effects at the initial dose tested (Preskorn et al., 2007). Additional studies will be required to further demonstrate the efficacy and safety of compounds selective for the NR2B receptor.
Other possible targets that could produce ketamine like effects also influence glutamate transmission. As discussed, ketamine-stimulation of mTOR signaling and antidepressant behavioral actions are dependent on glutamate-AMPA receptor activation. Drug targets that enhance glutamate transmission or that activate AMPA receptors could also produce rapid and efficacious antidepressant actions. Regulation of glutamate transmission has been an area of interest for drug development for several years, and includes cognitive enhancing agents for neurodegenerative disorders and medications for schizophrenia, as well as depression. Two major targets are presynaptic metabotropic glutamate type 2/3 receptors (mGlu2/3) that regulate glutamate release and postsynaptic AMPA receptors (Figure 3).
Figure 3.
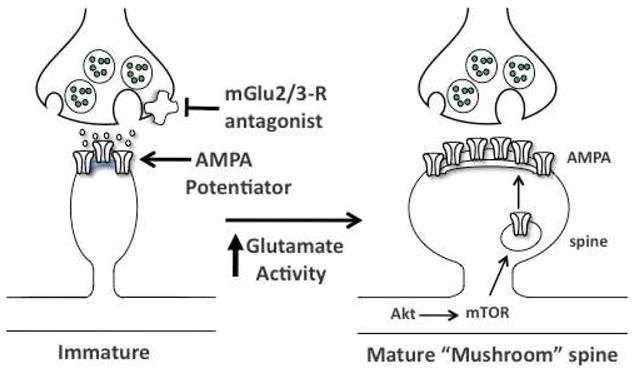
Targets for induction of synaptogenesis and ketamine-like effects. Potential targets that could produce ketamine like effects include presynaptic mGlu2/3 receptor antagonists and postsynaptic positive AMPA receptor potentiating agents. Both would lead to increased glutamate transmission and AMPA receptor activation, and thereby stimulate mTOR signaling and increase synaptogenesis. See text for details.
The mGlu2/3 receptors are located on presynaptic terminals and negatively regulate glutamate release. Antagonists of mGlu2/3 receptors are reported to have antidepressant actions in acute behavioral models (Pałucha-Poniewiera et al, 2010; Pilc et al, 2007). Positive AMPA receptor modulating agents have the ability to enhance AMPA receptor function without direct receptor activation (Arai and Kessler, 2007). AMPA receptor potentiating drugs are reported to increase LTP, learning and memory, and BDNF (Arai and Kessler, 2007; Arai et al, 2000), providing further evidence that these drugs could lead to activation of mTOR. There is also evidence that AMPA receptor potentiating drugs have antidepressant actions (Bai et al, 2003). Studies are currently underway to determine if mGlu2/3 antagonists or AMPA receptor potentiating drugs stimulate mTOR signaling, increase synaptogenesis, and produce rapid antidepressant behavioral actions in the CUS model of depression.
When developing these drugs it is important to recognize that increased glutamate neurotransmission could also have deleterious effects. For example stress acutely increases glutamate (Moghaddam, 2002), and sustained elevation of glutamate transmission could produce neurotoxic effects. Studies to determine optimal levels of glutamate transmission for a therapeutic response, the relevant brain circuits to target, and safety of these treatments must be determined before developing such agents for the widespread treatment of depression and other psychiatric illnesses.
6. Summary and future directions
The actions of NMDA receptor antagonists on mTOR signaling and on the density and function of spine synapses represents a fundamental shift in our understanding of the mechanisms underlying rapid acting, efficacious antidepressant treatments. The ability of ketamine to increase synaptogenesis could thereby rapidly reverse the structural deficits resulting from chronic stress exposure that are thought to contribute to depressive symptoms. Sustained induction of negative regulators of BDNF-ERK signaling, such as MKP-1, could also contribute to the atrophy of neurons and decreased volume of limbic brain regions in depression.
The mTOR translational system can be influenced, both positively and negatively by a variety of neurotransmitter, endocrine, and metabolic signaling pathways, and could thereby serve as a nexus for control of synaptogenesis. This complex regulatory system raises the possibility that there are additional negative regulators of mTOR signaling and synaptogenesis that play a role in neuronal atrophy caused by stress exposure, an area that is under active investigation. Conversely, pathways that enhance mTOR signaling could also be targeted for novel therapeutic approaches for the treatment of depression, although the ubiquitous expression and function of mTOR raises the possibility of side effects. Nevertheless, novel targets that influence glutamate transmission, BDNF-mTOR signaling, and synaptogenesis offer promise for the development of safer, rapid acting and efficacious antidepressant agents for the treatment of depression.
Clinical studies demonstrate that ketamine produces a rapid antidepressant response.
Ketamine rapidly increases synaptogenesis in rodent prefrontal cortex.
Ketamine-induction of synaptogenesis requires mTOR signaling.
Ketamine rapidly reverses synaptic loss caused by stress and depression.
Ketamine results in a functional reconnection of neurons.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arai A, Kessler M. Pharmacology of ampakine modulators: from AMPA receptors to synapses and behavior. Curr Drug Targets. 2007;8:583–602. doi: 10.2174/138945007780618490. [DOI] [PubMed] [Google Scholar]
- Arai A, Kessler M, Rogers G, Lynch G. Effects of the potent ampakine CX614 on hippocampal and recombinant AMPA receptors: interactions with cyclothiazide and GYKI 52466. Mol Pharmacol. 2000;58:802–813. doi: 10.1124/mol.58.4.802. [DOI] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM. NMDA receptor blockade at rest triggers rapid behavioural anatidepressant responses. Nature. 2011 doi: 10.1038/nature10130. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Behrens M, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Banasr M, Valentine GW, Li XY, Gourley S, Taylor J, Duman RS. Chronic stress decreases cell proliferation in adult ceregral cortex of rat: reversal by antidepressant treatment. Biol Psych. 2007;62:496–504. doi: 10.1016/j.biopsych.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Berman R, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiat. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Beurel E, Jope RS. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3. J Neuroinflamm. 2009;11:6–9. doi: 10.1186/1742-2094-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham C, Worley PF, Moore MJ, Guzowski JF. The immediate early gene arc/arg3.1: regulation, mechanisms, and function. J Neurosci. 2008;28:11760–11767. doi: 10.1523/JNEUROSCI.3864-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueller J, Aflab M, Sen S. BDNF Val66Met allele is associated with reduced hippocampal volume in healthy subjects. Biol Psych. 2006;59:812–815. doi: 10.1016/j.biopsych.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Casey B, Glatt CE, Tottenham N, Soliman F, Bath K, Amso D, Altemus M, Pattwell S, Jones R, Levita L, McEwen B, Magariños AM, Gunnar M, Thomas KM, Mezey J, Clark AG, Hempstead BL, Lee FS. Brain-derived neurotrophic factor as a model system for examining gene by environment interactions across development. Neurosci. 2009;164:108–120. doi: 10.1016/j.neuroscience.2009.03.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Leraci A, Khan T, Siao C-J, Herrara DG, Toth M, Yang C, McEwen BS, Hampstead BL, Lee FS. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary C, Linde JA, Hiscock KM, Hadas I, Belmaker RH, Agam G, Flaisher-Grinberg S, Einat H. Antidepressive-like effects of rapamycin in animal models: Implications for mTOR inhibition as a new target for treatment of affective disorders. Brain Res Bull. 2008;76:469–473. doi: 10.1016/j.brainresbull.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Clemens J, Nimmo WS, Grant IS. Bioavailability, pharmacokinetics, and analgesic activity of ketamine in humans. J Pharm Sci. 1982;71:539–42. doi: 10.1002/jps.2600710516. [DOI] [PubMed] [Google Scholar]
- Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, Kammerer WA, Quezado Z, Luckenbaugh DA, Salvadore G, Machado-Vieira R, Manji HK, Zarate CA., Jr A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psych. 2010;67:793–802. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diazgranados N, Ibrahim LA, Brutsche NE, Ameli R, Henter ID, Luchenbaugh DA, Machado-Vieira R, Zarate CA., Jr Rapid resolution of suicidal ideation after a single infusion of an N-methyl-D-asparate antagonist in patients with treatment-resistant major depressive disorder. 2010b doi: 10.4088/JCP.09m05327blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets W, Furey ML. Replication of scopolamine's antidepressant efficacy in major depressive disorder: A randomized, placebo-controlled clinical trial. Biol Psych. 2010;67:432–438. doi: 10.1016/j.biopsych.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman R, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psych. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Duric V, Banasr M, Licznerski P, Schmidt H, Stockmeier C, Simen A, Newton SS, Duman RS. Negative regulator of MAP kinase is increased in depression and is necessary and sufficient for depressive behavior. Nat Med. 2010;16:1328–1332. doi: 10.1038/nm.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Conley RR, Pandey GM. ERK MAP kinase signaling in post-mortem brain of suicide subjects: differential regulation of upstream Raf kinases Raf-1 and B-Raf. Mol Psych. 2006;11:86–98. doi: 10.1038/sj.mp.4001744. [DOI] [PubMed] [Google Scholar]
- Egan M, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Frodl T, Schaub A, Banac S, Charypar M, Jager M, Kummler P, Bottlender R, Zetzsche T, Leinsinger G, Reiser M, Moller HJ, Meisenzahl EM. Reduced hhippocampal vollume correlates with executive dysfunctioning in major depression. J Psych Neurosci. 2006;31:316–323. [PMC free article] [PubMed] [Google Scholar]
- Frodl T, Schule C, Schmitt G, Born C, Baghai T, Zill P, Bottlender R, Rupprecht R, Bondy B, Reiser M, Moller H-J, Meisenzahl EM. Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depressio. Arch Gen Psych. 2007;64:410–416. doi: 10.1001/archpsyc.64.4.410. [DOI] [PubMed] [Google Scholar]
- Gatt J, Nemeroff CB, Dobson-Stone C, Pauyl RH, Bryant RA, Schofield PR, Gordon E, Kemp AH, Williams LM. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psych. 2009;14:681–95. doi: 10.1038/mp.2008.143. [DOI] [PubMed] [Google Scholar]
- Hoeffer C, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey K, Camps M, Rommel C, Mackay CR. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov. 2007;6:391–403. doi: 10.1038/nrd2289. [DOI] [PubMed] [Google Scholar]
- Jourdi H, Hsu YT, Zhou M, Qin Q, Bi X, Baudry M. Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J Neurosci. 2009;29:8688–8697. doi: 10.1523/JNEUROSCI.6078-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman J, Yang BZ, Douglas-Palumberi H, Grasso D, Lipschitz D, Houshyar S, Krystal JH, Gelernter J. Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol Psychiatry. 2006;59:673–680. doi: 10.1016/j.biopsych.2005.10.026. [DOI] [PubMed] [Google Scholar]
- Kessels H, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler R, Berlund P, Demler O, Jin R, Koretz D, Merikangas KR. The epidemiology of major depressive disorder: results for the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- Keyse S, Emslie EA. Oxidative stress and heat shock induce a human gene encoding a protein-tyrosine phosphatase. Nature. 1992;359:644–647. doi: 10.1038/359644a0. [DOI] [PubMed] [Google Scholar]
- Kim J, Stewart R, Kim SW, Yang SJ, Shin IS, Kim YH, Yoon JS. Interactions Between Life Stressors and Susceptibility Genes (5-HTTLPR and BDNF) on Depression in Korean Elders. Biol Psych. 2007;62:423–428. doi: 10.1016/j.biopsych.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin GL, Beautrais AL. A preliminary naturalistic study of low-dose ketamine for depression and suicide ideation in the emergency department. Int J Neuropsychophamracol. 2010 doi: 10.1017/S1461145711000629. ebup ahead of print. [DOI] [PubMed] [Google Scholar]
- Li X, Jope RS. Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacol. 2010b;35:2143–2154. doi: 10.1038/npp.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lee BY, Liu RJ, Banasr M, Dwyer J, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Liu R0-J, Dwyer J, Banasr M, Lee B, Son J, Li X-Y, Aghajanian G, Duman RS. Glutamate N-methyl-D-aspartate Receptor Antagonists Rapidly Reverse Behavioral and Synaptic Deficits Caused by Chronic Stress Exposure. Biol Psych. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RJ, Aghajanian GK. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. PNAS. 2008;105:359–364. doi: 10.1073/pnas.0706679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston M, Atas E, Meller A, Sonenberg N. Mechanisms governing the control of mRNA translation. Phys Biol. 2010;7:021001. doi: 10.1088/1478-3975/7/2/021001. [DOI] [PubMed] [Google Scholar]
- Macqueen G, Yucel K, Taylor VH, Macdonald K, Joffe R. Posterior hippocampal volumes are associated with remission rates in patients with major depressive disorder. Biol Psych. 2008;64:880–883. doi: 10.1016/j.biopsych.2008.06.027. [DOI] [PubMed] [Google Scholar]
- Maeng S, Zarate CA, Jr, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psych. 2008;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- McEwen B. Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol. 2008;583:174–185. doi: 10.1016/j.ejphar.2007.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B. Stress activation of glutamate neurotransmission in the prefrontal cortex: implications for dopamine-associated psychiatric disorders. Biol Psych. 51:75–87. doi: 10.1016/s0006-3223(01)01362-2. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2912–2917. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pałucha-Poniewiera A, Wierońska JM, Brański P, Stachowicz K, Chaki S, Pilc A. On the mechanism of the antidepressant-like action of group II mGlu receptor antagonist, MGS0039. Psychopharm. 2010;212:523–535. doi: 10.1007/s00213-010-1978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezawas L, Verchinski BA, Mattay VS. The brain-derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J Neurosci. 2004;24:10099–10102. doi: 10.1523/JNEUROSCI.2680-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilc A, Chaki S, Nowak G, Witkin JM. Mood disorders: regulation by metabotropic glutamate receptors. Biochem Pharmacol. 2007;75:997–1006. doi: 10.1016/j.bcp.2007.09.021. [DOI] [PubMed] [Google Scholar]
- Preskorn S, Baker B, Omo K, Kolluri S, Menniti FS, Landen JW. A placebo-controlled trial of the NR2B subunit specific NMDA antagonist CP-101,606 plus paroxetine for treatment resistant depression (TRD). Paper presented at: APA.2007. [Google Scholar]
- Seta KA, Kim R, Kim HW, Millhorn DE, Beitner-Johnson D. Hypoxia- induced regulation of MAPK phosphatase-1 as identified by subtractive suppression hybridization and cDNA microarray analysis. J Biol Chem. 2001;276:44405–44412. doi: 10.1074/jbc.M103346200. [DOI] [PubMed] [Google Scholar]
- Shansky R, Morrison JH. Stress-induced dendritic remodeling in the medial prefrontal cortex: effects of circuit, hormones and rest. Brain Res. 2009:108–13. doi: 10.1016/j.brainres.2009.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slipczuk L, Bekinschtein P, Katche C, Cammarota M, Izquierdo I, Medina JH. BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLoS One. 2009;23:e6007. doi: 10.1371/journal.pone.0006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, Nawa H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci. 2004;24:9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi M, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz B, McGrath PJ, Shores-Wilson K, Biggs MM, Balasubramani GK, Fava M STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psych. 2006;163:28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- Wilner P. Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiol. 2005;52:90–110. doi: 10.1159/000087097. [DOI] [PubMed] [Google Scholar]
- Yoshihara Y, De Roo M, Muller D. Dendritic sppine formation and stablization. Curr Opin Neurobiol. 2009;19:146–153. doi: 10.1016/j.conb.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Zarate C, Singh J, Manji HK. Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol Psych. 2006;59:1006–1020. doi: 10.1016/j.biopsych.2005.10.021. [DOI] [PubMed] [Google Scholar]