

Abstract
In vascular tissues, hydrogen sulphide (H2S) is mainly produced from L-cysteine by the cystathionine gamma-lyase (CSE) enzyme. Recent studies show that administration of H2S to endothelial cells in culture stimulates cell proliferation, migration and tube formation. In addition, administration of H2S to chicken chorioallantoic membranes stimulates blood vessel growth and branching. Furthermore, in vivo administration of H2S to mice stimulates angiogenesis, as demonstrated in the Matrigel plug assay. Pathways involved in the angiogenic response of H2S include the PI-3K/Akt pathway, the mitogen activated protein kinase pathway, as well as ATP-sensitive potassium channels. Indirect evidence also suggests that the recently demonstrated role of H2S as an inhibitor of phosphodiesterases may play an additional role in its pro-angiogenic effect. The endogenous role of H2S in the angiogenic response has been demonstrated in the chicken chorioallantoic membranes, in endothelial cells in vitro and ex vivo. Importantly, the pro-angiogenic effect of vascular endothelial growth factor (but not of fibroblast growth factor) involves the endogenous production of H2S. The pro-angiogenic effects of H2S are also apparent in vivo: in a model of hindlimb ischaemia-induced angiogenesis, H2S induces a marked pro-angiogenic response; similarly, in a model of coronary ischaemia, H2S exerts angiogenic effects. Angiogenesis is crucial in the early stage of wound healing. Accordingly, topical administration of H2S promotes wound healing, whereas genetic ablation of CSE attenuates it. Pharmacological modulation of H2S-mediated angiogenic pathways may open the door for novel therapeutic approaches.
LINKED ARTICLES
This article is part of a themed issue on Vascular Endothelium in Health and Disease. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.164.issue-3
Keywords: blood vessels, endothelium, nitric oxide, kinases, cell differentiation, cell migration, cysteine, ischaemia
Hydrogen sulphide (H2S) is a colourless, flammable, water-soluble gas with the characteristic smell of rotten eggs. Until recently, H2S was viewed exclusively as a toxic gas and environmental hazard. However, a growing body of data accumulating over the last decade shows that H2S is also synthesized by mammalian tissues via two pyridoxal-5′-phosphate-dependent enzymes responsible for metabolism of L-cysteine: cystathionine beta-synthase (CBS) and cystathionine gamma-lyase (CSE), as well as by a recently identified third pathway involves the production from L-cysteine of H2S via the combined action of 3-mercaptopyruvate sulphurtransferase and cysteine aminotransferase. The distribution and regulation of H2S producing enzymes is complex and is discussed in separate reviews (Wang, 2003; Fiorucci et al., 2006; Szabo, 2007; Kimura, 2010).
The biological roles of endogenous H2S are multiple and rapidly expanding. These regulatory functions span the central and peripheral nervous system, the regulation of cellular metabolism, regulation of immunological/inflammatory responses and various aspects of cardiovascular biology. As far as the cardiovascular system, the principal enzyme involved in the formation of H2S is CSE, expressed in vascular endothelial cells, smooth muscle cells as well as cardiac myocytes. The cardiovascular regulatory roles of H2S include vasodilatation, vascular protection, regulation of blood pressure and many others (Szabo, 2007; Calvert et al., 2010; Elsey et al., 2010; Gadalla and Snyder, 2010; Predmore and Lefer, 2010).
As a gasotransmitter, H2S rapidly travels through cell membranes without utilizing specific transporters and exerts a host of biological effects on a variety of biological targets resulting in a variety of biological responses. Similarly to the other two gasotransmitters [nitric oxide (NO) and carbon monoxide], many of the biological responses to H2S follow a bell-shaped dose–response: the effects of H2S range from physiological, cytoprotective effects (which occur at low concentrations) to cytotoxic effects (which are generally only apparent at higher concentrations). Depending on the experimental system studied, the molecular mechanisms of the biological actions of H2S include antioxidant effects, both via direct chemical reactions with various oxidant species, as well as via increased cellular glutathione levels via activation/expression of gamma-glutamylcysteine synthetase; modulation of intracellular caspase and kinase pathways; stimulatory effects on the production of cAMP and modulation of intracellular calcium levels (Wang, 2003; Fiorucci et al., 2006; Szabo, 2007; Calvert et al., 2010; Elsey et al., 2010; Gadalla and Snyder, 2010; Kimura, 2010; Predmore and Lefer, 2010). A distinct pharmacological effect of H2S relates to the opening of potassium-opened ATP channels (KATP channels) (Zhao et al., 2001; Cheng et al., 2004; Yang et al., 2005; Siebert et al., 2008). Some of the beneficial therapeutic actions of H2S – including some of the vasodilatory effect as well as some of the preconditioning and cardiac protective effects – have been attributed to this molecular target of H2S, as the protective effects of H2S can be prevented by pretreatment of the cells, tissues or animals with KATP channel inhibitors such as glibenclamide.
H2S simulates endothelial cell proliferation and migration
The phenomenon that H2S stimulates endothelial cell proliferation and migration (essential, initial components of the angiogenic response) has been reported by two independent groups (Table 1), either in transformed endothelial cells, or in primary endothelial cells (Cai et al., 2007; Papapetropoulos et al., 2009). Additional studies, utilizing the Roche XCelligence system also demonstrate a concentration-dependent increase in cell index (a conductance-based composite measure, reflecting cell number, adherence and growth) (Li et al., 2006; Ozsvári et al., 2010) in response to H2S in cultured human umbilical vein endothelial cells (HUVEC) in vitro (Figure 1). Based on in vitro studies with exogenously applied H2S, it was concluded that the concentrations of H2S sufficient to induce angiogenesis are in the low micromolar (i.e. the physiological) concentration concentration range (Cai et al., 2007; Papapetropoulos et al., 2009). Nevertheless, it should be pointed out that the absolute (‘free’ and/or ‘bound, biologically available’) concentration of H2S in the blood and physiological fluids is highly dependent on the method of detection used, and there is still a considerable disagreement in the literature on this point (Whitfield et al., 2008; Olson, 2009; Toombs et al., 2010; Wintner et al., 2010). The pro-angiogenic effect of H2S is further evidenced by the increase in tube-like structure formation in endothelial cells in in vitro Matrigel assays, where increases in tube length and increases in the number of branching points were demonstrated (Figure 2) (Cai et al., 2007; Papapetropoulos et al., 2009). The magnitude of the response (% increase in tube formation) induced by H2S in the Matrigel assay was approximately 50%, an effect which is comparable to what was previously reported with NO donors, activators of soluble guanylyl cyclase (sGC), or with the phosphodiesterase (PDE) 5 inhibitor sildenafil in the same assay (Shimizu et al., 2004; Pyriochou et al., 2006; 2007a,b;). Additional in vitro data relevant in the context of angiogenesis include data demonstrating that in the RF/6A cells H2S also increases adhesion (by approximately 20% at the maximum of the dose–response) (Cai et al., 2007). In addition to direct pro-angiogenic effects, H2S has also been shown to induce angiogenesis indirectly: via release of vascular endothelial growth factor (VEGF) from hypoxic smooth muscle cells, and subsequent VEGF-mediated responses in the endothelial cells (Liu et al., 2010a).
Table 1.
Studies demonstrating the effect of H2S of endothelial cell proliferation, migration, tube formation and angiogenesis in vitro
| Cell type | H2S concentration (µM) | Time course (h) | Effect | Reference |
|---|---|---|---|---|
| RF/6A transformed endothelial cell line | 1–50 | 24 | An up to 20% increase in proliferation, as assessed by the BrdU method. | Cai et al., 2007 |
| HUVEC | 6–600 | 48 | An up to 100% increase in proliferation, as measured by counting of trypsinized cells in a haemocytometer. | Papapetropoulos et al., 2009. |
| RF/6A transformed endothelial cell line | 1–50 | 24 | An up to 30% increase in migration, as assessed by the Transwell method. | Cai et al., 2007 |
| HUVEC | 6–600 | 48 | An up to sixfold increase in migration, as assessed by the Transwell method. | Papapetropoulos et al., 2009. |
H2S, hydrogen sulphide; HUVEC, human umbilical vein endothelial cells.
Figure 1.
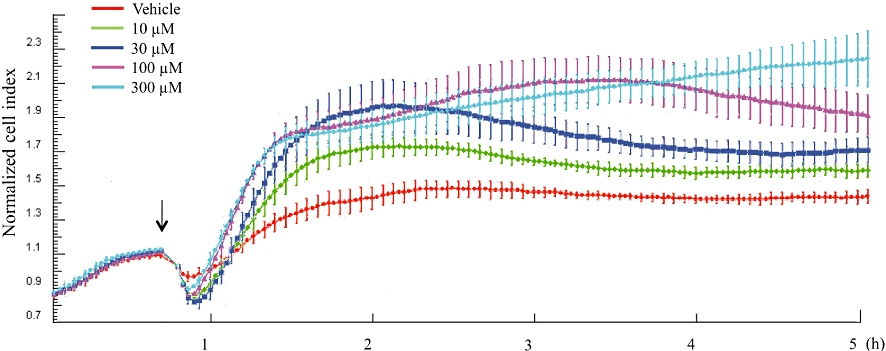
Concentration-dependent increase in the cell index by hydrogen sulphide (H2S) in human umbilical vein endothelial cells, as measured by the XCelligence cell-microelectronic sensing technique method. Cells were cultured in gelatin pretreated 96-well E-plates. Cells were attached and grown overnight and subjected to vehicle or various concentrations of H2S (10–300 µM) and cell index was continuously recorded over 5 h. Please note the concentration-dependent increase in cell index in response to H2S (10–300 µM). At a higher concentration tested (1000 µM) (not shown), the stimulatory effect of H2S was less pronounced that with 300 µM, consistent with the type of bell-shaped concentration-response curve often seen with H2S. Responses shown are the mean ± SEM of n = 6 wells for each condition.
Figure 2.

Increase by hydrogen sulphide (H2S) of tube formation in the Matrigel assay in cultured human umbilical vein endothelial cells in vitro. Cells were plated at a density of 15 000 cells/well in 96-well plates, precoated with 50 µL of growth factor-reduced Matrigel in the presence of H2S (60 µM) or vehicle, in complete medium. Following a 24 h-incubation, tube formation was quantified by image analysis software (Scion Image Release Beta 4.0.2) and expressed as a percentage of control. H2S treatment significantly (P < 0.05) increased tube formation.
H2S simulates angiogenesis in vivo
The chicken chorioallantoic membrane provides a simple and predictive model for investigating the regulatory processes of angiogenesis. Using this method, we have observed that H2S induces a potent and concentration-dependent increase in the length and complexity of the vascular network (Figure 3) (Papapetropoulos et al., 2009). The Matrigel plug assay provides another, rather reliable and predictable method to assess the angiogenic potential of test compounds. Moore and colleagues have used this assay in a mouse model with success, and demonstrated that intraperitoneal administration of NaHS (a H2S donor), for 7 days at 10–50 µmol·kg−1·day−1) increased neovascularization, cellular infiltration and haemoglobin content, demonstrating a pro-angiogenic effect in the mouse in vivo (Cai et al., 2007). It is noteworthy that at higher doses, the H2S donor failed to promote angiogenesis in the same assay (Cai et al., 2007), underlining the general theme already mentioned in the Introduction section with respect to the bell-shaped biological dose–responses frequently noted with H2S.
Figure 3.

Exogenously applied and endogenously produced hydrogen sulphide (H2S) promotes angiogenesis in the chicken chorioallantoic membrane. (A) Membranes were treated with H2S (240 pmol·egg−1) or the H2S synthesis inhibitor dl-propylargylglycine (PAG) (300 mmol·egg−1) or the combination of the two agents for 48 h and vascular network length and branching were determined. Note the enhancement of angiogenesis in response to H2S, the inhibition of angiogenesis by PAG, and the lack of effect of PAG on the pro-angiogenic effect of H2S. n = 36–45; *P < 0.05 versus vehicle. Panel (B) shows representative photomicrographs. BCA, beta-cyano-L-alanine.
Signalling pathways involved in the pro-angiogenic effect of H2S
The cellular signalling pathways involved in the angiogenic effect of H2S have been studied, in some detail, in endothelial cell models in vitro. Exposure of endothelial cells to donors of H2S has been shown to activate multiple signalling pathways with established roles in neovascularization. NaHS administration activates the PI-3K/Akt axis (Cai et al., 2007; Papapetropoulos et al., 2009) while exposure to Na2S enhances the phosphorylation of members of the mitogen activated protein kinase (MAPK) pathway [extracellular signal regulated kinase (ERK)1/2 and p38)] (Papapetropoulos et al., 2009), with time courses that are different (Figure 4). Activation of PI-3K/Akt has been shown to regulate tube-like structure formation in RF/6A cells (Cai et al., 2007), while activation of MAPK was shown to play a key role in migration in HUVEC (Papapetropoulos et al., 2009).
Figure 4.

Time-course showing the effect of hydrogen sulphide (H2S) (60 µM) on a number of signalling pathways involved in the pro-angiogenic effect of H2S in vascular endothelial cells. Please note the differential time-courses of the response. ERK, extracellular signal regulated kinase.
The KATP channels mediate, at least in part, the cellular action of H2S (Zhao et al., 2001). It was recently shown that H2S promotes KATP channel opening through interaction with Cys 6 and Cys26, both of which reside on the N-terminal region of the SUR subunit of the KATP channel complex (Jiang et al., 2010). To determine whether activation of KATP channels contributes to the angiogenic actions of H2S, we pretreated endothelial cells with sulphonylurea-type inhibitors and monitored their ability to migrate. In these experiments glibenclamide-treated cultures exhibited a markedly reduced migratory response to H2S (Papapetropoulos et al., 2009). The observation that glibenclamide blocked p38 phosphorylation indicates that KATP channels are upstream of MAPK pathways in the H2S-triggered angiogenic pathway. However, it is still unclear whether KATP channel opening is required for PI-3K/Akt activation by H2S. Furthermore, although H2S induces hsp27 phosphorylation (Papapetropoulos et al., 2009); siRNA inactivation of this protein inhibits the pro-angiogenic effect of H2S (Papapetropoulos et al., 2009). Cells pretreated with the p38 MAP kinase inhibitor SB203580 showed reduced H2S-induced hsp27 phosphorylation, suggesting that H2S enhances endothelial cells (EC) migration through a KATP channel/p38/hsp27 pathway. An interesting side observation of the above-mentioned series of studies was that KATP channel activation, on its own, is able to stimulate angiogenesis in HUVEC: incubation of cells with the KATP channel opener SG209, induced a concentration-dependent migratory response, indicating that K+ efflux per se can drive EC motility (Papapetropoulos et al., 2009).
The exact modes of interactions of the above-mentioned effectors of H2S-induced angiogenesis remains to be delineated in future studies. In particular, it remains unknown what the downstream effectors of MAPK and PI-3K pathways are that contribute to angiogenesis. It has been proposed that part of the downstream effectors include survivin and integrin α2 and integrin β1 (Cai et al., 2007). In addition, the up-regulation of integrin α2 and integrin β1 was also shown in the same experimental system. However, the causative role of these changes in the pro-angiogenic effect of H2S has not yet been definitively tested. When considering potential downstream effector pathways, it should also be noted that hypoxia inducible factor-1 (a transcription factor with a central role in the angiogenic response) was also shown to be activated by H2S (Budde and Roth, 2010; Liu et al., 2010b). A cartoon summarizing our current knowledge of the H2S-induced pro-angiogenic pathways and some of the potential interactions between them is shown in Figure 5.
Figure 5.

Signalling pathways involved in the pro-angiogenic effect of hydrogen sulphide (H2S) in vascular endothelial cells. ERK, extracellular signal regulated kinase; KATP, ATP-sensitive potassium channel.
Role of endogenous H2S in angiogenesis
We recently demonstrated that exposure of endothelial cells to VEGF increases the production of H2S and that endogenously produced H2S participates in the angiogenic signalling of VEGF (Papapetropoulos et al., 2009). The VEGF-induced and the H2S-induced endothelial cell migration response are not additive (Figure 6). This may be consistent with the notion that the two responses, at least in part, involve similar downstream mechanisms. Indeed, subsequent studies showed that endogenously produced H2S is a key mediator of VEGF-induced angiogenesis (Papapetropoulos et al., 2009). This conclusion is based on the finding that pharmacological inhibition, genetic deletion or silencing of CSE, a major H2S-producing enzyme in the endothelium, reduces migration and sprouting of endothelial cells in vitro (Figure 7). Further evidence for the importance of this pathway in VEGF-induced angiogenesis comes from the ex vivo mouse aorta sprouting assay, where the VEGF-induced angiogenesis was markedly suppressed in aortic rings of the CSE-/- mice (Figure 8) (Papapetropoulos et al., 2009).
Figure 6.

Incubation with vascular endothelial growth factor (VEGF) and hydrogen sulphide (H2S) does not produce an additive effect on endothelial migration. Endothelial cells were allowed to migrate for 4 h in response to VEGF (20 ng·mL−1), H2S (60 µM) or a combination of the two. n = 5; *P < 0.05 versus vehicle.
Figure 7.

H2S is required for vascular endothelial growth factor (VEGF)-stimulated, but not fibroblast growth factor (FGF)-stimulated endothelial cells (EC) migration; role of KATP channels in the response. The EC were serum starved overnight. Cells were then treated with the KATP channel inhibitors glibenclamide (10 µM) or 5-HD (100 µM) for 30 min. Cells were then trypsinized, placed in transwells and allowed to migrate for 4 h in the presence of vehicle, VEGF (20 ng·mL−1) (A) or FGF-2 (10 ng·mL−1) (B). n = 5; *P < 0.05 versus vehicle and #P < 0.05 versus VEGF. In the experiments shown in (C), EC were treated with the cystathionine gamma-lyase inhibitor dl-propylargylglycine (PAG) (3 mM) for 30 min. Cells were then placed in transwells and allowed to migrate for 4 h in the presence of vehicle or VEGF (20 ng·mL−1). n = 5; *P < 0.05 versus vehicle and $P < 0.05 versus VEGF.
Figure 8.

The angiogenic actions of vascular endothelial growth factor (VEGF) are regulated by endogenously produced hydrogen sulphide (H2S) in the in vitro aortic ring angiogenesis assay. Aortic ring explants from cystathionine gamma-lyase (CSE) wild-type (WT) or knockout (KO) mice were incubated in the presence or absence of VEGF (20 ng·mL−1). Representative photomicrographs are presented in (A), and quantification of the response is shown in the right panel (B). Please note the substantially lower number of new microvessels in the CSE-deficient groups. From Papapetropoulos et al. 2009, reproduced with permission.
H2S is not a universal requirement for all angiogenic growth factor signalling; for example, inhibition of CSE did not affect fibroblast growth factor-induced migration (Figure 7). The dependence of VEGF signalling on H2S might be due to the fact that VEGF increases intracellular calcium levels (Papapetropoulos et al., 1997) that will, in turn, promote a calmodulin-dependent increase in CSE activity (Yang et al., 2008). As inhibition of endogenously produced H2S reduces p38 and ERK1/2 phosphorylation, it is most likely that H2S production in response to VEGF lies upstream and is required for MAPK activation. Similar to the situation with exogenously applied H2S, the pro-angiogenic VEGF/H2S response also involves the activation of KATP channels: inhibition of these channels suppresses VEGF-induced angiogenesis (whereas FGF-induced angiogenesis does not involve KATP channels) (Figure 7) (Papapetropoulos et al., 2009).
There have been significant advances in the pharmacological modulation and therapeutic exploitation of angiogenesis, both in the field of tumour biology. In both cases, inhibition of angiogenesis is of therapeutic significance, as it induces, respectively, tumour hypoxia (Folkman, 2006; Ruegg and Mutter, 2007; Niu and Chen, 2010; Reynolds, 2010) and inhibition of unwanted overproliferation of blood vessels (Frank, 2009; Nicholson and Schachat, 2010). Based on the current results one can speculate that pharmacological inhibition of H2S biosynthesis may have the potential of affecting the same responses, either on its own, or in combination with other angiogenesis inhibitory approaches.
H2S/NO interactions in angiogenesis
NO, the ‘original’ gasotransmitter is a well-known pro-angiogenic mediator that exerts its effects primarily via the sGC/cyclic GMP (cGMP) pathway (Papapetropoulos et al., 1997; Morbidelli et al., 2003; Pyriochou et al., 2006; 2007a,b; Morbidelli et al., 2010). Over the last decade, a multitude of studies have investigated the complex relationship between NO and H2S in various experimental models. The interactions of these two gasotransmitters depend on the experimental system used, but include direct interactions, followed by the formation of nitrosothiols (Whiteman et al., 2006), release of NO from nitrite or nitrosothiols by H2S (Grossi, 2009; Tomaskova et al., 2009) synergistic vasorelaxant effects (Hosoki et al., 1997) and many others (Liu and Bian, 2010; Yong et al., 2010).
It should be noted that in addition to H2S, NO has also been shown to lie upstream of ERK1/2 activation in VEGF-treated cells (Parenti et al., 1998) and to contribute to many of its angiogenic properties (Zachary and Gliki, 2001). Careful analysis of the results in the literature reveals that simultaneous production of both H2S and NO may be required to drive the migratory response to VEGF; generation of either NO or H2S alone is not sufficient for the angiogenic response. For example, inhibition of NO production blunts the VEGF-stimulated migration in cultured EC (Ziche et al., 1997; Dimmeler et al., 2000) and so does CSE inhibition by dl-propylargylglycine (Papapetropoulos et al., 2009). These observations, when considered together, suggest that the two gasotransmitters may act in a concerted manner targeting the same pathway, and not additively through parallel pathways, to promote migration. One possible mechanism for such an orchestrated action requiring both H2S and NO to enhance VEGF migration might relate to the ability of H2S to inhibit PDE activity. We recently observed that H2S increases cGMP levels in smooth muscle cells by inhibiting cGMP-degrading PDE (Bucci et al., 2010). If a similar mechanism operates in EC, increased production of NO after VEGF exposure would stimulate sGC to produce cGMP; at the same time increased production of H2S would inhibit PDE activity allowing for a transient increase in cGMP levels that triggers the activation of downstream pathways (Figure 9). Indeed, H2S-stimulated migration can be blocked by cGMP-dependent protein kinase inhibition (A. Papapetropoulos and C. Szabó, unpubl. obs.). The interactions of NO and H2S in the context of angiogenesis remain to be studied in further detail. According to conflicting reports in the literature, this latter pathway may (Parenti et al., 1998) or may not (Breslin et al., 2003) be stimulated by NO in endothelial cells.
Figure 9.

A proposed pathway of the interaction between nitric oxide (NO) and hydrogen sulphide (H2S) in endothelial cells during angiogenesis. Vascular endothelial growth factor (VEGF) receptor activation increases NO production, leading to soluble guanylyl cyclase activation and increased cGMP synthesis. At the same time VEGF receptor activation triggers the generation of increased amounts of H2S; H2S inhibits cGMP-degrading phosphodiesterases leading to a further elevation of cGMP that in turn activates cGMP-dependent protein kinase and promotes angiogenesis. Because H2S increases the levels of intracellular calcium, VEGF-stimulated H2S production might enhance NO production. The relationship between these pathways and the canonical pathway of VEGF receptor activation-induced stimulation of the intracellular signalling axis of Raf, MAK and extracellular signal regulated kinase (ERK)1/2 remains to be delineated in future studies. PDE, phosphodiesterase; sGC, soluble guanylyl cyclase.
An alternative, but not mutually exclusive mode of H2S and NO interaction might involve intracellular calcium. Exposure to H2S was reported to increase endothelial calcium levels (Bauer et al., 2010), which would lead to eNOS activation and increased NO release. In line with the above mentioned interactions between NO and H2S, the protective actions of H2S in cardiac arrest and in intestinal injury is lost in eNOS knockout mice (Minamishima et al., 2009; Yusof et al. 2009).
H2S as a promoter of wound healing and therapeutic angiogenesis
In a model of cutaneous burn injury and wound healing we have demonstrated that topical administration of a H2S-saturated physiological solution significantly enhances the closure of the wound (Figure 10) (Papapetropoulos et al., 2009). Consistent with these observations, the wound healing response in CSE-/- mice is significantly delayed, when compared to the response in wild-type animals (Papapetropoulos et al., 2009). These observations may lay the groundwork for therapeutic approaches for the topical delivery of H2S in order to promote wound healing in burns or in other conditions. Since the topical delivery of gaseous H2S is challenging, one may envision approaches involving H2S donors or precursors and/or possibly approaches around the therapeutic local overexpression of H2S-producing enzymes. Polysulphides (including the ones that are present in garlic, such as diallyl disulphide and diallyl trisulphide) have recently been shown to produce H2S in cells via a reaction that involves glutathione (Benavides et al., 2007). Although such compounds may be interesting as potential therapeutic stimulators of H2S-mediated angiogenesis, it must be noted that several publications have reported that compounds of this class do not stimulate angiogenesis, but in fact, they inhibit it (Xiao et al., 2006; Thejass and Kuttan, 2007). The reason for this unexpected response remains to be investigated in future studies.
Figure 10.

Hydrogen sulphide (H2S) promotes re-epithelialization in burn wounds. Rats received a 30% total body surface area dorsal full-thickness scald burn under deep anaesthesia. Animals were treated daily with subcutaneous injections of vehicle, or H2S (50 µg·cm−2·day−1) at four equally spaced sites in the transition zone between burn eschar and healthy tissue. Planimetric measurement of the wound surface and re-epithelialization, as well as the ratio of wound contraction was performed. n = 6; *P < 0.05 versus control.
The healing of gastric ulcers is another process in which angiogenesis plays an important role (Szabo et al., 1999; Tarnawski, 2005). Wallace has demonstrated in a rat model of gastric ulceration (induced experimentally by topical administration of acetic acid) that H2S donors of two different chemical classes (as well as the H2S precursor L-cysteine) improved the resolution of the ulcers (Wallace et al., 2009). Although angiogenic responses were not measured in this study, it is conceivable that a pro-angiogenic effect of H2S may have contributed to its therapeutic effects. It is also interesting to note that there was an up-regulation of endogenous H2S production during the healing response (Wallace et al., 2009); this endogenous H2S may serve as a potential mechanism to accelerate wound healing. We speculate that some of the benefit of H2S donors seen in enhancing the resolution of colitis in rat models (Wallace et al., 2009) may also have involved, among others, a pro-angiogenic response.
Therapeutic angiogenesis is also of substantial importance in the context of organ ischaemia, or the reperfusion of previously ischaemic organs. Several in vivo studies began characterizing the effects of H2S administration in this context. In a rat model of chronic hindlimb ischaemia induced by unilateral ligation of the femoral artery the therapeutic intraperitoneal administration of the H2S donor NaHS (at 10–200 µmol·kg−1·day−1 for 28 days) was tested (Wang et al., 2010). The results (Figure 11) showed that at the lower doses used, H2S therapy significantly improved capillary density, angiographic scores, and these effects correlated with an improvement in hindlimb blood flow (Wang et al., 2010). Interestingly (and, again, underlying the bell-shaped dose–responses to H2S), higher doses of the H2S donor were not found to be effective in this model. Part of the response to H2S has been attributed to an up-regulation of VEGF production and an increase in the phosphorylation of the VEGF receptor (Wang et al., 2010). These findings are somewhat in line with the in vitro model of hypoxic angiogenesis discussed earlier, where conditioned media of smooth muscle cells treated with H2S contained significantly increased levels of VEGF, which, in turn, stimulated angiogenesis in endothelial cells (Liu et al., 2010a).
Figure 11.

Hydrogen sulphide (H2S) promotes collateral vessel formation and regional blood flow after femoral artery occlusion in the rat hind limb ischaemia model. (A) Representative post-mortem angiograms obtained 4 weeks after surgery. There was more collateral vessel formation in the ischaemic left hindlimb of the rats treated with NaHS at a dose of 100 µmol·kg−1·day−1. Arrow denotes the site of ligation at the femoral artery. The arrowhead indicates the typical ‘corkscrew’ appearance of collateral vessels. (B) Quantitative analysis of collateral vessel development was performed by measuring the total length of the contrast-opacified vessels. The angiographic score was significantly greater in the rats receiving NaHS (50 and 100 µmol·kg−1·day−1) than in the control animals (*P < 0.05). Data represent the mean ± SEM of three to five experiments in each group. (C) Blood flow measured with microsphere assay. The regional blood flow in ischaemic limb was standardized to tissue weight and is represented as the ratio of fluorescence intensity in the ischaemic hind limb to that of the contralateral nonischaemic hind limb in each animal. NaHS treatment (20 and 50 µmol·kg−1·day−1) significantly improved regional blood flow in the ischaemic limb (*P < 0.05). Data represent the mean ± SEM of seven to nine experiments (from Wang et al., 2010; reproduced with permission).
Consistent with the effects of H2S donation in hindlimb ischaemia, a recent study reported on the pro-angiogenic effect of H2S supplementation in the ischaemic rat heart. Permanent ligation of the left anterior descending artery was performed; H2S was administered as NaHS once a day at 56 µmol·kg−1 intravenously. Functional and histological evaluations, performed 42 days later, revealed that H2S therapy improved cardiac function (assessed by electrocardiography), and these alterations were associated with a significant increase in capillary density in the myocardium (Liu et al., 2010a).
Limitations, conclusions and future directions
A substantial limitation of the current state-of-the-art with respect to the role of H2S in angiogenesis is that it is primarily based on in vitro studies in reductionist models, such as endothelial cell proliferation, migration and tube formation. While these models do have substantial validity, they clearly do not mimic the complex phenomenon of angiogenesis in vivo. There are also important distinctions that can be made between the processes of vasculogenesis, angiogenesis and arteriogenesis. These complex phenomena are tightly regulated in a spatial and temporal manner, and involve the degradation of extracellular matrix, the proliferation and migration of endothelial cells towards angiogenic cues and the establishment of tubular structures, the basis of new blood vessels (Silvestre et al., 2008; De Val and Black, 2009; Eble and Niland, 2009). Much additional work needs to be done to delineate the exact role of H2S in each of these processes. While there are no published data on the potential effects of H2S on matrix degradation, we hypothesize (based on in vitro findings) that H2S may stimulate proliferation, migration and tube formation of endothelial cells in vivo. However, this remains to be proven in carefully designed future in vivo studies.
Another limitation of the current state-of-the art is that the role of KATP channels in angiogenesis is based on pharmacological studies alone. In fact, most of the work investigating the role of endothelial cell KATP channels is based on pharmacological inhibitors of limited selectivity and specificity (e.g. Broadhead et al., 2004; Tajima et al., 2008; Figura et al., 2009; Grossini et al., 2009), as opposed to studies knocking down or inactivating specific channel subunits. Therefore, the relative importance of sarcolemmal versus mitochondrial channels in the observed effects remains to be established. Indeed, one must point out that the type of KATP channel subunits present in endothelial cells have not yet been characterized in full detail (Yoshida et al., 2004; Morrissey et al., 2005; Malester et al., 2007) and there are only a few reports that provide electrophysiological evidence of KATP channels in endothelial cells (Adams and Hill, 2004; Jackson, 2005) and some of the endothelial responses to KATP channel openers have been attributed to indirect, gap junction-transmitted hyperpolarization from the underlying smooth muscle cells (White and Hiley, 2000; Takano et al., 2004). Because in the current studies, endothelial cells in culture were used, such transmission is not possible, but nevertheless the exact localization and nature of the KATP channels involved will require substantial additional work. Likewise, the potential contribution to the angiogenic responses of other types of potassium channels activated by H2S (Cheang et al., 2010; Tang et al., 2010; Zuidema et al., 2010) remains to be elucidated in further studies.
In conclusion, recently emerging work demonstrates the pro-angiogenic role of exogenously applied and endogenously produced H2S. Some of the intracellular signalling pathways have been delineated, and additional pathways are likely to be identified in the future. It is noteworthy that recent studies suggest a potential interaction of H2S with the NO pathway in the context of angiogenesis. Both pharmacological supplementation/stimulation of H2S and pharmacological inhibition of H2S may have potential therapeutic applications. Experimental data support the view that H2S donation may be of therapeutic relevance for wound healing and post-ischaemic therapy. In addition, one can hypothesize that H2S inhibition may be potentially applicable to inhibition of tumour blood supply and/or the hyperproliferative response of the blood vessels in the diabetic eye. However, these latter possibilities are, at the present time, are only based on in vitro findings, and remain to be tested in vivo.
Acknowledgments
The work of C. S. is supported by a grant from the National Institutes of Health (NIH R01 GM060915) and by a grant from the Shriners Burns Hospitals (#8661).
Glossary
Abbreviations
- CAM
chicken chorioallantoic membrane
- CBS
cystathionine beta-synthase
- cGMP
cyclic guanosine monophosphate
- CSE
cystathionine gamma-lyase
- ERK
extracellular signal regulated kinase
- H2S
hydrogen sulphide
- hsp27
heat shock protein 27
- HUVEC
human umbilical vein endothelial cells
- KATP
ATP-sensitive potassium channel
- MAPK
mitogen activated protein kinase
- NO
nitric oxide
- PAG
dl-propylargylglycine
- PDE
phosphodiesterase
- sGC
soluble guanylyl cyclase
- VEGF
vascular endothelial growth factor
Conflict of interest
C. S. is a shareholder of Ikaria Inc, a for-profit organization involved in therapeutic applications of H2S.
References
- Adams DJ, Hill MA. Potassium channels and membrane potential in the modulation of intracellular calcium in vascular endothelial cells. J Cardiovasc Electrophysiol. 2004;15:598–610. doi: 10.1046/j.1540-8167.2004.03277.x. [DOI] [PubMed] [Google Scholar]
- Bauer CC, Boyle JP, Porter KE, Peers C. Modulation of Ca(2+) signalling in human vascular endothelial cells by hydrogen sulfide. Atherosclerosis. 2010;209:374–380. doi: 10.1016/j.atherosclerosis.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Benavides GA, Squadrito GL, Mills RW, Patel HD, Isbell TS, Patel RP, et al. Hydrogen sulfide mediates the vasoactivity of garlic. Proc Natl Acad Sci U S A. 2007;104:17977–17982. doi: 10.1073/pnas.0705710104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslin JW, Pappas PJ, Cerveira JJ, Hobson RW, 2nd, Durán WN. VEGF increases endothelial permeability by separate signaling pathways involving ERK-1/2 and nitric oxide. Am J Physiol Heart Circ Physiol. 2003;284:H92–H100. doi: 10.1152/ajpheart.00330.2002. [DOI] [PubMed] [Google Scholar]
- Broadhead MW, Kharbanda RK, Peters MJ, MacAllister RJ. KATP channel activation induces ischemic preconditioning of the endothelium in humans in vivo. Circulation. 2004;110:2077–2082. doi: 10.1161/01.CIR.0000144304.91010.F0. [DOI] [PubMed] [Google Scholar]
- Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C, et al. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol. 2010;30:1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- Budde MW, Roth MB. Hydrogen sulfide increases hypoxia-inducible factor-1 activity independently of von Hippel-Lindau tumor suppressor-1 in C. elegans. Mol Biol Cell. 2010;21:212–217. doi: 10.1091/mbc.E09-03-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. 2007;76:29–40. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- Calvert JW, Coetzee WA, Lefer DJ. Novel insights into hydrogen sulfide-mediated cytoprotection. Antioxid Redox Signal. 2010;12:1203–1217. doi: 10.1089/ars.2009.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheang WS, Wong WT, Shen B, Lau CW, Tian XY, Tsang SY, et al. 4-aminopyridine-sensitive K+ channels contributes to NaHS-induced membrane hyperpolarization and relaxation in the rat coronary artery. Vascul Pharmacol. 2010;53:94–98. doi: 10.1016/j.vph.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol. 2004;287:H2316–H2323. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]
- De Val S, Black BL. Transcriptional control of endothelial cell development. Dev Cell. 2009;16:180–195. doi: 10.1016/j.devcel.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Dernbach E, Zeiher AM. Phosphorylation of the endothelial nitric oxide synthase at ser-1177 is required for VEGF-induced endothelial cell migration. FEBS Lett. 2000;477:258–262. doi: 10.1016/s0014-5793(00)01657-4. [DOI] [PubMed] [Google Scholar]
- Eble JA, Niland S. The extracellular matrix of blood vessels. Curr Pharm Des. 2009;15:1385–1400. doi: 10.2174/138161209787846757. [DOI] [PubMed] [Google Scholar]
- Elsey DJ, Fowkes RC, Baxter GF. Regulation of cardiovascular cell function by hydrogen sulfide (H(2)S) Cell Biochem Funct. 2010;28:95–106. doi: 10.1002/cbf.1618. [DOI] [PubMed] [Google Scholar]
- Figura M, Chilton L, Liacini A, Viskovic MM, Phan V, Knight D, et al. Blockade of K(ATP) channels reduces endothelial hyperpolarization and leukocyte recruitment upon reperfusion after hypoxia. Am J Transplant. 2009;9:687–696. doi: 10.1111/j.1600-6143.2009.02553.x. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Distrutti E, Cirino G, Wallace JL. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology. 2006;131:259–271. doi: 10.1053/j.gastro.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Folkman J. Antiangiogenesis in cancer therapy – endostatin and its mechanisms of action. Exp Cell Res. 2006;312:594–607. doi: 10.1016/j.yexcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Frank RN. Treating diabetic retinopathy by inhibiting growth factor pathways. Curr Opin Investig Drugs. 2009;10:327–335. [PubMed] [Google Scholar]
- Gadalla MM, Snyder SH. Hydrogen sulfide as a gasotransmitter. J Neurochem. 2010;113:14–26. doi: 10.1111/j.1471-4159.2010.06580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossi L. Hydrogen sulfide induces nitric oxide release from nitrite. Bioorg Med Chem Lett. 2009;19:6092–6094. doi: 10.1016/j.bmcl.2009.09.030. [DOI] [PubMed] [Google Scholar]
- Grossini E, Molinari C, Caimmi PP, Uberti F, Vacca G. Levosimendan induces NO production through p38 MAPK, ERK and Akt in porcine coronary endothelial cells: role for mitochondrial K(ATP) channel. Br J Pharmacol. 2009;156:250–261. doi: 10.1111/j.1476-5381.2008.00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation. 2005;12:113–127. doi: 10.1080/10739680590896072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Tang G, Cao K, Wu L, Wang R. Molecular mechanism for H(2)S-induced activation of K(ATP) channels. Antioxid Redox Signal. 2010;12:1167–1178. doi: 10.1089/ars.2009.2894. [DOI] [PubMed] [Google Scholar]
- Kimura H. Hydrogen sulfide: from brain to gut. Antioxid Redox Signal. 2010;12:1111–1123. doi: 10.1089/ars.2009.2919. [DOI] [PubMed] [Google Scholar]
- Li HB, Ge YK, Zhang L, Zheng XX. Astragaloside IV improved barrier dysfunction induced by acute high glucose in human umbilical vein endothelial cells. Life Sci. 2006;79:1186–1193. doi: 10.1016/j.lfs.2006.03.041. [DOI] [PubMed] [Google Scholar]
- Liu YH, Bian JS. Bicarbonate-dependent effect of hydrogen sulfide on vascular contractility in rat aortic rings. Am J Physiol Cell Physiol. 2010;299:C866–C872. doi: 10.1152/ajpcell.00105.2010. [DOI] [PubMed] [Google Scholar]
- Liu X, Pan L, Wang X, Gong Q, Deng H, Zheng H, et al. Hydrogen Sulfide Improves Cardiac Function through Anti-inflammation and Neovascularization in a Rat Model of Heart Failure. Biol Pharm Bull. 2010a in press. [Google Scholar]
- Liu X, Pan L, Zhuo Y, Gong Q, Rose P, Zhu Y. Hypoxia-inducible factor-1alpha is involved in the pro-angiogenic effect of hydrogen sulfide under hypoxic stress. Biol Pharm Bull. 2010b;33:1550–1554. doi: 10.1248/bpb.33.1550. [DOI] [PubMed] [Google Scholar]
- Malester B, Tong X, Ghiu I, Kontogeorgis A, Gutstein DE, Xu J, et al. Transgenic expression of a dominant negative K(ATP) channel subunit in the mouse endothelium: effects on coronary flow and endothelin-1 secretion. FASEB J. 2007;21:2162–2172. doi: 10.1096/fj.06-7821com. [DOI] [PubMed] [Google Scholar]
- Minamishima S, Bougaki M, Sips PY, Yu JD, Minamishima YA, Elrod JW, et al. Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3-dependent mechanism in mice. Circulation. 2009;120:888–896. doi: 10.1161/CIRCULATIONAHA.108.833491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morbidelli L, Donnini S, Ziche M. Role of nitric oxide in the modulation of angiogenesis. Curr Pharm Des. 2003;9:521–530. doi: 10.2174/1381612033391405. [DOI] [PubMed] [Google Scholar]
- Morbidelli L, Pyriochou A, Filippi S, Vasileiadis I, Roussos C, Zhou Z, et al. The soluble guanylyl cyclase inhibitor NS-2028 reduces vascular endothelial growth factor-induced angiogenesis and permeability. Am J Physiol Regul Integr Comp Physiol. 2010;298:R824–R832. doi: 10.1152/ajpregu.00222.2009. [DOI] [PubMed] [Google Scholar]
- Morrissey A, Rosner E, Lanning J, Parachuru L, Dhar Chowdhury P, Han S, et al. Immunolocalization of KATP channel subunits in mouse and rat cardiac myocytes and the coronary vasculature. BMC Physiol. 2005;5:1. doi: 10.1186/1472-6793-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson BP, Schachat AP. A review of clinical trials of anti-VEGF agents for diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2010;248:915–930. doi: 10.1007/s00417-010-1315-z. [DOI] [PubMed] [Google Scholar]
- Niu G, Chen X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr Drug Targets. 2010;11:1000–1017. doi: 10.2174/138945010791591395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KR. Is hydrogen sulfide a circulating ‘gasotransmitter’ in vertebrate blood? Biochim Biophys Acta. 2009;1787:856–863. doi: 10.1016/j.bbabio.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Ozsvári B, Puskás LG, Nagy LI, Kanizsai I, Gyuris M, Madácsi R, et al. A cell-microelectronic sensing technique for the screening of cytoprotective compounds. Int J Mol Med. 2010;25:525–530. doi: 10.3892/ijmm_00000373. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A, García-Cardeña G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2009;106:21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti A, Morbidelli L, Cui XL, Douglas JG, Hood JD, Granger HJ, et al. Nitric oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal-regulated kinase1/2 activation in postcapillary endothelium. J Biol Chem. 1998;273:4220–4226. doi: 10.1074/jbc.273.7.4220. [DOI] [PubMed] [Google Scholar]
- Predmore BL, Lefer DJ. Development of hydrogen sulfide-based therapeutics for cardiovascular disease. J Cardiovasc Transl Res. 2010;3:487–498. doi: 10.1007/s12265-010-9201-y. [DOI] [PubMed] [Google Scholar]
- Pyriochou A, Beis D, Koika V, Potytarchou C, Papadimitriou E, Zhou Z, et al. Soluble guanylyl cyclase activation promotes angiogenesis. J Pharmacol Exp Ther. 2006;319:663–671. doi: 10.1124/jpet.106.108878. [DOI] [PubMed] [Google Scholar]
- Pyriochou A, Vassilakopoulos T, Zhou Z, Papapetropoulos A. cGMP-dependent and -independent angiogenesis-related properties of nitric oxide. Life Sci. 2007a;81:1549–1554. doi: 10.1016/j.lfs.2007.09.014. [DOI] [PubMed] [Google Scholar]
- Pyriochou A, Zhou Z, Koika V, Petrou C, Cordopatis P, Sessa WC, et al. The phosphodiesterase 5 inhibitor sildenafil stimulates angiogenesis through a protein kinase G/MAPK pathway. J Cell Physiol. 2007b;211:197–204. doi: 10.1002/jcp.20929. [DOI] [PubMed] [Google Scholar]
- Reynolds AR. Potential relevance of bell-shaped and u-shaped dose-responses for the therapeutic targeting of angiogenesis in cancer. Dose Response. 2010;8:253–284. doi: 10.2203/dose-response.09-049.Reynolds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegg C, Mutter N. Anti-angiogenic therapies in cancer: achievements and open questions. Bull Cancer. 2007;94:753–762. [PubMed] [Google Scholar]
- Shimizu S, Kageyama M, Yasuda M, Sasaki D, Naito S, Yamamoto T, et al. Stimulation of in vitro angiogenesis by nitric oxide through the induction of transcription factor ETS-1. Int J Biochem Cell Biol. 2004;36:114–122. doi: 10.1016/s1357-2725(03)00170-5. [DOI] [PubMed] [Google Scholar]
- Siebert N, Cantré D, Eipel C, Vollmar B. H2S contributes to the hepatic arterial buffer response and mediates vasorelaxation of the hepatic artery via activation of K(ATP) channels. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1266–G1273. doi: 10.1152/ajpgi.90484.2008. [DOI] [PubMed] [Google Scholar]
- Silvestre JS, Mallat Z, Tedgui A, Lévy BI. Post-ischaemic neovascularization and inflammation. Cardiovasc Res. 2008;78:242–249. doi: 10.1093/cvr/cvn027. [DOI] [PubMed] [Google Scholar]
- Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- Szabo S, Gombos Z, Sandor Z. Growth factors in gastrointestinal diseases. BioDrugs. 1999;12:27–41. doi: 10.2165/00063030-199912010-00004. [DOI] [PubMed] [Google Scholar]
- Tajima M, Ishizuka N, Saitoh K, Sakagami H. Nicorandil enhances the effect of endothelial nitric oxide under hypoxia-reoxygenation: role of the KATP channel. Eur J Pharmacol. 2008;579:86–92. doi: 10.1016/j.ejphar.2007.10.074. [DOI] [PubMed] [Google Scholar]
- Takano H, Dora KA, Spitaler MM, Garland CJ. Spreading dilatation in rat mesenteric arteries associated with calcium-independent endothelial cell hyperpolarization. J Physiol. 2004;556:887–903. doi: 10.1113/jphysiol.2003.060343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Wu L, Wang R. Interaction of hydrogen sulfide with ion channels. Clin Exp Pharmacol Physiol. 2010;37:753–763. doi: 10.1111/j.1440-1681.2010.05351.x. [DOI] [PubMed] [Google Scholar]
- Tarnawski AS. Cellular and molecular mechanisms of gastrointestinal ulcer healing. Dig Dis Sci. 2005;50:S24–S33. doi: 10.1007/s10620-005-2803-6. [DOI] [PubMed] [Google Scholar]
- Thejass P, Kuttan G. Inhibition of angiogenic differentiation of human umbilical vein endothelial cells by diallyl disulfide (DADS) Life Sci. 2007;80:515–521. doi: 10.1016/j.lfs.2006.09.045. [DOI] [PubMed] [Google Scholar]
- Tomaskova Z, Cacanyiova S, Benco A, Kristek F, Dugovicova L, Hrbac J, et al. Lipids modulate H(2)S/HS(-) induced NO release from S-nitrosoglutathione. Biochem Biophys Res Commun. 2009;390:1241–1244. doi: 10.1016/j.bbrc.2009.10.128. [DOI] [PubMed] [Google Scholar]
- Toombs CF, Insko MA, Wintner EA, Deckwerth TL, Usansky H, Jamil K, et al. Detection of exhaled hydrogen sulphide gas in healthy human volunteers during intravenous administration of sodium sulphide. Br J Clin Pharmacol. 2010;69:626–636. doi: 10.1111/j.1365-2125.2010.03636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, Vong L, McKnight W, Dicay M, Martin GR. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology. 2009;137:569–578. doi: 10.1053/j.gastro.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Wang R. The gasotransmitter role of hydrogen sulfide. Antioxid Redox Signal. 2003;5:493–501. doi: 10.1089/152308603768295249. [DOI] [PubMed] [Google Scholar]
- Wang MJ, Cai WJ, Li N, Ding YJ, Chen Y, Zhu YC. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid Redox Signal. 2010;12:1065–1077. doi: 10.1089/ars.2009.2945. [DOI] [PubMed] [Google Scholar]
- White R, Hiley CR. Hyperpolarisation of rat mesenteric endothelial cells by ATP-sensitive K(+) channel openers. Eur J Pharmacol. 2000;397:279–290. doi: 10.1016/s0014-2999(00)00271-5. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M, et al. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem Biophys Res Commun. 2006;343:303–310. doi: 10.1016/j.bbrc.2006.02.154. [DOI] [PubMed] [Google Scholar]
- Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1930–R1937. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P, et al. A monobromobimane-based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br J Pharmacol. 2010;160:941–957. doi: 10.1111/j.1476-5381.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Li M, Herman-Antosiewicz A, Antosiewicz J, Xiao H, Lew KL, et al. Diallyl trisulfide inhibits angiogenic features of human umbilical vein endothelial cells by causing Akt inactivation and down-regulation of VEGF and VEGF-R2. Nutr Cancer. 2006;55:94–107. doi: 10.1207/s15327914nc5501_12. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Yang G, Jia X, Wu L, Wang R. Activation of KATP channels by H2S in rat insulin-secreting cells and the underlying mechanisms. J Physiol. 2005;569:519–531. doi: 10.1113/jphysiol.2005.097642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong QC, Hu LF, Wang S, Huang D, Bian JS. Hydrogen sulfide interacts with nitric oxide in the heart: possible involvement of nitroxyl. Cardiovasc Res. 2010;88:482–489. doi: 10.1093/cvr/cvq248. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Feig J, Ghiu IA, Artman M, Coetzee WA. Native KATP Channels in Human Coronary Artery Endothelial Cells consist of a Heteromultimeric Complex of Kir6.1, Kir6.2, and SUR2B Subunits. J Mol Cell Cardiol. 2004;37:857–869. doi: 10.1016/j.yjmcc.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Yusof M, Kamada K, Kalogeris T, Gaskin FS, Korthuis RJ. Hydrogen sulfide triggers late-phase preconditioning in postischemic small intestine by an NO- and p38 MAPK-dependent mechanism. Am J Physiol Heart Circ Physiol. 2009;296:H868–H876. doi: 10.1152/ajpheart.01111.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachary I, Gliki G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res. 2001;49:568–581. doi: 10.1016/s0008-6363(00)00268-6. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziche M, Morbidelli L, Choudhuri R, Zhang HT, Donnini S, Granger HJ, et al. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J Clin Invest. 1997;99:2625–2634. doi: 10.1172/JCI119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuidema MY, Yang Y, Wang M, Kalogeris T, Liu Y, Meininger CJ, et al. Antecedent hydrogen sulfide elicits an anti-inflammatory phenotype in postischemic murine small intestine: role of BK channels. Am J Physiol Heart Circ Physiol. 2010;299:H1554–H1567. doi: 10.1152/ajpheart.01229.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]