

Abstract
BACKGROUND AND PURPOSE
Controlling vascular tone involves K+ efflux through endothelial cell small- and intermediate-conductance calcium-activated potassium channels (KCa2.3 and KCa3.1, respectively). We investigated the expression of these channels in astrocytes and the possibility that, by a similar mechanism, they might contribute to neurovascular coupling.
EXPERIMENTAL APPROACH
Transgenic mice expressing enhanced green fluorescent protein (eGFP) in astrocytes were used to assess KCa2.3 and KCa3.1 expression by immunohistochemistry and RT-PCR. KCa currents in eGFP-positive astrocytes were determined in situ using whole-cell patch clamp electrophysiology. The contribution of KCa3.1 to neurovascular coupling was investigated in pharmacological experiments using electrical field stimulation (EFS) to evoke parenchymal arteriole dilatation in FVB/NJ mouse brain slices and whisker stimulation to evoke changes in cerebral blood flow in vivo, measured by laser Doppler flowmetry.
KEY RESULTS
KCa3.1 immunoreactivity was restricted to astrocyte processes and endfeet and RT-PCR confirmed astrocytic KCa2.3 and KCa3.1 mRNA expression. With 200 nM [Ca2+]i, the KCa2.1-2.3/KCa3.1 opener NS309 increased whole-cell currents. CyPPA, a KCa2.2/KCa2.3 opener, was without effect. With 1 µM [Ca2+]i, the KCa3.1 inhibitor TRAM-34 reduced currents whereas apamin (KCa2.1-2.3 blocker) had no effect. CyPPA also inhibited currents evoked by NS309 in HEK293 cells expressing KCa3.1. EFS-evoked Fluo-4 fluorescence confirmed astrocyte endfoot recruitment into neurovascular coupling. TRAM-34 inhibited EFS-evoked arteriolar dilatation by 50% whereas charybdotoxin, a blocker of KCa3.1 and the large-conductance KCa channel, KCa1.1, inhibited dilatation by 82%. TRAM-34 reduced the cortical hyperaemic response to whisker stimulation by 40%.
CONCLUSION AND IMPLICATIONS
Astrocytes express functional KCa3.1 channels, and these contribute to neurovascular coupling.
LINKED ARTICLES
This article is part of a themed issue on Vascular Endothelium in Health and Disease. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.164.issue-3
Keywords: astrocyte, brain slice, KCa2.3, KCa3.1, electrical field stimulation, laser Doppler flowmetry, neurovascular coupling, CyPPA, NS309
Introduction
Astrocytes are uniquely positioned to act as signalling intermediates between active neurones and local intracerebral arterioles: astrocyte endfoot processes ensheath intracerebral blood vessels (Simard et al., 2003), while at the same time, other processes from the same astrocytes interact with local synapses (Kang et al., 1998; Ventura and Harris, 1999). This structural arrangement led to the speculation, and subsequent demonstration, that astrocytes detect neuronal activity and match this to local arteriolar diameter changes, thus enabling increased blood flow to the active brain region to satisfy additional oxygen and nutrient demand (a process termed neurovascular coupling; Zonta et al., 2003). Indeed, it is now known that astrocytes respond to neuronal stimulation with increased [Ca2+]i (Porter and McCarthy, 1996; Kang et al., 1998; Zonta et al., 2003), which then stimulates the liberation of a number of vasoactive mediators including prostaglandins, epoxyeicosatrienoic acids (EETs) and K+ ions (see Koehler et al., 2009 for a recent review).
Many similarities exist between astrocytic and endothelial cell regulation of vascular tone. Indeed, the vasomodulatory factors described above are also involved in the endothelial regulation of vascular diameter in small arteries and arterioles of the periphery (Félétou and Vanhoutte, 2006). Of particular interest to the present study, vasomodulatory K+ signalling between the endothelial cells and myocytes of small arteries is mediated by the activation of endothelial small- and intermediate-conductance calcium-activated potassium channels (KCa2.3 and KCa3.1 respectively; Burnham et al., 2002; Bychkov et al., 2002; Taylor et al., 2003; Wölfle et al., 2009) and K+ released through these channels accumulates in the myo-endothelial space, causing an increase in the extracellular K+ concentration (Edwards et al., 1998). This increased extracellular K+ can dilate the arteriole by activating myocyte Type 2 and/or Type 3 Na+/K+ ATPases and KIR2.1 channels, which results in hyperpolarization of the myocyte and leads to smooth muscle relaxation (Quayle et al., 1993; Knot et al., 1996; Bradley et al., 1999; Edwards et al., 1999; Zaritsky et al., 2000; Weston et al., 2002).
In astrocytes it is known that large-conductance calcium-activated potassium channels (KCa1.1), which are present on astrocyte endfeet contacting intracerebral blood vessels (Price et al., 2002), mediate a portion of K+-induced dilatation of parenchymal arterioles by increasing extracellular K+ leading to activation of myocyte KIR channels (Filosa et al., 2006; Girouard et al., 2010). However, at present little is known of the expression of other KCa channels in astrocytes. Armstrong et al. (2005) identified the KCa2.3 protein in astrocytes of the rat supraoptic nucleus, but it is not known whether this channel is functional at the level of the cell membrane. Furthermore, there are no data on the expression of KCa3.1 in astrocytes. Therefore, the aim of the present study was to investigate the functional expression of KCa2.3 and KCa3.1 channels in astrocytes and, if present, to assess whether they contribute to neurovascular coupling. This report presents novel immunohistochemical, molecular and electrophysiological evidence in favour of the functional expression of KCa3.1 but not KCa2.3 channels in astrocytes in the mouse brain, and provides evidence supporting the involvement of KCa3.1 in neurovascular coupling.
Methods
Mice
Male and female mice, genetically modified to express enhanced green fluorescent protein (eGFP) in astrocytes and belonging to the strain TgN(GFAP-EGFP)GFEC-Fki (herein referred to as eGFP mice; Nolte et al., 2001), and male FVB/NJ mice (background strain of eGFP mice) and C57Bl/6 mice were housed on a 12 h light : dark cycle and given ad libitum access to food and water. All animal procedures were approved by the local ethical committees (The University of Manchester Ethical Review Committee, The University of Vermont Institutional Animal Care and Use Committee, Boehringer Ingelheim Ethical Review Committee) and complied with UK Home Office regulations (Animals Scientific Procedures Act 1986) and in all experiments mice used were killed with an overdose of isoflurane or pentobarbitone.
Immunofluorescence
Periodate-lysine-paraformaldehyde fixative (PLP) was prepared according to the method of McLean and Nakane (1974). Transverse sections (500 µm) of the cerebral cortices of >25-day-old eGFP mice were fixed for 30 min in PLP at room temperature then cryoprotected in 30% sucrose in PBS at 4°C for 1–2 days. Cortical slices were frozen in Tissue-Tek OCT® (Thermofisher Scientific, UK) and 20–30 µm sections were cut and stored at −20°C until required.
Prior to staining, the tissue was permeablized with 0.25% Triton X-100 (Dow Chemical Co. Ltd) in PBS for 10 min, then blocked with 10% (v v-1) normal goat serum in water for 1 h at room temperature. Rabbit polyclonal anti-KCa3.1 antibody (Alomone Labs, Israel) was applied at 1:100 dilution in blocking solution for 18 h at room temperature, followed by 1:100 goat anti-rabbit Texas-Red® conjugated secondary antibody (Invitrogen, UK) for 40 min at room temperature. Slides were then mounted in Vectashield® with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, UK) and visualized.
Image acquisition
Confocal images were collected using a Nikon C1 confocal on an upright 90i microscope equipped with a 60x/1.40 NA Plan Apo objective. DAPI, eGFP and Texas Red were excited at 405 nm, 488 nm and 543 nm respectively. When acquiring 3D optical stacks the confocal software (EZ-C1, Nikon, UK) was used to section the tissue at 1 µm intervals along the z-axis.
Western blotting
Whole-brain tissue was homogenized in protein extraction buffer (in mM: Tris-HCl 20, sucrose 0.25, EDTA 5, EGTA 10, dithiothreitol 10, phenylmethanesulphonylfluoride 1, plus 50 µg·mL−1 leupeptin) using a Tissue Tearor Homogeniser (Biospec Products, USA), then placed on a rotating platform overnight at 4°C.
Before use, sample protein concentrations were quantified using the Bradford method (Bradford, 1976). Protein lysate (30 µg) was separated using SDS-PAGE on a 10% gel under reducing conditions, then transferred to a PVDF membrane (Bio-Rad Laboratories, UK) by electrophoresis for 90 min at 80 V. Non-specific binding sites were blocked with 2% BSA in tris-buffered saline containing 0.1% Tween (TTBS) for 1 h at room temperature. The membrane was then incubated with primary antibody diluted 1:1000 in TTBS overnight at 4°C followed by exposure to 1:5000 horseradish peroxidase-conjugated anti-rabbit secondary antibody (Promega, UK) in TTBS for 1 h at room temperature. The blot was then developed on X-ray film using an ECL Western blotting detection system (ECL Plus, GE Healthcare, UK). All incubations and washes were performed with constant agitation.
RT-PCR
Total astrocyte RNA was extracted from populations of freshly isolated eGFP-positive astrocytes (for detailed information regarding cell separation and FACS, see Appendix S1) using an RNeasy microkit (Qiagen, UK) and treated with DNase I (Qiagen, UK). Purified RNA was reverse transcribed using a Sensiscript RT kit (Qiagen, UK) with random primers (Invitrogen, UK). Primers used for cDNA amplification are listed in Table 1. For PCR reactions, Ex Taq DNA polmerase (Lonza, UK) and a Techne TC-512 thermal cycler (Fisher Scientific, UK, cycling conditions: 10 min 94°C hot start, followed by 40 cycles of 15 s at 94°C and 1 min at 60°C) were employed. PCR products were visualized by separation on a 2.5% agarose gel containing 0.5 µg·mL−1 ethidium bromide. Products yielding a band of the appropriate molecular weight were purified using a QIAquick Spin Kit (Qiagen, UK) and sequenced using an ABI Prism 3000 genetic analyser at the DNA sequencing facility of the University of Manchester.
Table 1.
Primers used in PCR reactions
| Target | GenBank number | Primer sequence (sense followed by antisense) | Product size (bp) |
|---|---|---|---|
| KCa2.3 | 158854043 | 5′-TGGATGAAGACCCCAAGTGT-3′ | 261 |
| 5′-ACAGACCAGGATGGACAAGC-3′ | |||
| KCa3.1 | 141803303 | 5′-GCCATGCTCCTGCGTCTCTACCT-3′ | 139 |
| 5′-TCATGTACAGCTTGGCCACGAACC-3′ | |||
| GFAP | 196115301 | 5′-GAGGCAGAAGCTCCAAGATG-3′ | 470 |
| 5′-GCTCTAGGGACTCGTTCGTG-3′ | |||
| NFh | 124286810 | 5′-GAGGAGTGGTTCCGAGTGAG-3′ | 346 |
| 5′-GCTTTCTGTAAGCGGCAATC-3′ |
GFAP, glial fibrillary acidic protein; NFh, neurofilament H.
Brain slice preparation and patch clamp electrophysiology
The brain slice preparation protocol used in this study followed many of the procedures previously set out by Debanne and co-workers (2008). Briefly, eGFP mice were killed by isoflurane overdose and then decapitated. Mice were killed with anaesthetics because we found that cervical dislocation led to significant damage to the hindbrain and rear of the cortex.
The brain was quickly removed into ice-cold artificial cerebrospinal fluid (aCSF, containing in mM: NaCl 124, KCl 5, CaCl2 2, MgSO4 2, NaH2PO4 1.25, NaHCO3 26, glucose 10, osmolality ∼300 mOsm·kg−1) that had been equilibrated with 95% oxygen, 5% carbon dioxide for 20 min prior. Slices (150–200 µm) were prepared using a Leica VT 1000S vibratome (Leica, Germany) and then stored at 35°C for 1 h in oxygenated aCSF to recover. After this recovery step, slices were maintained at room temperature until required, for up to 10 h.
Whole-cell currents were amplified with an HEKA EPC-9 patch clamp amplifier (Lambrecht/Pfalz, Germany). Data acquisition and storage were carried out with TIDA 5.05 software. Microelectrodes were pulled from filamented borosilicate glass (1.5 mm outer diameter, 0.84 mm inner diameter; World Precision Instruments) using a Flaming-Brown P-97 puller (Sutter, Novato, CA, USA), and had a resistance of 4–6 MΩ when filled with pipette solution containing in mM: ATP 5, KCl 144, MgCl2 1.2, HEPES 10, CaCl2 3.3 and EGTA 5 (200 nM free calcium; calculated using WebmaxC Standard software), or CaCl2 4.5 and EGTA 5 (1 µM free calcium; pH set to 7.2 with KOH, osmolality ∼310 mOsm·kg−1). Slices were continuously perfused with oxygenated aCSF, and brightly fluorescing, process-bearing astrocytes near the slice surface were selected for experiments. The holding potential was −80 mV. After a minimum equilibration period of 10 min, cells with stable currents were subjected to repeated 1 s duration voltage ramps from −150 mV to +50 mV, separated by a period of 10 s, and drug application. Time-matched control experiments were also performed. Approximately 50% of patch attempts yielded a stable recording. Electrophysiological experiments were performed at room temperature (20–22°C).
Details of HEK 293 cell culture and electrophysiology can be found in Appendix S2.
EFS-evoked neurovascular coupling
The methods used for these studies were essentially as described by Girouard et al. (2010). The aCSF used in these experiments contained 3 mM KCl. Briefly, brain slices from 21- to 40-day-old male FVB/NJ mice were prepared as described above and were placed immediately in aCSF containing 10 µM Fluo-4-acetoxymethyl (Fluo-4) and 2.5 µg·mL−1 pluronic F-127 (both from Invitrogen, USA) for 60 min at 29°C in an oxygenated (95% O2, 5% CO2) and humidified chamber. These conditions selectively load astrocyte endfeet (Girouard et al., 2010) and enable monitoring of endfoot calcium as an indicator of astrocyte recruitment into neurovascular coupling. After this loading period, slices were washed once with aCSF and stored in fresh, oxygenated aCSF at room temperature until required.
During an experiment, arteriolar diameter and astrocyte endfoot fluorescence were imaged simultaneously using a Bio-Rad radiance 2100 MP two-photon laser scanning microscope coupled to a titanium : sapphire laser (140-fs pulses, 1.5 W) and an Olympus BX51WI upright microscope equipped with a 20x/0.95 NA Olympus XLUMPlan FI water dipping objective. A maximal digital zoom was applied in each experiment using the LaserSharp software (Biorad, USA), which was used for data collection. Arteriolar diameter was imaged using a transmitted light detector and infrared differential image contrast microscopy. Fluo-4 fluorescence was excited at 820 nm and collected with a 575/150 nm bandpass filter.
A slice was transferred to the recording chamber and continuously perfused with oxygenated aCSF at 37°C in the presence of 125 nM of the thromboxane receptor agonist 9,11-dideoxy-11α,9α-epoxymethanoPGF2α (U46619) to mimic physiological tone. Penetrating parenchymal arterioles arising from pial arteries were selected for study. For each vessel, 20 control images were collected over a period of 15 s, allowing the offline measurement of baseline diameter. Following this, EFS was applied through a pair of parallel platinum wires placed on either side of the vessel of interest. EFS consisted of a 50 Hz alternating square pulse (0.3 ms duration) for 3 s, at 10–20 V. These conditions evoke neuronal activity within the slice, which sets in motion neurovascular coupling (Zonta et al., 2003; Girouard et al., 2010). A further 70 images were collected over the 47 s immediately after the onset of EFS, to determine the effect on diameter and endfoot calcium.
Pharmacological studies were performed as paired experiments in which vessel diameter in response to EFS was measured before and after a 20 min incubation with aCSF containing drugs. This experimental paradigm was used to ensure that the astrocyte endfeet covering the vessel of interest were exposed to the desired concentration of drug and to enable the study of the initial phase of the neurovascular response to EFS, during which vasoactive substances are released from the astrocyte endfeet. Time-matched control experiments in the presence of U46619 alone were also performed. At the end of each experiment, 200 µM papaverine in calcium-free aCSF was applied for 10 min to allow determination of maximal vessel diameter, which was normalized to 100% for statistical analysis.
Laser Doppler flowmetry
Cortical functional hyperaemia in response to contralateral whisker stimulation was recorded in vivo using laser Doppler flowmetry of cerebral blood flow (CBF) in the mouse somatosensory cortex as described previously, with modifications (Niwa et al., 2000; Girouard et al., 2010). Surgical plane anaesthesia was induced in 2- to 3-month-old male C57Bl/6 mice with 5% isoflurane and maintained with 2% isoflurane for the duration of the surgical portion of the protocol. Mice were then administered chloralose (50 mg·kg−1) and urethane (750 mg·kg−1) i.p. for maintenance of anaesthesia after withdrawal of isoflurane. Body temperature was recorded with a rectal probe and maintained at 37°C by a servo-controlled heating pad. A femoral arterial catheter was implanted for blood pressure recording and arterial blood gas measurement. An endotracheal catheter was inserted for mechanical ventilation (SAR-830; CWE Inc., USA). The head was immobilized in a stereotaxic frame, a 2 × 2 mm craniotomy performed over the somatosensory cortex, and the underlying dura removed. The cranial window was superfused with aCSF composed of (in mM): NaCl 125, KCl 3, NaHCO3 26, NaH2PO4 1.25, CaCl2 2, MgCl2 1, glucose 4. At the conclusion of the surgical preparation, isoflurane was withdrawn. Depth of anaesthesia was assessed throughout the experiment by monitoring the blood pressure and reflex responses to tail pinch. A laser Doppler flow probe (Perimed, Sweden) was placed over the cranial window for CBF measurement. Arterial pressure and CBF were recorded using computerized data-acquisition software (PowerLab, AD Instruments, USA). Following a 30 min equilibration period, contralateral vibrissae (cut to 10 mm length) were stimulated at a frequency of 4 Hz for 1 min with a total deflection of 4 mm using a piezoelectric actuator with a 70 mm light-weight metal extension coupled to a programmable waveform generator and an amplifier (Piezo Master; Viking Industrial Products, USA; Andermann et al., 2004). TRAM-34 (10 µM) was delivered locally via the cranial window superfusate for 25 min prior to a second vibrissal stimulation.
Data and statistical analysis
Data and statistical analysis was performed using Prism 5.0 software (GraphPad, San Diego, CA, USA) and results are given as mean ± SEM; n indicates the number of cells or vessels from which measurements were made and the number of animals used for each set of experiments is also noted. Data were analysed using the statistical tests noted in each Figure legend. The level of significance in each test is also noted in each Figure, P < 0.05 was considered significant.
For electrophysiological data, raw whole-cell control currents at 0 mV were routinely normalized to 100%. For neurovascular coupling experiments, vessel diameter measurements were performed offline using ImageJ software. The 90 images collected throughout each experiment were first corrected for movement in the x and y planes using the rigid body algorithm of the TurboReg plugin. Following this, diameter was measured in pixels at at least 3 points along the vessel before and after EFS, so that the mean diameter change of each vessel could be calculated. At least two images before EFS, taken ∼5 s apart, were analysed in this way to ensure that the vessel diameter did not change in the absence of stimulation. Movies S1–S4 were prepared using the ImageFusion program, courtesy of Adrian Bonev and Mark Nelson. For in vivo data, CBF was expressed as % increase relative to the resting level of perfusion measured in arbitrary perfusion units. Zero values were obtained after the heart was stopped with an overdose of isoflurane.
Drugs and chemicals
All drugs and chemicals were purchased from Sigma Aldrich, unless otherwise stated. NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime) and CyPPA (cyclohexyl-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-pyrimidin-4-yl]-amine) were synthesized at the Department of Medicinal Chemistry at Boehringer Ingelheim (Biberach, Germany) according to the methods described in Strøbaek et al. (2004) and Hougaard et al. (2007), respectively. All drugs were dissolved in dimethyl sulphoxide, except for apamin which was dissolved in aCSF.
All drug and molecular target nomenclature conform to the British Journal of Pharmacology's Guide to Receptors and Channels, 4th Edition (Alexander et al., 2009).
Results
Immunofluorescence
Anti-KCa3.1 antibody characterization
The anti-KCa3.1 antibody used in this study has been used by others to successfully identify the KCa3.1 protein in the endothelial cells of the rat mesenteric artery (Sandow et al., 2006) and was selected after characterization using immunofluorescence and Western blotting, see Figure 1D. Immunofluorescence revealed immunoreactivity for the KCa3.1 protein in the endothelium but not smooth muscle or adventitia, which corroborates previously published data (Edwards et al., 1998; Bychkov et al., 2002; Sandow et al., 2006). Based on cDNA sequence (GenBank accession number: 141803303) the molecular weight for the mouse KCa3.1 channel was calculated as 48 kDa. Western blots using mouse brain tissue revealed a single band of an appropriate size using this antibody (inset, Figure 1D).
Figure 1.
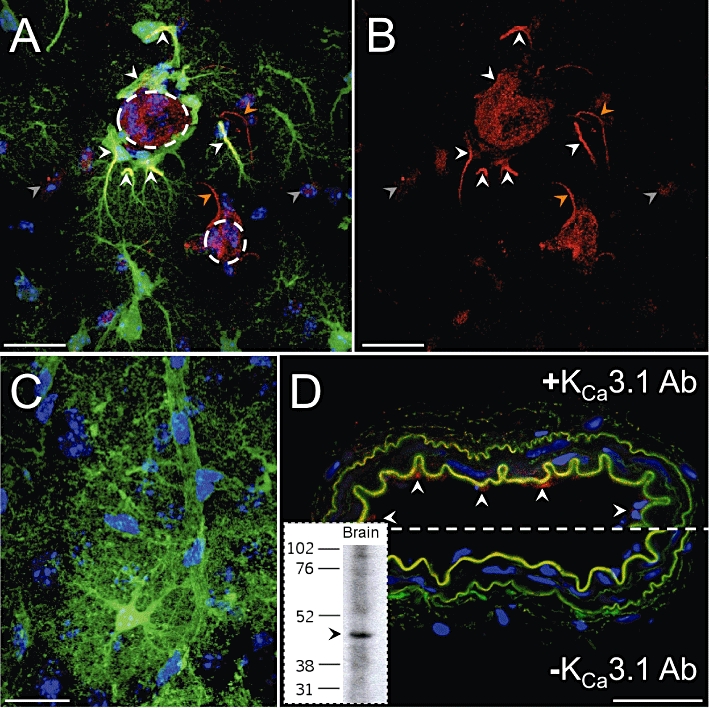
Immunolocalization of the KCa3.1 channel in eGFP-positive astrocytes in a 20 µm brain slice section. (A) Three-dimensionally rendered stack of 20 confocal images taken at 1 µm intervals through the z-axis. White arrowheads highlight anti-KCa3.1 immunoreactivity (red) along the processes and endfeet of astrocytes (green) contacting an intracerebral arteriole (large dashed circle) resulting in a yellow colour indicating co-localization. The red, process-like immunoreactivity which is not co-localized with green processes (orange arrowheads) may correspond to astrocytes which expressed a level of eGFP below the threshold for detection in these images. Another arteriole (small dashed circle) is also visible and some cells of the arterioles are stained by the anti-KCa3.1 antibody. The cell body of another cell type (grey arrowheads) also showed some low-level immunoreactivity. (B) As image (A) but with blue (DAPI) and green colours removed to clearly demonstrate the anti-KCa3.1 staining. (C) Negative control in which omission of the primary antibody indicates the absence of non-specific secondary antibody reactivity. (D) Anti-KCa3.1 antibody characterization in the mouse mesenteric artery. Top: strong anti-KCa3.1 immunoreactivity (red) highlights endothelial cells (arrowheads). The internal and external elastic laminae (green autofluorescence) appear ruffled due to the absence of intraluminal pressure. Bottom: negative control. Inset: Western blot of eGFP mouse brain with the anti-KCa3.1 antibody. A single band of approximately 48 kDa was observed. Scale bars: (A–C) 10 µm; (D) 30 µm. Images are representative of experiments in at least three animals.
KCa3.1 localizes to astrocytic processes and endfeet
The KCa2.3 protein has previously been identified in rat supraoptic nucleus astrocytes using immunohistochemical methods (Armstrong et al., 2005). We therefore used immunohistochemistry to investigate the expression of the KCa3.1 protein in eGFP mouse astrocytes. eGFP expression allowed detailed visualization of individual astrocytes and their extensive processes. Using the anti-KCa3.1 antibody, strong immunoreactivity was observed on the processes and endfeet of astrocytes contacting intracerebral vessels, see Figure 1A–C. KCa3.1 immunoreactivity was also occasionally observed in the processes and cell bodies of a non-eGFP-positive cell type. These observations may correspond with astrocytes that express a low level of eGFP (Nolte et al., 2001; Nimmerjahn et al., 2004), which was not detectable using the confocal settings in these experiments. Alternatively, they may indicate a different, unidentified cell type. Vessels were also stained by the anti-KCa3.1 antibody, which may be due to expression of this protein in endothelial cells.
RT-PCR
Freshly isolated eGFP-positive astrocytes express KCa2.3 and KCa3.1 mRNA
RT-PCR was used to verify the expression of KCa2.3 and KCa3.1 channels in populations of eGFP-positive astrocytes isolated by FACS. Using four separate FACS-isolated astrocyte mRNA samples, PCR products separated by electrophoresis revealed bands of the expected molecular weight for KCa2.3 (261 bp, n = 3), KCa3.1 (139 bp, n = 3) and glial fibrillary acidic protein (GFAP, a cytoskeletal marker of astrocytes) (470 bp, n = 4), but not the neuronal marker NFh (346 bp), thus demonstrating the expression of KCa2.3 and KCa3.1 mRNA in populations of pure mouse astrocytes. See Figure 2. Sequencing confirmed the molecular identities of these PCR products (Figure S1).
Figure 2.

Astrocytes sorted by FACS contained message for the expression of GFAP but not the neuronal marker NFh indicating that the sorted astrocyte populations were not contaminated with neurones. Using these samples, mRNA for KCa2.3 and KCa3.1 was identified (n = 3 each in four FACS-isolated mRNA samples). M, marker ladder; N, no-template control; +, sample following reverse transcription; −, sample without reverse transcription.
Electrophysiology
A subpopulation of neocortical astrocytes express KCa3.1, but not KCa2.1-2.3 current
Currents in green fluorescent astrocytes, patch-clamped in the whole-cell configuration, had a reversal potential of −70 ± 1 mV (n = 50) and were of variable peak amplitude (2–5 nA at 0 mV). In control eGFP-positive astrocytes, there was an 8 ± 2% run-down of ramp-induced whole-cell currents over the duration of the experiment (approximately 10.5 min) in the presence of 1 µM [Ca2+]i (n = 9, three animals) and 13 ± 5% in the presence of 200 nM [Ca2+]i (n = 10, four animals). With [Ca2+]i set to 200 nM to prime KCa channels for opening, 14 cells (from three animals) were challenged with 500 nM NS309, an opener of KCa2.1-2.3 and KCa3.1 channels, which evoked a current increase of 183 ± 43% at 0 mV in eight cells. The remaining six cells were classed as unresponsive to NS309 and their current after drug application showed a 1 ± 2% rundown. In all responsive cells, the NS309 response took several minutes to occur. Under these same conditions, 30 µM CyPPA, an opener of both KCa2.2 and KCa2.3, did not evoke current and over the time-course of the experiment there was a 4 ± 2% current decrease (n = 9, two animals), similar to that of time matched controls (see Figure 3).
Figure 3.

Effects of openers of KCa2.1-2.3 and KCa3.1 in eGFP-positive astrocytes patch clamped in the whole-cell configuration. (A) Typical (time-matched) control time-course indicating currents at the 0 mV point of voltage ramps plotted as a function of time. (B) Typical time-course showing an increase in outward current ∼7 min after application of NS309 [arrowheads indicate time points of the current-voltage relationships shown in (C)]. (C) Current-voltage relationship (1 s voltage ramp) showing selected full traces from the cell described in (B) under control conditions and after exposure to NS309. (D) Typical time course showing lack of induction of current by CyPPA [arrowheads indicate the time points at which the current-voltage relationships shown in (E) were obtained under control conditions and after exposure to CyPPA]. (F) Mean (±SEM) currents at 0 mV normalized to initial control current level. In the presence of 200 nM [Ca2+]i NS309 (500 nM; n = 8) evoked a significant current increase whereas CyPPA (30 µM) was without effect (n = 9) (one-way anova with post hoc Bonferroni's multiple comparison test ***P < 0.0001).
In eGFP-positive astrocytes with 1 µM [Ca2+]i (to activate any KCa channels present), nine out of 16 cells (from three animals) challenged with 1 µM TRAM-34 (a selective KCa3.1 blocker) responded with a 41 ± 6% inhibition of whole-cell potassium currents. The remaining seven cells showed no significant difference to time-matched controls, and current declined by 10 ± 2% over the course of the experiment. In a separate set of experiments application of 100 nM apamin, a selective KCa2.1-2.3 channel blocker, had no effect on currents and cells showed a mean current decrease of 7 ± 4% (n = 7, two animals; see Figure 4).
Figure 4.
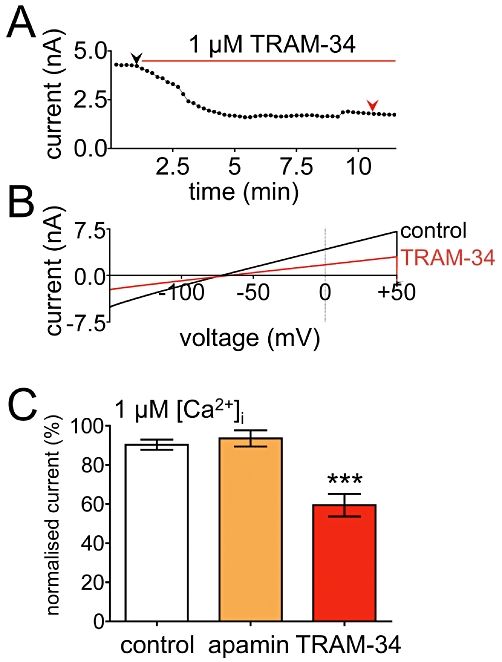
Effects of inhibitors of KCa2.1-2.3 and KCa3.1 on whole-cell currents of eGFP-positive astrocytes. (A) Typical time-course showing inhibition of outward currents by TRAM-34. Currents correspond to the 0 mV point of voltage ramps plotted as a function of time [arrowheads indicate the time points at which the full traces shown in (B) were derived under control conditions and after TRAM-34]. (C) Mean (±SEM) currents at 0 mV during voltage ramps. In the presence of 1 µM [Ca2+]i, 100 nM apamin was without effect (n = 7) whereas 1 µM TRAM-34 (n = 9) evoked a significant current decrease (one-way anova with post hoc Bonferroni's multiple comparison test ***P < 0.0001).
CyPPA inhibits NS309-evoked currents in a HEK293 cell line stably overexpressing KCa3.1
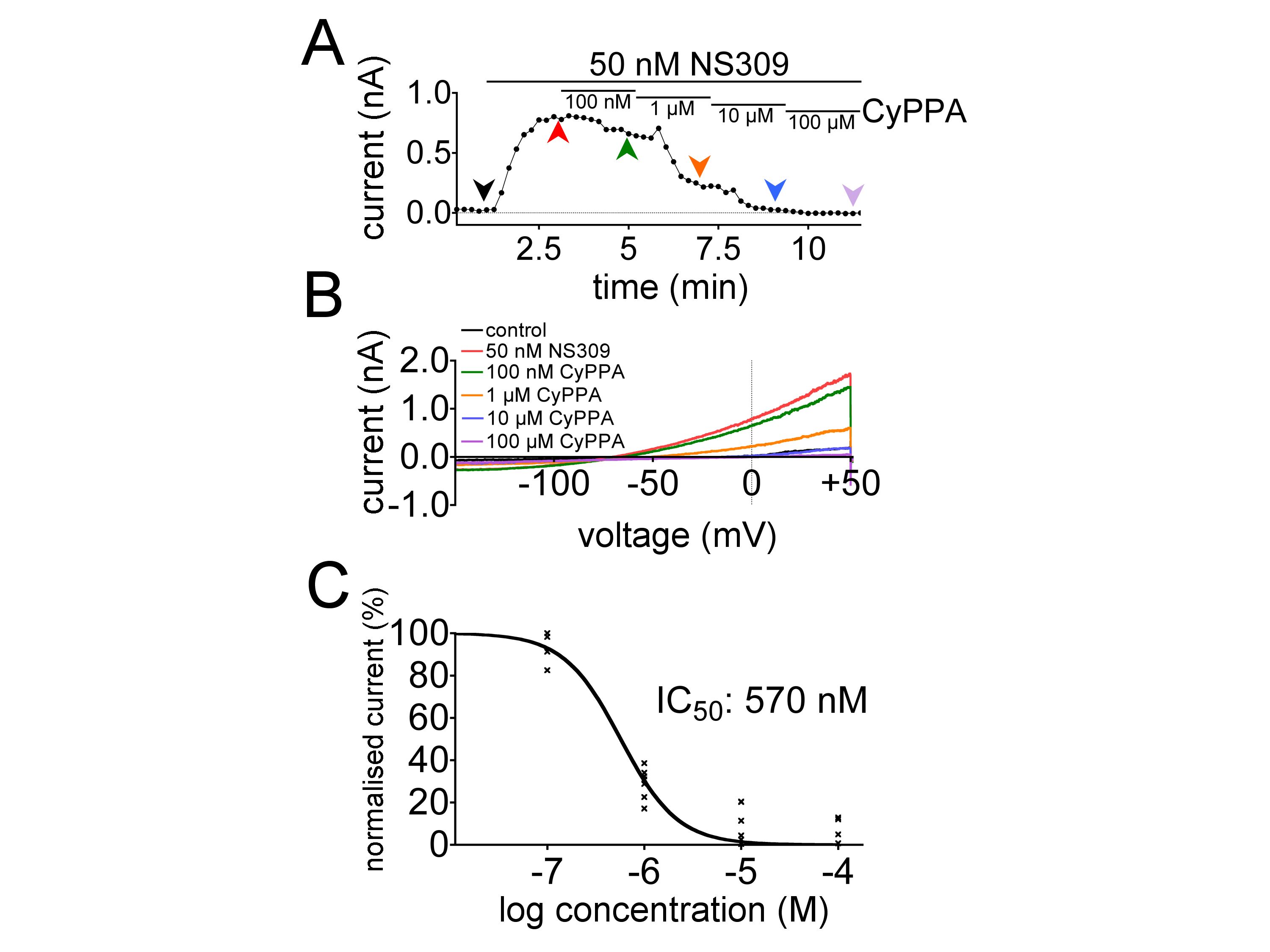
In response to some unexpected observations we tested whether CyPPA, a selective opener of KCa2.2 and KCa2.3, gains its selectivity in part through blockade of KCa3.1 by using a HEK 293 cell line stably overexpressing the human KCa3.1 channel. In the presence of 200 nM intracellular calcium and 50 nM NS309 to evoke KCa3.1 currents, CyPPA (100 nM–100 µM) inhibited NS309-evoked currents with an IC50 of 570 nM (n = 7; see Figure S2).
KCa3.1 contributes to neurovascular coupling
After confirming that astrocytes express functional KCa3.1 channels, which appear to be localized to astrocyte processes and endfeet around blood vessels, we tested the hypothesis that this channel contributes to the process of neurovascular coupling. Increases in astrocyte endfoot calcium observed upon application of EFS confirmed astrocyte recruitment into neurovascular coupling. Under control conditions, the presence of 125 nM U46619 constricted parenchymal arterioles by 48 ± 3%, giving them a mean diameter of 5 ± 1 µm (n = 16). EFS evoked a mean diameter increase of 28 ± 3% in control vessels (n = 16). Time-matched control experiments indicated that the mean magnitude of the diameter change evoked by EFS was unchanged after 20 min (n = 4, two animals; P > 0.05, data not shown).
In paired experiments, perfusion of slices for 20 min with aCSF containing 1 µM TRAM-34 reduced the magnitude of EFS-evoked vasodilatation by 50% (Figure 5A–C and Movies S1 and S2), leaving a residual EFS-evoked diameter increase of 12 ± 3% (n = 5, three animals). Perfusion of slices for 20 min with aCSF containing 300 nM charybdotoxin (a blocker of both KCa3.1 and KCa1.1) reduced the magnitude of EFS-evoked vasodilatation by 82%, leaving a residual EFS-evoked diameter increase of 5 ± 4% (n = 6, four animals; see Figure 5D–F and Movies S3 and S4).
Figure 5.
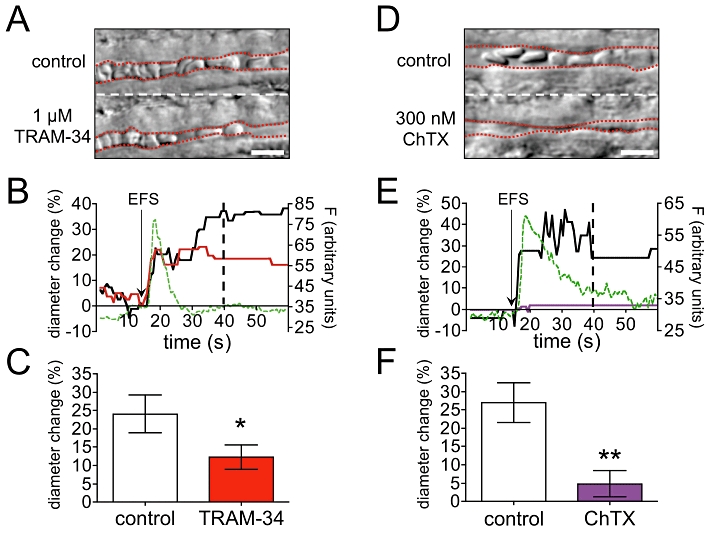
TRAM-34 and charybdotoxin (ChTX) significantly reduce EFS-evoked diameter increases in parenchymal arterioles. (A) Example vessel showing post-EFS diameter under control conditions (top) and after 20 min 1 µM TRAM-34 incubation (bottom). Red dashed lines: lumen edges. (B) Time course of diameter and endfoot fluorescence changes for the vessel shown in (A); 1 µM TRAM-34 (red) reduced the magnitude of the EFS-evoked diameter increase. The increase in astrocyte endfoot fluorescence (F, green dashed line) closely matched the onset of vasodilatation. Black arrow: EFS application. Dashed black line: time point of the images shown in (A). (C) Mean (±SEM) EFS-evoked diameter changes in five paired experiments where 1 µM TRAM-34 reduced the magnitude of vasodilatation by 50% (paired t-test, *P = 0.0168). (D) As (A) but bottom image shows EFS-evoked diameter after 20 min 300 nM ChTX incubation. Red dashed lines: lumen edges. (E) Time course of diameter and endfoot fluorescence changes for the vessel shown in (D); 300 nM ChTX (purple) almost abolished the EFS-evoked diameter increase. The increase in astrocyte endfoot fluorescence (F, green dashed line) closely matched the onset of vasodilatation. Black arrow: EFS application. Dashed black line: time point of the images shown in (D). (F) Mean (±SEM) EFS-evoked diameter changes in 6 paired experiments; 300 nM ChTX reduced EFS-evoked vasodilatation by 82% (paired t test, **P = 0.0024). Scale bars in (A) and (D): 10 µm.
Incubation with either 1 µM TRAM-34 (n = 5, three animals) or 300 nM charybdotoxin (n = 5, four animals) for 20 min had no overall effect on basal vessel tone prior to EFS, and had no effect on the relative increase in astrocyte endfoot calcium evoked by EFS (P > 0.05, data not shown).
In in vivo experiments measuring changes in CBF using laser Doppler flowmetry, whisker stimulation evoked a 23 ± 5% increase in CBF (n = 6). TRAM-34 (10 µM) reduced whisker stimulation evoked increases in CBF by 40%, leaving a residual increase of 14 ± 2% (n = 6), see Figure 6.
Figure 6.

TRAM-34 significantly reduced whisker stimulation-evoked increases in cerebral blood flow (CBF) in vivo. (A) Example experiment showing increases in CBF evoked by 60 s whisker stimulation under control conditions and after 25 min 10 µM TRAM-34. PU, arbitrary perfusion units. (B) Mean (±SEM) whisker stimulation-evoked CBF changes in six paired experiments; 10 µM TRAM-34 reduced the CBF response by 40% (paired t test, **P = 0.0022).
Discussion and conclusions
Previous studies (Filosa et al., 2006; Girouard et al., 2010) have identified K+ efflux through astrocytic KCa1.1 channels as a mediator of parenchymal arteriole relaxation in the process of neurovascular coupling. In the present study, these observations have been extended to show that astrocytic KCa3.1 channels are also involved in neurovascular coupling in mouse brain.
KCa3.1 mRNA was detected in preparations of freshly-isolated astrocytes and, using immunohistochemistry, the KCa3.1 α subunit protein was found to be localized within astrocytic processes and endfeet. In electrophysiological experiments, a substantial increase in whole-cell current was generated on exposure to the KCa2.1-2.3 and KCa3.1 opener NS309 (Strøbaek et al., 2004). As neither the KCa2.2- and 2.3-selective opener CyPPA (Hougaard et al., 2007) nor the KCa2.1-2.3 blocker apamin had any effect on astrocyte currents, it is concluded that the activation of only KCa3.1 channels contributed to the observed NS309-induced increases in whole-cell current. This conclusion is supported by data showing that TRAM-34, a selective blocker of KCa3.1, inhibited astrocytic whole-cell currents.
In a very recent study focusing on the activation of rat astrocytic KCa1.1 channels by EETs, it was reported that TRAM-34 did not reduce astrocytic potassium currents (Higashimori et al., 2010). However, in these experiments, the astrocytes were dialysed with a calcium chelator (0.2 mM EGTA), and therefore it is not surprising that TRAM-34 was ineffective. In contrast, in our experiments, astrocytes were dialysed with intracellular calcium of 0.2 and 1.0 µM; additionally both blockers and activators of KCa3.1 were tested.
In the present study, mRNA for the KCa2.3 α subunit was also detected and the presence of this KCa channel has previously been reported in astrocytes from the rat supra-optic nucleus (Armstrong et al., 2005). However, no evidence of functional KCa2.3 channels at the level of the cell membrane was obtained in the present investigation. This may indicate that KCa2.3 has an intracellular role in astrocytes and/or that it is not inserted into the plasma membrane, although differences in the species and anatomical locations studied should also be taken into account when interpreting these results.
CyPPA was the first selective activator of KCa2.2 and KCa2.3 to be described (Hougaard et al., 2007). In preliminary electrophysiological experiments on mouse astrocytes in which NS309 was applied after CyPPA, no response was observed to either drug (TA Longden, unpublished observations). It was therefore possible that the imidazole ring of CyPPA (which is a common feature of KCa3.1 blockers such as clotrimazole and TRAM-34; Wulff et al., 2000) had conferred KCa3.1 inhibitory properties on CyPPA. Experiments were thus carried out to determine whether the apparent selectivity of CyPPA for KCa2.2 and KCa2.3 could be in part attributed to blockade of KCa3.1. This seems to be the case, as our Supporting information shows that CyPPA (in the nanomolar range) inhibited NS309 responses in HEK 293/KCa3.1 cells. This action of CyPPA occurred at a concentration considerably lower than its EC50 concentrations for opening KCa2.2 and KCa2.3 (5.6 µM and 14 µM respectively; Hougaard et al., 2007), an indication that CyPPA will concomitantly block KCa3.1 while opening KCa2.2 and 2.3 channels.
The response of mouse astrocytes to NS309 and TRAM-34 was heterogeneous, with some cells (56–67% depending on the drug employed) showing a pharmacological response typical of KCa3.1 and others not responding to drug application. Concentrations of 500 nM NS309 and 1 µM TRAM-34 (respective EC50/IC50 at KCa3.1: 10 nM and 20 nM; Strøbaek et al., 2004; Wulff et al., 2007) were used in slice experiments to ensure that an adequate concentration of drug reached the cell of interest, despite potential penetration problems in this complex tissue. A possible explanation for the observed heterogeneity is that functional KCa3.1 channels are only expressed in a subpopulation of dividing astrocytes, as KCa3.1 has been implicated in the process of mitosis (Khanna et al., 1999; Jensen et al., 2001; Wulff et al., 2004; Grgic et al., 2005). However, in experiments attempting to co-localize astrocytic KCa3.1 immunoreactivity with the cellular proliferation marker Ki67, Ki67 was only very rarely co-localized with KCa3.1 (TA Longden, unpublished data) suggesting an alternative role for this channel in astrocytes. Another possible explanation is that control of intracellular calcium by pipette calcium was variable, and therefore, KCa3.1 channel activity and drug response were variable. This is a distinct possibility, given observations of spontaneous calcium fluctuations in astrocytes (Aguado et al., 2002; Beck et al., 2004), as well as their large size and extensive processes. We also observed a slow onset of channel activation by NS309 that was very robust, with the sudden appearance of channel activity never occurring before ∼6 min. A delay with NS309 has previously been described (Pedarzani et al., 2005), a phenomenon that might be due to poor penetration of NS309 into the slice or across the cell membrane.
KCa3.1 channels play an important role in endothelium-dependent vasodilatation (Félétou and Vanhoutte, 2006; Edwards et al., 2010) suggesting that these channels in astrocytes might fulfil a similar role by stimulating cerebral arteriole vasodilatation. In mouse brain-slice experiments in which neurovascular coupling was initiated by electrical field stimulation, Fluo-4 fluorescence confirmed the recruitment of astrocyte endfeet into neurovascular coupling (see Movies S1–S4). We found that the time-scale of the endfoot fluorescence increase almost exactly matched the onset of vessel dilatation, which is in general agreement with other studies that have specifically investigated astrocyte calcium signalling (Takano et al., 2006; Winship et al., 2007; Girouard et al., 2010). Further comment on astrocyte endfoot calcium dynamics is beyond the scope of the present study, as calcium measurements were intended solely to confirm astrocyte recruitment into the neurovascular coupling process.
The magnitude of parenchymal arteriole dilatation in response to EFS was halved by the presence of TRAM-34. This compound is known to also block an unidentified non-selective cation channel, which markedly reduced lipophosphatidylcholine-induced calcium rises in microglia (Schilling and Eder, 2007). Under our experimental conditions, TRAM-34 inhibited a calcium-activated potassium current in astrocytes (Figure 3), without any indication of inhibition of a non-selective cation channel. Furthermore, the relative increase in endfoot calcium in response to EFS was unaffected by TRAM-34 (data not shown) suggesting that the inhibitory effect of TRAM-34 on neurovascular coupling does not arise from its ability to block non-selective cation channels. Moreover, charybdotoxin, a scorpion toxin which blocks both KCa3.1 and KCa1.1, reduced EFS-evoked neurovascular coupling by more than 80%, which is consistent with a combined block of KCa3.1 and KCa1.1 channels. From these data we conclude that KCa3.1 (as well as KCa1.1; Filosa et al., 2006; Girouard et al., 2010) contributes significantly to the process of neurovascular coupling. In these experiments, it was not possible to demonstrate unequivocally that it was the astrocytic KCa3.1 channel that contributed to neurovascular coupling under slice conditions. Indeed, endothelial KCa3.1 channels exert significant control over parenchymal arteriolar diameter (Hannah et al., 2010). However, given the immunohistochemical, molecular and electrophysiological evidence already discussed, a prominent role for astrocytic KCa3.1 channels seems to provide a simple and consistent explanation for these experimental observations. Charybdotoxin had a greater inhibitory effect on arteriolar dilatation than TRAM-34. This is not surprising because most neurovascular coupling mechanisms (nitric oxide, prostaglandins, EETs and K+ release) rely on either vascular myocyte or astrocyte KCa1.1 channels to bring about vasodilatation (Filosa et al., 2006; Girouard et al., 2010). If electrical field stimulation recruits several or all of these vasodilator mechanisms, the greater inhibitory effect of charybdotoxin compared with that of TRAM-34 is easy to understand. It is perhaps also possible that differential penetration of charybdotoxin and TRAM-34 into the slice may have resulted in a different efficacy for each drug. However, as charybdotoxin is much larger than the low molecular weight, lipophilic TRAM-34, the greater blocking effect of charybdotoxin is best explained by a combined blocking action of this toxin at both KCa1.1 and KCa3.1, whereas TRAM-34 solely inhibits KCa3.1 under these conditions.
The findings of our in situ slice experiments are further enhanced by data demonstrating that TRAM-34 reduced whisker stimulation-evoked increases in CBF by 40% in vivo. This evidence is in agreement with the hypothesis that IKCa is involved in neurovascular coupling and suggests that KCa3.1 participates in this process under physiological conditions.
The mechanism(s) by which KCa3.1 influences neurovascular coupling remains to be elucidated. KCa3.1 may contribute to the increase in extracellular K+, which currently is attributed to KCa1.1 activation and activates K+ efflux from myocyte KIR channels (Filosa et al., 2006; Girouard et al., 2010). However, preliminary data suggested that TRAM-34 still exerts a blocking effect in the presence of barium, a KIR blocker (TA Longden, unpublished observations). Thus, the astrocytic KCa3.1 might instead be linked to a myocyte Na+/K+ ATPase, as has been described in the periphery (Dora et al., 2008; Harno et al., 2008; Weston et al., 2010), or the endothelial KCa3.1 could even be recruited into the neurovascular coupling process (Hannah et al., 2010). Such an engagement of endothelial KCa3.1 channels in neurovascular coupling would probably require endothelial activation by an unknown factor released from the astrocytic processes. Future investigations will address the mechanism through which KCa3.1 contributes to neurovascular coupling.
In conclusion, this report presents novel immunohistochemical, molecular and electrophysiological evidence, which demonstrate the functional expression of KCa3.1 channels in mouse astrocytes contacting the vasculature. No evidence was found for the functional expression of KCa2.1-2.3 channels in these cells. Our results support a novel role of KCa3.1 channels in neurovascular coupling.
Acknowledgments
This study was supported by a BBSRC CASE Studentship, in association with Boehringer Ingelheim Pharma GmbH & Co KG, and by the Totman Medical Research Trust Fund, National Institutes of Health grants P20-RR-16435, P01-HL-095488, R01-HL-044455, R01-HL-098243 and T32 HL07944. The neurovascular coupling investigation was also supported by the British Pharmacological Society's Schachter Award. eGFP mice were a kind gift of the laboratories of Alexej Verkhratsky (University of Manchester, UK) and Frank Kirchhoff (Max Planck Institute, Göttingen, Germany) and HEK 293/KCa3.1 cells were kindly provided by Matthew Burnham (University of Manchester, UK). The authors gratefully acknowledge Robert Fernandez, Ian Johnson, Simon Moussaud, Doris Linn, Carsten Hecker, Fabrice Dabertrand and Adrian Bonev for their superb methodological assistance and helpful discussions. Three of us (T. L., G. E., A. H. W.) acknowledge the special role of Bastian Hengerer (Boehringer Ingelheim) during Case Studentship negotiations.
Glossary
Abbreviations
- [Ca2+]i
concentration of intracellular calcium
- aCSF
artificial cerebrospinal fluid
- CBF
cerebral blood flow
- CyPPA
cyclohexyl-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-pyrimidin-4-yl]-amine
- EETs
epoxyeicosatrienoic acids
- eGFP
enhanced green fluorescent protein
- Fluo-4
Fluo-4-acetoxymethyl ester
- KCa1.1
large-conductance calcium-activated potassium channel
- KCa2.X
small-conductance calcium-activated potassium channel subtype X
- KCa3.1
intermediate-conductance calcium-activated potassium channel
- KIR
inwardly rectifying potassium channel
- NS309
6,7-dichloro-1H-indole-2,3-dione 3-oxime
- TRAM-34
1-[(2- chlorophenyl)diphenyl-methyl]-1H-pyrazole
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Sequence alignment of KCa2.3, KCa3.1 and GFAP PCR products with their corresponding GenBank sequences using ClustalW2 software. For each row, the top line of bases shows the GenBank sequence and the bottom line shows the sequence of the relevant PCR product. Asterisks denote a match. (A) Sequences for the KCa2.3 PCR 5′ product (top panel) and 3′ product (bottom panel) aligned with GenBank sequence 158854043. (B) Sequences for the KCa3.1 PCR 5′ product (top panel) and 3′ product (bottom panel) aligned with GenBank sequence 141803303. (D) Sequences for the GFAP PCR 5′ product (top panel) and 3′ product (bottom panel) aligned with GenBank sequence 196115301.
{kind=link}
Figure S2 Inhibitory effect of CyPPA on currents evoked by 50 nM NS309 in HEK 293/KCa3.1 cells. (A) Typical time course showing inhibition of NS309-evoked currents at 0 mV caused by CyPPA. Coloured arrowheads indicate time points at which the current-voltage traces in (B) were derived under control conditions (black), in the presence of 50 nM NS309 (red) and in the presence of 50 nM NS309 plus increasing concentrations of CyPPA (green, 100 nM; orange, 1 µM; blue, 10 µM; violet, 100 µM). (C) Concentration-response curve for CyPPA in the presence of 50 nM NS309. CyPPA (100 nM–100 µM) inhibited NS309-evoked currents with an IC50 of 570 nM (n = 7).
{kind=link}
Movie S1 Application of a control EFS pulse prior to TRAM-34 incubation.
Movie S2 Application of EFS after 20 min TRAM-34 incubation.
Movie S3 Application of a control EFS pulse prior to charybdotoxin incubation.
Movie S4 Application of EFS after 20 min charybdotoxin incubation.
Appendix S1 Cell separation and fluorescence activated cell sorting.
Appendix S2 HEK 293/KCa3.1 cell culture and patch clamp electrophysiology.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aguado F, Espinosa-Parrilla JF, Carmona MA, Soriano E. Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ. J Neurosci. 2002;22:9430–9444. doi: 10.1523/JNEUROSCI.22-21-09430.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann ML, Ritt J, Neimark MA, Moor CI. Neural correlates of vibrissa resonance; band pass and somatotopic representation of high frequency stimuli. Neuron. 2004;42:451–463. doi: 10.1016/s0896-6273(04)00198-9. [DOI] [PubMed] [Google Scholar]
- Armstrong W, Rubrum A, Teruyama R, Bond C, Adelman J. Immunocytochemical localization of small-conductance, calcium-dependent potassium channels in astrocytes of the rat supraoptic nucleus. J Comp Neurol. 2005;491:175–185. doi: 10.1002/cne.20679. [DOI] [PubMed] [Google Scholar]
- Beck A, Nieden RZ, Schneider HP, Deitmer JW. Calcium release from intracellular stores in rodent astrocytes and neurons in situ. Cell Calcium. 2004;35:47–58. doi: 10.1016/s0143-4160(03)00171-4. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Bradley KK, Jaggar JH, Bonev AD, Heppner TJ, Flynn ER, Nelson MT, et al. Kir2.1 encodes the inward rectifier potassium channel in rat arterial smooth muscle cells. J Physiol. 1999;515:639–651. doi: 10.1111/j.1469-7793.1999.639ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham M, Bychkov R, Félétou M, Richards GR, Vanhoutte PM, Weston A, et al. Characterization of an apamin-sensitive small-conductance Ca2+-activated K+ channel in porcine coronary artery endothelium: relevance to EDHF. Br J Pharmacol. 2002;135:1133–1143. doi: 10.1038/sj.bjp.0704551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bychkov R, Burnham M, Richards G, Edwards G, Weston A, Félétou M, et al. Characterization of a charybdotoxin-sensitive intermediate conductance Ca2+-activated K+ channel in porcine coronary endothelium: relevance to EDHF. Br J Pharmacol. 2002;137:1346–1354. doi: 10.1038/sj.bjp.0705057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Boudkkazi S, Campanac E, Cudmore RH, Giraud P, Fronzaroli-Molinieres L, et al. Paired-recordings from synaptically coupled cortical and hippocampal neurons in acute and cultured brain slices. Nat Protoc. 2008;3:1559–1568. doi: 10.1038/nprot.2008.147. [DOI] [PubMed] [Google Scholar]
- Dora KA, Gallagher NT, McNeish A, Garland C. Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res. 2008;102:1247–1255. doi: 10.1161/CIRCRESAHA.108.172379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland C, Weston A. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- Edwards G, Gardener MJ, Félétou M, Brady G, Vanhoutte PM, Weston A. Further investigation of endothelium-derived hyperpolarizing factor (EDHF) in rat hepatic artery: studies using 1-EBIO and ouabain. Br J Pharmacol. 1999;128:1064–1070. doi: 10.1038/sj.bjp.0702916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Félétou M, Weston AH. Endothelium-derived hyperpolarizing factors and associated pathways: a synopsis. Pflügers Arch. 2010;459:863–879. doi: 10.1007/s00424-010-0817-1. [DOI] [PubMed] [Google Scholar]
- Félétou MF, Vanhoutte PM. EDHF The Complete Story. New York, NY: CRC Taylor & Francis; 2006. [Google Scholar]
- Filosa J, Bonev A, Straub S, Meredith A, Wilkerson M, Aldrich R, et al. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. 2006;9:1397–1403. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- Girouard H, Bonev A, Hannah RM, Meredith A, Aldrich R, Nelson M. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. PNAS. 2010;107:3811–3816. doi: 10.1073/pnas.0914722107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grgic I, Eichler I, Heinau P, Si H, Brakemeier S, Hoyer J, et al. Selective blockade of the intermediate-conductance Ca2+-activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler Thromb Vasc Biol. 2005;25:704–709. doi: 10.1161/01.ATV.0000156399.12787.5c. [DOI] [PubMed] [Google Scholar]
- Hannah RM, Dunn KM, Bonev A, Nelson MT. Endothelial SKCa and IKCa channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J Cereb Blood Flow Metab. 2010;31:1175–1186. doi: 10.1038/jcbfm.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harno E, Edwards G, Geraghty AR, Ward DT, Dodd RH, Dauban P, et al. Evidence for the presence of GPRC6A receptors in rat mesenteric arteries. Cell Calcium. 2008;44:210–219. doi: 10.1016/j.ceca.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Higashimori H, Blanco VM, Tuniki VR, Falck J, Filosa J. Role of epoxyeicosatrienoic acids as autocrine metabolites in glutamate-mediated K+ signaling in perivascular astrocytes. Am J Physiol. 2010;299:C1068–C1078. doi: 10.1152/ajpcell.00225.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hougaard C, Eriksen B, Jorgensen S, Johansen T, Dyhring T, Madsen L, et al. Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+-activated K+ channels. Br J Pharmacol. 2007;151:655–665. doi: 10.1038/sj.bjp.0707281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BS, Strøbaek D, Olesen SP, Christophersen P. The Ca2+-activated K+ channel of intermediate conductance: a molecular target for novel treatments? Curr Drug Targets. 2001;2:401–422. doi: 10.2174/1389450013348173. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Khanna R, Chang MC, Joiner WJ, Kaczmarek LK, Schlichter LC. hSK4/hIK1, a calmodulin-binding KCa channel in human T lymphocytes. Roles in proliferation and volume regulation. J Biol Chem. 1999;274:14838–14849. doi: 10.1074/jbc.274.21.14838. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Zimmermann PA, Nelson MT. Extracellular K+-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels. J Physiol. 1996;492:419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler RC, Roman RJ, Harder DR. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009;32:160–169. doi: 10.1016/j.tins.2008.11.005. [DOI] [PubMed] [Google Scholar]
- McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J Histochem Cytochem. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods. 2004;1:31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- Niwa K, Araki E, Morham SG, Ross ME, Iadecola C. Cyclooxygenase-2 contributes to functional hyperaemia in whisker-barrel cortex. J Neurosci. 2000;20:763–770. doi: 10.1523/JNEUROSCI.20-02-00763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte C, Matyash M, Pivneva T, Schipke CG, Ohlemeyer C, Hanisch UK, et al. GFAP promoter-controlled EGFP-expressing transgenic mice: a tool to visualize astrocytes and astrogliosis in living brain tissue. Glia. 2001;33:72–86. [PubMed] [Google Scholar]
- Pedarzani P, McCutcheon JE, Rogge G, Jensen BS, Christophersen P, Hougaard C, et al. Specific enhancement of SK channel activity selectively potentiates the afterhyperpolarizing current IAHP and modulates the firing properties of hippocampal pyramidal neurons. J Biol Chem. 2005;280:41404–41411. doi: 10.1074/jbc.M509610200. [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DL, Ludwig JW, Mi H, Schwarz TL, Ellisman MH. Distribution of rSlo Ca2+-activated K+ channels in rat astrocyte perivascular endfeet. Brain Res. 2002;956:183–193. doi: 10.1016/s0006-8993(02)03266-3. [DOI] [PubMed] [Google Scholar]
- Quayle JM, McCarron JG, Brayden JE, Nelson MT. Inward rectifier K+ currents in smooth muscle cells from rat resistance-sized cerebral arteries. Am J Physiol. 1993;265:C1363–C1370. doi: 10.1152/ajpcell.1993.265.5.C1363. [DOI] [PubMed] [Google Scholar]
- Sandow S, Neylon C, Chen M, Garland C. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels (KCa) and connexins: possible relationship to vasodilator function? J Anat. 2006;209:689–698. doi: 10.1111/j.1469-7580.2006.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling T, Eder C. TRAM-34 inhibits non-selective cation channels. Pflugers Arch. 2007;454:559–563. doi: 10.1007/s00424-007-0232-4. [DOI] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254–9262. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strøbaek D, Teuber L, Jørgensen TD, Ahring PK, Kjaer K, Hansen RS, et al. Activation of human IK and SK Ca2+-activated K+ channels by NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime) Biochim Biophys Acta. 2004;1665:1–5. doi: 10.1016/j.bbamem.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, et al. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Taylor MS, Bonev A, Gross TP, Eckman DM, Brayden JE, Bond C, et al. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res. 2003;93:124–131. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci. 1999;19:6897–6906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston A, Richards GR, Burnham M, Félétou M, Vanhoutte PM, Edwards G. K+-induced hyperpolarization in rat mesenteric artery: identification, localization and role of Na+/K+-ATPases. Br J Pharmacol. 2002;136:918–926. doi: 10.1038/sj.bjp.0704787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston A, Porter EL, Harno E, Edwards G. Impairment of endothelial SK channels and of downstream hyperpolarizing pathways in mesenteric arteries from spontaneously hypertensive rats. Br J Pharmacol. 2010;160:836–843. doi: 10.1111/j.1476-5381.2010.00657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winship IR, Plaa N, Murphy TH. Rapid astrocyte calcium signals correlate with neuronal activity and onset of the hemodynamic response in vivo. J Neurosci. 2007;27:6268–6272. doi: 10.1523/JNEUROSCI.4801-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wölfle SE, Schmidt VJ, Hoyer J, Köhler R, de Wit C. Prominent role of KCa3.1 in endothelium-derived hyperpolarizing factor-type dilations and conducted responses in the microcirculation in vivo. Cardiovasc Res. 2009;82:476–483. doi: 10.1093/cvr/cvp060. [DOI] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan M, Chandy K. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. PNAS. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Knaus HG, Pennington M, Chandy KG. K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity. J Immunol. 2004;173:776–786. doi: 10.4049/jimmunol.173.2.776. [DOI] [PubMed] [Google Scholar]
- Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier J, Shakkottai V. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem. 2007;14:1437–1457. doi: 10.2174/092986707780831186. [DOI] [PubMed] [Google Scholar]
- Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circ Res. 2000;87:160–166. doi: 10.1161/01.res.87.2.160. [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.