

Abstract
BACKGROUND AND PURPOSE
Regression of left ventricular hypertrophy by moxonidine, a centrally acting sympatholytic imidazoline compound, results from a sustained reduction of DNA synthesis and transient stimulation of DNA fragmentation. Because apoptosis of cardiomyocytes may lead to contractile dysfunction, we investigated in spontaneously hypertensive rats (SHR), time- and dose-dependent effects of in vivo moxonidine treatment on cardiac structure and function as well as on the inflammatory process and signalling proteins involved in cardiac cell survival/death.
EXPERIMENTAL APPROACH
12 week old SHR received moxonidine at 0, 100 and 400 µg·kg−1·h−1, s.c., for 1 and 4 weeks. Cardiac function was evaluated by echocardiography; plasma cytokines were measured by elisa and hearts were collected for histological assessment of fibrosis and measurement of cardiac proteins by Western blotting. Direct effects of moxonidine on cardiac cell death and underlying mechanisms were investigated in vitro by flow cytometry and Western blotting.
KEY RESULTS
After 4 weeks, the sub-hypotensive dose of moxonidine (100 µg) reduced heart rate and improved global cardiac performance, reduced collagen deposition, regressed left ventricular hypertrophy, inhibited Akt and p38 MAPK phosphorylation, and attenuated circulating and cardiac cytokines. The 400 µg dose resulted in similar effects but of a greater magnitude, associated with blood pressure reduction. In vitro, moxonidine inhibited norepinephrine-induced neonatal cardiomyocyte mortality but increased fibroblast mortality, through I1-receptor activation and differential effects on downstream Akt and p38 MAPK.
CONCLUSIONS AND IMPLICATIONS
While the antihypertensive action of centrally acting imidazoline compounds is appreciated, new cardiac-selective I1-receptor agonists may confer additional benefit.
Keywords: left ventricular hypertrophy, echocardiography, apoptosis, flow cytometry, cardiomyocytes, p38 MAPK, Akt, cytokines
Introduction
The hypertensive heart undergoes several structural and functional alterations, including increased left ventricular mass, abnormal left ventricular texture, function and geometry, and impaired coronary reserve. Left ventricular hypertrophy (LVH), present in all patients with severe hypertension, is linked to unfavourable prognosis. A variety of deleterious consequences of cardiovascular diseases, with conditions such as coronary heart disease, stroke, congestive heart failure and sudden death, are aggravated by LVH. It is not surprising therefore that numerous therapeutic approaches have been pursued with the aim of regressing or preventing LVH and, hence, reducing cardiovascular risk (Pierdomenico et al., 2008).
Many genetic, haemodynamic, neurohormonal and growth factors, including sympathetic factors, play an important role in left ventricular remodelling (Grassi et al., 2009). In human hypertension, LVH correlates with increased cardiac noradrenaline spillover (Schlaich et al., 2003). Exposure to high concentrations of noradrenaline causes cardiomyocyte hypertrophy and loss as well as expansion of the interstitial fibroblast compartment and enhanced collagen deposition. noradrenaline stimulates the β-adrenoceptor pathway that includes activation of oxidative stress and the pro-inflammatory cytokines, TNF-α, IL-1β (Communal et al., 1998; Fu et al., 2004; Neri et al., 2007) and IL-6 (Leicht et al., 2003).
Moxonidine is a centrally acting antihypertensive imidazoline compound that reduces excessive sympathetic tone and peripheral vascular resistance, accompanied by reduced plasma noradrenaline concentration (Wenzel et al., 1998). Moxonidine treatment in patients and rats results in regression of cardiac hypertrophy (Haczynski et al., 2001; Paquette et al., 2008; Mukaddam-Daher et al., 2009), restoring the myocardial structure and improving the coronary flow reserve (Mitrovic et al., 2001). We have reported that the anti-hypertrophic effect of moxonidine in hearts from spontaneously hypertensive rats (SHR), results from a sustained reduction of DNA synthesis and transient stimulation of DNA fragmentation and apoptotic proteins Bax and caspase3, occurring after 1 week but subsiding by 4 weeks of treatment. Cardiomyocyte apoptosis plays a causal role in the development of heart failure through progressive left ventricular wall thinning (Liao et al., 2004). Inhibition of cardiomyocyte apoptosis attenuates contractile dysfunction in heart failure (Foo et al., 2005). Accordingly, if moxonidine-stimulated apoptosis occurred in cardiomyocytes, cardiac dysfunction would ensue. In addition, a sub-hypotensive dose of moxonidine tended to offset some of the favourable effects of the angiotensin receptor blocker, eprosartan, on cardiac function in stroke-prone SHR (Mukaddam-Daher et al., 2009), suggesting that higher doses may be detrimental. These studies therefore investigated, in SHR, the time- and dose-dependent effects of chronic moxonidine treatment on cardiac structure and function as well as on the inflammatory process and signalling proteins involved in cardiac cell survival/death. In this respect, studies revealed pressure-dependent and -independent improvement of cardiac function and regression of LVH and fibrosis by moxonidine. Together with our previous finding that imidazoline I1-receptors, which selectively mediate the actions of moxonidine in brainstem medulla, are also expressed in the heart (El-Ayoubi et al., 2002), these results led us to suggest that, in addition to central actions, the control of LVH by moxonidine may, at least in part, include direct effects on the heart. However, the participation of cardiac I1-receptors in LVH regression in whole animal studies may be masked by the antihypertensive and sympatholytic effects of the drug. Therefore; the direct effect of moxonidine on cardiac cell growth and death and the implicated signalling proteins were investigated in vitro, on cardiomyocytes and fibroblasts in culture, in the absence of confounding mechanisms.
Methods
For further details of the methods please see Appendix S1.
Animals
All animal care and experimental procedures for this study were approved by the Institutional Committee for Animal Protection, according to the Canadian Guidelines. SHR and normotensive Wistar-Kyoto (WKY) rats, purchased from Charles River (St. Constant, Quebec, Canada), were investigated at 12–13 weeks of age, when hypertension and LVH are established. The animals were housed at 22°C, maintained on a 12 h light-dark cycle, fed Purina Rat Chow (Ralston Purina) and had free access to tap water. They were allowed at least 3 days before experimentation.
Experimental protocols
Cardiac structure and function were analysed by transthoracic echocardiography, as previously described (Mukaddam-Daher et al., 2009). Then, the rats were randomly assigned to treatment with moxonidine (0, 100 or 400 µg·kg−1·h−1), for 1 and 4 weeks, via Alzet osmotic minipumps (model 2ML1 and 2ML4, Alzet Corporation Cupertino, CA, USA), implanted subcutaneously at the neck area, under isoflurane anaesthesia, as previously described (Paquette et al., 2008; Mukaddam-Daher et al., 2009). The low and high concentrations of moxonidine (sub-hypotensive and antihypertensive respectively) were chosen from previous studies showing regression of LVH in hypertensive rats (Paquette et al., 2008; Mukaddam-Daher et al., 2009).
Rats were weighed and inspected twice per week. Food and water intake and 2 × 24 h urine output (metabolic cages) were measured after 4 weeks of treatment. Echocardiographic examination was repeated after 4 weeks of treatment. At the end of the experiment (after 1 and 4 weeks), intra-carotid blood pressure was recorded under 2% isoflurane anaesthesia. Then, blood and hearts were collected for later measurements of cytokines, molecular determinations and histopathological examination, as previously described (Paquette et al., 2008; Mukaddam-Daher et al., 2009).
Histopathological examination
Cross-sections of heart ventricles were stored in neutral buffered 4% formalin. After ethanol dehydration and embedding in paraffin, 5 µm sections were obtained and stained with haematoxylin-phloxine-saffron for cell surface measurements, or with Masson's Trichrome for measurement of collagen deposition. Microscopic visualization and photographs were obtained and measurements performed by a blinded investigator, using computer software (Micro ImageJ1.38x, NIH, USA).
Plasma and cardiac cytokines
Plasma inflammatory cytokines (IL-1β, IL-6 and TNF-α) and anti-inflammatory cytokine (IL-10) were measured by Bio-Plex 200 System (Bio-Rad Laboratories, ON, Canada). IL-1β (Pierce/Thermo scientific, Rockford, IL, USA) and TNF-α (Invitrogen, Camarillo, CA, USA) were also measured in left ventricles, by elisa.
Cardiac cell culture, flow cytometry
Cardiomyocytes and fibroblasts were isolated from neonatal (1–2 days old) rat hearts using the Neonatal Cardiomyocyte Isolation System (Worthington Biochemical Corp, Lakewood, NJ, USA), following the manufacturer's instructions. Cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum, then serum deprived (DMEM + 0.1% fetal bovine serum) for 24 h to induce quiescence, before incubation with DMEM alone or with increasing concentrations of noradrenaline (10−7 to 10−4 mol·L−1) without and with moxonidine at 10−7 and 10−5 mol·L−1 concentrations. Cell death was measured after 24 and 48 h, by flow cytometry after the addition of propidium iodide. In addition, cells were incubated with the above treatments for 15 min, then collected for measurement of Akt and MAPK phosphorylation, by Western blot. The participation of imidazoline I1-receptors (nomenclature follows Alexander et al., 2009) was identified using the I1-receptor antagonist, AGN192403 (10−5 mol·L−1), added 15 min before addition of treatment products.
Western blot analysis
Cardiac proteins were measured by Western blotting using antibodies recognizing the following antigens: total and phospho-specific Akt at Ser473 (1:1000), total and phospho-specific ERK1/2 at Thr202/Tyr204 (1:1000), total and phospho-specific p38 MAPK at Thr180/Tyr182 (1:1000), total and phospho-specific c-Jun N-terminal kinase (JNK) at Thr183-Tyr185 (1:1000) (Cell signalling Technology, Inc, MA, USA), α-smooth muscle actin (α-SMA, 1:1000) and anti-GAPDH (1:10 000) (Sigma-Aldrich, ON, Canada). Imidazoline I1-receptor protein expression was identified in cardiomyocytes and fibroblasts using antibody raised against the murine form, nischarin (1:1000, BD Biosciences, ON, Canada). Densitometric measurements of bands was performed using ImageQuant 5.1.
Statistical analysis
All data obtained from SHR were compared with corresponding normotensive WKY rats and moxonidine-treated SHR. Data obtained from cultured cells were compared with corresponding DMEM-, noradrenaline- or noradrenaline + moxonidine-treated cells, as required. Comparisons were performed by one-way anova (and non-parametric) test followed by Neuman–Keuls multiple comparison test, using the computer program PRISM 4.0. Statistical significance was taken as P < 0.05. Data are reported as mean ± SEM.
Materials
The sources of the compounds used were as follows: noradrenaline (L-(-)-norephinephrine (+)-bitartrate salt monohydrate) from Sigma-Aldrich Canada Ltd., Oakville, Ontario, Canada; AGN192403 (2-endo-amino-3-exo-isopropylbicyclo[2.2.1]heptane) from Cedarlane Laboratories, Burlington, Ontario, Canada; isoflurane, from Abbott Laboratories, Saint-Laurent, Quebec, Canada.
Results
Physical and haemodynamic parameters
The effect of moxonidine on biomechanical stress in the hypertensive heart was investigated in SHR and compared with normotensive WKY rats. The SHR had higher blood pressure, LVH (left ventricular mass normalized to tibia length) and hypertrophied cardiomyocytes (Table 1), as well as higher interstitial and perivascular collagen volume ( Figure 1). Moxonidine (100 µg) lowered blood pressure and heart rate after 1 week, and by 4 weeks it lowered heart rate, LVH and collagen deposition, without significant changes in blood pressure. Moxonidine at 400 µg reduced blood pressure, heart rate and LVH, at 1 and 4 weeks and collagen deposition after 4 weeks of treatment (Table 1, Figure 1). Moxonidine did not alter 24 h volume and electrolyte excretion in these rats (data not shown).
Table 1.
Physical and haemodynamic parameters after 1 and 4 weeks of treatment
| SHR + moxonidine (µg·kg−1·h−1) 1 week | SHR + moxonidine (µg·kg−1·h−1) 4 weeks | ||||||
|---|---|---|---|---|---|---|---|
| Haemodynamic and physical parameters | WKY | 0 | 100 | 400 | 0 | 100 | 400 |
| MAP (mm Hg) | 94 ± 3 | 167 ± 1* | 147 ± 4‡ | 128 ± 5‡ | 160 ± 3* | 152 ± 5 | 130 ± 7‡ |
| Heart rate (bpm) | 337 ± 7 | 335 ± 8 | 313 ± 8 | 277 ± 14† | 338 ± 10 | 282 ± 11† | 265 ± 8‡ |
| LVM (mg) | 455 ± 13 | 551 ± 4* | 527 ± 25 | 488 ± 8‡ | 595 ± 6* | 571 ± 9† | 535 ± 10‡ |
| LVH (mg·mm−1) | 10.1 ± 0.3 | 12.3 ± 0.1* | 11.8 ± 0.6 | 11.0 ± 0.2‡ | 13.1 ± 0.1* | 12.6 ± 0.2† | 11.9 ± 0.2‡ |
| CSA (µm) | 407 ± 4 | 479 ± 2* | 444 ± 6‡ | 435 ± 5† | 565 ± 2* | 522 ± 5‡ | 488 ± 3‡ |
P < 0.05 versus WKY
P < 0.05
P < 0.01 versus corresponding vehicle.
CSA: cross-sectional area; LVH, left ventricular hypertrophy; LVM: left ventricular mass; MAP, mean arterial pressure; SHR, spontaneously hypertensive rats; WKY, Wistar-Kyoto.
Figure 1.
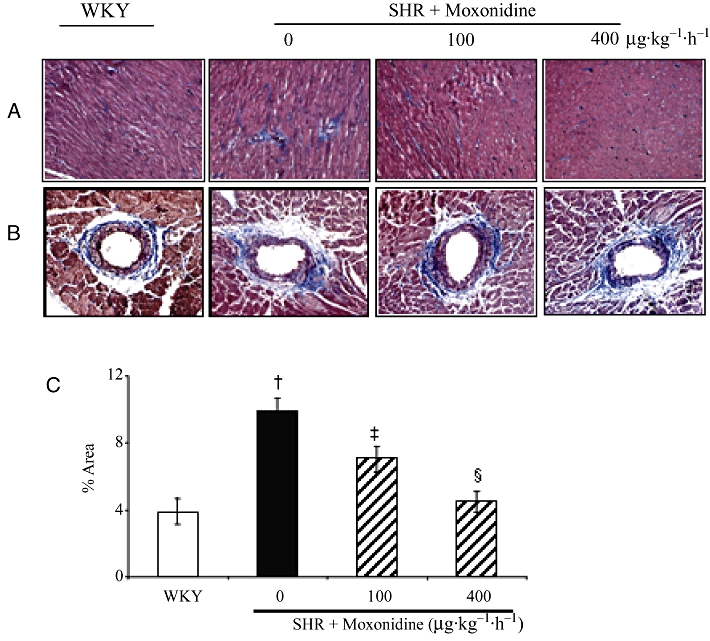
Representative photomicrographs of Masson's Trichrome stained heart sections of spontaneously hypertensive rats (SHR) and Wistar-Kyoto (WKY) rats with and without moxonidine treatment. (A) Interstitial, (B) perivascular collagen deposition (in blue) and (C) percentage of interstitial and perivascular collagen deposition in total area. n = 4–6 rats per group; †P < 0.01 versus WKY; ‡P < 0.05 versus vehicle; §P < 0.01 versus vehicle. Magnification 200×.
The structure and function of the left ventricle, evaluated by echocardiography (Table 2), showed that there was no significant difference in systolic functions, ejection fraction and fractional shortening among the WKY and SHR groups. Anterior wall and left ventricular posterior wall tended to increase, but relative wall thickness and left ventricular end systolic diameter were significantly higher in SHR, indicating LVH. There was no significant difference among the WKY and the three SHR groups in the ejection fraction and fractional shortening of the left ventricle, indicating maintained systolic function (Table 2). The diastolic and global functions of the heart were compromised in SHR, evidenced by delayed isovolumic relaxation time (IVRT) and higher left ventricular myocardial performance index (LVMPI) (Table 2). All systolic and diastolic function parameters were not altered within the study duration in vehicle-treated rats. However, the two concentrations of moxonidine increased left ventricular ejection time and E wave deceleration time, and reduced E wave deceleration rate, in comparison to corresponding vehicle-treated SHR (Figure 2) or pretreatment values (not shown). In addition, 400 µg moxonidine lowered IVRT (Figure 2), and reduced relative wall thickness as well as LVMPI (Table 2), denoting decreased hypertrophy and improved global cardiac performance.
Table 2.
Echocardiographic measurements after 4 weeks of treatment
| WKY | SHR + moxonidine (µg·kg−1·h−1) | |||
|---|---|---|---|---|
| Echocardiographic parameters | 0 | 0 | 100 | 400 |
| ESD (mm) | 2.3 ± 0.0 | 2.7 ± 0.1 | 2.8 ± 0.2 | 2.9 ± 0.1 |
| EDD (mm) | 5.8 ± 0.1 | 5.6 ± 0.1 | 6.0 ± 0.0 | 5.8 ± 0.0 |
| AW (mm) | 1.7 ± 0.1 | 1.8 ± 0.1 | 1.7 ± 0.0 | 1.7 ± 0.1 |
| PW (mm) | 2.1 ± 0.1 | 2.3 ± 0.0 | 2.2 ± 0.1 | 2.1 ± 0.1 |
| RWT | 0.72 ± 0.04 | 0.83 ± 0.03* | 0.68 ± 0.06† | 0.68 ± 0.07† |
| ESV (mL) | 0.04 ± 0.00 | 0.06 ± 0.00* | 0.07 ± 0.01 | 0.08 ± 0.01 |
| EDV (mL) | 0.69 ± 0.04 | 0.63 ± 0.02 | 0.76 ± 0.04‡ | 0.70 ± 0.03 |
| Fractional shortening (%) | 60.4 ± 0.9 | 52.8 ± 0.7‡ | 54.1 ± 1.4 | 50.7 ± 1.8 |
| Ejection fraction (%) | 92.4 ± 0.4 | 91.3 ± 0.4 | 91.0 ± 0.7 | 88.5 ± 1.2 |
| Stroke index (mL·100 g−1) | 0.39 ± 0.02 | 0.29 ± 0.01* | 0.36 ± 0.02‡ | 0.35 ± 0.02‡ |
| Cardiac index (mL·min−1·100 g−1) | 124 ± 8 | 98 ± 6* | 98 ± 8 | 87 ± 5 |
| LVMPI | 0.17 ± 0.02 | 0.32 ± 0.02* | 0.31 ± 0.02 | 0.26 ± 0.02† |
P < 0.05 versus WKY
P < 0.05
P < 0.01 versus corresponding vehicle.
AW, anterior wall; EDD, end diastolic diameter; EDV, end diastolic volume; ESD, end systolic diameter; ESV, end systolic volume; LVMPI, left ventricular myocardial performance index; PW, posterior wall; RWT, relative wall thickness; SHR, spontaneously hypertensive rats; WKY, Wistar-Kyoto.
Figure 2.
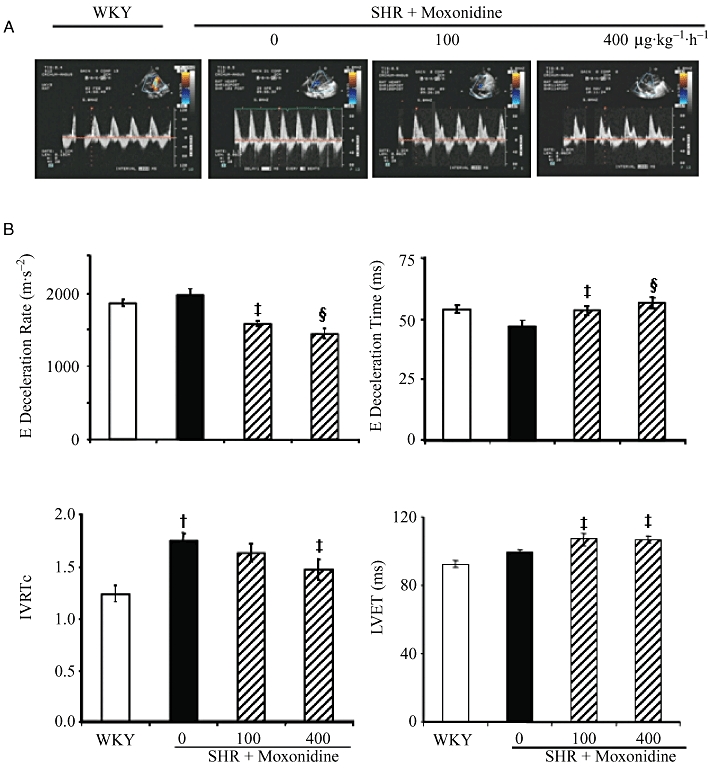
(A) Representative echo-Doppler images of diastolic function parameters in WKY and SHR. (B) Bargraphs represent data obtained after 4 weeks of study. n = 10–11 rats per group. †P < 0.01 versus WKY; ‡P < 0.05, §P < 0.01 versus vehicle. IVRTc, isovolumic relaxation time corrected for heart rate; LVET, left ventricular ejection time.
Cytokines
Inflammatory and anti-inflammatory cytokines were measured in plasma and hearts of SHR with and without moxonidine treatment and compared with WKY rats. Figure 3 shows that compared with WKY, circulating TNF-α and IL-6 levels were reduced by treatment, but IL-1β levels were elevated in SHR and were further stimulated by moxonidine after 1 week, then tended to decrease after 4 weeks of treatment. Plasma IL-10 was not altered by hypertension or treatment. In contrast, left ventricular IL-1β (28 ± 1 pg·mg−1 protein) which was not altered by hypertension, was significantly decreased (14 ± 1 and 17 ± 2 pg·mg−1 protein; P < 0.01) after 4 weeks of treatment with both moxonidine concentrations respectively. Left ventricular TNF-α (25 ± 5 pg·mg−1 protein) was not altered by hypertension or treatment (Figure S1).
Figure 3.
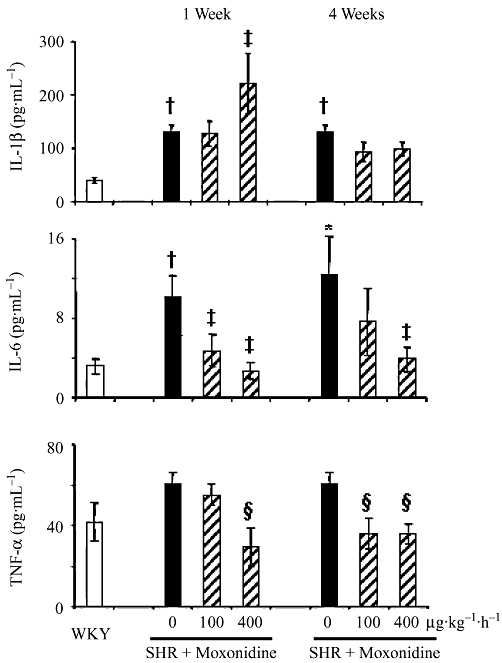
Effect of moxonidine treatment on plasma cytokines. n = 9–16 rats per group. *P < 0.05, †P < 0.01 versus WKY rats; ‡P < 0.05; §P < 0.01 versus vehicle.
Cardiac proteins
To further elucidate the underlying molecular mechanism involved in signal transduction responsible for cardioprotection by moxonidine, we investigated activation of Akt and the MAPK pathway in SHR and WKY rat hearts. Figure 4 and Figure S2 show that the protein expression of P-Akt/Akt, P-ERK/ERK and P-p38/p38 were enhanced in SHR left ventricles. Akt and p38 (Figure 4), but not ERK phosphorylation (Figure S2) were reduced by the two concentrations of moxonidine after 1 and 4 weeks of treatment. Neither hypertension nor treatment had an effect on total or phosphorylated JNK (Figure S2). Figure 4 also shows that moxonidine reduced the protein expression of α-SMA, a marker of fibroblast differentiation into contractile and hyper-secretory myofibroblasts (Petrov et al., 2002).
Figure 4.
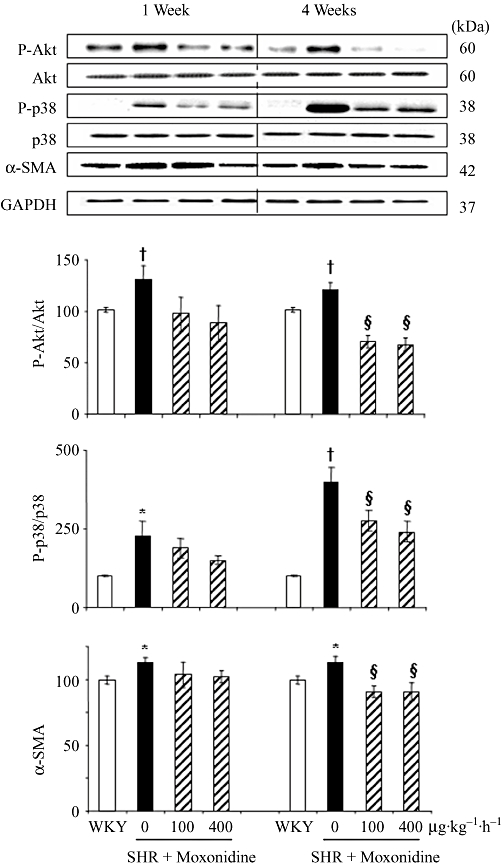
Western blot analysis of the effect of treatments on left ventricular Akt and p38 phosphorylation at 1 and 4 weeks of treatment. Column graphs depict the ratios of phospho-Akt to total Akt, phospho-p38 to total p38, and α-smooth muscle actin (α-SMA), compared to GAPDH (loading control) and presented as per cent of the corresponding WKY values (100%). n = 6–10 rats per group. *P < 0.05, †P < 0.01 versus WKY; §P < 0.01 versus vehicle.
Cardiomyocyte and fibroblast mortality and the underlying mechanisms
Western blot analysis revealed that neonatal rat cardiomyocytes and fibroblasts express imidazoline receptors (Figure S3). Cardiomyocytes and fibroblasts also express α- and β-adrenoceptors whose activation by noradrenaline stimulates cell growth and death. Cardiomyocytes and fibroblasts were exposed to increasing concentrations of noradrenaline, for 24 and 48 h. Analysis by flow cytometry showed that noradrenaline at 10−7 to 10−4 mol·L−1 increased cardiomyocyte mortality in a concentration- and time-dependent manner (data not shown) and that these effects were prevented in cells co-incubated with moxonidine at 10−7 and 10−5 mol·L−1, indicating a protective action of moxonidine in cardiomyocytes (Figure 5). Moxonidine abolished noradrenaline-induced p38 MAPK phosphorylation, while maintaining noradrenaline-induced Akt phosphorylation (Figure 6). In contrast, incubation of fibroblasts with increasing concentrations of noradrenaline resulted in less cell death (reflecting proliferation), an effect prevented upon co-incubation with moxonidine (Figure 7). Moxonidine also abolished noradrenaline-induced p38 MAPK and Akt phosphorylation (Figure 8). The effects of moxonidine on cell death/protection and downstream signalling were opposed by the antagonist, AGN192403, demonstrating imidazoline I1-receptor-mediated actions.
Figure 5.
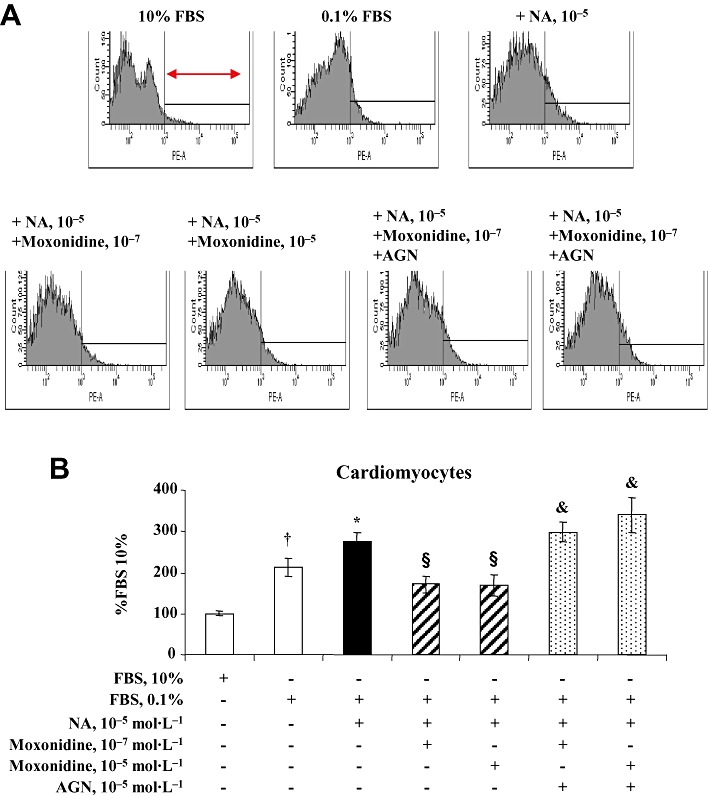
(A) Representative flow cytometry and propidium iodide staining depicting total mortality of neonatal rat cardiomyocytes in culture. Cells on the right (arrow) represent the percentage of cell death out of total number of cells measured. (B) Bargraph represents cardiomyocyte mortality after 48 h incubation in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), and in conditions of starvation (DMEM + 0.1% FBS) alone or in addition to noradrenaline (NA) without and with co-incubation with moxonidine at 10−7 and 10−5 mol·L−1, without and with and I1-receptor antagonist, AGN 192403 (AGN) at 10−5 mol·L−1. Data presented as per cent FBS 10%. n = 8–12 wells per treatment, from five independent cultures. †P < 0.01 versus 10% FBS; *P < 0.05 versus 0.1% FBS; §P < 0.01 versus NA; &P < 0.05 versus corresponding NA + moxonidine.
Figure 6.
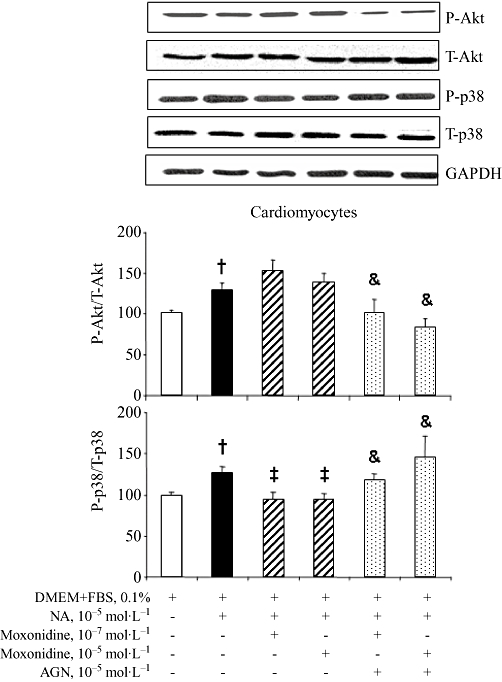
(A) Representative bands of total and phosphorylated Akt and p38 MAPK detection by Western blot in cultured neonatal rat ventricular cardiomyocytes incubated with noradrenaline (NA) alone, or upon co-incubation with moxonidine at 10−7 and 10−5 mol·L−1, without and with I1-receptor antagonist, AGN 192403 (AGN) at 10−5 mol·L−1. (B) Bargraph represents ratios of phospho-Akt to total Akt and phospho-p38 to total p38, compared with GAPDH (loading control) and presented as per cent corresponding untreated cells. n = 10–12 wells per treatment from three independent cultures. †P < 0.01 versus Dulbecco's modified Eagle's medium (DMEM) + fetal bovine serum (FBS), 0.1%; ‡P < 0.05 versus NA; &P < 0.05 versus corresponding NA + moxonidine.
Figure 7.
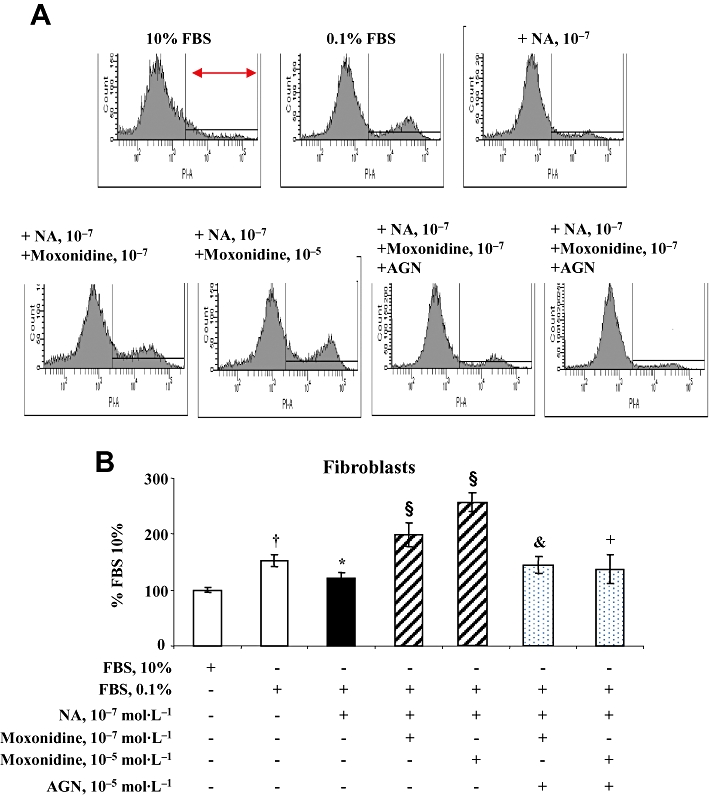
(A) Representative flow cytometry and propidium iodide staining depicting total mortality of neonatal rat cardiac fibroblasts in culture. Cells on the right represent the percentage of cell death out of total number of cells measured. (B) Bargraph represents fibroblast mortality after 48 h incubation in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), and in conditions of starvation (DMEM + 0.1% FBS), noradrenaline (NA) alone, or upon co-incubation with moxonidine at 10−7 and 10−5 mol·L−1, without and with I1-receptor antagonist, AGN 192403 (AGN) at 10−5 mol·L−1. Data presented as per cent FBS 10%. n = 8–12 wells per treatment, from five independent cultures. †P < 0.01 versus 10% FBS; *P < 0.05 versus 0.1% FBS; §P < 0.01 versus NA; &P < 0.05, +P < 0.01 versus corresponding NA + moxonidine.
Figure 8.
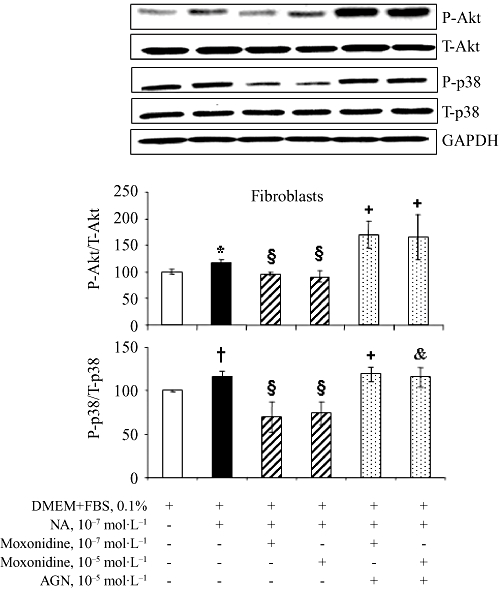
(A) Representative bands of total and phosphorylated Akt and p38 MAPK detection by Western blot in cultured neonatal rat ventricular fibroblasts incubated with noradrenaline (NA) alone, or upon co-incubation with moxonidine at 10−7 and 10−5 mol·L−1, without and with I1-receptor antagonist, AGN 192403 (AGN) at 10−5 mol·L−1. (B) Bargraph represents ratios of phospho-Akt to total Akt and phospho-p38 to total p38, compared with GAPDH (loading control) and presented as per cent corresponding untreated cells. n = 10–12 wells per treatment from three independent cultures.*P < 0.05, †P < 0.01 versus Dulbecco's modified Eagle's medium (DMEM); §P < 0.01 versus NA; &P < 0.05, +P < 0.01 versus corresponding NA + moxonidine. FBS, fetal bovine serum.
Discussion
Studies were performed in SHR, a genetic model of hypertension that shares many common features of human essential hypertension, including increased activity of the sympathetic nervous system and cardiac noradrenaline spillover and myocardial inflammation (Kai et al., 2005; Nonaka-Sarukawa et al., 2008; Kumar et al., 2009). These studies reveal that 1 month treatment with moxonidine improves diastolic function and global cardiac performance in hypertensive rat hearts. The effects were associated with regression of cardiac hypertrophy and fibrosis, attenuation of cardiac IL-1β and circulating TNF-α and IL-6 as well as inhibition of Akt and p38 MAPK phosphorylation. In vitro studies point to a possible contribution of cardiac imidazoline I1-receptors in the structural improvement, protecting against cardiomyocyte death while selectively stimulating loss of fibroblasts.
Echocardiographic/Doppler measurements revealed that systolic function was preserved in SHR but diastolic function was reduced, as shown by reduced left ventricle deceleration and ejection time and delayed relaxation time. Diastolic dysfunction, an early manifestation of cardiac dysfunction in hypertensive patients, correlates with fibrosis and energetic functional changes in myocytes (Verma and Solomon, 2009) and independently correlates with cardiovascular risk and total mortality (Schillaci et al., 2002).
Moxonidine, at both doses, did not affect systolic function but improved diastolic function parameters, even after correction to heart rate changes. The higher dose attenuated the delayed relaxation (IVRT corrected for heart rate) and reduced LVMPI, a calculated index, which includes systolic and diastolic cardiac effects independently from heart rate influence. The improved cardiac performance, which includes reduced stiffness and improved relaxation, may be due to the decrease in blood pressure and inhibition of sympathetic nerve activity and subsequent noradrenaline release by the higher dose of moxonidine (Van Kerckhoven et al., 2000). However, the sub-hypotensive dose, which is lower than the concentration that did not reduce plasma noradrenaline levels in a rat model of myocardial infarction-induced heart failure (Van Kerckhoven et al., 2000), also improved cardiac parameters. The effects were associated with attenuated LVH and collagen accumulation and substantial reduction in left ventricular IL-1β and circulating TNF-α and IL-6. The cytokines, IL-1β, IL-6 and TNF-α are implicated in the pathogenesis of cardiac hypertrophy and cardiomyocyte apoptosis, in vivo and in vitro (Bozkurt et al., 1998; Dhingra et al., 2007) and in stimulation of cardiac fibroblast proliferation and differentiation into activated myofibroblasts, which produce large amounts of collagens (Petrov et al., 2002; Melendez et al., 2010).
Hypertension-associated neurohormones, growth factors and cytokines activate MAPK signalling cascade and the phosphatidylinositol 3-kinase (PI3K)/Akt pathway. MAPKs, including ERK, p38 and JNK have distinct functions: ERK1/2 activation results in cell proliferation and survival responses, whereas JNKs and p38 are implicated in inflammation and cell growth, differentiation, migration and apoptosis (Wang et al., 1998). Akt (or PKB), a serine/threonine kinase with potent anti-apoptotic action in vitro and in vivo, is a downstream target of PI3K and is linked to hyperplasia and hypertrophy of cardiomyocytes and increased proliferation and collagen synthesis in fibroblasts (Colombo et al., 2003; Oudit et al., 2004; Heineke and Molkentin, 2006). MAPK and Akt activation were evaluated in WKY and SHR hearts with and without moxonidine treatment. Our results showed no change in total or phosphorylated JNK but higher levels of phosphorylated ERK, p38 and Akt in SHR than WKY hearts. Previous studies have shown LVH and cardiac fibrosis, in association with elevated p38 and Akt phosphorylation in several models of experimental and genetic hypertension (Behr et al., 2001; Liang et al., 2006; Bao et al., 2007; Bartha et al., 2009; Soesanto et al., 2009; Esposito et al., 2010). Activation of p38 in cardiomyocytes leads to a rapid onset of lethal cardiomyopathy associated with cardiomyocyte hypertrophy, interstitial fibrosis and contractile dysfunction (Liao et al., 2004; Streicher et al., 2010). p38 is activated by cytokines and, in turn, it is involved in the release of pro-inflammatory cytokines (Fotheringham et al., 2004; Li et al., 2005), a mechanism by which p38 contributes to myocyte cell death (Dhingra et al., 2007). On the other hand, ERK and Akt activation reduces cardiomyocyte death and protects against ischaemia-reperfusion injury (Fujio et al., 2000; Liu et al., 2004), but sustained activation of Akt signalling results in progression from adaptive to maladaptive hypertrophy and fibrosis (Taniyama et al., 2005).
Our results showed that moxonidine treatment reduceds the hypertension-associated activation of Akt and pro-apoptotic p38, without affecting anti-apoptotic ERK. Such changes could contribute to attenuation of hypertension-induced myocardial hypertrophy and fibrosis as well as improved cardiac performance. These results are in agreement with studies showing a protective effect of p38-specific inhibitor SB203580, regressing LVH (Behr et al., 2001; Bao et al., 2007) and others reporting improved cardiac performance by treatments that reduced p38 levels (Liang et al., 2006; Bartha et al., 2009) as well as studies showing in SHR hearts that attenuation of high Akt phosphorylation, by blocking mammalian target of rapamycin (mTOR) downstream of Akt, reduces LVH (Soesanto et al., 2009).
The reduction in p38 MAPK and Akt may be indirect effects mediated by central inhibition of noradrenaline and subsequent cytokine release (Communal et al., 1998; Leicht et al., 2003; Fu et al., 2004; Neri et al., 2007), direct effects on cytokine signalling, or even downstream effects of moxonidine opposing the actions of noradrenaline, by activating cardiac imidazoline I1-receptors (El-Ayoubi et al., 2002). While further studies are required to evaluate these mechanisms, in vitro studies on cultured cardiac myocytes and fibroblasts show that moxonidine may act directly on cardiac cells, exerting opposite effects on noradrenaline-induced p38 MAPK and Akt activation in cardiomyocytes and fibroblasts, and consequently reducing cardiomyocyte mortality but stimulating fibroblast mortality. Together, these effects may, at least in part, contribute to LVH control and consequently, improved cardiac function.
In conclusion, these studies show that the benefits of moxonidine extend beyond blood pressure reduction. The previously observed early transient apoptotic effect of moxonidine (Paquette et al., 2008), does not lead to deterioration of cardiac function. On the contrary, it may be therapeutic apoptosis that targets a subpopulation of susceptible cells (deBlois et al., 2005) very likely macrophages and myofibroblasts; thus, reducing cytokine secretion and collagen accumulation. This anti-inflammatory effect, in addition to moxonidine-induced attenuated DNA and protein synthesis (Paquette et al., 2008), may attenuate cardiac hypertrophy and fibrosis and improve cardiac function. Importantly, imidazoline I1-receptors in the heart can control cardiac cell death/survival in the absence of central and haemodynamic contributions. Future new drugs or procedures, specifically targeting heart I1-receptors, may prevent the development of cardiac remodelling and heart failure. While the antihypertensive action of centrally acting imidazoline compounds is appreciated, new cardiac-selective imidazoline receptor agonists with pathway-selective properties may confer additional benefit.
Acknowledgments
Moxonidine was kindly provided by Solvay Pharmaceuticals, GMBH.
Glossary
Abbreviations
- α-SMA
α smooth muscle actin
- AGN192403
(2-endo-amino-3-exo-isopropylbicyclo[2.2.1]heptane)
- AW
anterior wall
- EDD
end diastolic diameter
- EDV
end diastolic volume
- ESD
end systolic diameter
- ESV
end systolic volume
- IVRTc
isovolumic relaxation time corrected for heart rate
- LVET
left ventricular ejection time
- LVETc
left ventricular ejection time corrected for heart rate
- LVH
left ventricular hypertrophy
- LVM
left ventricular mass
- MAP
mean arterial pressure
- PW
posterior wall
- RWT
relative wall thickness
Funding
This work was supported by grants from the Canadian Institutes of Health Research (MOP-82708) and the Heart and Stroke Foundation of Canada (to S. M. D.).
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effect of moxonidine treatment during 4 weeks on left ventricular (LV) cytokines, interleukin 1-beta (IL-1β) and tumour necrosis factor-alpha (TNF-α). n = 7–9 each; §P < 0.01 versus vehicle. SHR, spontaneously hypertensive rats.
Figure S2 Western blot analysis of the effect of treatments on left ventricular ERK1/2 and JNK phosphorylation at 1 and 4 week treatment. Column graph depicts the ratio of phospho-ERK to ERK and p-JNK to JNK, normalized to GAPDH (loading control) and presented as per cent Wistar-Kyoto (WKY) (100%). *P < 0.05 versus WKY. n = 6–10 rats/group. SHR, spontaneously hypertensive rats.
Figure S3 Western blot analysis of imidazoline receptor (nischarin) in left ventricular neonatal cardiomyocytes and fibroblasts (second passage) and GAPDH (loading control). A total of 20 µg protein was loaded.
Appendix S1 Detailed methods.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, Behm DJ, Nerurkar SS, Ao Z, Bentley R, Mirabile RC, et al. Effects of p38 MAPK Inhibitor on angiotensin II-dependent hypertension, organ damage, and superoxide anion production. J Cardiovasc Pharmacol. 2007;49:362–368. doi: 10.1097/FJC.0b013e318046f34a. [DOI] [PubMed] [Google Scholar]
- Bartha E, Solti I, Kereskai L, Lantos J, Plozer E, Magyar K, et al. PARP inhibition delays transition of hypertensive cardiopathy to heart failure in spontaneously hypertensive rats. Cardiovasc Res. 2009;83:501–510. doi: 10.1093/cvr/cvp144. [DOI] [PubMed] [Google Scholar]
- Behr TM, Nerurkar SS, Nelson AH, Coatney RW, Woods TN, Sulpizio A, et al. Hypertensive end-organ damage and premature mortality are p38 mitogen-activated protein kinase-dependent in a rat model of cardiac hypertrophy and dysfunction. Circulation. 2001;104:1292–1298. doi: 10.1161/hc3601.094275. [DOI] [PubMed] [Google Scholar]
- deBlois D, Tea B-S, Beaudry D, Hamet P. Regulation of therapeutic apoptosis: a potential target in controlling hypertensive organ damage. Can J Physiol Pharmacol. 2005;83:29–41. doi: 10.1139/y05-001. [DOI] [PubMed] [Google Scholar]
- Bozkurt B, Kribbs SB, Clubb FJ, Jr, Michael LH, Didenko VV, Hornsby PJ, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-α promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–1391. doi: 10.1161/01.cir.97.14.1382. [DOI] [PubMed] [Google Scholar]
- Colombo F, Gosselin H, El-Helou V, Calderone A. Beta-adrenergic receptor-mediated DNA synthesis in neonatal rat cardiac fibroblasts proceeds via a phosphatidylinositol 3-kinase dependent pathway refractory to the antiproliferative action of cyclic AMP. J Cell Physiol. 2003;195:322–330. doi: 10.1002/jcp.10251. [DOI] [PubMed] [Google Scholar]
- Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. 1998;98:1329–1334. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- Dhingra S, Sharma AK, Singla DK, Singal PK. p38 and ERK1/2 MAPKs mediate the interplay of TNF-alpha and IL-10 in regulating oxidative stress and cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007;293:3524–3531. doi: 10.1152/ajpheart.00919.2007. [DOI] [PubMed] [Google Scholar]
- El-Ayoubi R, Gutkowska J, Regunathan S, Mukaddam-Daher S. Imidazoline receptors in the heart: characterization, distribution, and regulation. J Cardiovasc Pharmacol. 2002;39:875–883. doi: 10.1097/00005344-200206000-00013. [DOI] [PubMed] [Google Scholar]
- Esposito G, Perrino C, Schiattarella GG, Belardo L, di Pietro E, Franzone A, et al. Induction of mitogen-activated protein kinases is proportional to the amount of pressure overload. Hypertension. 2010;55:137–143. doi: 10.1161/HYPERTENSIONAHA.109.135467. [DOI] [PubMed] [Google Scholar]
- Foo RS, Mani K, Kitsis RN. Death begets failure in the heart. J Clin Invest. 2005;115:565–571. doi: 10.1172/JCI24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotheringham JA, Mayne MB, Grant JA, Geiger JD. Activation of adenosine receptors inhibits tumor necrosis factor-alpha release by decreasing TNF-alpha mRNA stability and p38 activity. Eur J Pharmacol. 2004;497:87–95. doi: 10.1016/j.ejphar.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Fu YC, Chi CS, Yin SC, Hwang B, Chiu YT, Hsu SL. Norepinephrine induces apoptosis in neonatal rat cardiomyocytes through a reactive oxygen species-TNF alpha-caspase signalling pathway. Cardiovasc Res. 2004;62:558–567. doi: 10.1016/j.cardiores.2004.01.039. [DOI] [PubMed] [Google Scholar]
- Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia reperfusion injury in mouse heart. Circulation. 2000;101:660–667. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Quarti-Trevano F, Dell'Oro R, Arenare F, Spaziani D, et al. Sympathetic and baroreflex cardiovascular control in hypertension-related left ventricular dysfunction. Hypertension. 2009;53:205–209. doi: 10.1161/HYPERTENSIONAHA.108.121467. [DOI] [PubMed] [Google Scholar]
- Haczynski J, Spring A, Przewlocka-Kosmala M, Flasinski J. Effect of moxonidine on left ventricular hypertrophy in hypertensive patients. J Clin Basic Cardiol. 2001;4:61–65. [Google Scholar]
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Kai H, Kuwahara F, Tokuda K, Imaizumi T. Diastolic dysfunction in hypertensive hearts: roles of perivascular inflammation and reactive myocardial fibrosis. Hypertens Res. 2005;28:483–490. doi: 10.1291/hypres.28.483. [DOI] [PubMed] [Google Scholar]
- Kumar S, Seth S, Jaiswal A, Enjamoori R, Dinda AK, Ray R, et al. Chronic beta-adrenergic activation-induced left ventricular systolic dysfunction is associated with systemic release of TNF-alpha and IL-1-beta in rats. Pharmacol Rep. 2009;61:870–876. doi: 10.1016/s1734-1140(09)70143-4. [DOI] [PubMed] [Google Scholar]
- Leicht M, Briest W, Zimmer HG. Regulation of norepinephrine-induced proliferation in cardiac fibroblasts by interleukin-6 and p42/p44 mitogen activated protein kinase. Mol Cell Biochem. 2003;243:65–72. doi: 10.1023/a:1021655023870. [DOI] [PubMed] [Google Scholar]
- Li M, Georgakopoulos D, Lu G, Hester L, Kass DA, Hasday J, et al. p38 MAP kinase mediates inflammatory cytokine induction in cardiomyocytes and extracellular matrix remodeling in heart. Circulation. 2005;111:2494–2502. doi: 10.1161/01.CIR.0000165117.71483.0C. [DOI] [PubMed] [Google Scholar]
- Liang Q, Elson AC, Gerdes AM. p38 MAP kinase activity is correlated with angiotensin II type 1 receptor blocker-induced left ventricular reverse remodeling in spontaneously hypertensive heart failure rats. J Card Fail. 2006;12:479–486. doi: 10.1016/j.cardfail.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Liao Y, Asakura M, Takashima S, Ogai A, Asano Y, Shintani Y, et al. Celiprolol, a vasodilatory beta-blocker, inhibits pressure overload-induced cardiac hypertrophy and prevents the transition to heart failure via nitric oxide-dependent mechanisms in mice. Circulation. 2004;110:692–699. doi: 10.1161/01.CIR.0000137831.08683.E1. [DOI] [PubMed] [Google Scholar]
- Liu HR, Gao F, Tao L, Yan WL, Gao E, Christopher TA, et al. Antiapoptotic mechanisms of benidipine in the ischemic/reperfused heart. Br J Pharmacol. 2004;142:627–634. doi: 10.1038/sj.bjp.0705847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225–231. doi: 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic V, Hamel M, Miric M, Thormann J, Hamm C. Effect of the imidazoline receptor agonist moxonidine on hemodynamics, coronary circulation, metabolic ischemia markers and the neurohumoral system in patients with essential hypertension. Effects of moxonidine on coronary circulation. Z Kardiol. 2001;90:953–963. doi: 10.1007/s003920170066. [DOI] [PubMed] [Google Scholar]
- Mukaddam-Daher S, Menaouar A, Paquette P-A, Jankowski M, Gutkowska J, Gillis M-A, et al. Hemodynamic and cardiac effects of chronic eprosartan and moxonidine therapy in Stroke Prone Spontaneous Hypertensive Rats. Hypertension. 2009;53:775–781. doi: 10.1161/HYPERTENSIONAHA.108.126524. [DOI] [PubMed] [Google Scholar]
- Neri M, Cerretani D, Fiaschi AI, Laghi PF, Lazzerini PE, Maffione AB, et al. Correlation between cardiac oxidative stress and myocardial pathology due to acute and chronic norepinephrine administration in rats. J Cell Mol Med. 2007;11:156–170. doi: 10.1111/j.1582-4934.2007.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka-Sarukawa M, Okada T, Ito T, Yamamoto K, Yoshioka T, Nomoto T, et al. Adeno-associated virus vector-mediated systemic interleukin-10 expression ameliorates hypertensive organ damage in Dahl salt-sensitive rats. J Gene Med. 2008;10:368–374. doi: 10.1002/jgm.1166. [DOI] [PubMed] [Google Scholar]
- Oudit GY, Sun H, Kerfant BG, Crackower MA, Penninger JM, Backx PH. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J Mol Cell Cardiol. 2004;37:449–471. doi: 10.1016/j.yjmcc.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Paquette P-A, Duguay D, El-Ayoubi R, Menaouar A, Danalache B, Gutkowska J, et al. Control of left ventricular mass by moxonidine involves reduced DNA synthesis and enhanced DNA fragmentation. Br J Pharmacol. 2008;153:459–467. doi: 10.1038/sj.bjp.0707588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov VV, Fagard RH, Lijnen PJ. Stimulation of collagen production by transforming growth factor-β1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension. 2002;39:258–263. doi: 10.1161/hy0202.103268. [DOI] [PubMed] [Google Scholar]
- Pierdomenico SD, Lapenna D, Cuccurullo F. Regression of echocardiographic left ventricular hypertrophy after 2 years of therapy reduces cardiovascular risk in patients with essential hypertension. Am J Hypertens. 2008;21:464–470. doi: 10.1038/ajh.2008.2. [DOI] [PubMed] [Google Scholar]
- Schillaci G, Pasqualini L, Verdecchia P, Vaudo G, Marchesi S, Porcellati C, et al. Prognostic significance of left ventricular diastolic dysfunction in essential hypertension. J Am Coll Cardiol. 2002;39:2005–2011. doi: 10.1016/s0735-1097(02)01896-x. [DOI] [PubMed] [Google Scholar]
- Schlaich MP, Kaye DM, Lambert E, Sommerville M, Socratous S, Esler MD. Relation between cardiac sympathetic activity and hypertensive left ventricular hypertrophy. Circulation. 2003;108:560–565. doi: 10.1161/01.CIR.0000081775.72651.B6. [DOI] [PubMed] [Google Scholar]
- Soesanto W, Lin HY, Hu E, Lefler S, Litwin SE, Sena S, et al. Mammalian target of rapamycin is a critical regulator of cardiac hypertrophy in spontaneously hypertensive rats. Hypertension. 2009;54:1321–1327. doi: 10.1161/HYPERTENSIONAHA.109.138818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streicher JM, Ren S, Herschman H, Wang Y. MAPK-activated protein kinase-2 in cardiac hypertrophy and cyclooxygenase-2 regulation in heart. Circ Res. 2010;106:1434–1443. doi: 10.1161/CIRCRESAHA.109.213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniyama Y, Ito M, Sato K, Kuester C, Veit K, Tremp G, et al. Akt3 overexpression in the heart results in progression from adaptive to maladaptive hypertrophy. J Mol Cell Cardiol. 2005;38:375–385. doi: 10.1016/j.yjmcc.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Van Kerckhoven R, van Veen TA, Boomsma F, Saxena PR, Schoemaker RG. Chronic administration of moxonidine suppresses sympathetic activation in a rat heart failure model. Eur J Pharmacol. 2000;397:113–120. doi: 10.1016/s0014-2999(00)00232-6. [DOI] [PubMed] [Google Scholar]
- Verma A, Solomon SD. Diastolic dysfunction as a link between hypertension and heart failure. Med Clin North Am. 2009;93:647–664. doi: 10.1016/j.mcna.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Wang Y, Huang S, Sah VP, Ross J, Jr, Brown JH, Han J, et al. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem. 1998;273:2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- Wenzel RR, Spieker L, Qui S, Shaw S, Luscher TF, Noll G. I1-imidazoline agonist moxonidine decreases sympathetic nerve activity and blood pressure in hypertensives. Hypertension. 1998;32:1022–1027. doi: 10.1161/01.hyp.32.6.1022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.