

Abstract
BACKGROUND AND PURPOSE
β-Adrenoceptor stimulation causes pronounced vasodilatation associated with smooth muscle hyperpolarization. Although the hyperpolarization is known to reflect KATP channel activation, it is not known to what extent it contributes to vasodilatation.
EXPERIMENTAL APPROACH
Smooth muscle membrane potential and tension were measured simultaneously in small mesenteric arteries in a wire myograph. The spread of vasodilatation over distance was assessed in pressurized arteries following localized intraluminal perfusion of either isoprenaline, adrenaline or noradrenaline.
KEY RESULTS
Isoprenaline stimulated rapid smooth muscle relaxation associated at higher concentrations with robust hyperpolarization. Noradrenaline or adrenaline evoked a similar hyperpolarization to isoprenaline if the α1-adrenoceptor antagonist prazosin was present. With each agonist, glibenclamide blocked hyperpolarization without reducing relaxation. Focal, intraluminal application of isoprenaline, noradrenaline or adrenaline during block of α1-adrenoceptors evoked a dilatation that spread along the entire length of the isolated artery. This response was endothelium-dependent and inhibited by glibenclamide.
CONCLUSIONS AND IMPLICATIONS
Hyperpolarization is not essential for β-adrenoceptor-mediated vasodilatation. However, following focal β-adrenoceptor stimulation, this hyperpolarization underlies the ability of vasodilatation to spread along the artery wall. The consequent spread of vasodilatation is dependent upon the endothelium and likely to be of physiological relevance in the coordination of tissue blood flow.
LINKED ARTICLES
This article is part of a themed issue on Vascular Endothelium in Health and Disease. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.164.issue-3
Keywords: β-adrenoceptor, endothelium, hyperpolarization, cAMP, adrenaline, noradrenaline, isoprenaline, adenylyl cyclase, β-adrenoceptor blockers, KATP channels
Introduction
It is now nearly a century since Dale first revealed the very powerful dilator effect catecholamines can evoke in the vasculature (Dale, 1913). This effect is mediated by β-adrenoceptors, and for a long time, it was thought that β2-adrenoceptors were responsible (Lands et al., 1967). However, studies using β-adrenoceptor-selective knock-out mice in combination with wire myography enabled pharmacological studies in much smaller blood vessels and revealed that β1 receptors are the perdominant subtype in the peripheral vasculature. Furthermore, in small resistance arteries from both the mouse and the rat β-adrenoceptor vasorelaxation appears to be entirely explained by β1-adrenoceptors (Chruscinski et al., 2001; Briones et al., 2005).
As well as causing pronounced vasorelaxation, the stimulation of vascular β-adrenoceptors is also associated with smooth muscle hyperpolarization (Holman et al., 1968; Somlyo et al., 1970; Prehn and Bevan, 1983). Hyperpolarization, like vasorelaxation, follows the activation of adenylyl cyclase and as such can be mimicked with forskolin (Nakashima and Vanhoutte, 1995; Fujii et al., 1999). It appears to reflect the opening of KATP channels, as it is sensitive to block with glibenclamide but resistant to blockers for other types of K-channel (Nakashima and Vanhoutte, 1995; Fujii et al., 1999).
However, although hyperpolarization due to β-adrenoceptor activation is associated with vasorelaxation, it is unclear to what extent this activation contributes to the functional response, particularly with the physiological agonists, noradrenaline and adrenaline. Also, as previous studies linking β1-adrenoceptors to vasorelaxation in small resistance arteries have not measured membrane potential changes, it is unclear if subtypes other than the β1-adrenoceptor might contribute. Finally, hyperpolarization can spread axially through the wall of arteries to evoke distant dilatation, an effect mediated by the endothelium and shown to improve blood flow in a range of arteries. These include the rat small mesenteric artery and in response to the KATP activator, levcromakalim (Takano et al., 2004). The possibility that β-adrenoceptors activated by circulating agonists might initiate the spread of physiologically relevant dilatation was therefore investigated in pressurized mesenteric arteries.
Methods
Measurement of smooth muscle cell tension and membrane potential
Male Wistar rats (200–250 g) were killed following procedures defined by the Animals (Scientific Procedures) Act 1986, UK (Schedule 1 procedure). Segments of mesenteric artery (2 mm long) from a third-order branch (∼300 µm o.d.) were mounted in a Mulvany-Halpern myograph (model 400A, DMT Aarhus, Denmark) in Krebs solution containing (mM) NaCl, 118.0; NaCO3, 25; KCl, 3.6; MgSO4·7H2O, 1.2; KH2PO4, 1.2; glucose, 11.0; CaCl2, 2.5 and gassed with 95 % O2 and 5 % CO2 at 37°C. Following equilibration, arteries were set to a resting tension equivalent to that generated at 0.9 times the diameter of the vessel at 70 mmHg. Unless otherwise stated, only arteries with functional endothelium were used, assessed by >95% relaxation to 1 µM ACh. All experiments were performed in the presence of the nitric oxide (NO) synthase inhibitor, Nω-nitro-l-arginine methyl ester (100 µM l-NAME).
Sharp glass microelectrodes were used to measure the membrane potential of individual smooth muscle cells (filled with 2 M KCl, tip resistances 70–100 MΩ), as previously described (Garland and McPherson, 1992). Rapid negative deflections in membrane potential were observed upon cell impalement. The membrane potential was recorded via a pre-amplifier (Neurolog system, Digitimer Ltd, Welwyn Garden City, UK) linked to a MacLab data acquisition system (AD Instruments Model 4e, usually at 100 Hz). Drugs were added directly to the 10 mL bath and mixed by gassing.
In some experiments, phenylephrine (PE) was used at a concentration (c. 3 µM) that evoked a stable level of depolarization and contraction (c. 50% maximum). Simultaneous changes in membrane potential and tension were then assessed to (cumulative) agonist concentrations and inhibitors preincubated with the tissue for at least 20 min before agonist application.
Preparation of arteries for pressure myography
A segment of mesenteric artery (c. 3 mm) was cut to include a side branch and then triple cannulated with glass pipettes and positioned near the base of a 2 mL temperature-regulated chamber (Warner Instruments, Welwyn Garden City, UK). The chamber was placed on the stage of an inverted microscope (IX 70, Olympus, Cambridge, UK) with attached confocal scanning unit (FV500, Olympus, Tokyo, Japan) and continuously superfused at 2 mL·min−1 with 3-morpholinopropane-1-sulphonic acid (MOPS) buffer containing (mM) NaCl 145.0, KCl 4.7, CaCl2·2H2O 2.0, MgSO4·7H2O 1.17, Mops 2.0, NaH2PO4·H2O 1.20, glucose 5.0, pyruvate 2.0, EDTA 0.02 NaOH 2.75, adjusted to pH 7.40 ± 0.02 as previously described (Takano et al., 2004; Mather et al., 2005; Winter and Dora, 2007). In some experiments, arteries were cannulated at each end without including a side branch (data in Figure 4). After 30 min superfusion at 37°C, artery reactivity was assessed by contraction to 3 µM PE followed by endothelium-dependent relaxation to 1 µM ACh. Only vessels relaxing >95% were subsequently used.
Figure 4.
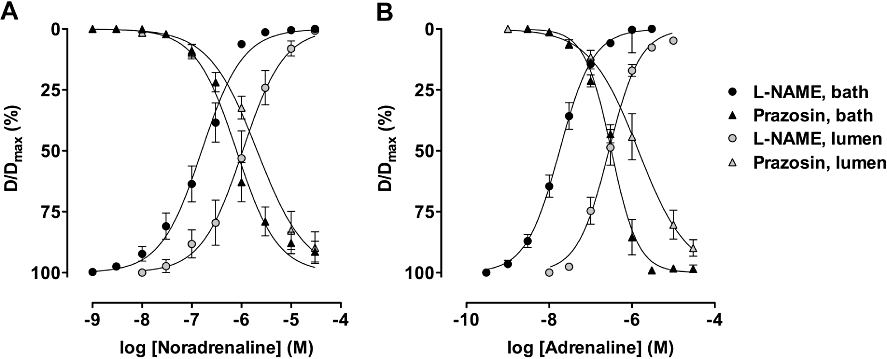
Concentration–response curves to noradrenaline or adrenaline in pressurized mesenteric arteries. To assess vasodilatation, the arteries were precontracted with U46619 in the presence of 1 µM prazosin. Noradrenaline (A) or adrenaline (B) were each applied cumulatively, either via the artery lumen (n = 6 for each agonist with or without prazosin) or directly into the tissue bath (n = 9 and 7 with and without prazosin for noradrenaline, and n = 4 and 6 for adrenaline). Both agonists were more potent when applied via the tissue bath to evoke vasoconstriction or vasorelaxation.
Luminal application of agonists
Inflow into the upstream end of the artery (feed branch) was kept constant at 1 µL·min−1 using a syringe pump system (BeeHive®, Bioanalytical Systems, West Lafayette, IN, USA). Using a second syringe pump connected to one branch of the bifurcation (branch 1), agonist solutions were perfused through the lumen of branch 1 for at least 2 min at 2 µL·min−1. Branch 2 of the bifurcation was connected to an open-ended, gravity-fed reservoir and served as the outlet for both pumped solutions. Pumping vehicle alone had no effect on arterial diameter (not shown). In all experiments, the passage of agonist-containing solution was monitored visually by including 0.1 µM carboxyfluorescein. Arteries were visualized (excitation 488 nm, emission 505 nm) to enable simultaneous fluorescence and bright-field imaging, with a 4×/0.13 NA objective (UplanFl, Olympus, Tokyo, Japan). Images were recorded with Fluoview software (Olympus, Branson, MO, USA) at 1 Hz. For all measurements of spreading dilatation, artery outer diameter was measured offline using motion analysis software (MetaMorph, Universal Imaging, West Chester, PA, USA). This enabled simultaneous analysis of multiple, calibrated distances along the artery wall and direct comparisons of local dilatation to spreading dilatation for a single application of agonist, which is not possible with higher magnification objectives. The resolution of the system was 5 µm (equivalent to one pixel), c. 1.5% of the maximum diameter of arteries. Fluorescence intensity was also measured offline simultaneously at multiple positions in the lumen of arteries, which was temporally matched to diameter measurements.
Drugs and reagents
All drugs used were purchased from Sigma Chemical Company (Poole, UK) except iberiotoxin (Latoxan, Rosans, France). Stock solutions were prepared in MilliQ water, with the exception of U46619, glibenclamide and forskolin, which were dissolved in DMSO; iberiotoxin, which was dissolved in physiological buffer; and noradrenaline and adrenaline where the stock solution included 0.1 mM ascorbic acid. The stock solutions were all 10 mM, except L-NAME (0.1 M) and iberiotoxin (50 µM). Immediately prior to use in experiments, drugs were diluted in physiological buffer, and arteries were incubated with inhibitors for a minimum of 20 min prior to stimulating responses.
Data analysis
For wire myography, results are summarized as means ± SEM of n replicates. Statistical comparisons were made using Mann–Whitney test or one-way anova with Bonferroni's post test, and P < 0.05 was taken as statistically significant. With spreading dilatation, data are summarized as means ± SEM of n arteries, one per animal. Statistical comparisons were made using the Kruskal–Wallis test followed by Dunn's multiple-comparison post test, with P < 0.05 considered statistically significant. The vasorelaxation evoked by each agonist was expressed either as the % of maximum PE contraction (wire myograph, T/Tmax), or in pressurized arteries as a % of maximum diameter (D/Dmax), 100% being the maximum diameter in each experiment. Concentration–response curves and histograms were prepared using Prism v5.0 software (GraphPad Software, San Diego, CA, USA).
Results
Simultaneous measurements of smooth muscle Em and tension
Smooth muscle resting potential of −52.9 ± 1.3 mV was reduced with PE to −38.9 ± 0.9 mV, with an associated contraction of 11.4 ± 0.6 mN (n = 18). Isoprenaline (1 nM–10 µM) then evoked concentration-dependent hyperpolarization to an Emax of −62.5 ± 2.0 mV (ΔEm 1 µM: −23.9 ± 1.9 mV, n = 15), associated with complete reversal of the PE-evoked contraction. Isoprenaline more potently evoked relaxation (pEC50 7.9 ± 0.1) than hyperpolarization (pEC50 7.1 ± 0.1, n = 18) (Figure 1). The hyperpolarization but not relaxation was inhibited by the presence of 5 µM glibenclamide (ΔEm 1 µM: −8.6 ± 1.7 mV, n = 5), while 0.1 µM iberiotoxin had no effect (ΔEm 1 µM: −28.6 ± 3.4 mV, n = 8). These data were all obtained in the presence of 100 µM L-NAME, which did not affect either the hyperpolarization or relaxation to isoprenaline, although removal of the endothelium did reduce the potency with which isoprenaline evoked relaxation (pEC50: 7.4 ± 0.1, n = 7), but not hyperpolarization (pEC50: 7.1 ± 0.1; ΔEm 1 µM: −22.9 ± 0.4 mV, n = 3). Similar hyperpolarization (against PE) followed increasing concentrations of forskolin (10 nM–10 µM), to an Emax of −60.5 ± 2.9 mV (ΔEm 10 µM: −21.9 ± 3.8 mV, n = 5). This hyperpolarization was effectively abolished in the presence of glibenclamide (ΔEm 10 µM: −2.2 ± 0.7 mV, n = 3).
Figure 1.
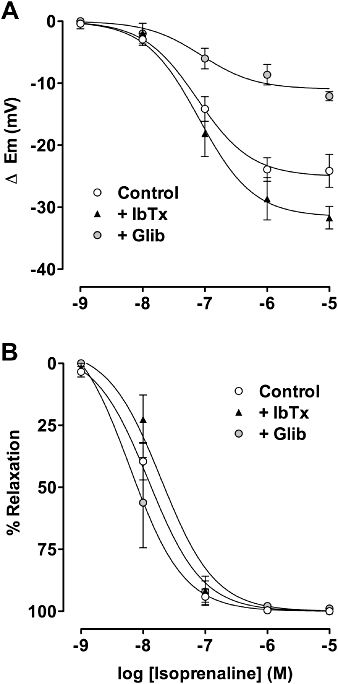
Concentration-response curves to isoprenaline in mesenteric arteries precontracted with phenylephrine. Simultaneous measurement of smooth muscle membrane potential (A) and tension (B) in control arteries (n = 18) and arteries exposed to either 0.1 µM iberiotoxin or 5 µM glibenclamide. The NO-synthase inhibitor 100 µM L-NAME was present in all experiments. NO, nitric oxide.
Effect of adrenaline and noradrenaline on Em and tension
Noradrenaline (10 nM–3 µM) progressively depolarized mesenteric smooth muscle cells to a maximum of 17.7 ± 2.6 mV (pEC50 6.6 ± 0.2, n = 8), from a resting potential of −54.2 ± 2.6 mV. This depolarization was associated with maximum contraction of 12.1 ± 1.1 mN, with qualitatively similar data obtained using adrenaline (pEC50 7.0 ± 0.1; n = 4) (Figure 2A, B, D, E). Both the depolarization and the associated contraction were blocked in the presence of the α1-adrenoceptor antagonist prazosin (1 µM). Noradrenaline or adrenaline now evoked a robust hyperpolarization (maximum of 26.9 ± 1.2 mV and 25.3 ± 3.0 mV, n = 4 and 5 respectively), with pEC50 values of 6.0 ± 0.1 and 6.1 ± 0.1. In both cases, hyperpolarization was abolished in the presence of 5 µM glibenclamide. In the presence of ongoing contraction to U46619, both catecholamines also stimulated smooth muscle relaxation (Figure 2C, F). In this series, the pEC50 values for noradrenaline and adrenaline were 6.2 ± 0.1 and 6.6 ± 0.04 respectively. In contrast to hyperpolarization, relaxation was not modified by glibenclamide.
Figure 2.
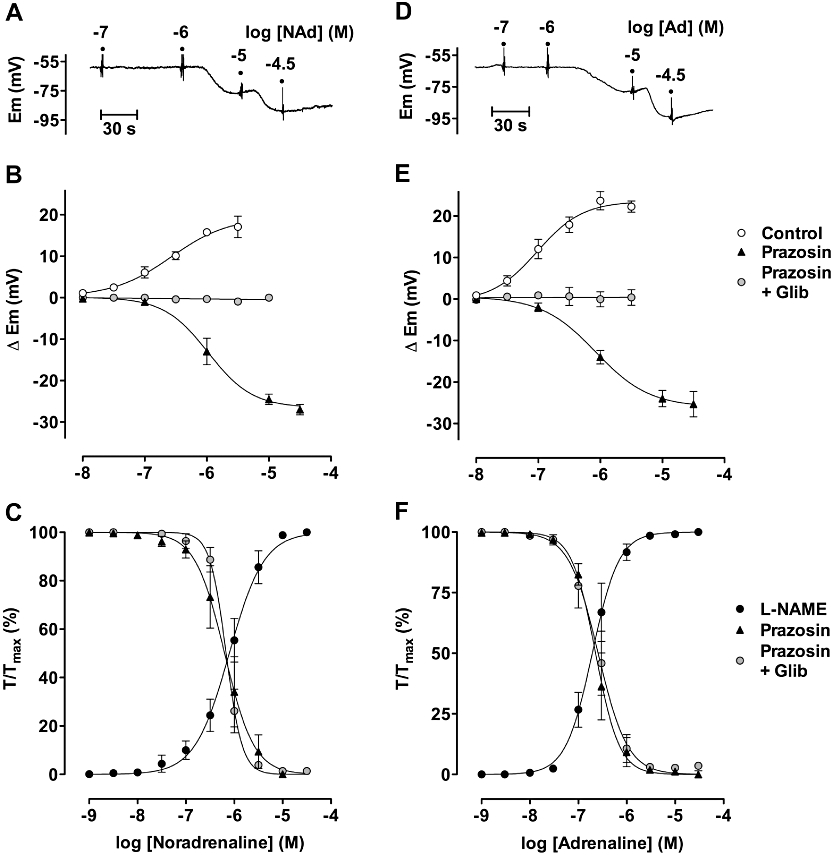
Concentration–responses of mesenteric arteries to noradrenaline (A–C) and adrenaline (D–F). Panels A and D show hyperpolarization recorded in the presence of 1 µM prazosin. These data are summarized in panels B and E (n = 4–11 in each case), which also show the mean depolarization (n = 11) obtained in the absence of prazosin and block of hyperpolarization with 5 µM glibenclamide (in the presence of 1 µM prazosin, n = 7 in each case). Panels C and F summarize contraction and relaxation to noradrenaline or adrenaline (n = 14 and 6 respectively). Contraction was blocked in the presence of 1 µM prazosin when, in the presence of contraction to U46619, each catecholamine stimulated relaxation that was insensitive to 5 µM glibenclamide across the entire range of concentrations (n = 6 in each case).
Effect of other adrenoceptor agonists and some antagonists on Em
Dobutamine (β1 selective agonist) or salbutamol (β2 selective agonist) each hyperpolarized the resting membrane potential ( Figure 3A). The Emax in each case was 10.7 ± 1.6 mV (from a r.m.p. of −52.6 ± 0.7 mV, n = 13) and 14.1 ± 1.5 mV (from −55.9 ± 1.1 mV, n = 7). The β3-ligand, BRL 37344, in concentrations up to 30 µM did not alter membrane potential, apart from a few mV depolarization (n = 7).
Figure 3.
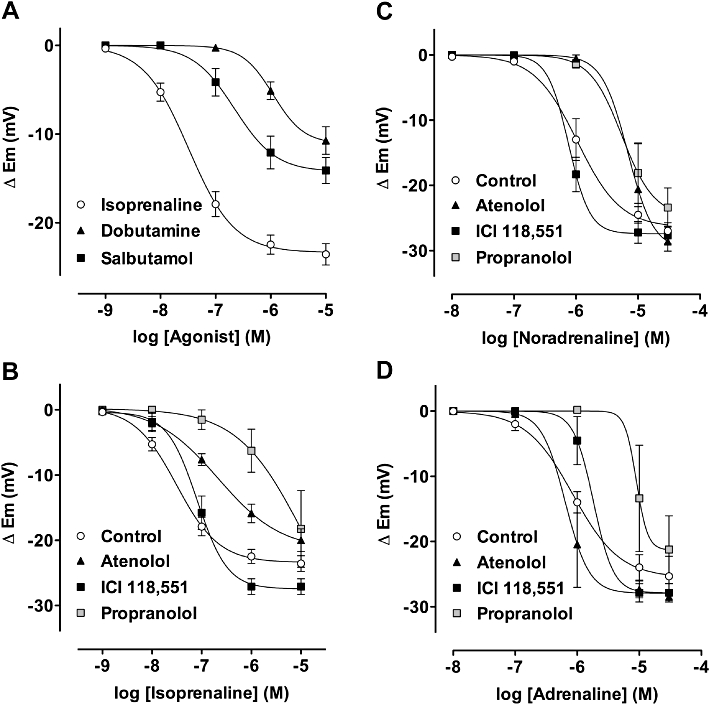
Summarized data showing the average change in smooth muscle membrane potential to β-adrenoceptor agonists and to agonists with various antagonists present. (A) Hyperpolarization to increasing concentrations of isoprenaline (n = 23), dobutamine and salbutamol (n = 13 and 7 respectively). (B) Hyperpolarization to increasing concentrations of isoprenaline in the presence of 1 µM atenolol (n = 3, P < 0.001), 100 nM ICI 118,551 (n = 4, N.S.) or 100 nM propranolol (n = 4, P < 0.001) (C) to noradrenaline in the presence of either 1 µM atenolol (n = 3, P < 0.001), 100 nM ICI 118,551 (n = 6, N.S.) or 100 nM propranolol (n = 6, P < 0.001) (D) to adrenaline in the presence of either 1 µM atenolol (n = 4, N.S.), 100 nM ICI 118,551 (n = 4, N.S.) or 100 nM propranolol (n = 4, P < 0.001). N.S., not significant.
Data with propranolol (non-selective) and β1 (atenolol) and β2 (ICI 118 551) selective adrenoceptor antagonists are also shown in Figure 3B, C and D. Atenolol 1 µM and 100 nM propranolol right-shifted the concentration–response curve for hyperpolarization to isoprenaline and noradrenaline (isoprenaline pEC50 7.5 ± 0.1, shifted to 6.7 ± 0.2 and 5.3 ± 0.8, respectively, n = 4; noradrenaline pEC50 6.0, shifted to 5.2 ± 0.2 and 5.3 ± 0.2, respectively, n = 6). With adrenaline, only propranolol was effective (pEC50 6.1, shifted to 5.0, n = 4). However, the combined presence of 1 µM atenolol and 100 nM ICI 118 551 did shift the hyperpolarization curve to the right (not shown, pEC50c. 5.5, n = 3).
Spreading vasodilatation to adrenoceptor agonists in pressurized mesenteric arteries
Noradrenaline or adrenaline evoked either contraction or relaxation, whether applied into the lumen of pressurized mesenteric arteries or directly to the tissue bath (see Figure 4). To evoke relaxation, the agonists were applied cumulatively to arteries submaximally contracted with U46619 and in the presence of 1 µM prazosin. From these curves, a submaximal (for vasodilatation) concentration of adrenaline and noradrenaline (1 µM c. 50% dilatation) was selected for subsequent experiments in triple-cannulated (allowing intraluminal local application of agonists) and pressurized, mesenteric arteries (see Figure 5A). Submaximal contraction to U46619 and the subsequent local infusion of 1 µM isoprenaline, noradrenaline or adrenaline evoked a marked local dilatation (in branch 1) followed by spread of dilatation upstream and against the direction of the intraluminal perfusate. Significant dilatation was observed along the entire length of the isolated artery, which at 2.5 mm upstream represented 36.5 ± 5.2% (n = 8), 27.2 ± 4.6% (n = 7) and 19.3 ± 7.7% (n = 7) for each of the catecholamines (Figure 5B–D), respectively. In each case, dilatation did not spread from the local site by more than 0.5 mm (1 mm in the case of adrenaline) in the presence of 10 µM glibenclamide (n = 5 in each case). Furthermore, destroying the endothelium by infusing an air bubble through the artery (but bypassing branch 1) blocked the spreading but not the local dilatation (in branch 1) to isoprenaline (n = 4). Local infusion of 1 µM ACh into branch 1 also evoked a robust spreading dilatation (around 20% at 2.5 mm upstream) that was unaffected by the presence of glibenclamide (not shown).
Figure 5.
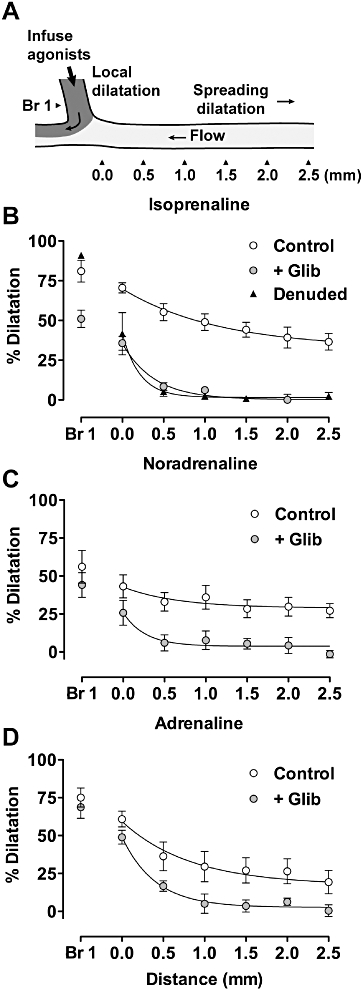
Spreading vasodilatation to catecholamines in triple-cannulated, pressurized mesenteric arteries partially contracted with U46619 and with 1 µM prazosin present. (A) Each agonist was infused into branch 1 (branch 1, tracked with carboxyfluorescein) during established flow in the artery lumen. Dilatation was then measured at defined points along the pressurized artery as indicated (top panel). (B–D) Summarized data showing average dilatation to 1 µM isoprenaline (B, n = 8), 1 µM noradrenaline (C, n = 7) 1 µM or adrenaline (D, n = 5). In each case, dilatation was abolished with 10 µM glibenclamide (n = 5, B–D) and in addition, with isoprenaline, after removal of the endothelium (B, n = 4).
Discussion
Overall, there are two key findings in the present study. First, in spite of the fact that catecholamines can evoke a robust smooth muscle hyperpolarization through β-adrenoceptor stimulation, this response is not a prerequisite for vasorelaxation. Second, β-mediated hyperpolarization evoked by the localized application of catecholamines can initiate vasodilatation that then spreads along the artery wall to distant sites not exposed to β-adrenoceptor agonists.
β-Mediated smooth muscle hyperpolarization is not the primary cause of vasodilatation
Although it has been clear for some time that stimulation of vascular β-adrenoceptors causes smooth muscle hyperpolarization, it was not clear until now that the hyperpolarization per se is not important for the associated relaxation. In rat small mesenteric arteries, hyperpolarization can cause complete vasorelaxation, so as such β-adrenoceptor-linked hyperpolarization might potentially be a very important means to ensure vasodilatation (Waldron and Garland, 1994). The lack of clarity regarding the importance of β-adrenoceptor-linked hyperpolarization for vasorelaxation reflects the fact that previous studies have measured changes in membrane potential or tension independently, and that quite apart from evoking hyperpolarization β-adrenoceptors also link to activate multiple cellular pathways leading to vasorelaxation. What is clear is that β-adrenoceptor-evoked hyperpolarization can be inhibited by glibenclamide, indicating a central role for KATP channels (Nakashima and Vanhoutte, 1995; Fujii et al., 1999). However, when assessed against dilatation/relaxation in different vascular preparations, the effects of glibenclamide are quite variable. In arterioles, glibenclamide increased myogenic tone and partially inhibited dilatation to either isoprenaline or forskolin (Jackson, 1993). Similarly, in cerebral arteries, vasodilatation evoked by noradrenaline acting on β1-adrenoceptors, or by forskolin, was only partially inhibited (Kitazono et al., 1993). While in the perfused superior mesenteric vascular bed, dilatation appeared to reflect the activity of both β1- and β2-adrenoceptors and was only slightly reduced by glibenclamide (Randall and McCulloch, 1995). Glibenclamide did not, however, alter vasorelaxation to isoprenaline in isolated mesenteric arteries (White et al., 2001). Our data confirm and extend significantly the latter. Measuring membrane potential and tension simultaneously, they show that glibenclamide effectively abolished hyperpolarization, while at the same time and under the same experimental conditions, vasorelaxation was unaffected. Thus, hyperpolarization evoked by β-adrenoceptors is not essential for the vasorelaxation, although, if all other routes to relaxation through cAMP were somehow to be blocked, presumably hyperpolarization would then be able to support relaxation.
Routes to vasodilatation via β-adrenoceptors
β-Adrenoceptors are Gs coupled and as such stimulate an increase in cAMP and the activation of protein kinase A (PKA). One consequence is the activation of KATP channels, resulting from PKA phosphorylation of sites in both the pore-forming and regulatory subunits of the channel (Quinn et al., 2004; Shi et al., 2007; Yang et al., 2008). This regulation depends on the colocalization within caveolae of KATP channels with adenylyl cyclase and alongside other key signalling proteins, such as PKA tethered by A-kinase anchoring proteins (Feliciello et al., 2001; Sampson et al., 2004). Since endogenous dilators such as CGRP and adenosine also activate KATP channels via Gs/PKA (Quayle et al., 1994; Kleppisch and Nelson, 1995; Wellman et al., 1998), it has been suggested that a key physiological control mechanism for regulating vascular tone with both vasodilators and vasoconstrictors is the targeted modulation of the basal activity of KATP channels (Quayle et al., 1997; Sampson et al., 2007). However, KATP channels are only one cellular target for PKA. Smooth muscle relaxation reflects changes in both calcium handling and sensitivity, mechanisms that are clearly able to underpin relaxation to catecholamines effectively in the mesenteric artery, without reliance on changes in KATP activity. cAMP also directly activates exchange proteins (Epac), which also have a range of cellular effects, some of which may influence tone. In rat aortic smooth muscle cells, Epac links to regulate KATP channel activity. However, this pathway, which appears to involve the activation of calcineurin, serves to inhibit rather than increase KATP activity (Purves et al., 2009). So whether or not Epac is active and has any physiologically relevant influence during stimulation of β-adrenoceptors in smooth muscle from small resistance arteries remains to be seen.
β-Adrenoceptors initiate spreading or conducted vasodilatation
What is clear from the present study is that β-mediated hyperpolarization can initiate spreading dilatation. In general, agonists that evoke hyperpolarization in arteries or arterioles cause dilatation that spreads over a considerable distance, particularly in the microcirculation. This is the case whether the hyperpolarization is initiated in the endothelial or the smooth muscle cells, and spread can occur in either direction along the vessel wall (Dora, 2010). Physiologically, upstream vasodilatation is extremely important in order significantly to increase local tissue blood flow. This reflects the fact that vascular resistance is a function of both vessel diameter and length. Spreading dilatation appears to be due to current spread from the initiating region to adjacent cells and may involve amplification to sustain spread (Emerson and Segal, 2000; 2001; Emerson et al., 2002). As such, the cellular mechanism(s) responsible for spread can be, and most likely is, distinct from that responsible for the local ‘triggering’ hyperpolarization. For example, in mesenteric arteries, spreading dilatation can be evoked by the local activation of KCa channels, yet spread of dilatation along the endothelium is not associated with a measurable increase in endothelial cell calcium (Takano et al., 2004).
Conduction along the vessel wall is sustained by the endothelium. The cells in the endothelium are linked together by large gap junctions and aligned longitudinally, so each one traverses many smooth muscle cells. This arrangement clearly facilitates conduction and the ensuing vasodilatation (Dora, 2010; Garland et al., 2011). Small mesenteric arteries display spreading vasodilatation that also depends on an extant endothelium. In these vessels, spreading dilatation can be evoked with hyperpolarizing agonists that include the KATP activator, levcromakalim (Takano et al., 2004; Winter and Dora, 2007). KATP channels are found on the smooth muscle not the endothelial cells in the mesenteric artery, so the hyperpolarization evoked by levcromakalim passes to the endothelium via myoendothelial gap junctions, before spreading longitudinally (Takano et al., 2004). The fact that we now show catecholamines to cause spreading dilatation by an action on β-adrenoceptors suggests a physiologically relevant role for this pathway. Interestingly, in humans, β-adrenoceptor stimulation in vivo augments hyperaemic blood flow and improves vasodilatation in the forearm after transient ischaemic occlusion (Delehanty et al., 1999). In other species, reactive hyperaemia in the hindlimb vascular bed in part involves the opening of vascular KATP channels, which may well implicate a similar mechanism (Minkes et al., 1995). On the other hand, if catecholamines do have a significant role controlling local blood flow via β-adrenoceptor-mediated vascular hyperpolarization, this mechanism may be disrupted in patients taking β-blockers, particularly as the receptor responsible seems to be the β1-adrenoceptor.
One drawback to the suggestion that catecholamines may induce significant spread of dilatation in vivo is the fact these agents also stimulate α1-adrenoceptors leading to smooth muscle depolarization and contraction. In the present study, we routinely blocked these receptors, so while our data may help in part to explain the antihypertensive action of α1-adrenoceptor antagonists such as prazosin, how relevant will they be when these agents are not present? Interestingly, in preliminary experiments, we have also observed spreading dilatation in the absence of prazosin. Adrenaline (1 µM) caused local constriction when infused into branch 1, but this was followed by emerging dilatation of c. 25% next to the side branch (equivalent to 0 mm in Figure 5) and sustained at about 16% by 2 mm upstream in arteries partially constricted with U46619. The dilatation was blocked by propranolol. One possible explanation is that while branch 1 develops an α1-mediated vasoconstriction to adrenaline, the simultaneous activation of β-adrenoceptors generates sufficient cAMP to activate KATP channels in adjacent cells affecting hyperpolarization that then spreads along the endothelium. The precise mechanism responsible is, however, the subject of ongoing studies.
β-Adrenoceptor subtypes and vasodilatation
Finally, as regards the β-adrenoceptor responsible for the action of catecholamines in rat small mesenteric arteries, previous studies have not assessed this with regard to membrane hyperpolarization. However, Briones et al., (2005) have provided a very comprehensive study of β-adrenoceptor relaxation in the mesenteric artery, using a range of agonists and antagonists and complemented by direct visualization of the receptors. β-Adrenoceptors are mainly but not exclusively localized to the smooth muscle layers, and the agonist/antagonist profile suggests vasorelaxation is due to activation of β1-adrenoceptors. Our hyperpolarization data are consistent with this work, as they indicate a predominant route to hyperpolarization through β1-adrenoceptors. However, they also raise the possibility that β2-adrenoceptors may be involved. The β2-adrenoceptor agonist, salbutamol, evoked hyperpolarization with a pEC50 of 6.6 (and threshold of 0.1 µM). Although this value is slightly higher than expected for an action through β2-adrenoceptors (pEC50 usually between 7 and 8), it was lower than the pEC50 of 5.5 for vasorelaxation (Briones et al., 2005). In addition, the β2-adrenoceptor antagonist ICI 118,551 right-shifted the concentration–response curve for hyperpolarization to adrenaline, but only if atenolol was also present. This could be explained by a parallel effect of adrenaline on both β1- and β2-adrenoceptors, each leading to hyperpolarization in the mesenteric artery. However, if this is the case, the effect through β2-adrenoceptors is certainly inferior to β1-receptors. β3-Adrenoceptors do not appear to be present or functional in the mesenteric artery, as the β3-adrenoceptor agonist BRL37344 did not stimulate hyperpolarization. This conclusion is supported by the lack of any β3-adrenoceptor vasorelaxation in the study by Briones et al., (2005). Finally, it has been suggested that endothelial cell β1-adrenoceptors contribute significantly to vasorelaxation by mediating the release of NO (Graves and Poston, 1993). However, the NO-synthase inhibitor L-NAME did not affect either hyperpolarization or vasorelaxation, although physical destruction of the endothelium attenuated the latter. A similar profile was previously reported in the mesenteric artery with a range of vasodilators, in addition to β-adrenoceptor agonists by Briones et al., (2005), who concluded that destruction of the endothelium removed a constitutive dilator influence, rather than a specific link between β-adrenoceptors and the generation of NO.
So in summary, we have shown for the first time that the robust vascular hyperpolarization that follows the stimulation of β-adrenoceptors is not necessary for vasorelaxation. This functional effect can be sustained by other targets of the cAMP signalling pathway operating in parallel. However, localized intraluminal application of catecholamines does cause widespread vascular dilatation that is initiated by β-adrenoceptor-linked hyperpolarization and reflects activation of KATP channels. This spreading or conducted vasodilatation appears to pass along the artery wall by way of the endothelium and may represent an important physiological mechanism for regulating blood flow locally.
Acknowledgments
This research was supported by a Programme Grant from the Wellcome Trust. KAD is a BHF Senior Basic Science Research Fellow, CJG holds a Royal Society-Wolfson Research Merit Award and FJ-A was supported by the Ministerio de Educación y Ciencia. PY was supported by an ORS award.
Glossary
Abbreviations
- BRL 37344
(±)-(R*,R*)-[4-[2-[[2-(3-chlorophenyl)-2-hydroxyethyl]amino]propyl]phenoxy]acetic acid sodium hydrate
- ICI 118,551
(±)-erythro-(S*,S*)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol hydrochloride
- KATP
ATP-sensitive K+-channels
- l-NAME
Nω-nitro-l-arginine methyl ester
- MOPS
3-morpholinopropane-1-sulphonic acid
- PE
phenylephrine
- U46619
(Z)-7-[(1S,4R,5R,6S)-5-[(E,3S)-3-hydroxyoct-1-enyl]-3-oxabicyclo[2.2.1]heptan-6-yl]hept-5-enoic acid
Conflict of interest
The authors have no conflict of interest.
References
- Briones AM, Daly CJ, Jiménez-Altayó F, Martinez-Revelles S, Gonzalez JM, McGrath JC, et al. Direct demonstration of beta1- and evidence against beta2- and beta3-adrenoceptors, in smooth muscle cells of rat small mesenteric arteries. Br J Pharmacol. 2005;146:679–691. doi: 10.1038/sj.bjp.0706369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chruscinski A, Brede ME, Meinel L, Lohse MJ, Kobilka BK, Hein L. Differential distribution of beta-adrenergic receptor subtypes in blood vessels of knockout mice lacking beta(1)- or beta(2)-adrenergic receptors. Mol Pharmacol. 2001;60:955–962. doi: 10.1124/mol.60.5.955. [DOI] [PubMed] [Google Scholar]
- Dale HH. On the action of ergotoxine; with special reference to the existence of sympathetic vasodilators. J Physiol. 1913;46:291–300. doi: 10.1113/jphysiol.1913.sp001592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delehanty J, Liang CS, Odorisi ML. Effects of dobutamine on ischemic vasodilation of the forearm in patients with severe congestive heart failure. J Card Fail. 1999;5:25–30. doi: 10.1016/s1071-9164(99)90021-0. [DOI] [PubMed] [Google Scholar]
- Dora KA. Coordination of vasomotor responses by the endothelium. Circ J. 2010;74:226–232. doi: 10.1253/circj.cj-09-0879. [DOI] [PubMed] [Google Scholar]
- Emerson GG, Segal SS. Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ Res. 2000;86:94–100. doi: 10.1161/01.res.86.1.94. [DOI] [PubMed] [Google Scholar]
- Emerson GG, Segal SS. Electrical activation of endothelium evokes vasodilation and hyperpolarization along hamster feed arteries. Am J Physiol Heart Circ Physiol. 2001;280:H160–H167. doi: 10.1152/ajpheart.2001.280.1.H160. [DOI] [PubMed] [Google Scholar]
- Emerson GG, Neild TO, Segal SS. Conduction of hyperpolarization along hamster feed arteries: augmentation by acetylcholine. Am J Physiol Heart Circ Physiol. 2002;283:H102–H109. doi: 10.1152/ajpheart.00038.2002. [DOI] [PubMed] [Google Scholar]
- Feliciello A, Gottesman ME, Avvedimento EV. The biological functions of A-kinase anchor proteins. J Mol Biol. 2001;308:99–114. doi: 10.1006/jmbi.2001.4585. [DOI] [PubMed] [Google Scholar]
- Fujii K, Onaka U, Goto K, Abe I, Fujishima M. Impaired isoproterenol-induced hyperpolarization in isolated mesenteric arteries of aged rats. Hypertension. 1999;34:222–228. doi: 10.1161/01.hyp.34.2.222. [DOI] [PubMed] [Google Scholar]
- Garland CJ, McPherson GA. Evidence that nitric oxide does not mediate the hyperpolarization and relaxation to acetylcholine in the rat small mesenteric artery. Br J Pharmacol. 1992;105:429–435. doi: 10.1111/j.1476-5381.1992.tb14270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland CJ, Hiley CR, Dora KA. EDHF: spreading the influence of the endothelium. Br J Pharmacol. 2011;164:839–852. doi: 10.1111/j.1476-5381.2010.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves J, Poston L. Beta-adrenoceptor agonist mediated relaxation of rat isolated resistance arteries: a role for the endothelium and nitric oxide. Br J Pharmacol. 1993;108:631–637. doi: 10.1111/j.1476-5381.1993.tb12853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman ME, Kasby CB, Suthers MB, Wilson JA. Some properties of the smooth muscle of rabbit portal vein. J Physiol. 1968;196:111–132. doi: 10.1113/jphysiol.1968.sp008498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson WF. Arteriolar tone is determined by activity of ATP-sensitive potassium channels. Am J Physiol. 1993;265:H1797–H1803. doi: 10.1152/ajpheart.1993.265.5.H1797. [DOI] [PubMed] [Google Scholar]
- Kitazono T, Faraci FM, Heistad DD. Effect of norepinephrine on rat basilar artery in vivo. Am J Physiol. 1993;264:H178–H182. doi: 10.1152/ajpheart.1993.264.1.H178. [DOI] [PubMed] [Google Scholar]
- Kleppisch T, Nelson MT. Adenosine activates ATP-sensitive potassium channels in arterial myocytes via A2 receptors and cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1995;92:12441–12445. doi: 10.1073/pnas.92.26.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lands AM, Arnold A, McAuliff JP, Luduena FP, Brown TG., Jr Differentiation of receptor systems activated by sympathomimetic amines. Nature. 1967;214:597–598. doi: 10.1038/214597a0. [DOI] [PubMed] [Google Scholar]
- Mather S, Dora KA, Sandow SL, Winter P, Garland CJ. Rapid endothelial cell-selective loading of connexin 40 antibody blocks endothelium-derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ Res. 2005;97:399–407. doi: 10.1161/01.RES.0000178008.46759.d0. [DOI] [PubMed] [Google Scholar]
- Minkes RK, Santiago JA, McMahon TJ, Kadowitz PJ. Role of K+ATP channels and EDRF in reactive hyperemia in the hindquarters vascular bed of cats. Am J Physiol. 1995;269:H1704–H1712. doi: 10.1152/ajpheart.1995.269.5.H1704. [DOI] [PubMed] [Google Scholar]
- Nakashima M, Vanhoutte PM. Isoproterenol causes hyperpolarization through opening of ATP-sensitive potassium channels in vascular smooth muscle of the canine saphenous vein. J Pharmacol Exp Ther. 1995;272:379–384. [PubMed] [Google Scholar]
- Prehn JL, Bevan JA. Facial vein of the rabbit. Intracellularly recorded hyperpolarization of smooth muscle cells induced by beta-adrenergic receptor stimulation. Circ Res. 1983;52:465–470. doi: 10.1161/01.res.52.4.465. [DOI] [PubMed] [Google Scholar]
- Purves GI, Kamishima T, Davies LM, Quayle JM, Dart C. Exchange protein activated by cAMP (Epac) mediates cAMP-dependent but protein kinase A-insensitive modulation of vascular ATP-sensitive potassium channels. J Physiol. 2009;587:3639–3650. doi: 10.1113/jphysiol.2009.173534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quayle JM, Bonev AD, Brayden JE, Nelson MT. Calcitonin gene-related peptide activated ATP-sensitive K+ currents in rabbit arterial smooth muscle via protein kinase A. J Physiol. 1994;475:9–13. doi: 10.1113/jphysiol.1994.sp020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- Quinn KV, Giblin JP, Tinker A. Multisite phosphorylation mechanism for protein kinase A activation of the smooth muscle ATP-sensitive K+ channel. Circ Res. 2004;94:1359–1366. doi: 10.1161/01.RES.0000128513.34817.c4. [DOI] [PubMed] [Google Scholar]
- Randall MD, McCulloch AI. The involvement of ATP-sensitive potassium channels in beta-adrenoceptor-mediated vasorelaxation in the rat isolated mesenteric arterial bed. Br J Pharmacol. 1995;115:607–612. doi: 10.1111/j.1476-5381.1995.tb14975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson LJ, Hayabuchi Y, Standen NB, Dart C. Caveolae localize protein kinase A signaling to arterial ATP-sensitive potassium channels. Circ Res. 2004;95:1012–1018. doi: 10.1161/01.RES.0000148634.47095.ab. [DOI] [PubMed] [Google Scholar]
- Sampson LJ, Davies LM, Barrett-Jolley R, Standen NB, Dart C. Angiotensin II-activated protein kinase C targets caveolae to inhibit aortic ATP-sensitive potassium channels. Cardiovasc Res. 2007;76:61–70. doi: 10.1016/j.cardiores.2007.05.020. [DOI] [PubMed] [Google Scholar]
- Shi Y, Wu Z, Cui N, Shi W, Yang Y, Zhang X, et al. PKA phosphorylation of SUR2B subunit underscores vascular KATP channel activation by beta-adrenergic receptors. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1205–R1214. doi: 10.1152/ajpregu.00337.2007.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AV, Haeusler G, Somlyo AP. Cyclic adenosine monophosphate: potassium-dependent action on vascular smooth muscle membrane potential. Science. 1970;169:490–491. doi: 10.1126/science.169.3944.490. [DOI] [PubMed] [Google Scholar]
- Takano H, Dora KA, Spitaler MM, Garland CJ. Spreading dilatation in rat mesenteric arteries associated with calcium-independent endothelial cell hyperpolarization. J Physiol. 2004;556:887–903. doi: 10.1113/jphysiol.2003.060343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron GJ, Garland CJ. Contribution of both nitric oxide and a change in membrane potential to acetylcholine-induced relaxation in the rat small mesenteric artery. Br J Pharmacol. 1994;112:831–836. doi: 10.1111/j.1476-5381.1994.tb13154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman GC, Quayle JM, Standen NB. ATP-sensitive K+ channel activation by calcitonin gene-related peptide and protein kinase A in pig coronary arterial smooth muscle. J Physiol. 1998;507:117–129. doi: 10.1111/j.1469-7793.1998.117bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R, Bottrill FE, Siau D, Hiley CR. Protein kinase A-dependent and -independent effects of isoproterenol in rat isolated mesenteric artery: interactions with levcromakalim. J Pharmacol Exp Ther. 2001;298:917–924. [PubMed] [Google Scholar]
- Winter P, Dora KA. Spreading dilatation to luminal perfusion of ATP and UTP in rat isolated small mesenteric arteries. J Physiol. 2007;582:335–347. doi: 10.1113/jphysiol.2007.135202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shi Y, Guo S, Zhang S, Cui N, Shi W, et al. PKA-dependent activation of the vascular smooth muscle isoform of KATP channels by vasoactive intestinal polypeptide and its effect on relaxation of the mesenteric resistance artery. Biochim Biophys Acta. 2008;1778:88–96. doi: 10.1016/j.bbamem.2007.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]