

Abstract
Our view of the endothelium was transformed around 30 years ago, from one of an inert barrier to that of a key endocrine organ central to cardiovascular function. This dramatic change followed the discoveries that endothelial cells (ECs) elaborate the vasodilators prostacyclin and nitric oxide. The key to these discoveries was the use of the quintessentially pharmacological technique of bioassay. Bioassay also revealed endothelium-derived hyperpolarizing factor (EDHF), particularly important in small arteries and influencing blood pressure and flow distribution. The basic idea of EDHF as a diffusible factor causing smooth muscle hyperpolarization (and thus vasodilatation) has evolved into one of a complex pathway activated by endothelial Ca2+ opening two Ca2+-sensitive K+-channels, KCa2.3 and KCa3.1. Combined application of apamin and charybdotoxin blocked EDHF responses, revealing the critical role of these channels as iberiotoxin was unable to substitute for charybdotoxin. We showed these channels are arranged in endothelial microdomains, particularly within projections towards the adjacent smooth muscle, and close to interendothelial gap junctions. Activation of KCa channels hyperpolarizes ECs, and K+ efflux through them can act as a diffusible ‘EDHF’ stimulating Na+/K+-ATPase and inwardly rectifying K-channels. In parallel, hyperpolarizing current can spread from the endothelium to the smooth muscle through myoendothelial gap junctions upon endothelial projections. The resulting radial hyperpolarization mobilized by EDHF is complemented by spread of hyperpolarization along arteries and arterioles, effecting distant dilatation dependent on the endothelium. So the complexity of the endothelium still continues to amaze and, as knowledge evolves, provides considerable potential for novel approaches to modulate blood pressure.
LINKED ARTICLES
This article is part of a themed issue on Vascular Endothelium in Health and Disease. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.164.issue-3
Keywords: EDHF, endothelium, NO, prostacyclin, vasodilatation, potassium channels, gap-junctions, KCa2.3, KCa3.1, connexins, hyperpolarization
This review was inspired by the 2009 JR Vane Lecture, delivered by the first author in the Summer of that year at the Edinburgh BPS meeting. It tracks our perspective of the key discoveries and the development of understanding of the pathway commonly referred to as endothelium-derived hyperpolarizing factor or EDHF. As such, it is probably not as all encompassing as many reviews, but provides more of a personal view of the evolution of this field over the last 20 or so years.
After 30 years, it still seems quite remarkable to think that until circa 1980, the endothelium was considered to be merely an inert lining, covering the luminal surface of blood vessels. Much like Teflon®, endothelial cells (ECs) were assumed simply to present a non-stick surface, enabling them to separate the flowing blood from the pro-aggregatory constituents of the sub-intimal layers of the vascular wall. This view changed dramatically with Robert Furchgott's discovery of endothelium-derived relaxing factor (EDRF), reported in a seminal paper (Furchgott and Zawadzki, 1980) that formed the basis for the later award of the 1998 Nobel Prize in Physiology or Medicine. The prize was awarded to Furchgott, along with Louis Ignarro and Ferid Murad, for ‘their discoveries concerning nitric oxide (NO) as a signalling molecule in the cardiovascular system’. However, as we shall see, NO represents just one of several key processes by which the endothelium regulates the smooth muscle cells (SMCs) that form the major part of the wall of most blood vessels.
It is of course interesting to speculate how long it might have taken without this discovery before we appreciated the crucial role the endothelium plays in controlling both the distribution of blood and pressure within the circulation. In all likelihood, not very long at all, because an active role for the endothelium had already been recognized in the UK by John Vane, Salvador Moncada and colleagues. During the 1970s, they discovered that the anti-aggregating properties of this cellular monolayer reflected its ability to elaborate a novel arachidonic acid derivative that was also a potent vasodilator. They initially termed this short-lived derivative prostaglandin X (PGX), though we of course now know it as prostacyclin (PGI2). The discovery played a significant part in securing the 1982 Nobel Prize in Physiology or Medicine for Vane, together with Bergström and Samuelsson. So as such, 1982 could well be considered to represent the first Nobel Prize for the discovery of an endothelium-derived mediator. Indeed, by 1990, the secretory activity of the endothelium was sufficiently appreciated for Anggard (1990) to propose that it should be considered ‘the largest endocrine gland in the body’. Vane showed clearly that this was an appropriate description in his Croonian Lecture delivered to the Royal Society a few years later (Vane, 1994). Vane's very significant contributions, including the discovery of PGI2, have been elegantly reviewed recently by Rod Flower, one of his distinguished colleagues during that exciting period of pharmacological discovery in the 1970s (Flower, 2006).
Of great interest to pharmacologists is of course the fact that the discovery of both PGI2 and EDRF, and thus the award subsequently of two Nobel prizes, relied on the classic pharmacological approach of bioassay elegantly developed by Sir John Gaddum. He once famously remarked ‘The pharmacologist has been a “jack of all trades”, borrowing from physiology, biochemistry, pathology, microbiology, and statistics – but he has developed one technique of his own and that is the technique of bioassay …’ (Gaddum, 1964). Vane very cleverly adapted Gaddum's ‘superfusion’ bioassay to provide the cascade bioassay. Carefully selected tissues were arranged in two series, to allow the parallel assay of injected samples and/or the active components in fluid taken directly from the outflow of a perfused organ (Vane, 1964). This approach enabled first the discovery of thromboxane A2 and, subsequently, the demonstration that arterial microsomes can metabolise PGG2 to a labile product with both vasodilator and platelet anti-aggregatory properties. This was PGX, which once the structure was elucidated was given the trivial name prostacyclin (Whittaker et al., 1976). Shortly afterwards, it was realized that the site of vascular synthesis was in fact the endothelium (Bunting et al., 1977; Moncada et al., 1977; Weksler et al., 1977). A more detailed overview of these major discoveries can be obtained in Moncada et al. (1976), Gryglewski et al. (1976) and Flower (2006).
Of course, Furchgott's discovery of EDRF also depended entirely on the use of bioassay, or perhaps more accurately the judicious interpretation of what was his rather unexpected data. Having designed an experimental protocol to investigate β-adrenoceptor-evoked relaxation in isolated aortic rings, the story is that his technician mixed up the protocol. The result was that a robust relaxation was obtained to muscarinic agonists. At the time this was a very surprising outcome, because although muscarinic agonists were known as potent vasodilators in vivo, in vitro they either did not affect vascular preparations or caused vasoconstriction. Furchgott quickly realized that an absence of dilator ability in vitro correlated with damage to the endothelium during tissue isolation. Like Vane, he then modified the bioassay technique, this time sandwiching together artery strips, one with and one without endothelium, to show that smooth muscle relaxation to muscarinic agonists was due to the release of a diffusible dilator from the endothelium, which he named EDRF. The rest, as they say, is history. By 1988, EDRF had been identified as NO and the fact that NO serves as a fundamental biological signalling molecule throughout the body was widely recognized.
A third endothelium-derived dilator: the discovery of EDHF
Kuriyama's group in Japan had shown, rather oddly, that muscarinic stimulation of rabbit and guinea pig arteries could evoke vascular hyperpolarization associated with vasoconstriction (Kuriyama and Suzuki, 1978; Kitamura and Kuriyama, 1979). After Furchgott's discovery of the endothelium as the primary target for muscarinic agonists, Bolton showed that smooth muscle hyperpolarization to carbachol also depended on the integrity of this monolayer of cells, and suggested that muscarinic stimulation might cause hyperpolarization by releasing a factor from the ECs (Bolton et al., 1984). The coincident contractile action reported by Kuriyama et al. could then be explained simply by the fact that, in the species they studied, the muscarinic receptors were present on endothelial and vascular SMCs, with stimulation of both receptor populations generating a predominantly contractile response.
Two absolutely fundamental questions arose from considering these observations together. First, as both EDRF and hyperpolarization originated in the endothelium, was the latter simply a facet of the former? Second, how important was the smooth muscle hyperpolarization in terms of vascular physiology; was it simply a pharmacological curiosity?
Both questions were cleverly addressed by Chen et al., and presented first at a meeting in Melbourne in 1987, then in a classic paper that truly marked the beginning of the EDHF field (Chen et al., 1988). Why was this paper so seminal? Because through the use of microelectrodes, they were able to provide the first direct demonstration indicating endothelium-dependent hyperpolarization to be distinct from EDRF. By 1987, it was clear that EDRF was in fact NO or an NO-releasing entity (see Furchgott, 1998). At that time, the pharmacological tools available to probe the effects of NO were fairly limited, not least by the fact that NO synthase had yet to be identified (see Schmidt et al., 1988a,b; Bredt and Snyder, 1990; Forstermann et al., 1990), so of course inhibitors for this enzyme remained unavailable for a few years (Moore et al., 1990; Rees et al., 1990). However, methylene blue (which blocks guanylyl cyclase and scavenges NO) and haemoglobin (which binds NO) were available (Martin et al., 1985), and neither altered endothelium-dependent hyperpolarization in the rat aorta and pulmonary artery. Nor did they depress 86Rb efflux, a marker of K+ movement. In contrast, smooth muscle relaxation (of tone induced with noradrenaline) was inhibited by well over 50% while cGMP accumulation was effectively abolished. Furthermore, none of these responses were altered by indomethacin. Not surprisingly then, the major conclusion was ‘the properties of this [hyperpolarizing] agent suggest it should be designated endothelium-derived hyperpolarizing factor (EDHF).’ Although this work did not provide any evidence for a discrete factor per se, the use of bioassay again proved crucial. Félétou and Vanhoutte (1988) reported smooth muscle hyperpolarization in endothelium-denuded small coronary arteries, but only when these arteries were co-incubated with femoral artery segments with an intact endothelium. Furthermore, although this study did not suggest that hyperpolarization might be due to anything other than NO/EDRF, it did reveal that hyperpolarization could be blocked with ouabain. As we discuss later, it transpires in some arteries that Na+/K+-ATPase is a target for K+ acting as an EDHF.
While Chen et al. (1988) clearly showed that endothelium-dependent hyperpolarization was distinct from the action of EDRF, the strong and predominant vasodilator influences of the latter seemed to argue against the possibility that EDHF was actually an important physiological entity. The concept of a new endogenous relaxing agent elaborated by the endothelium had been developed in a ‘Current Awareness’ article in Trends in Pharmacological Sciences by Taylor and Weston (1988), and they speculated that as hyperpolarization presumably caused vasodilatation by closing voltage-dependent Ca2+ channels, ‘Such a process is likely to be very important in small arterioles which are very dependent on Ca2+ influx during contraction’. This proved to be a very insightful suggestion. In small resistance arteries, neither hyperpolarization nor relaxation to ACh were reduced by oxyhaemoglobin, indomethacin or nitro-L-arginine (Garland and McPherson, 1992). So hyperpolarization could completely relax pre-constricted resistance arteries. By this time, the use of the Mulvany–Halpern wire myograph made it possible to attempt simultaneous recording of smooth muscle membrane potential and tension, so data could be generated from a single artery segment under the same experimental conditions, avoiding any differences due to the stretch applied to the muscle cells, a concern when these parameters were recorded independently.
So the EDHF phenomenon came to be recognized as smooth muscle relaxation in blood vessels, evoked (usually by the endothelium-specific agonist, ACh) in the presence of inhibitors of cyclooxygenases and NO synthase. Given the technical difficulties associated with microelectrode recording in vascular tissue, it is not really surprising that, in spite of the quite considerable literature generated in this area, the vast majority of studies simply assess EDHF indirectly, by measuring vascular relaxation in the presence of a contractile agent (such as an α-adrenoceptor or thromboxane receptor agonist). Thus, although many papers refer to EDHF, they overlook the fundamental importance of recording the membrane potential changes that defined the pathway. So in effect, most studies were not investigating the consequences of hyperpolarization but of repolarization. This distinction has actually proved to be crucial when coming to understand the complexity of the mechanisms that are responsible for initiating and spreading endothelium-dependent hyperpolarization. Although the acronym EDHF has been used most widely to denote the phenomenon of endothelium-dependent hyperpolarization (EDH), it is perhaps pertinent at this stage to point out that, especially in smaller vessels, there is very good evidence a factor may not necessarily be involved. As such, EDH is really a more accurate description of what is a key pathway for vasodilatation.
ECs SKCa (KCa2.3) and IKCa (KCa3.1) are the basis of EDH
The next major step in defining the EDH pathway was the discovery that hyperpolarization reflected the activation of two types of K+-channel, the small and intermediate conductance Ca2+-sensitive K+ channels (KCa) – SKCa (KCa2.3) and IKCa (KCa3.1)(nomenclature follows Alexander et al., 2009). Furthermore, and probably equally as important, these channels were shown to be located on the endothelium, rather than serving as a smooth muscle target for a diffusible endothelium-derived factor or EDHF.
Although K+ efflux was clearly a fundamental, defining feature of the EDHF response (Chen et al., 1988), it took quite some time to identify the underlying K+-channels. With hindsight, it is clear that in large part this was because two types of K+-channel are involved. EDHF-mediated responses were quickly shown to be insensitive to glibenclamide, excluding a role for ATP-sensitive K+-channels (KATP) (Garland and McPherson, 1992; Plane and Garland, 1993). A key role for voltage-gated K+-channels (KV) also seemed extremely unlikely, as the EDHF response relied on pronounced and sustained hyperpolarization. This was confirmed when EDHF-like relaxation was shown to be insensitive to KV block (Adeagbo and Triggle, 1993). However, KCa did seem to have some role, as relaxation was reduced by either the non-selective blocker tetrabutylammonium or the specific SKCa (KCa2.3) channel blocker apamin (Adeagbo and Triggle, 1993; Holzmann et al., 1994). Taking this further, by recording both hyperpolarization and relaxation simultaneously, we found not only that apamin but also that charybdotoxin slightly reduced both responses. We assumed at that stage charybdotoxin must be blocking the large-conductance KCa (BKCa: KCa1.1) channels known to be present on the SMCs, and reasoned that these data indicated a role for both SKCa and BKCa channels. Suspecting parallel signalling, we applied both of these toxins in combination, and for the first time obtained a block of EDHF hyperpolarization and the associated relaxation (Waldron and Garland, 1994). This proved to be a seminal observation. It was confirmed soon after by Zygmunt and Hogestatt (1996), who crucially extended the observation to show that iberiotoxin was not able to substitute for charybdotoxin. As well as BKCa, charybdotoxin was known to block the intermediate form of KCa (IKCa: KCa3.1) channels, thus it appeared to be that these channels, along with the SKCa channels, were involved in the EDHF response.
It was generally assumed, wrongly as it turns out, that both types of K+-channel must be located on the smooth muscle, and thus subject to activation by a diffusible EDHF released from the endothelium. At that time, it was clear from the work of Mark Nelson and others, that vascular smooth muscle contained BKCa channels, and we were able to demonstrate this with high concentrations of NO, which activated BKCa channels in mesenteric artery SMCs. However, we could find no evidence for SKCa channels (Mistry and Garland, 1998a,b;). Around this time, Gillian Edwards and Arthur Weston in Manchester published a critical appraisal of the EDHF field, and with the benefit of hindsight it is really quite remarkable how accurate the insights provided in this article have proved to be. Not least was the suggestion that perhaps the KCa-channels might actually reside on the endothelium rather than the SMCs. They also suggested that K+ ion efflux through these channels may serve to activate muscle Na+/K+-ATPase and/or inwardly rectifying K+-channels (KIR) to cause hyperpolarization, making K+ ion the elusive EDHF (Edwards and Weston, 1998). This period marked the start of an exciting collaboration between two of us (CJG and KAD) and the Manchester group, with a number of major outcomes. First, intracellular recording directly from endothelial cells in situ revealed that ACh evoked hyperpolarization in these cells, and this response could be blocked by apamin and charybdotoxin in combination. The simplest explanation was that the KCa resides on the ECs. This of course made sense when one remembers that M3 muscarinic receptors signal via inositol trisphosphate (InsP3), so are able to provide the signal for activation of the Ca2+-sensitive, but voltage-insensitive, KCa. Thus, we concluded that the SKCa and IKCa channels were present on the endothelium not the SMCs, a finding which was subsequently confirmed by us and others. The second major outcome of our joint studies was to provide direct evidence suggesting that, after efflux from these channels, K+ acts as an EDHF (Edwards et al., 1998).
K+– a humoral EDHF
The concept of a local increase in extracellular K+ serving to evoke vasodilatation was not in itself novel. Small increases in [K+]o had been shown directly to cause vasodilatation by Dawes (1941) and, in the cerebral circulation, similar increases were suggested to underlie, at least in part, the close coupling between nerve activity and blood flow that had first been defined in 1890 by Roy and Sherrington (Roy and Sherrington, 1890; Kuschinsky et al., 1972). The mechanism responsible is explained by observations in pressurized coronary and cerebral arteries, where increases in extracellular K+ of <20 mM (in total) stimulated smooth muscle hyperpolarization and relaxation by increasing inwardly rectifying K+-channel (KIR) conductance and activating Na+/K+-ATPase (McCarron and Halpern, 1990; Knot et al., 1996). Completely novel now was the discovery that K+ released from the ECs into the artery wall could evoke hyperpolarization, and thus relaxation, of the adjacent SMCs. Using isolated mesenteric and hepatic arteries from the rat, we observed that both smooth muscle hyperpolarization and relaxation to either exogenous K+ or EDHF (activated by stimulating selectively the endothelium with ACh) were of similar magnitude and sensitivity to inhibition with a combination of Ba2+ and ouabain, employed to block KIR channels and Na+/K+-ATPase. However, in contrast to the SMCs, the EC hyperpolarization to ACh was not sensitive to Ba2+ and ouabain (Figure 1). This observation suggested that the release of K+ through SKCa and IKCa channels was activating KIR channels and Na+/K+-ATPase to evoke hyperpolarization. Furthermore, during EC stimulation with ACh, a K+-selective electrode inserted into the proximity of the endothelial-smooth muscle interface recorded an increase in [K+]o of around 10 mM. The increase was sufficient to explain the measured EDHF hyperpolarization and relaxation (Edwards et al., 1998). However, despite the importance of these observations, everything was not completely clear-cut. The small arteries used in this study were the rat hepatic and mesenteric arteries, and while the EDHF response in the former was blocked with Ba2+ and ouabain, in the latter a significant component persisted. At the time, we concluded that the persistent relaxation must reflect an additional pathway, and suggested that myoendothelial gap junctions (MEGJs) between endothelial and SMCs may be responsible. We subsequently proved this is, in fact, the case and this work is discussed next (Mather et al., 2005).
Figure 1.
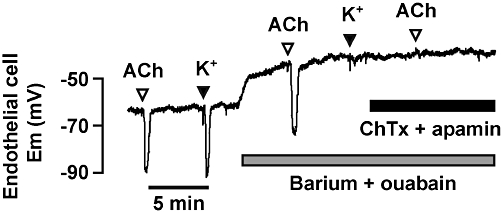
Hyperpolarization of impaled endothelial cells to bolus additions of ACh (10 µM) and K+ (5 mM) in isolated, pinned and superfused rat hepatic arteries. The combination of barium (30 µM, to block KIR channels) and ouabain (1 mM, to inhibit the Na+/K+-ATPase) fully blocked hyperpolarization to K+, but not to ACh. In separate experiments, this combination blocked smooth muscle cell hyperpolarization to ACh (not shown). The KCa channel blockers charybdotoxin (ChTx; 100 nM) and apamin (100 nM), abolished both endothelial cell (shown) and smooth muscle cell (not shown) hyperpolarization to ACh. First published in Edwards et al. (1998).
The concept of endothelium-derived K+ acting as a hyperpolarizing factor has been subsequently supported by studies in arteries from a range of species and locations, and including human arteries (Chrissobolis et al., 2000; McGuire et al., 2002; Bussemaker et al., 2003; Nelli et al., 2003; McNeish et al., 2005). However, this was not immediately the case. Following the initial suggestion in Edwards et al. (1998), a number of laboratories were unable to replicate arterial relaxation by raising [K+]o, which of course is an absolute requirement if this ion really is an endogenous hyperpolarizing factor (Edwards et al., 1999; 2000; Quignard et al., 1999). Critically, this failure to evoke vasorelaxation to [K+]o included the rat mesenteric resistance artery (Doughty et al., 2000; Lacy et al., 2000), one of the vessels used in Edwards et al. (1998), and the internal carotid and small coronary arteries. In the latter, the EDHF response appears mediated predominately through MEGJs, while in the studies using the mesenteric artery, the important concept of functional or physiological antagonism was overlooked in the experimental design.
The latter point was demonstrated by examining the ability of elevated [K+]o to relax and repolarize rat small mesenteric arteries during either submaximal or maximal stimulation with phenylephrine. The mesenteric arteries used did not spontaneously develop tone, so to study both hyperpolarization (or more accurately repolarization) and the resulting relaxation, the α1-adrenoceptor agonist was applied before K+, to evoke smooth muscle depolarization and contraction. As the phenylephrine concentration increased towards maximal, repolarization and relaxation to [K+]o was blocked (Dora and Garland, 2001). The explanation was simply that stimulation of the muscle α1-adrenoceptors was increasing the efflux of K+ through iberiotoxin-sensitive BKCa channels, with the result that K+ accumulated in the extracellular space and saturated the Na+/K+-ATPase activity. This saturation then effectively blocked responses to any additional (exogenous) K+ ion. When efflux from the smooth muscle was reduced, in the presence of iberiotoxin, then the functional antagonism was removed and exogenous K+ ion was again able to evoke repolarization/hyperpolarization and relaxation, or in pressurized arteries increase diameter (Dora et al., 2002). The extracellular accumulation of K+ that follows smooth muscle stimulation, and underlies the physiological antagonism, was termed the ‘K+-cloud’ by Edwards and Weston (Weston et al., 2002).
Gap junctions – a physical link between muscle and endothelium
At the same time as the K+ story was developing, Tudor Griffith's group in Cardiff proposed that electrical coupling through MEGJs could explain NO-independent, endothelium-dependent, relaxations in rabbit iliac artery (Taylor et al., 1998). This was despite earlier suggestions that the gap junction inhibitors, halothane and 1-heptanol, had no effect on responses ascribed to EDHF (Zygmunt and Hogestatt, 1996). The presence of gap junctions between cells in the vascular wall had been recognized for many years (Rhodin, 1967), and a key role for gap junctions as a means to enable conducted vasodilatation to ACh had been clearly demonstrated in arterioles of the microcirculation by Segal and Duling (1989). This group then used intracellular microelectrodes to demonstrate directly that close electrical coupling existed between the smooth muscle and the ECs, suggesting that this coupling enabled hyperpolarization to spread along arterioles and affect dilatation (Xia et al., 1995). Furthermore, they identified connexin 40 and 43 (Cx40 and Cx43) as integral components of the gap junctions (Little et al., 1995).
That gap junctions could underpin EDHF responses was supported by whole-cell patch clamp experiments in arterioles. These showed that hyperpolarization generated by ACh in the endothelium of intact vessels was, to all intents and purposes, identical in these cells and the adjacent SMCs (Yamamoto et al., 1999). However, after blocking gap junctions with 18β-glycyrrhetinic acid, ACh only induced an outward current in the ECs. Furthermore, removing extracellular Ca2+ or adding charybdotoxin reduced the EC hyperpolarization, supporting the suggestion that hyperpolarization of the smooth muscle was purely indirect. However, the functional significance of gap junctions, just like K+, seemed to vary between arteries and was not apparent in every preparation (Bény and Schaad, 2000). One possibility was that the functional role of MEGJs might increase in importance as vessels decreased in size. If this were true, it might then explain the fact that EDHF-evoked relaxation increases in importance as artery diameter decreases (Garland et al., 1995; Shimokawa et al., 1996).
This hypothesis gained considerable support from a morphological study that attempted to quantify muscle–endothelial and endothelial–endothelial gap junctions in the rat mesenteric arterial bed. MEGJs were indeed present in this artery and, at <100 nm in diameter, small compared with the homocellular gap junctions between the ECs. Importantly, they also appeared to be present in larger numbers in the distal compared with the proximal regions of the mesenteric arterial bed. These observations were of course consistent with a functional role of increasing importance as artery size decreased (Sandow and Hill, 2000). The larger gap junctions between the ECs were clearly pentalaminar and distributed extensively throughout the mesenteric arterial bed. These observations have not been extended to other beds to allow confirmation, or not, of the general principle, presumably in large part because of the extremely laborious nature of this type of research. However, Sandow et al. (2004) did show that the incidence of MEGJs decreased in the saphenous artery of rats as they matured to adulthood, and that this decrease was accompanied by a loss of EDHF-dependent relaxation to ACh. Again, evidence consistent with an important functional role for MEGJs.
MEGJs appear as thickenings of the membranes of the two adjacent cell types, where they come into close apposition (Rhodin, 1967). In the rat, they occur on projections of the ECs which pass through the internal elastic lamina (IEL; Figure 2), although not all projections seem to possess a junction (Sandow and Hill, 2000). Incidentally, it is presumably this anchoring that had made it so difficult to remove endothelium from perfused rat arterial beds in early studies of EDRF, effective removal requiring the use of either detergent (Randall and Hiley, 1988), an air bubble (Atkinson et al., 1994) or Staphylococcus aureusα-toxin (Laher et al., 1995) physically to destroy the cells, rather than collagenase simply to dislodge them from the vessel wall, as in small rabbit vessels (Furchgott et al., 1987). The gap junctions are formed by connexins located in the membrane of the connecting cells, proteins with four trans-membrane domains which come together as hexamers to make up a hemichannel (a connexon) in each of the coupled cells (see Johnstone et al., 2009). In the vasculature, connexons can be formed from Cx37, Cx40, Cx43 and/or Cx45, the number indicating the molecular mass of the protein. For a detailed overview of gap junctions in EDH, readers are referred to a recent review (de Wit and Griffith, 2010).
Figure 2.
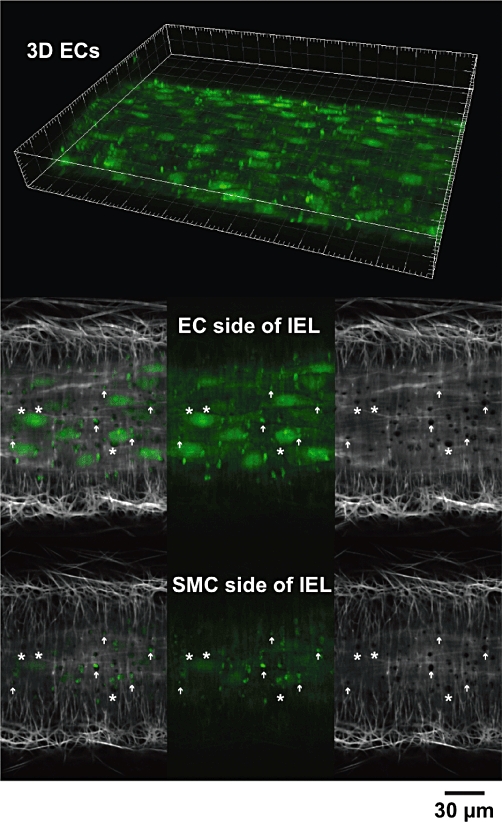
Confocal micrographs of endothelial cells (ECs) in an artery. The fluorescent Ca2+ indicator Oregon Green 488 BAPTA-1 was loaded into ECs (green) of a pressurized rat mesenteric artery. Serial images through the z-axis allowed 3D reconstruction of the artery wall (top) where bright EC projections were clearly visible. Frame major ticks 20 µm. Following visualization of the internal elastic lamina (IEL, grey scale) with Alexa 633 hydrazide, single image planes collected 2 µm apart at the EC and smooth muscle cell (SMC) side of the IEL (lower panels) showed the projections passed towards the SMCs (arrows). These projections are sites where myoendothelial gap junctions may form. Note that not all holes were associated with EC staining (asterisks) (P. Yarova, unpubl. data).
The involvement by MEGJs in the EDHF response, minor or not, of course removes any absolute reliance on a diffusible factor as the underlying mechanism. So, as mentioned earlier, because hyperpolarization is initiated in the endothelium, a more accurate and up-to-date descriptor for this route to vasodilatation is EDH rather than EDHF. Unraveling the functional contribution of gap junctions in EDH dilatation, and identifying if individual connexins are interchangeable, or if they are restricted in any contribution they may make, has not been at all easy. This is largely because the pharmacological tools available to uncouple gap junctions invariably have other effects. As already mentioned, Cx40 appears important in vascular gap junctions and was hypothesized to play a role in the MEGJs underlying EDHF-mediated responses. To probe a potential functional role, we developed a technique selectively to load the endothelium of pressurized resistance arteries intracellularly with antibodies (by pinocytosis). Using this approach, we found that anti-Cx40 antibody blocked EDHF dilatation but only in arteries undergoing a robust pre-constriction to phenylephrine sufficient to block the parallel action of K+ as an EDHF (Mather et al., 2005). Under similar conditions, antibodies to the intracellular carboxy-terminal region of Cx40 (residues 340–358) or a mimetic peptide directed at the cytoplasmic loop (Cx40; residues 130–140) were each effective in this regard (Figure 3). However, antibodies against intracellular regions of Cx37 or Cx43, or mimetic peptide for the intracellular loop region of Cx37, were inactive. Simultaneous intra- and extraluminal incubation of arteries with inhibitory peptides targeted to extracellular regions of EC connexins (43Gap 26, 40Gap 27, and 37,43Gap 27) also failed to modify the EDHF response. The functional significance of Cx40 was strengthened by the fact that it could actually be visualized (by immunohistochemistry) within the end of EC projections.
Figure 3.
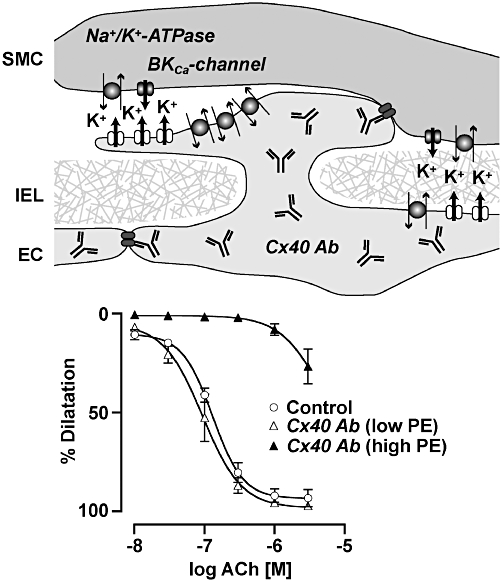
Antibodies targeted to connexin 40 (Cx40 Ab) can inhibit endothelium-dependent hyperpolarization-mediated dilatation responses. Cx40 Abs were loaded into endothelial cells (ECs) of pressurized rat mesenteric arteries and inhibited cell–cell coupling via Cx40 expressed at both EC–EC and myoendothelial gap junctions. An effect against ACh was only observed when the concentration of phenylephrine (PE) used to generate tone was high. This may be due to an ability of a true endothelium-dependent hyperpolarizing factor such as K+ to independently act of electrical coupling via the smooth muscle cell (SMC) Na+/K+-ATPase. At high concentrations, PE activates BKCa-channels which can prevent further activation of the Na+/K+-ATPase, possibly due to a ‘K+ cloud’. Modified from Mather et al. (2005). IEL, internal elastic lamina.
Overall, our data described a mechanism for EDH-mediated dilatation whereby hyperpolarization generated in the ECs diverged along two paths, one mediated by K+ efflux and the other by hyperpolarization conducted to the muscle through MEGJs and critically involving Cx40. In arteries undergoing submaximal constriction, both pathways contribute to dilatation, but as constriction intensifies so does the ‘density’ of the extracellular K+ cloud. The result is block of the ability of K+ released by the endothelium to serve as an EDHF, by saturating its target effector. At this point, MEGJs alone sustain EDH vasodilatation. Therefore, together these two pathways explain the ‘EDHF’ response in the mesenteric artery.
Differential activation of 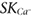 and IKCa and the development of a concept of endothelial membrane microdomains
and IKCa and the development of a concept of endothelial membrane microdomains
Interestingly, and perhaps very importantly, the individual contributions that EC SKCa and IKCa channels make to EDH can vary. Although both of these K+-channels, are of course, central to EDH, in isolated mesenteric arteries simultaneous measurements of tension and membrane potential showed that SKCa channels alone were responsible for hyperpolarization (from circa −55 mV) (Figure 4). However, input from IKCa channels also became apparent once arteries were stimulated with phenylephrine, used to develop the (depolarization and) contraction necessary to evoke endothelium-dependent relaxation (Crane et al., 2003). Selective, differential activation of KCa channels might be explained by the intracellular Ca2+ sensitivity of the two channel types and/or access to cytoplasmic Ca2+, or perhaps reflect a separate and distinct subcellular localization in each case. Clearly, there is some division of signalling within ECs, because, for example, although both NO synthase and KCa channels are activated by Ca2+, not all agents inducing endothelium-dependent relaxation activate both NO- and EDHF-mediated relaxation: these agents include abnormal cannabidiol and virodhamine which act at a receptor for cannabinoid-like agents in the endothelium (Ho and Hiley, 2003; 2004;).
Figure 4.
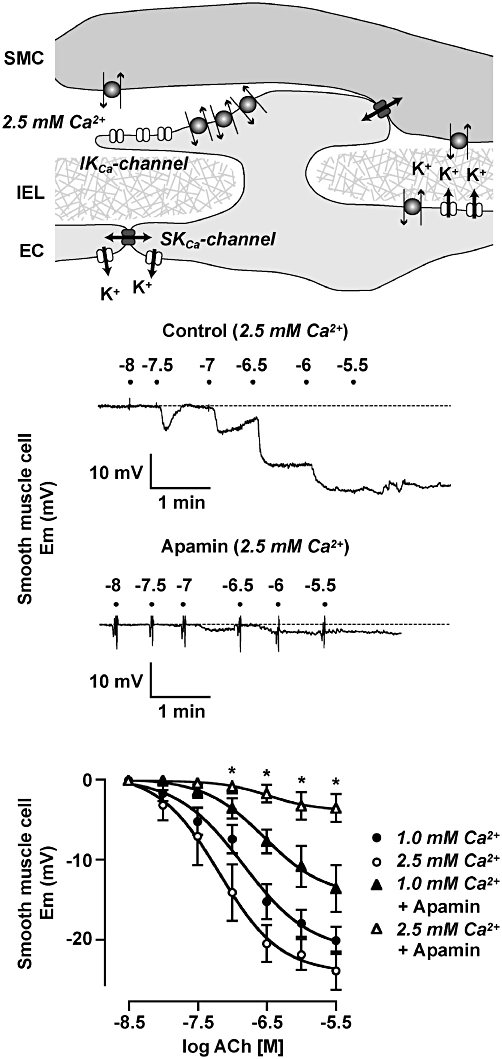
Role for SKCa channels in endothelium-dependent hyperpolarization responses depends on the extracellular Ca2+ concentration. In rat mesenteric arteries, SKCa (KCa2.3) channels are highly expressed at endothelial cell (EC) borders and to a limited extent, in the EC projections through the internal elastic lamina (IEL). In contrast, IKCa (KCa3.1) channels appear confined to the EC projection microdomain (top panel). The hyperpolarization to ACh when extracellular [Ca2+] is held at 2.5 mM is fully blocked by apamin (middle panels), but not so when extracellular Ca2+ is reduced to 1 mM (bottom panel). Modified from Crane et al. (2003); Dora et al. (2008). SMC, smooth muscle cell.
As far as EDH in the rat small mesenteric artery is concerned, it turns out to be the case that the differential activation correlates with the restriction of IKCa (KCa3.1) channels to the endothelial projections, the projections passing through holes in the IEL towards the smooth muscle. In marked contrast to KCa3.1, SKCa (KCa2.3) channels are distributed throughout the EC membrane, but are observed in particularly high concentration close to the relatively large EC–EC gap junctions (Sandow et al., 2006; Dora et al., 2008) (Figure 4). Also, in marked contrast to KCa3.1, the KCa2.3 channels are located in caveolae (Absi et al., 2007). The concentration of KCa3.1 channels in the endothelial projections seems to be part of a complex signalling microdomain, as the projections and their immediate vicinity also contain high levels of both Na+/K+-ATPase and the Cx37 and Cx40 that are integral to the MEGJs (Mather et al., 2005; Dora et al., 2008). But the true level of complexity may well be far greater. For example, endothelial projections within cerebral arteries contain transient receptor potential (TRP) A1, a Ca2+-permeable, non-selective, cation channel which is activated to generate EDH by electrophilic compounds such as the irritants acrolein and mustard oil (Earley et al., 2009). On the other hand, in mesenteric arteries, calcium-sensing receptors (CaSR) co-localize with KCa3.1 channels, outside the caveolae (Absi et al., 2007; Saliez et al., 2008).
Caveolae are flask-shaped indentations (or caves) covering the entire cell surface. They serve to anchor proteins, including both G protein-coupled and other receptors (such as InsP3, receptors), with proteins such as KCa2.3, TRPC1 and TRPV4 channels (Lockwich et al., 2000), thus forming highly organized, signalling microdomains (Rath et al., 2009; Chidlow and Sessa, 2010). They seem crucial for EDH-mediated vasodilatation, as dilatation is absent in mice with the caveolin-1 gene deleted. The deficiency has been linked with TRPV4 activity, rather than to any direct effect on KCa channels, although the latter was not assessed (Saliez et al., 2008). Interestingly, this particular gene knockout also reduced Cx40, Cx43 and Cx37 expression, suggesting that gap junction formation may be compromised, and thus the integration of electrical events within the blood vessel wall (see next).
Restriction of KCa3.1 channels to the relatively narrow EC projections might hinder the access of EC cytoplasmic Ca2+ necessary for channel activation, while perhaps facilitating a signal for activation derived from the smooth muscle. To investigate this possibility, we imaged [Ca2+]i changes within the EC projections in mesenteric arteries mounted in a wire myograph, but we were unable to resolve any difference in [Ca2+]i increases following EC stimulation with ACh and during simultaneous smooth muscle stimulation with phenylephrine (Dora et al., 2008). The ECs display spontaneous Ca2+ events, and these have been termed ‘pulsars’. Interestingly, they appear to align with EC projections, and KCa3.1 channels are basally activated by these spontaneous events, suppressing smooth muscle membrane potential by around 8 mV. As well as KCa3.1 channels, InsP3-sensitive Ca2+ stores are also concentrated in the projections and ACh stimulates a circa 2.5-fold increase in pulsar frequency (Ledoux et al., 2008).
Although there appeared to be no difference in the evoked [Ca2+]i during stimulation with ACh in the absence or presence of phenylephrine, IKCa-mediated hyperpolarization could be detected alongside that due to SKCa, if extracellular [Ca2+]o was reduced from 2.5 mM to 1 mM (Figure 4). Furthermore, the normal recruitment of IKCa channels during stimulation of the endothelium with ACh, while phenylephrine was stimulating the smooth muscle, was lost if Ca2+ entry into the muscle was reduced with voltage-dependent Ca2+ channel blockers (Dora et al., 2008). So, the localized dynamics of [Ca2+]o in the intercellular space, around EC projections, may be a fundamental physiological controlling mechanism in both the elaboration and transmission of EDH. This mechanism may well involve the CaSR shown by Arthur Weston and colleagues to be localized within the endothelium and, most importantly, outside caveolae and in close association with KCa3.1 channels (Weston et al., 2005). These receptors respond to changes in [Ca2+]o within the physiological range, and link to activate IKCa channels (Weston et al., 2005) and cause vasorelaxation (Ohanian et al., 2005). Finally, IKCa channels are also subject to control by cAMP, as forskolin suppressed hyperpolarization through these channels (Dora et al., 2008).
Does EC KCa preferentially link to separate vasodilatation mechanisms?
The fact that EDH is generated by the activity of two types of KCa channels, and that in the rat small mesenteric artery, KCa3.1 channels are only found on the EC projections, raises the possibility that the transmission of EDH to the muscle via either K+ or MEGJs may be linked to one type of KCa channel. This seems to be the case. Uncoupling gap junctions with carbenoxolone caused IKCa channels alone to underlie EDH. Furthermore, the ensuing vasorelaxation could be inhibited with ouabain, suggesting that IKCa channel activity is linked, presumably through changes in extracellular K+ concentration, to the activation of smooth muscle Na+/K+-ATPase. On the other hand, SKCa-mediated hyperpolarization appeared mainly to affect relaxation through MEGJs and involve preferential activation of KIR channels. Using a selective activator for SKCa channels, N-cyclohexyl-N-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-4-pyrimidinamine, Weston et al. (2010) demonstrated that the hyperpolarization evoked by this agent was blocked with either apamin or Ba2+ and was therefore consistent with this suggestion.
Other endothelium-dependent pathways involving hyperpolarization
In the years following the discovery of ‘EDHF’, a number of candidate molecules have been proposed as diffusible EDHFs. A recent review (Edwards et al., 2010) summarizes the key candidates and highlights a critical point: the majority of these agents do not show sensitivity to block with a combination of apamin and charybdotoxin (or TRAM-34) and are invariably sensitive to agents targeting smooth muscle K+-channels. For example, epoxyeicosatetraenoic acids (EETs) (Campbell et al., 1996) activate smooth muscle BKCa channels and NO acts principally via KATP channels (Tare et al., 1990; Garland and McPherson, 1992). The role of EETs in EDHF vasorelaxation has, in particular, been the subject of quite considerable investigation and the reader is directed to an extensive and recent review of this literature by Campbell and Fleming (2010). The 2010 review by G. Edwards, M. Félétou and A. Weston proposes a helpful classification of hyperpolarizing pathways, or more usually EDH-like relaxation, defining them as either classical or non-classical. The former show sensitivity to block with apamin and TRAM-34, while being resistant to block with apamin and iberiotoxin. The non-classical group describes agents that are EDHFs, and play a role in evoking or facilitating smooth muscle hyperpolarization in/under specified circumstances (Edwards et al., 2010). The non-classical group does not preclude signalling via EC projection microdomains and, for some of the more hydrophobic EDHFs, this may actually be crucial in terms of reducing diffusion distance.
Spreading the influence: conducted vasodilatation
So far we have focused on that hyperpolarization (or EDH) originating in the endothelium that passes radially into the artery wall to evoke vasodilatation. However, hyperpolarization can also move more generally through the artery wall, to affect a response first described by August Krogh (Krogh, 1920) as spreading or conducted vasodilatation. As well as the endothelium, hyperpolarization can of course be initiated directly in smooth muscle by mechanisms independent of either SKCa or IKCa. channels Whether originating in the smooth muscle or the endothelium, hyperpolarization can then spread axially to evoke vasodilatation. As such, it is able to translate focal dilatation into the more widespread drop in vascular resistance that is necessary to increase blood flow. For agents to achieve significant increases in tissue blood flow, any increase in artery diameter must significantly reduce vascular resistance. If dilatation only occurred focally, it would be unlikely to do this, because resistance is in part a function of the vessel length. However, if dilatation can also spread upstream, significant blood flow increases will follow (Kurjiaka and Segal, 1995; Dora et al., 2000).
Conducted vasodilatation has been most extensively studied in the microcirculation, where there appears to be a correlation between agonists that open K+-channels and an ability to reliably evoke spreading dilatation (Delashaw and Duling, 1991). Thus, the signal for spreading dilatation is thought simply to reflect the passage of current to adjacent cells, that is, cells not directly stimulated by the agonist (Dora et al., 2000; Emerson and Segal, 2000). Passage can occur bi-directionally along the length of the vessel and is intrinsic to the wall of the artery. Furthermore, it relies on the endothelium and is not dependent on either nerves or a change in blood flow (Delashaw and Duling, 1991; Rivers, 1997; Dora et al., 2000; Emerson and Segal, 2000; Segal, 2005; Winter and Dora, 2007). Studies in exteriorized microvascular beds in situ (Dora et al., 2000), and arterioles isolated from those vascular beds, have used the endothelium-dependent agonist ACh to evoke robust responses following application to the outside of arterioles using micropipettes.
In skeletal muscle, spreading or ascending dilatation also occurs in larger arteries (Hilton, 1959) and we demonstrated recently that it can also be sustained in small resistance arteries, such as the rat mesenteric artery, which have three to five layers of muscle, suggesting that the phenomenon is of relevance across the vasculature (Takano et al., 2004; Winter and Dora, 2007).
Importance of the endothelium in spreading dilatation
Orientation favours a role for ECs as the conduit for spread of hyperpolarization: they align along the principal axis of the vessel while the muscle cells are arranged circularly. This seems indeed to be the case, as activating the KATP channels, which are restricted to the SMCs in rat mesenteric arteries (White and Hiley, 2000; Takano et al., 2004; Dora et al., 2008) showed that the SMCs are not coupled sufficiently to enable dilatation to spread over long distances (Takano et al., 2004) and, secondly, that the endothelium is the conduit for electrical coupling (Haas and Duling, 1997; Yamamoto et al., 1999; Emerson and Segal, 2000; Winter and Dora, 2007). The way in which the hyperpolarization is elicited is not crucial in determining the subsequent spread, only in initiating cell–cell coupling, because activation of either endothelial or smooth muscle K+-channels causes this response. However, some agonists may stimulate additional signalling pathways that may either augment (e.g. by cAMP; Popp et al., 2002) or reduce (e.g. by α1-adrenoceptor stimulation, protein kinase C; Haug et al., 2003; Bao et al., 2004) the ability of hyperpolarization to spread between the vascular cells.
Spreading dilatation following luminal perfusion of agonists
Circulating vasoactive agonists are crucially involved in the physiological control of regional blood flow, yet quite remarkably only very few studies attempt to apply agonists into the artery lumen when studying vascular reactivity. The consideration of extra- versus intra-luminal agonist application is of course crucial when probing the mechanism of action of agonists with opposing effects through the endothelium and on the SMCs, for example, purinergic (such as ATP) and adrenergic (such as adrenaline) agonists. We have addressed this issue by developing a novel approach with isolated and pressurized small arteries. By triple-cannulation of small arteries (the third cannula being inserted into a side branch), it is possible to infuse agonists luminally into a restricted downstream region. In this way, any spreading dilatation that is evoked can be quantified easily, as it spreads upstream against the direction of intraluminal fluid flow. Using this approach, luminal perfusion of either ATP or UTP was shown to stimulate both local and spreading dilatations (Winter and Dora, 2007). As all the evidence available is consistent with an essential role for the endothelium, in enabling such vascular responses to be coordinated effectively, any dysfunction in this monolayer, for example in conditions such as diabetes, obesity and hypertension, will markedly disrupt the ability of blood vessels to control local tissue perfusion.
What mechanisms sustain spreading dilatation: another signalling circuit?
As spreading dilatation appears to be completely reliant on membrane hyperpolarization, any agonist able to stimulate hyperpolarization will potentially evoke this response. Spread will rely on both homo- and hetero-cellular gap junctions to enable the necessary current spread. So a key question is whether current passing between cells of the arterial wall decays passively, or does an amplification mechanism exist to sustain dilatation. The available evidence in isolated, pressurized arteries supports a role for an undefined amplification process, as current appears to spread further than predicted by passive decay. This was elegantly demonstrated in cannulated hamster retractor small arteries (one to two layers of smooth muscle), where the decay of injected current was faster than the decay of hyperpolarization to ACh, despite the fact both induced a similar local (initiating) hyperpolarization (length constants 1.2 and 1.9 mm, respectively; Emerson et al., 2002). Therefore, the intercellular spread of current via gap junctions appears in some way to be sustained. One ion channel that appears central in this response is KIR (Rivers et al., 2001; Goto et al., 2004; Jantzi et al., 2006). However, a widespread role for this channel does not seem likely, as Ba2+ (which inhibits KIR channels) had no effect on spreading dilatation to either ACh or levcromakalim in rat mesenteric arteries (Takano et al., 2004). However, the Na+/K+-ATPase activated by modest increases in extracellular [K+] (Edwards et al., 1998) may play some role. This raises the possibility that the release of K+ from vascular cells during hyperpolarization may act as an amplification mechanism, with a distinct parallel to its role as an EDHF (Edwards et al., 1998). This is supported by the expression of both KIR and KCa2.3 channels at the borders of ECs (Dora et al., 2008), at sites resembling that of inter- EC gap junctions (Kansui et al., 2004; Winter and Dora, 2007). It is also highly likely that an isoform of the ATPase is also expressed within this space. Thus, release of K+ through any of these channels could serve to act on adjacent channels, pumps and/or cells to amplify hyperpolarization.
In addition, there are other candidates that may act together with or in parallel to K+. Given its central importance in vascular function, NO might play a role (Budel et al., 2003; Uhrenholt et al., 2007) although inhibition of NO synthase does not appear to reduce spreading dilatation (Oishi et al., 2001; Domeier and Segal, 2007; Winter and Dora, 2007). We have previously shown that EC [Ca2+]o does not detectably increase in response to local (McSherry et al., 2005; 2006;) or spreading (Takano et al., 2004) hyperpolarization in either rat mesenteric or cremasteric arteries. However, recent evidence shows that the presence of tone can unmask a detectable, slow, conducted intercellular Ca2+ wave, both in vitro (Uhrenholt et al., 2007) and in vivo (Tallini et al., 2007), although it is important to note that this wave is not a requirement for the rapid spread of hyperpolarization and associated dilatation (Takano et al., 2004; Tallini et al., 2007).
EDHF in disease
It is clear that EDH is a prominent regulatory mechanism in the vasculature and, as EC dysfunction appears to be an early feature in cardiovascular disease (Vanhoutte et al., 2009), the question arises as to the extent of involvement of hyperpolarizing responses. Although the role of NO signalling has been extensively studied in disease, fewer data are available with regard to the effects of cardiovascular disease or vascular injury on EDH. However, modifying the expression levels of EC SKCa channels in a transgenic model (SK3T/T) clearly demonstrated that decreasing or increasing these endothelial cell channels caused commensurate changes in blood pressure (Taylor et al., 2003). These observations were extended by studies involving targeted deletion of KCa3.1 channels, and also KCa3.1/SK3 channel double knock out animals, to show significant rises in mean arterial blood pressure (Si et al., 2006; Brahler et al., 2009). Thus, openers of these channels may represent a novel therapeutic approach to hypertension (Grgic et al., 2009). Most recently, electrophysiological studies have revealed that hyperpolarization through caveolae-based SKCa channel activation was compromised in mesenteric arteries from 12 to 16-week-old animals spontaneously hypertensive rats, while IKCa channel pathways were unaltered (Weston et al., 2010). However, although the hyperpolarization linked to IKCa channel activation was not reduced at this age, it appeared that the channel protein level was decreasing. So perhaps, in the longer term, hyperpolarization through this EC channel is also disrupted. Clearly, much more research is needed to define the extent and means by which EDH is altered by cardiovascular disease, and in a range of disease models.
Conclusion
Since the discovery of EDHF in the 1980s, clear and sustained progress has been made in defining the fundamental mechanisms responsible for generating EC hyperpolarization, or EDH. Our understanding of how EDH then transfers radially into the adjacent smooth muscle, and axially along arteries to evoke distant vasodilatation, has similarly increased. These responses together serve to integrate changes in tissue blood flow. Perhaps the most important key steps along the way have been to recognize that hyperpolarization arises from activation of SKCa and IKCa channels, and that both channel types reside in the endothelium, but within different microdomains. Future challenges will be to unravel the complexity and local regulation of these membrane microdomains, and to show how they may be disrupted by disease and possibly modulated to improve or sustain physiological function.
Acknowledgments
The Wellcome Trust and the British Heart Foundation support work in our laboratories. CJG holds a Royal Society-Wolfson Research Merit Award. KAD is a British Heart Foundation Senior Basic Science Research Fellow.
Glossary
Abbreviations
- BKCa
large conductance Ca2+-sensitive K+-channels
- CaSR
calcium-sensing receptors
- Cx
connexin
- CyPPA
N-cyclohexyl-N-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-4-pyrimidinamine
- EDH
endothelium-dependent hyperpolarization
- EDRF
endothelium-derived relaxing factor
- EET
epoxyeicosatetraenoic acid
- EDHF
endothelium-dependent hyperpolarizing factor
- IKCa
intermediate conductance Ca2+-sensitive K+-channels
- InsP3
inositol trisphosphate
- KCa
Ca2+-sensitive K+ channels
- NO
nitric oxide
- KATP
ATP-sensitive K+-channels
- KIR
inwardly-rectifying K+-channels
- Kv
voltage-gated K+-channels
- MEGJ
myoendothelial gap junctions
- Na+/K+-ATPase
sodium and potassium dependent adenosine triphosphatase
- PGI2
prostaglandin I2
- SKCa
small conductance Ca2+-sensitive K+-channels
- TRP
transient receptor potential
Conflict of interest
The authors have no conflicts of interest.
References
- Absi M, Burnham MP, Weston AH, Harno E, Rogers M, Edwards G. Effects of methyl beta-cyclodextrin on EDHF responses in pig and rat arteries; association between SK(Ca) channels and caveolin-rich domains. Br J Pharmacol. 2007;151:332–340. doi: 10.1038/sj.bjp.0707222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeagbo AS, Triggle CR. Varying extracellular [K+]: a functional approach to separating EDHF- and EDNO-related mechanisms in perfused rat mesenteric arterial bed. J Cardiovasc Pharmacol. 1993;21:423–429. [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn.) 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggard EE. The endothelium – the body's largest endocrine gland? J Endocrinol. 1990;127:371–375. doi: 10.1677/joe.0.1270371. [DOI] [PubMed] [Google Scholar]
- Atkinson J, Tatchum-Talom R, Capdeville-Atkinson C. Reduction of endothelial function with age in the mesenteric arterial bed of the normotensive rat. Br J Pharmacol. 1994;111:1184–1188. doi: 10.1111/j.1476-5381.1994.tb14870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Altenberg GA, Reuss L. Mechanism of regulation of the gap junction protein connexin 43 by protein kinase C-mediated phosphorylation. Am J Physiol Cell Physiol. 2004;286:C647–C654. doi: 10.1152/ajpcell.00295.2003. [DOI] [PubMed] [Google Scholar]
- Bény JL, Schaad O. An evaluation of potassium ions as endothelium-derived hyperpolarizing factor in porcine coronary arteries. Br J Pharmacol. 2000;131:965–973. doi: 10.1038/sj.bjp.0703658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton TB, Lang RJ, Takewaki T. Mechanisms of action of noradrenaline and carbachol on smooth muscle of guinea-pig anterior mesenteric artery. J Physiol. 1984;351:549–572. doi: 10.1113/jphysiol.1984.sp015262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, Kaistha BP, et al. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation. 2009;119:2323–2332. doi: 10.1161/CIRCULATIONAHA.108.846634. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budel S, Bartlett IS, Segal SS. Homocellular conduction along endothelium and smooth muscle of arterioles in hamster cheek pouch: unmasking an NO wave. Circ Res. 2003;93:61–68. doi: 10.1161/01.RES.0000080318.81205.FD. [DOI] [PubMed] [Google Scholar]
- Bunting S, Moncada S, Vane JR. Antithrombotic properties of vascular endothelium. Lancet. 1977;2:1075–1076. doi: 10.1016/s0140-6736(77)91906-7. [DOI] [PubMed] [Google Scholar]
- Bussemaker E, Popp R, Binder J, Busse R, Fleming I. Characterization of the endothelium-derived hyperpolarizing factor (EDHF) response in the human interlobar artery. Kidney Int. 2003;63:1749–1755. doi: 10.1046/j.1523-1755.2003.00910.x. [DOI] [PubMed] [Google Scholar]
- Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch. 2010;459:881–895. doi: 10.1007/s00424-010-0804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- Chen G, Suzuki H, Weston AH. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br J Pharmacol. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidlow JH, Jr, Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc Res. 2010;86:219–225. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrissobolis S, Ziogas J, Chu Y, Faraci FM, Sobey CG. Role of inwardly rectifying K+ channels in K+-induced cerebral vasodilatation in vivo. Am J Physiol Heart Circ Physiol. 2000;279:H2704–H2712. doi: 10.1152/ajpheart.2000.279.6.H2704. [DOI] [PubMed] [Google Scholar]
- Crane GJ, Gallagher NT, Dora KA, Garland CJ. Small and intermediate calcium-dependent K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J Physiol. 2003;553:183–189. doi: 10.1113/jphysiol.2003.051896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawes GS. The vaso-dilator action of potassium. J Physiol. 1941;99:224–238. doi: 10.1113/jphysiol.1941.sp003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delashaw JB, Duling BR. Heterogeneity in conducted arteriolar vasomotor response is agonist dependent. Am J Physiol. 1991;260:H1276–H1282. doi: 10.1152/ajpheart.1991.260.4.H1276. [DOI] [PubMed] [Google Scholar]
- Domeier TL, Segal SS. Electromechanical and pharmacomechanical signalling pathways for conducted vasodilatation along endothelium of hamster feed arteries. J Physiol. 2007;579:175–186. doi: 10.1113/jphysiol.2006.124529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora KA, Garland CJ. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am J Physiol Heart Circ Physiol. 2001;280:H2424–H2429. doi: 10.1152/ajpheart.2001.280.6.H2424. [DOI] [PubMed] [Google Scholar]
- Dora KA, Damon DN, Duling BR. Microvascular dilation in response to occlusion: a coordinating role for conducted vasomotor responses. Am J Physiol Heart Circ Physiol. 2000;279:H279–H284. doi: 10.1152/ajpheart.2000.279.1.H279. [DOI] [PubMed] [Google Scholar]
- Dora KA, Ings NT, Garland CJ. K(Ca) channel blockers reveal hyperpolarization and relaxation to K+ in rat isolated mesenteric artery. Am J Physiol Heart Circ Physiol. 2002;283:H606–H614. doi: 10.1152/ajpheart.01016.2001. [DOI] [PubMed] [Google Scholar]
- Dora KA, Gallagher NT, McNeish A, Garland CJ. Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res. 2008;102:1247–1255. doi: 10.1161/CIRCRESAHA.108.172379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughty JM, Boyle JP, Langton PD. Potassium does not mimic EDHF in rat mesenteric arteries. Br J Pharmacol. 2000;130:1174–1182. doi: 10.1038/sj.bjp.0703412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Gonzales AL, Crnich R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-activated K+ channels. Circ Res. 2009;104:987–994. doi: 10.1161/CIRCRESAHA.108.189530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Weston AH. Endothelium-derived hyperpolarizing factor – a critical appraisal. Prog Drug Res. 1998;50:107–133. doi: 10.1007/978-3-0348-8833-2_2. [DOI] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- Edwards G, Félétou M, Gardener MJ, Thollon C, Vanhoutte PM, Weston AH. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br J Pharmacol. 1999;128:1788–1794. doi: 10.1038/sj.bjp.0703009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Thollon C, Gardener MJ, Félétou M, Vilaine J, Vanhoutte PM, et al. Role of gap junctions and EETs in endothelium-dependent hyperpolarization of porcine coronary artery. Br J Pharmacol. 2000;129:1145–1154. doi: 10.1038/sj.bjp.0703188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Félétou M, Weston AH. Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch. 2010;459:863–879. doi: 10.1007/s00424-010-0817-1. [DOI] [PubMed] [Google Scholar]
- Emerson GG, Segal SS. Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ Res. 2000;86:94–100. doi: 10.1161/01.res.86.1.94. [DOI] [PubMed] [Google Scholar]
- Emerson GG, Neild TO, Segal SS. Conduction of hyperpolarization along hamster feed arteries: augmentation by acetylcholine. Am J Physiol Heart Circ Physiol. 2002;283:H102–H109. doi: 10.1152/ajpheart.00038.2002. [DOI] [PubMed] [Google Scholar]
- Félétou M, Vanhoutte PM. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br J Pharmacol. 1988;93:515–524. doi: 10.1111/j.1476-5381.1988.tb10306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower RJ. Prostaglandins, bioassay and inflammation. Br J Pharmacol. 2006;147:S182–S192. doi: 10.1038/sj.bjp.0706506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstermann U, Gorsky LD, Pollock JS, Schmidt HH, Heller M, Murad F. Regional distribution of EDRF/NO-synthesizing enzyme(s) in rat brain. Biochem Biophys Res Commun. 1990;168:727–732. doi: 10.1016/0006-291x(90)92382-a. [DOI] [PubMed] [Google Scholar]
- Furchgott RF. Nitric oxide: from basic research on isolated blood vessels to clinical relevance in diabetes. An R Acad Nac Med. 1998;115:317–331. [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Carvalho MH, Khan MT, Matsunaga K. Evidence for endothelium-dependent vasodilation of resistance vessels by acetylcholine. Blood Vessels. 1987;24:145–149. doi: 10.1159/000158689. [DOI] [PubMed] [Google Scholar]
- Gaddum JH. A perspective on pharmacology. In: Talalay P, Murnaghan JH, editors. Drugs in our Society. Baltimore, MD: Johns Hopkins Press; 1964. pp. 17–26. [Google Scholar]
- Garland JG, McPherson GA. Evidence that nitric oxide does not mediate the hyperpolarization and relaxation to acetylcholine in the rat small mesenteric artery. Br J Pharmacol. 1992;105:429–435. doi: 10.1111/j.1476-5381.1992.tb14270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland CJ, Plane F, Kemp BK, Cocks TM. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends Pharmacol Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- Goto K, Rummery NM, Grayson TH, Hill CE. Attenuation of conducted vasodilatation in rat mesenteric arteries during hypertension: role of inwardly rectifying potassium channels. J Physiol. 2004;561:215–231. doi: 10.1113/jphysiol.2004.070458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grgic I, Kaistha BP, Hoyer J, Kohler R. Endothelial Ca+-activated K+ channels in normal and impaired EDHF-dilator responses – relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol. 2009;157:509–526. doi: 10.1111/j.1476-5381.2009.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryglewski RJ, Bunting S, Moncada S, Flower RJ, Vane JR. Arterial walls are protected against deposition of platelet thrombi by a substance (prostaglandin X) which they make from prostaglandin endoperoxides. Prostaglandins. 1976;12:685–713. doi: 10.1016/0090-6980(76)90047-2. [DOI] [PubMed] [Google Scholar]
- Haas TL, Duling BR. Morphology favors an endothelial cell pathway for longitudinal conduction within arterioles. Microvasc Res. 1997;53:113–120. doi: 10.1006/mvre.1996.1999. [DOI] [PubMed] [Google Scholar]
- Haug SJ, Welsh DG, Segal SS. Sympathetic nerves inhibit conducted vasodilatation along feed arteries during passive stretch of hamster skeletal muscle. J Physiol. 2003;552:273–282. doi: 10.1113/jphysiol.2003.046284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton SM. A peripheral arterial conducting mechanism underlying dilatation of the femoral artery and concerned in functional vasodilatation in skeletal muscle. J Physiol. 1959;149:93–111. doi: 10.1113/jphysiol.1959.sp006327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WS, Hiley CR. Vasodilator actions of abnormal-cannabidiol in rat isolated small mesenteric artery. Br J Pharmacol. 2003;138:1320–1332. doi: 10.1038/sj.bjp.0705160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WS, Hiley CR. Vasorelaxant activities of the putative endocannabinoid virodhamine in rat isolated small mesenteric artery. J Pharm Pharmacol. 2004;56:869–875. doi: 10.1211/0022357023682. [DOI] [PubMed] [Google Scholar]
- Holzmann S, Kukovetz WR, Windischhofer W, Paschke E, Graier WF. Pharmacologic differentiation between endothelium-dependent relaxations sensitive and resistant to nitro-L-arginine in coronary arteries. J Cardiovasc Pharmacol. 1994;23:747–756. doi: 10.1097/00005344-199405000-00009. [DOI] [PubMed] [Google Scholar]
- Jantzi MC, Brett SE, Jackson WF, Corteling R, Vigmond EJ, Welsh DG. Inward rectifying potassium channels facilitate cell-to-cell communication in hamster retractor muscle feed arteries. Am J Physiol Heart Circ Physiol. 2006;291:H1319–H1328. doi: 10.1152/ajpheart.00217.2006. [DOI] [PubMed] [Google Scholar]
- Johnstone S, Isakson B, Locke D. Biological and biophysical properties of vascular connexin channels. Int Rev Cell Mol Biol. 2009;278:69–118. doi: 10.1016/S1937-6448(09)78002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansui Y, Fujii K, Nakamura K, Goto K, Oniki H, Abe I, et al. Angiotensin II receptor blockade corrects altered expression of gap junctions in vascular endothelial cells from hypertensive rats. Am J Physiol Heart Circ Physiol. 2004;287:H216–H224. doi: 10.1152/ajpheart.00915.2003. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Kuriyama H. Effects of acetylcholine on the smooth muscle cell of isolated main coronary artery of the guinea-pig. J Physiol. 1979;293:119–133. doi: 10.1113/jphysiol.1979.sp012881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Zimmermann PA, Nelson MT. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol. 1996;492(Pt 2):419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A. Studies on the capillariometer mechanism: I. The reaction to stimuli and the innervation of the blood vessels in the tongue of the frog. J Physiol. 1920;53:399–419. doi: 10.1113/jphysiol.1920.sp001887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama H, Suzuki H. The effects of acetylcholine on the membrane and contractile properties of smooth muscle cells of the rabbit superior mesenteric artery. Br J Pharmacol. 1978;64:493–501. doi: 10.1111/j.1476-5381.1978.tb17310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurjiaka DT, Segal SS. Conducted vasodilation elevates flow in arteriole networks of hamster striated muscle. Am J Physiol. 1995;269:H1723–H1728. doi: 10.1152/ajpheart.1995.269.5.H1723. [DOI] [PubMed] [Google Scholar]
- Kuschinsky W, Wahl M, Bosse O, Thurau K. Perivascular potassium and pH as determinants of local pial arterial diameter in cats. A microapplication study. Circ Res. 1972;31:240–247. doi: 10.1161/01.res.31.2.240. [DOI] [PubMed] [Google Scholar]
- Lacy PS, Pilkington G, Hanvesakul R, Fish HJ, Boyle JP, Thurston H. Evidence against potassium as an endothelium-derived hyperpolarizing factor in rat mesenteric small arteries. Br J Pharmacol. 2000;129:605–611. doi: 10.1038/sj.bjp.0703076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laher I, Thorin-Trescases N, Ding A, Laporte R, Osol G. alpha-Toxin perfusion: a new method for selective impairment of endothelial function in isolated vessels or intact vascular beds. Can J Phys Pharmacol. 1995;73:1669–1673. doi: 10.1139/y95-729. [DOI] [PubMed] [Google Scholar]
- Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, et al. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci USA. 2008;105:9627–9632. doi: 10.1073/pnas.0801963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little TL, Beyer EC, Duling BR. Connexin 43 and connexin 40 gap junctional proteins are present in arteriolar smooth muscle and endothelium in vivo. Am J Physiol. 1995;268:H729–H739. doi: 10.1152/ajpheart.1995.268.2.H729. [DOI] [PubMed] [Google Scholar]
- Lockwich TP, Liu X, Singh BB, Jadlowiec J, Weiland S, Ambudkar IS. Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J Biol Chem. 2000;275:11934–11942. doi: 10.1074/jbc.275.16.11934. [DOI] [PubMed] [Google Scholar]
- McCarron JG, Halpern W. Potassium dilates rat cerebral arteries by two independent mechanisms. Am J Physiol. 1990;259:H902–H908. doi: 10.1152/ajpheart.1990.259.3.H902. [DOI] [PubMed] [Google Scholar]
- McGuire JJ, Hollenberg MD, Andrade-Gordon P, Triggle CR. Multiple mechanisms of vascular smooth muscle relaxation by the activation of proteinase-activated receptor 2 in mouse mesenteric arterioles. Br J Pharmacol. 2002;135:155–169. doi: 10.1038/sj.bjp.0704469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeish AJ, Dora KA, Garland CJ. Possible role for K+ in endothelium-derived hyperpolarizing factor-linked dilatation in rat middle cerebral artery. Stroke. 2005;36:1526–1532. doi: 10.1161/01.STR.0000169929.66497.73. [DOI] [PubMed] [Google Scholar]
- McSherry IN, Spitaler MM, Takano H, Dora KA. Endothelial cell Ca2+ increases are independent of membrane potential in pressurized rat mesenteric arteries. Cell Calcium. 2005;38:23–33. doi: 10.1016/j.ceca.2005.03.007. [DOI] [PubMed] [Google Scholar]
- McSherry IN, Sandow SL, Campbell WB, Falck JR, Hill MA, Dora KA. A role for heterocellular coupling and EETs in dilation of rat cremaster arteries. Microcirculation. 2006;13:119–130. doi: 10.1080/10739680500466400. [DOI] [PubMed] [Google Scholar]
- Martin W, Villani GM, Jothianandan D, Furchgott RF. Selective blockade of endothelium-dependent and glyceryl trinitrate-induced relaxation by hemoglobin and by methylene blue in the rabbit aorta. J Pharmacol Exp Ther. 1985;232:708–716. [PubMed] [Google Scholar]
- Mather S, Dora KA, Sandow SL, Winter P, Garland CJ. Rapid endothelial cell-selective loading of connexin 40 antibody blocks endothelium-derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ Res. 2005;97:399–407. doi: 10.1161/01.RES.0000178008.46759.d0. [DOI] [PubMed] [Google Scholar]
- Mistry DK, Garland CJ. Characteristics of single, large-conductance calcium-dependent potassium channels (BKCa) from smooth muscle cells isolated from the rabbit mesenteric artery. J Membr Biol. 1998a;164:125–138. doi: 10.1007/s002329900399. [DOI] [PubMed] [Google Scholar]
- Mistry DK, Garland CJ. Nitric oxide (NO)-induced activation of large conductance Ca2+-dependent K+ channels (BK(Ca)) in smooth muscle cells isolated from the rat mesenteric artery. Br J Pharmacol. 1998b;124:1131–1140. doi: 10.1038/sj.bjp.0701940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Gryglewski R, Bunting S, Vane JR. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976;263:663–665. doi: 10.1038/263663a0. [DOI] [PubMed] [Google Scholar]
- Moncada S, Herman AG, Higgs EA, Vane JR. Differential formation of prostacyclin (PGX or PGI2) by layers of the arterial wall. An explanation for the anti-thrombotic properties of vascular endothelium. Thromb Res. 1977;11:323–344. doi: 10.1016/0049-3848(77)90185-2. [DOI] [PubMed] [Google Scholar]
- Moore PK, al-Swayeh OA, Chong NW, Evans RA, Gibson A. L-NG-nitro arginine (L-NOARG), a novel, L-arginine-reversible inhibitor of endothelium-dependent vasodilatation in vitro. Br J Pharmacol. 1990;99:408–412. doi: 10.1111/j.1476-5381.1990.tb14717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelli S, Wilson WS, Laidlaw H, Llano A, Middleton S, Price AG, et al. Evaluation of potassium ion as the endothelium-derived hyperpolarizing factor (EDHF) in the bovine coronary artery. Br J Pharmacol. 2003;139:982–988. doi: 10.1038/sj.bjp.0705329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohanian J, Gatfield KM, Ward DT, Ohanian V. Evidence for a functional calcium-sensing receptor that modulates myogenic tone in rat subcutaneous small arteries. Am J Physiol Heart Circ Physiol. 2005;288:H1756–H1762. doi: 10.1152/ajpheart.00739.2004. [DOI] [PubMed] [Google Scholar]
- Oishi H, Budel S, Schuster A, Stergiopulos N, Meister JJ, Bény JL. Cytosolic-free calcium in smooth-muscle and endothelial cells in an intact arterial wall from rat mesenteric artery in vitro. Cell Calcium. 2001;30:261–267. doi: 10.1054/ceca.2001.0233. [DOI] [PubMed] [Google Scholar]
- Plane F, Garland CJ. Differential effects of acetylcholine, nitric oxide and levcromakalim on smooth muscle membrane potential and tone in the rabbit basilar artery. Br J Pharmacol. 1993;110:651–656. doi: 10.1111/j.1476-5381.1993.tb13861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp R, Brandes RP, Ott G, Busse R, Fleming I. Dynamic modulation of interendothelial gap junctional communication by 11,12-epoxyeicosatrienoic acid. Circ Res. 2002;90:800–806. doi: 10.1161/01.res.0000015328.20581.d6. [DOI] [PubMed] [Google Scholar]
- Quignard JF, Félétou M, Thollon C, Vilaine JP, Duhault J, Vanhoutte PM. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br J Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall MD, Hiley CR. Detergent and methylene blue affect endothelium-dependent vasorelaxation and pressure/flow relations in rat blood perfused mesenteric arterial bed. Br J Pharmacol. 1988;95:1081–1088. doi: 10.1111/j.1476-5381.1988.tb11742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath G, Dessy C, Feron O. Caveolae, caveolin and control of vascular tone: nitric oxide (NO) and endothelium derived hyperpolarizing factor (EDHF) regulation. J Physiol Pharmacol. 2009;60(Suppl 4):105–109. [PubMed] [Google Scholar]
- Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodin JA. The ultrastructure of mammalian arterioles and precapillary sphincters. J Ultrastruct Res. 1967;18:181–223. doi: 10.1016/s0022-5320(67)80239-9. [DOI] [PubMed] [Google Scholar]
- Rivers R. Conducted arteriolar dilations persist in the presence of nitroarginine. J Cardiovasc Pharmacol. 1997;30:309–312. doi: 10.1097/00005344-199709000-00006. [DOI] [PubMed] [Google Scholar]
- Rivers RJ, Hein TW, Zhang C, Kuo L. Activation of barium-sensitive inward rectifier potassium channels mediates remote dilation of coronary arterioles. Circulation. 2001;104:1749–1753. doi: 10.1161/hc4001.098053. [DOI] [PubMed] [Google Scholar]
- Roy CS, Sherrington CS. On the regulation of the blood-supply of the brain. J Physiol. 1890;11:85–158. 117. doi: 10.1113/jphysiol.1890.sp000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, Rezzani R, et al. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation. 2008;117:1065–1074. doi: 10.1161/CIRCULATIONAHA.107.731679. [DOI] [PubMed] [Google Scholar]
- Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ Res. 2000;86:341–346. doi: 10.1161/01.res.86.3.341. [DOI] [PubMed] [Google Scholar]
- Sandow SL, Goto K, Rummery NM, Hill CE. Developmental changes in myoendothelial gap junction mediated vasodilator activity in the rat saphenous artery. J Physiol. 2004;556:875–886. doi: 10.1113/jphysiol.2003.058669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow SL, Neylon CB, Chen MX, Garland CJ. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels (K(Ca)) and connexins: possible relationship to vasodilator function? J Anat. 2006;209:689–698. doi: 10.1111/j.1469-7580.2006.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HH, Klein MM, Niroomand F, Bohme E. Is arginine a physiological precursor of endothelium-derived nitric oxide? Eur J Pharmacol. 1988a;148:293–295. doi: 10.1016/0014-2999(88)90578-x. [DOI] [PubMed] [Google Scholar]
- Schmidt HH, Nau H, Wittfoht W, Gerlach J, Prescher KE, Klein MM, et al. Arginine is a physiological precursor of endothelium-derived nitric oxide. Eur J Pharmacol. 1988b;154:213–216. doi: 10.1016/0014-2999(88)90101-x. [DOI] [PubMed] [Google Scholar]
- Segal SS. Regulation of blood flow in the microcirculation. Microcirculation. 2005;12:33–45. doi: 10.1080/10739680590895028. [DOI] [PubMed] [Google Scholar]
- Segal SS, Duling BR. Conduction of vasomotor responses in arterioles: a role for cell-to-cell coupling? Am J Physiol. 1989;256:H838–H845. doi: 10.1152/ajpheart.1989.256.3.H838. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Yasutake H, Fujii K, Owada MK, Nakaike R, Fukumoto Y, et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol. 1996;28:703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, Grgic I, et al. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ Res. 2006;99:537–544. doi: 10.1161/01.RES.0000238377.08219.0c. [DOI] [PubMed] [Google Scholar]
- Takano H, Dora KA, Spitaler MM, Garland CJ. Spreading dilatation in rat mesenteric arteries associated with calcium-independent endothelial cell hyperpolarization. J Physiol. 2004;556:887–903. doi: 10.1113/jphysiol.2003.060343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallini YN, Brekke JF, Shui B, Doran R, Hwang SM, Nakai J, et al. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC GCaMP2 transgenic mice. Circ Res. 2007;101:1300–1309. doi: 10.1161/CIRCRESAHA.107.149484. [DOI] [PubMed] [Google Scholar]
- Tare M, Parkington HC, Coleman HA, Neild TO, Dusting GJ. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- Taylor SG, Weston AH. Endothelium-derived hyperpolarizing factor: a new endogenous inhibitor from the vascular endothelium. Trends Pharmacol Sci. 1988;9:272–274. doi: 10.1016/0165-6147(88)90003-x. [DOI] [PubMed] [Google Scholar]
- Taylor HJ, Chaytor AT, Evans WH, Griffith TM. Inhibition of the gap junctional component of endothelium-dependent relaxations in rabbit iliac artery by 18-alpha glycyrrhetinic acid. Br J Pharmacol. 1998;125:1–3. doi: 10.1038/sj.bjp.0702078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, et al. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res. 2003;93:124–131. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- Uhrenholt TR, Domeier TL, Segal SS. Propagation of calcium waves along endothelium of hamster feed arteries. Am J Physiol Heart Circ Physiol. 2007;292:H1634–H1640. doi: 10.1152/ajpheart.00605.2006. [DOI] [PubMed] [Google Scholar]
- Vane JR. The use of isolated organs for detecting active substances in the circulating blood. Br J Pharmacol Chemother. 1964;23:360–373. doi: 10.1111/j.1476-5381.1964.tb01592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR. The Croonian Lecture, 1993. The endothelium: maestro of the blood circulation. Philos Trans R Soc Lond B Biol Sci. 1994;343:225–246. doi: 10.1098/rstb.1994.0023. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM, Shimokawa H, Tang EH, Félétou M. Endothelial dysfunction and vascular disease. Acta Physiol. 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- Waldron GJ, Garland CJ. Effect of potassium channel blockers on L-NAME insensitive relaxations in rat small mesenteric artery. Can J Physiol Pharmacol. 1994;72(Suppl 1):11. [Google Scholar]
- Weksler BB, Marcus AJ, Jaffe EA. Synthesis of prostaglandin I2 (prostacyclin) by cultured human and bovine endothelial cells. Proc Natl Acad Sci USA. 1977;74:3922–3926. doi: 10.1073/pnas.74.9.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston AH, Richards GR, Burnham MP, Félétou M, Vanhoutte PM, Edwards G. K+-induced hyperpolarization in rat mesenteric artery: identification, localization and role of Na+/K+-ATPases. Br J Pharmacol. 2002;136:918–926. doi: 10.1038/sj.bjp.0704787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston AH, Absi M, Ward DT, Ohanian J, Dodd RH, Dauban P, et al. Evidence in favor of a calcium-sensing receptor in arterial endothelial cells: studies with calindol and Calhex 231. Circulation Res. 2005;97:391–398. doi: 10.1161/01.RES.0000178787.59594.a0. [DOI] [PubMed] [Google Scholar]
- Weston AH, Porter EL, Harno E, Edwards G. Impairment of endothelial SK channels and of downstream hyperpolarizing pathways in mesenteric arteries from spontaneously hypertensive rats. Br J Pharmacol. 2010;160:836–843. doi: 10.1111/j.1476-5381.2010.00657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R, Hiley CR. Hyperpolarisation of rat mesenteric endothelial cells by ATP-sensitive K(+) channel openers. Eur J Pharmacol. 2000;397:279–290. doi: 10.1016/s0014-2999(00)00271-5. [DOI] [PubMed] [Google Scholar]
- Whittaker N, Bunting S, Salmon J, Moncada S, Vane JR, Johnson RA, et al. The chemical structure of prostaglandin X (prostacyclin) Prostaglandins. 1976;12:915–928. doi: 10.1016/0090-6980(76)90126-x. [DOI] [PubMed] [Google Scholar]
- Winter P, Dora KA. Spreading dilatation to luminal perfusion of ATP and UTP in rat isolated small mesenteric arteries. J Physiol. 2007;582:335–347. doi: 10.1113/jphysiol.2007.135202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit C, Griffith TM. Connexins and gap junctions in the EDHF phenomenon and conducted vasomotor responses. Pflugers Arch. 2010;459:897–914. doi: 10.1007/s00424-010-0830-4. [DOI] [PubMed] [Google Scholar]
- Xia J, Little TL, Duling BR. Cellular pathways of the conducted electrical response in arterioles of hamster cheek pouch in vitro. Am J Physiol. 1995;269:H2031–H2038. doi: 10.1152/ajpheart.1995.269.6.H2031. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Imaeda K, Suzuki H. Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea-pig mesenteric arterioles. J Physiol. 1999;514:505–513. doi: 10.1111/j.1469-7793.1999.505ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Hogestatt ED. Role of potassium channels in endothelium-dependent relaxation resistant to nitroarginine in the rat hepatic artery. Br J Pharmacol. 1996;117:1600–1606. doi: 10.1111/j.1476-5381.1996.tb15327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]