

Abstract
BACKGROUND AND PURPOSE
Kaempferol, a dietary flavonoid and phyto-oestrogen, is known to have anti-inflammatory properties. Microglial activation has been implicated in various neurodegenerative diseases. Anti-inflammatory effects of kaempferol and the underlying mechanisms were investigated by using LPS-stimulated microglial BV2 cells.
EXPERIMENTAL APPROACH
Cell viability was measured using MTT and neutral red assays. elisa, Western blot, immunocytochemistry and electrophoretic mobility-shift assay were used to analyse NO, PGE2, TNF-α and IL-1β production, inducible NOS (iNOS), COX-2 expression and the involvement of signalling pathways such as toll-like receptor-4 (TLR4), MAPK cascades, PKB (AKT) and NF-κB. Accumulation of reaction oxygen species (ROS) was measured by nitroblue tetrazolium and 2′7′-dichlorofluorescein diacetate assay. Matrix metalloproteinase activity was investigated by zymography and immunoblot assay. Phagocytotic activity was assessed by use of latex beads.
KEY RESULTS
Kaempferol significantly attenuated LPS-induced NO, PGE2, TNF-α, IL-1β and ROS production and phagocytosis in a concentration-dependent manner. Kaempferol suppressed the expression of iNOS, COX-2, MMP-3 and blocked the TLR4 activation. Moreover, kaempferol inhibited LPS-induced NF-κB activation and p38 MAPK, JNK and AKT phosphorylation.
CONCLUSION AND IMPLICATIONS
Kaempferol was able to reduce LPS-induced inflammatory mediators through the down-regulation of TLR4, NF-κB, p38 MAPK, JNK and AKT suggesting that kaempferol has therapeutic potential for the treatment of neuroinflammatory diseases.
Keywords: kaempferol, lipopolysaccharide, neuroinflammation, microglia, mitogen-activated protein kinases, AKT, NF-κB
Introduction
Microglia is the resident immune surveillance cells of the central nervous system (CNS) and has been proposed to play an active role in host defence and tissue repair in the brain. A large body of evidence has suggested a strong association between microglia-mediated neuroinflammation and the pathogenesis of various neurological and neurodegenerative diseases such as brain ischaemia, Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease and amyotrophic lateral sclerosis (Nguyen et al., 2002; Block and Hong, 2005; Tansey and Goldberg, 2010). The hallmark of neuroinflammation is the activation of microglia (Kim et al., 2000; Walker and Lue, 2005). They readily became activated in response to abnormal stimulation, such as neurotoxins or injury. Activated microglia release a variety of pro-inflammatory cytokines (e.g. IL-1β, IL-6 and TNF-α), reactive oxygen species (ROS), NO and PGE2, which cause neuronal dysfunction and cell death, ultimately creating a vicious cycle (McGeer and McGeer, 1995; Liu et al., 2001; Lai and Todd, 2006).
MMPs consist of a large family of endopeptidases that have been demonstrated to play a critical role in inflammation by modulating inflammatory mediators such as cytokines and chemokines (Rosenberg, 2002; Parks et al., 2004; Manicone and McGuire, 2008). Regulation of MMPs expression and activation is very complex and tightly controlled. Several studies suggest that MMPs can either protect against or contribute to pathology in inflammatory processes. Since MMPs have been linked to axonal growth, myelin formation, angiogenesis and regeneration in the CNS (Yong et al., 2001; Agrawal et al., 2007), uncontrolled expression of MMPs may result in inflammation, demyelination, neurotoxicity and various CNS disorders including ischaemia, AD, PD, multiple sclerosis and malignant glioma (Rosenberg et al., 1996; Yong et al., 1998; Lorenzl et al., 2004; Yin et al., 2006).
Activation of microglia results in the induction of several intracellular signalling pathways. However, our knowledge of these signalling pathways that are stimulated during microglia activation and their modulatory mechanism is limited. Recent studies suggest a possible involvement of the activate members of the MAPK family and phosphoinositide 3-kinase (PI3K)/PKB (AKT) pathway (Cobb, 1999; Nikodemova et al., 2006). MAPKs including p38 MAPK, JNK and ERK1/2 have been suggested to be involved in inflammatory mediator production in response to a variety of stimuli including LPS (Guha and Mackman, 2001; Rao, 2001). Moreover, It has been reported that binding of LPS to toll-like receptor 4 (TLR4) on the surface of microglia leads to the activation of several signal transduction pathways that all lead to the activation of NF-κB, a transducer, to mediate amplifiers and effectors such as proinflammatory cytokines, chemokines and inducible enzymes including inducible NOS (iNOS) and COX-2 (Medzhitov, 2001; Okun et al., 2009; Glass et al., 2010). Nevertheless, the PI3K/AKT pathway is known to regulate cellular activation, inflammatory responses and apoptosis (Cantley, 2002). Recent studies have demonstrated that this pathway imposes a braking mechanism to limit the expression of pro-inflammatory mediators in LPS-treated microglia by inhibiting the JNK and p38 MAPK pathways (Guha and Mackman, 2002).
In recent years, considerable attention has focused on identifying naturally occurring neuroprotective substances that may be promising therapeutics for neurodegenerative disorders. Phyto-oestrogens are plant-derived polyphenolic non-steroidal compounds that have found to be beneficial in the prevention and treatment of cardiovascular diseases, osteoporosis, diabetes and obesity (Bhathena and Velasquez, 2002; Usui, 2006). Flavonoids are naturally occurring polyphenolic compounds that have attracted the attention of the scientific community because of their wide range of biological activities. Kaempferol (3, 4′, 5, 7, -tetrahydroxyflavone), a phyto-oestrogen, and one of the most common dietary flavonoids, is commonly found in tea, broccoli, grapefruit and other various plant sources (Olszewska, 2008). It is known to have anti-oxidative and anti-inflammatory properties. Previous studies have reported that kaempferol modulates iNOS, COX-2 expression in chang liver cells (García-Mediavilla et al., 2007), inhibits LPS-induced NO production and iNOS expression in activated macrophages (Hämäläinen et al., 2007), reduces LPS-induced COX-2 level in RAW 264.7 cells (Kim et al., 2008) and inhibits ROS production through the inhibition of iNOS and TNF-α protein expression in aged gingival tissues (Kim et al., 2007). Recently, Lee et al. (2010) has demonstrated that kaempferol inhibits ultraviolet B-induced COX-2 expression in mouse skin epidermal cells and Mahat et al. (2010) has shown, in a rat model of inflammation, that kaempferol's anti-inflammatory action is mediated by an effect on COX pathways via inhibition of NO production.
Nevertheless, there is no report on the effect of kaempferol against neuroinflammatory conditions. Therefore, this study was designed to investigate whether kaempferol exerts neuroprotection via inhibition of microglial activation and the subsequent neuroinflammation. It is notable that LPS-activated microglial cells have been suggested to be a good cellular model to test the efficacy of the potential of therapeutic compounds for neuroinflammatory disorders (Chao and Hu, 1994; Le et al., 2001). In the present study, we tried to explore the ability of kaempferol to inhibit the LPS-induced expression of iNOS, COX-2, MMPs and subsequent production of ROS, TNF-α, NO, PGE2 and IL-1β in BV2 microglial cells. We also attempted to elucidate the underlying mechanisms by investigating the involvement of TLR4, NF-κB, MAPKs and AKT.
Methods
Materials
Dulbecco's modified Eagel's medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco (Grand Island, NY, USA). LPS (055:B5), recombinant mouse interferon-γ (IFN-γ), kaempferol, 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT), nitroblue tetrazolium (NBT), Griess reagent, PD98059, SB203580 (4-(4-fluorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)-1H-imidazole), SP600125 (1, 9-pyrazoloanthrone, anthrapyrazolone) and 4′, 6-diamidino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich (St Louis, MO, USA). LY 294002 (2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride) and pyrrolidine dithiocarbamate (PDTC) and N-isobutyl-N-(4-methoxyphenylsulphonyl)-glycylhydroxamic acid (NNGH) were purchased from Calbiochem (San Diego, CA, USA). Primary antibodies iNOS, COX-2, pERK, ERK, pJNK, JNK, pP38, p38, NF-κB p65, IκB and Lamin B were purchased from Santa Cruz (Santa Cruz, CA, USA.) and pAKT and AKT were from Cell Signaling Technology (Beverley, CA, USA). IL-1βelisa kit was from Biolegend (San Diego, CA, USA), and TNF-α and PGE2 EIA kit were from Assay design (Ann Arbor, MI, USA).
Cell culture and treatment
The murine microglial BV2 cells, originally developed by Dr V. Bocchini at University of Perugia (Perugia, Italy; Blasi et al., 1990) were generously provided by Professor Hong Sung Chun at Chosun University (Gwangju, Korea). The cells were cultured in DMEM supplemented with 10% FBS, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin, and kept at 37°C in humidified 5% CO2/95% air. BV2 cells (passage 21) were subcultured every 3 days by trypsinization when growing up to 75% confluence. Kaempferol was dissolved in dimethyl sulphoxide (DMSO) to a final concentration of 0.01%. LPS was diluted in Hanks' balanced salt solution and BV2 cells were stimulated with 1.0 µg·mL−1 LPS for indicated time. In all experiments, cells were treated with the indicated concentrations of kaempferol 1 h before the addition of LPS.
Analysis of cell viability
Cell viability was measured by MTT and neutral red assays. For MTT assay, BV2 cells were seeded in 96-well plate at a density of 1 × 104 cell per well and incubated for 24 h prior to experimental treatments. The cells were then treated with different concentrations of kaempferol (10, 30, 50 and 100 µM). Similarly, cells were treated with kaempferol (10–100 µM) for 1 h then stimulated with 1.0 µg·mL−1 LPS (kaempferol + LPS group), with LPS alone and with vehicle only. After incubation for 24 h, the culture medium was then removed and MTT (0.5 mg·mL−1) was added in each well. Following an additional 4 h incubation at 37°C, 100 µL of DMSO was added in each well to dissolve formazan crystals. The absorbance was then measured at 540 nm using a VERSAmax micro plate reader (Molecular Devices, CA, USA). Wells without cells were used as blanks and were subtracted as background from each sample. Results are expressed as a percentage of control. For neutral red assay, BV2 cells were cultured overnight in 24-well plate at a density of 1 × 105 cells per well. The cells were then pretreated with kaempferol (10–100 µM) for 1 h before being stimulated with 1.0 µg·mL−1 LPS for 24 h. For determination of cell viability, cells were washed with PBS and incubated for 3 h in medium containing neutral red (100 µg·mL−1) as described previously (Richter-Landsberg and Besser, 1994). The damaged or dead cells lose the ability to retain neutral red. Cells were washed with PBS and the dye was extracted with a solution of acetic acid (1%)/methanol (50%) (300 µL per well). After agitating for 10 min, the aliquots were transferred to 96-well plate and the absorbance was measured at 540 nm in VERSAmax micro plate reader. After similar treatments, neutral red incubation and washing were carried out and pictured under a fluorescent microscope (Nikon, Eclipse TE 2000-U, Japan).
Measurement of NO production
Production of NO was assayed by measuring the levels of nitrite in culture medium as previously described (Green et al., 1982). BV2 cells were cultured overnight in 24-well plates at a density of 1 × 105 cells per well. Cells were pretreated with kaempferol (10–100 µM) for 1 h and stimulated with 1.0 µg·mL−1 LPS or 20 U·mL−1 of IFN-γ for 24 h. Similarly, in another experiment, SB203580 (2.5 µM); a selective p38 inhibitor, PD98059 (10 µM); a selective MEK/ERK inhibitor, SP600125 (10 µM); a selective JNK inhibitor, and PDTC (50 µM); a selective NF-κB inhibitor was added 1 h before treatment with LPS (1.0 µg·mL−1) while LY294002 (20 µM); a selective AKT inhibitor was added 30 min before LPS exposure. The culture supernatants were then reacted with an equal volume of Griess reagent (1% sulphanilamide, 0.1% naphthylethylenediamine dihydrochloride and 2% phosphoric acid) for 10 min at room temperature in the dark. Absorbances were measured using a microplate reader (VERSAmax micro plate reader, Molecular Devices, CA, USA) at 542 nm. Nitrite concentrations were calculated by using standard solution of sodium nitrite.
IL-1β, TNF-α and PGE2 measurement
BV2 cells were plated at a density of 1 × 105 cells per well in a 24-well plate. Cells were then pretreated with kaempferol (10–100 µM) for 1 h and stimulated with LPS for 24 h. Production of IL-1β, TNF-α and PGE2 in culture medium were measured by mouse IL-1βelisa kit, and TNF-α PGE2 EIA kit according to the procedures provided by the manufacturers. For detection of the involvement of MAPKs and AKT in LPS-induced PGE2 production, cells were pre-incubated with SB203580, a selective p38 inhibitor, PD98059, a selective MEK/ERK inhibitor, SP600125, a selective JNK inhibitor was added 1 h before treatment with LPS while LY294002, a selective AKT inhibitor was added 30 min before LPS exposure. Production of PGE2 in culture medium was measured.
Measurement of ROS production
Production of ROS was measured by two different methods; modified NBT assay and 2′7′-dichlorofluorescein diacetate (DCF-DA) assay. The modified NBT assay is based on the conversion of NBT to NBT diformazan by superoxide anion. DCF-DA assay is based on the ROS-dependent oxidation of DCF-DA to the highly fluorescent compound dichlorofluorescein (DCF). The cells (1 × 105 cell per well) were cultured in 96-well plates and then treated with 10–100 µM kaempferol for 1 h and then stimulated with 1.0 µg·mL−1 LPS for 24 h. For NBT assay, 0.1% NBT was added to the media at the end of the treatment periods. After incubation for 45 min at 37°C, the cells were washed twice with PBS, then once with methanol, and air-dried. The NBT deposited inside the cells was then dissolved with 240 µL of 2 M potassium hydroxide (KOH) and 280 µL of DMSO with gentle shaking for 10 min at room temperature. The dissolved NBT solution was then transferred to a 96-well plate and absorbance was measured using a microplate reader (VERSAmax micro plate reader, Molecular Devices, CA, USA) at 630 nm. Meanwhile, BV2 cells were cultured in 24-well plates at a density of 1 × 105 cells per well. After similar treatments, NBT incubation, washing and fixing with methanol were carried out. The cells were then monitored by a fluorescent microscope (Nikon, Eclipse TE 2000-U, Japan). For DCF-DA assay, medium was removed and cells were washed twice with PBS. After being washed, the cells were incubated with DCF-DA (10 µM) for 30 min at 37°C in the dark. Cellular fluorescence was measured in a fluorescence microplate reader (Spectra Max Gemini EM, Molecular Devices, Sunnyvale, CA, USA) at excitation wavelength 485 nm and emission wavelength 535 nm. The cells were also pictured by using fluorescent microscope (Nikon, Eclipse TE 2000-U, Japan).
Western blot analysis
BV2 cells were seeded in 100 mm cell culture plate at a density of 1 × 105 cells per plate. The treated cells were washed with ice-cold PBS and then were incubated with RIPA buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg·mL−1 leupeptin) for 30 min. Cells lysates were centrifuged at 17 700×g for 15 min, and the protein concentrations were determined by the bicinchoninic acid (BCA) method using bovine serum albumin as standard. Equal amounts of cell extracts were separated by electrophoresis using a 10.5% SDS-polyacrylamide gel and transferred to polyvinylidine difluoride (PVDF) membrane. After being blocked at room temperature in 5% non-fat dry milk with Tris-buffered saline Tween-20 (TBST) buffer (10 mM Tris-HCl, 150 mM NaCl and 0.1% Tween 20, pH 7.5) for 2 h, the membrane was incubated with primary antibody for iNOS (1:2000 dilution), COX-2 (1:1000 dilution), ERK, pERK, JNK, pJNK, P38 and pP38 (1:1000 dilution), AKT and pAKT (1:2000 dilution), MMP-3, MMP-9 (1:1000 dilution), Lamin B (1:1000) and Actin (1:2500 dilution) overnight at 4°C. Membranes were washed three times in TBST buffer, and incubated with horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature. To reveal the reaction bands, the membrane was reacted with WESTZOL (plus) Western blot detection system (Intron Biotechnology, Inc., Korea) and exposed on X-ray film (Fujifilm Corporation, Tokyo, Japan).
MMP zymography
MMP-3,9 activities in the culture medium were determined by 10% SDS-polyacrylamide gels containing casein/gelatin. BV2 cells were treated with 100 µM kaempferol for 1 h followed by stimulation with LPS for 24 h. After treatment, the culture medium was collected and centrifuged at 17 700×g for 5 min at 4°C to remove cells and debris. Cell mediums were mixed with SDS sample buffer and applied on the gel. After being run, the gels were incubated in the renaturing buffer (2.5% Triton X-100) for 45 min with gentle agitation at room temperature. After removal of the renaturing buffer, the gels were incubated in the developing buffer (50 mM Tris base, 40 mM HCl, 200 mM NaCl, 5 mM CaCl2 and 0.2% Briji 35), overnight at 37°C. After incubation, the gels were stained with staining buffer (30% methanol, 10% acetic acid and 0.5% w/v coomassie Brilliant Blue R-250) and destained with destaining buffer (10% methanol, 10% acetic acid and 80% distilled water). Area of proteinase activity was visualized as clear bands.
Immunocytochemistry
The BV2 cells were seeded into eight-well chamber slides and treated with kaempferol and LPS. Then, the cells were rinsed twice with PBS and fixed with 4% paraformaldehyde solution for 10 min at 4°C. The cells were rinsed with PBS and permeabilized with in 0.4% Triton X-100 for 20 min at room temperature. After three rinses with PBS, the permeabilized cells were blocked with 1% bovine serum albumin for 2 h at room temperature. The blocked cells were incubated with rabbit anti-NF-κB p65 primary antibody (1:200 dilutions) at 4°C overnight. After being washed three times with PBS, the cells were then incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit secondary antibody (1:400 dilutions) for 2 h at room temperature. After a wash, nuclei were counterstained with 1 µg·mL−1 DAPI solution for 15 min in dark. The cells were observed with fluorescent microscope (Nikon, Eclipse TE 2000-U, Japan) and photographed at 100× magnification.
Detection of NF-κB p65 translocation
BV2 cells were seeded in 100 mm cell culture plate at a density of 1 × 105 cells per plate. The cells were then incubated with LPS and/or kaempferol for 1 h. For detection of NF-κB p65 translocation, cells were rinsed with PBS and suspended in hypotonic buffer A [10 mM HEPES, pH 7.6, 10 mM KCl, 1 mM dithiothreitol (DTT), 0.1 mM EDTA and 0.5 mM PMSF] for 10 min 4°C. Cell lysates were centrifuged at 12 000×g for 2 min to separate into cytosolic and nuclear fractions. The supernatants containing cytosolic proteins were transferred to new tube. The pellet containing nuclei was resuspended in buffer C (20 mM HEPES, pH 7.6, 1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, 25% glycerol and 0.4 M NaCl) for 30 min at 4°C. The supernatants containing nuclei proteins were centrifuged at 12 000×g for 20 min. The protein concentrations were determined by the BCA method. The cytosolic and nuclei proteins were then subjected to Western blotting as mentioned above using anti-NF-κB p65 (1:1000 dilutions) and anti-IκBα (1:1000 dilutions) primary antibodies.
TLR4 protein expression assay
To detect TLR4 expression, BV2 cells were treated with 100 µM kaempferol for 1 h followed by stimulation with LPS for 24 h, then cytosol and cell membrane were prepared. For membrane extraction, cells were washed with Tris-buffered saline, and then exposed to ice-cold membrane separation buffer (0.25 M sucrose, 5 mM MgCl2, 2 mM EGTA, 2 mM EDTA, 10 mM Tris pH 7.5, 10 µg·mL−1 pepstatin, 10 µg·mL−1 leupeptin, 1 mM PMSF and 5 mM NaF). Cells were disrupted with sonication and then centrifuged at 1000×g for 5 min to remove unbroken cells and nuclei. Supernatant was removed to a fresh tube and centrifuged at 100 000×g for 1 h. The supernatant containing the cytosolic fraction was removed. The pellets were then resuspended in membrane separation buffer with 1% Triton X-100. The protein concentrations were determined by the BCA method and subjected to Western blotting as mentioned above using anti-TLR4 (1:800 dilution) antibody.
Microglia phagocytosis assay
BV2 cells were seeded in a 12-well plate. The cells were then pretreated with kaempferol for 1 h and stimulated with LPS for 24 h. To assess microglial phagocytotic activity, 1 µL·mL−1 latex beads (0.8 µM) were added into each well. After 2 h, the cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min at room temperature. The fixed cells were then rinsed with PBS and observed with fluorescent microscope (Nikon, Eclipse TE 2000-U, Japan) and photographed at 100× magnification. Cells containing more than 10 beads were counted as phagocytotic cells.
Electrophoretic mobility-shift assay (EMSA)
EMSA was performed using a DIG gel shift kit (Roche, Mannheim, Germany) according to the manufacturer's instructions. Briefly, BV2 cells were harvested and suspended in hypotonic solution (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT and 0.5 mM PMSF) including 0.5% Nonidet P-40 on ice for 10 min. Cells were centrifuged at 4420×g for 10 min at 4°C and the pellet (nuclear fraction) was collected. The nuclear fraction was resuspended in a buffer containing 20 mM HEPES, pH 7.9, 20% glycerol, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT and 1 mM PMSF, incubated on ice for 60 min with occasional gentle shaking, and centrifuged at 17 700×g for 25 min. The crude nuclear proteins in the supernatant were collected and stored at −70°C until used. Protein-DNA binding was carried out in a mixture containing 4 µL of a 5× binding buffer (1× concentration of 5% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 40 mM KCl, 40 mM Tris, pH 8.0) along with dI-dC and 20 µg nuclear proteins. For specific and non-specific competitor reactions, 100-fold excess of the appropriate unlabelled double-strand DNA was added: specific, AGTTGAGGGGACTTTCCCAGGC (NF-κB) and non-specific GTACGGAGTATCCAGCTCCGTAGCATGCAAATCCTGC (Oct 2A). Two microliter of DIG labelled NF-κB probe was added to each reaction and incubated for 20 min at room temperature. Samples were resolved on a 6% acrylamide gel in 0.5× Tris-borate-EDTA (TBE) buffer. The DNA was then electro-transferred to a positively charged nylon membrane. Anti-DIG antibody conjugated to alkaline phosphatase (1:10 000) was then added to the membrane. Disodium 3-(4-methoxyspiro {l, 2-dioxetane-3, 2'-(5'-chloro) tricyclo[3.3.1.13,7] decan}-4-yl) phenyl phosphate was used as a chemiluminescent substrate for alkaline phosphatase. The membrane was then exposed to an X-ray film at room temperature.
Microglia-conditioned media
The human dopaminergic neuronal cell line, SH-SY5Y, obtained from the American Type Culture Collection (Rockville, MD, USA) was plated at a density of 1 × 104 cell per well in 96-well plates and allowed to settle at 37°C for 24 h before replacement with conditioned media. BV2 cells were treated with different concentrations of kaempferol (10–100 µM) for 1 h then stimulated with LPS (1.0 µg·mL−1) for 24 h. The culture media were collected as conditioned media and clarified by centrifugation at 20 780×g for 5 min to remove cellular debris. The conditioned media were then transferred to SH-SY5Y cells and the cells were further incubated at 37°C for 24 h. Cell viability was measured by MTT assay as described above.
Statistical analysis
Each experiment was performed at least in triplicate. The data are expressed as the means ± SD Statistical significance was assessed with one-way ANOVA followed by a post hoc (Bonferroni) test for multiple group comparison. Differences with a P-value less than 0.05 were considered statistically significant.
Results
Effects of kaempferol and LPS on the survival of BV2 cells
We first examined the effects of kaempferol on the survival of microglial BV2 cells. Cells were treated with various concentrations of kaempferol (10, 30, 50 and 100 µM) for 24 h and cell survival was determined by MTT assay. As shown in Figure 1B, when exposed to kaempferol concentrations of 100 µM or lower, the viability of BV2 cells was the same as untreated control cells. In order to determine the effects of kaempferol on LPS-stimulated BV2 cells, cells were pretreated with kaempferol for 1 h followed by treatment with 1.0 µg·mL−1 LPS for 24 h. As shown Figure 1C, there were no significant changes in the survival ratio of cells when treated with LPS alone or with kaempferol. To further evaluate the effects of kaempferol on the survival of LPS-treated BV2 cells, cytotoxicity was quantified by the neutral red assay. This assay is based on the fact that compromised or dead cells lose the ability to retain the dye normally accumulating in lysosomes. The compounds did not have a significant cytotoxicity toward BV2 cells (Figure 1D). The effects of kaempferol and LPS on BV2 cell survival determined by neutral red assay was similar to that determined by MTT assay. These results were also confirmed by morphological observation (Figure 1E), indicating that both kaempferol and LPS did not affect the viability of microglial cells.
Figure 1.
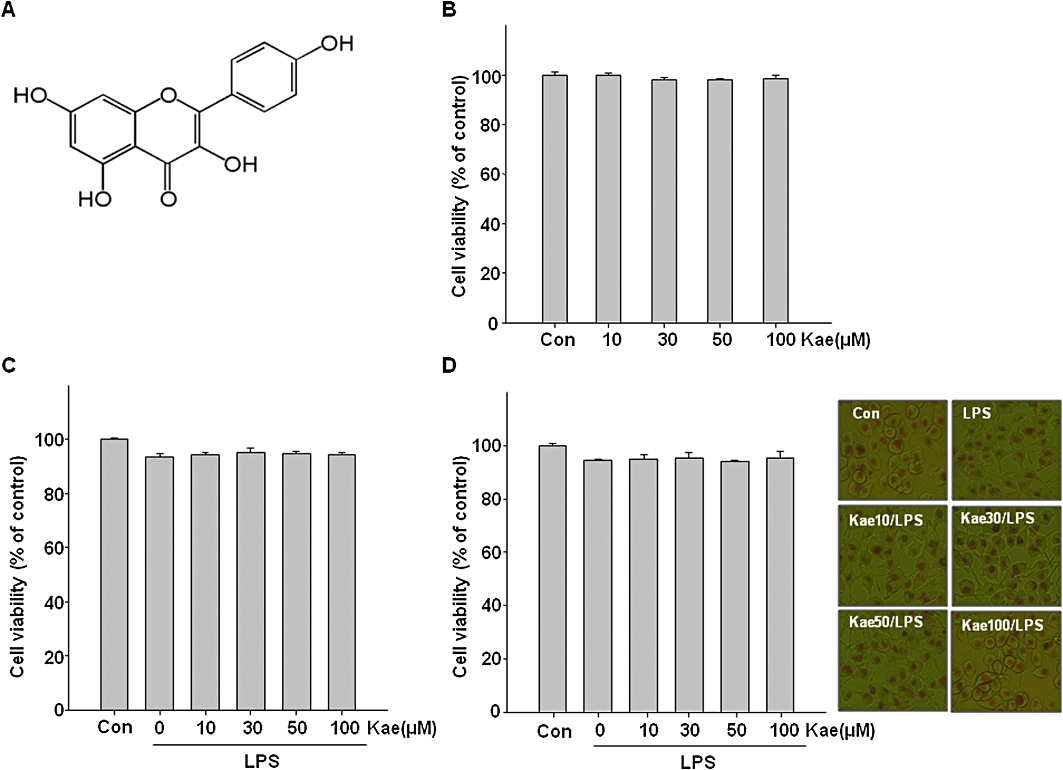
Effects of kaempferol on the survival of BV2 cells. (A) The chemical structure of kaempferol. (B) Cells were treated with 10–100 µM of kaempferol for 24 h, and cell viability was determined by MTT assay. Cells were treated with different concentrations of kaempferol (10–100 µM) for 1 h followed by LPS (1.0 µg·mL−1) treatment for 24 h. Cell viability was assessed by the MTT assay (C) and neutral red assay (D). Effects of LPS and kaempferol + LPS on BV2 cell morphology stained with neutral red (E). Morphological studies were conducted by phase-contrast microscopy. The data are represented as means ± SD of three independent experiments. Con, untreated control; Kae, kaempferol.
Kaempferol suppresses the production of NO, PGE2, IL-1β and TNF-α in LPS-stimulated BV2 cells and attenuates IFN-γ-induced NO production
Next, to investigate the anti-inflammatory effects of kaempferol on LPS-induced microglial activation, the BV2 cells were pretreated with various concentrations of kaempferol (10–100 µM) for 1 h and then stimulated with 1.0 µg·mL−1 LPS for another 24 h. The results show that exposure of BV2 cells to LPS increased the production of NO, PGE2, IL-1β and TNF-α by 8-, 10.9-, 6.2- and 9.7-fold over basal level, respectively (Figure 2A–C and D, column 2). However, these proinflammatory mediators were significantly inhibited by kaempferol in a concentration-dependent manner (Figure 2). Kaempferol at 100 µM remarkably decreased the levels of NO, PGE2, IL-1β and TNF-α to 63.4%, 76%, 63.5% and 71.6%, respectively. Further, we tested the effects of kaempferol on NO production in IFN-γ-stimulated BV2 cells. As shown in Figure 2A, IFN-γ induced NO production in BV2 cells and it was also inhibited in a concentration-dependent manner by kaempferol.
Figure 2.
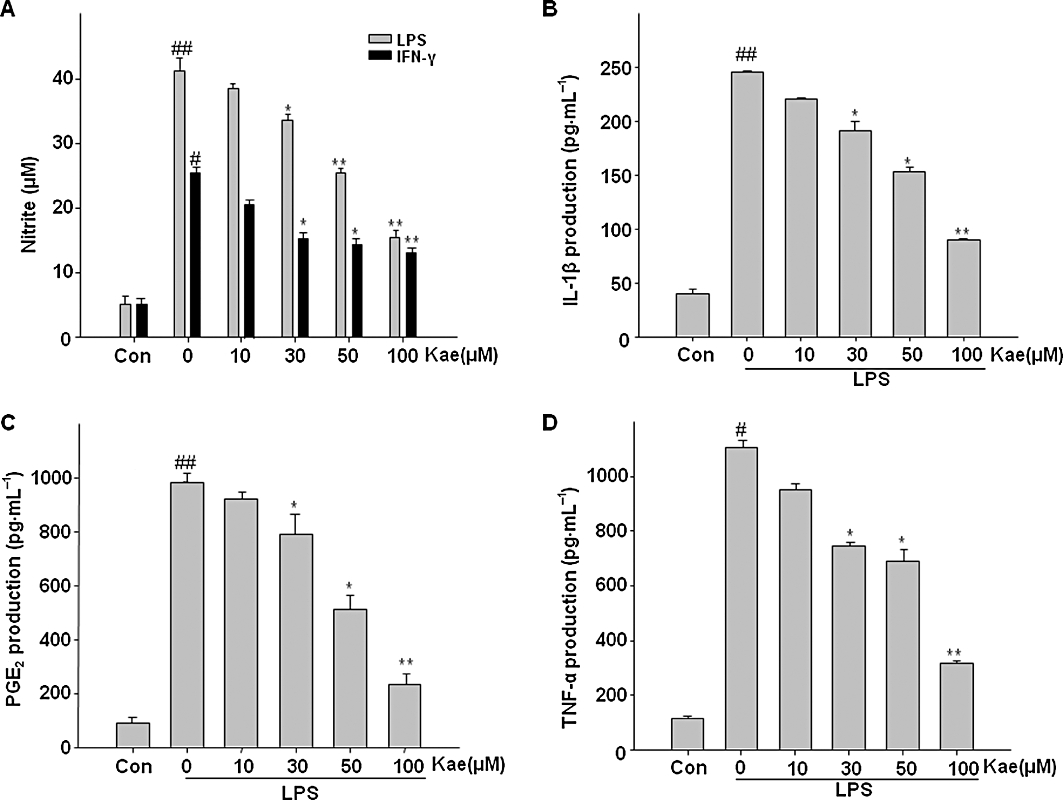
Kaempferol inhibited NO, IL-1β, PGE2 and TNF-α production in BV2 cells. Cells were pretreated with kaempferol (10–100 µM) for 1 h, then stimulated with LPS (1.0 µg·mL−1) or 20 U·mL−1 interferon- γ (IFN-γ) for 24 h. (A) The concentration of nitrite in the culture medium was determined by Griess reaction. (B) The production of IL-1β was monitored by elisa. Levels of PGE2 and TNF-α in culture medium were determined by EIA analysis (C,D). Each value is expressed as mean ± SD of at least three independent experiments. ##P < 0.01, compared with control group. *P < 0.05, **P < 0.01, compared with LPS or IFN-γ alone. Con, untreated control; Kae, kaempferol.
Kaempferol attenuates LPS-induced intracellular ROS production in BV cells
There is cumulative evidence that ROS are associated with various neurodegenerative disorders. Production of inflammatory mediators by the activated microglia is known to stimulate the release of ROS. To determine whether LPS-induced ROS production in BV2 cells is attenuated by kaempferol, we measured the formation of ROS in cells with NBT and DCF-DA assays. The results obtained by using the NBT assay showed that exposure of BV2 cells to LPS for 24 h increased NBT positive cells (Figure 3A, B, b). Notably, kaempferol pretreatment markedly inhibited the effect of LPS on BV2 cells (Figure 3B, c–f). The NBT reduction quantified assay showed that kaempferol reduced LPS-induced ROS production in BV2 cells in a concentration-dependent way (Figure 3A). The results of the DCF-DA assay (Figure 3C and D) also demonstrated that exposure of BV2 cells to LPS for 24 h caused a significant increase in intracellular ROS levels. However, pretreatment with kaempferol significantly suppressed the LPS-induced ROS production.
Figure 3.
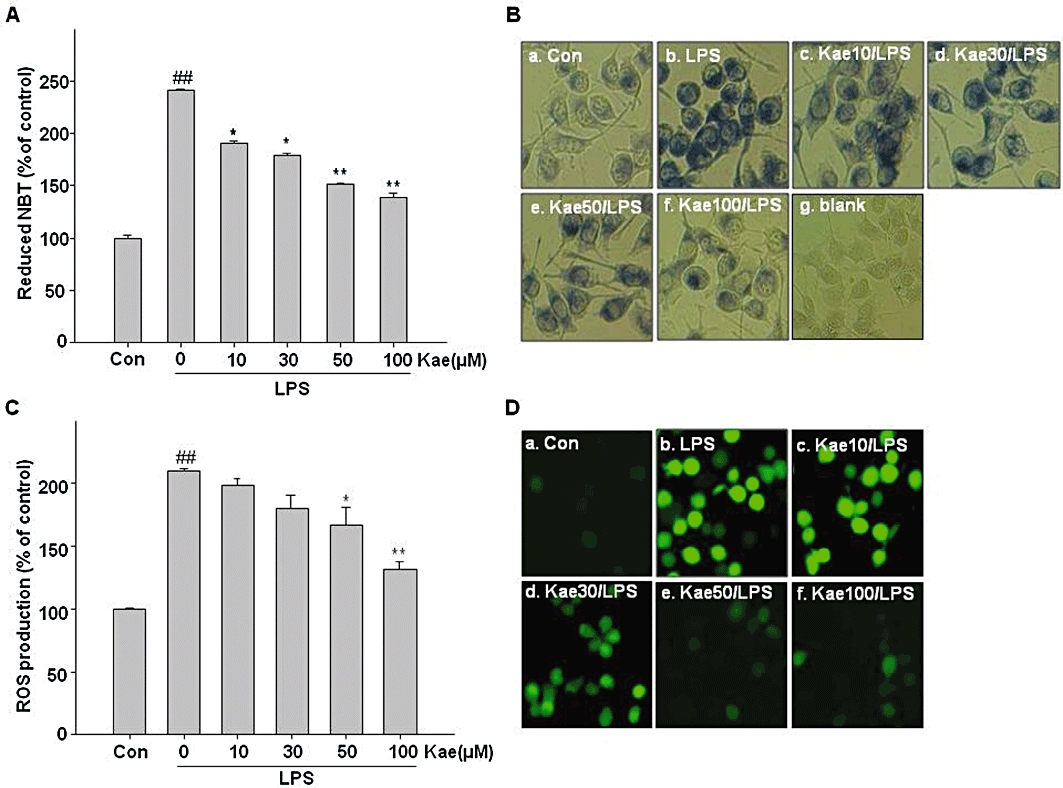
Effects of kaempferol on LPS-induced ROS formation. BV2 cells were treated with 10–100 µM kaempferol for 1 h, followed by vehicle or LPS (1.0 µg·mL−1) for 24 h. Intracellular reactive oxygen species (ROS) accumulation was measured by nitroblue tetrazolium (NBT) (A,B) and 2′7′-dichlorofluorescein diacetate (DCF-DA) (C,D) assay. (A) The NBT deposited inside the cells were dissolved and then the intracellular ROS level was quantified using a VERSAmax microplate reader at 630 nm. (B) The NBT positive cells were observed under fluorescent microscope. (C) Intracellular ROS accumulation was measured using the fluorescence probe DCF-DA. The fluorescence intensity was determined using a Spectra Max Gemini EM fluorometer at 485 nm excitation and 535 nm emission. (D) Cells were incubated with DCF-DA and accumulation of intracellular ROS was monitored by fluorescent microscope. Each value is expressed as mean ± SD of at least three independent experiments. ##P < 0.01, compared with control group. *P < 0.05, **P < 0.01, compared with LPS treated alone. Con, untreated control; Kae, kaempferol.
Kaempferol reduces LPS-induced microglial phagocytosis
Since phagocytosis is one of the markers of activated microglia, we investigated whether kaempferol prevents LPS-induced phagocytotic activity in the BV2 cells by a phagocytosis assay using latex beads. As shown in Figure 4, phagocytotic activity was significantly increased in cells treated with LPS (16-fold). However, kaempferol pretreatment markedly reduced the LPS-induced phagocytosis in concentration-dependent manner. These results clearly suggest a critical role of kaempferol in the regulation of phagocytotic activity of microglia.
Figure 4.
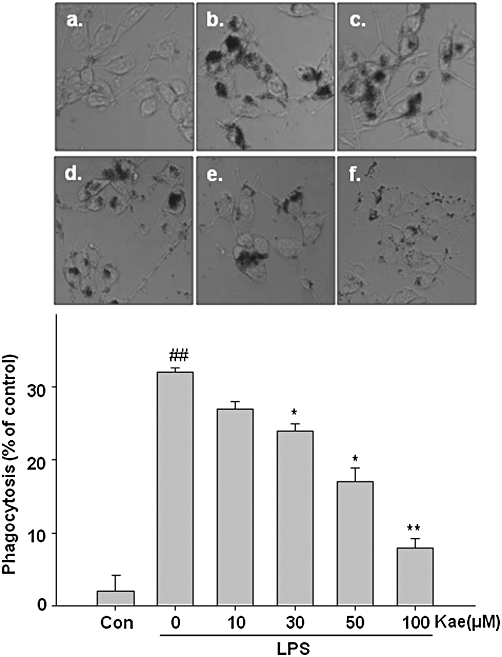
Effects of kaempferol on phagocytotic activity of LPS-activated BV2 cells. Cells were pretreated with kaempferol (10–100 µM) for 1 h, followed by LPS (1.0 µg·mL−1) for 24 h. (A) Phagocytosed beads were observed by fluorescent microscope. (B) The bead density was counted and quantified as percentage. (a) Control; (b) LPS alone; (c) 10 µM kaempferol + LPS; (d) 30 µM kaempferol + LPS; (e) 50 µM kaempferol + LPS; (f) 100 µM kaempferol + LPS. Each value is expressed as mean ± SD of at least three independent experiments. ##P < 0.01, compared with control group. *P < 0.05, **P < 0.01, compared with LPS treated alone. Con, untreated control; Kae, kaempferol.
Kaempferol inhibits the expression of iNOS and COX-2 in LPS-stimulated BV2 cells
We further examined whether kaempferol's inhibitory effect on NO and PGE2 production was associated with decreased iNOS and COX-2 expression in LPS-treated BV2 cells. To evaluate this effect, the levels of iNOS and COX-2 protein expression were measured, by Western blotting, after exposure of the BV cells to LPS (1.0 µg·mL−1) for 24 h with or without kaempferol (100 µM) pretreatment for 1 h. As shown in Figure 5, upon LPS treatment for 24 h, the expression of iNOS and COX-2 protein levels were increased significantly (4.2-fold and 6-fold respectively). However, these expressions were dramatically attenuated in BV2 cells pretreated with kaempferol.
Figure 5.
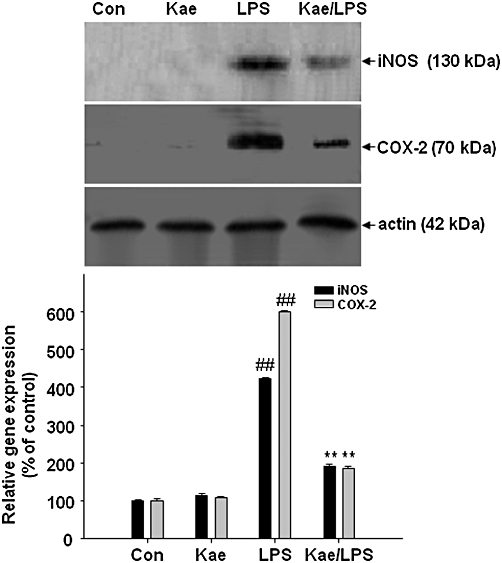
Inhibitory effects of kaempferol on the LPS-induced expression of iNOS and COX-2 in BV2 cells. BV2 cells were pretreated with 100 µM kaempferol for 1 h, followed by 1.0 µg·mL−1 LPS for 24 h. Expression of the iNOS and COX-2 were detected by immunoblot assay. Actin was used as an internal loading control. Results are expressed as mean ± SD of three independent experiments. ##P < 0.01, compared with control group. **P < 0.01, compared with LPS treated alone. Con, untreated control; Kae, kaempferol.
Kaempferol inhibits the expression of MMP-3, but not MMP-9, and inhibition of MMP-3 suppresses iNOS expression in LPS-stimulated BV2 cells
It is known that microglia bear a MMPs signature that is distinct from other cells (Nuttall et al., 2007). MMPs, particularly MMP-3 and –9, are emerging as important molecules in neuroinflammation and neuronal cell death. Inhibition of these molecules dramatically suppressed various inflammatory mediators and related signalling pathways (Woo et al., 2008). Moreover, it has been suggested that treatments that target multiple pathways may prove most beneficial (Byrnes et al., 2009). Although a number of paper have reported that the function of MMPs are multiple and complex, the role of MMPs in the activated microglia is little understood and the relative contribution of kaempferol to the modulation of MMPs activation is unclear. Therefore, the present study is designed to understand this mechanism, to explore the multifunction of kaempferol, and to highlight the possibility that MMPs inhibition may have the potential as therapeutics to decrease microglial inflammation in neurodegenerative diseases. First, we examined whether kaempferol might down-regulate MMP-3 and -9 expressions in activated microglia. The exposure of BV2 cells to LPS alone at 1 µg·mL−1 for 24 h resulted in a dramatic increase in MMP-3, and -9 expression as detected by zymography (Figure 6A) and Western blot analysis (Figure 6B). Interestingly, pretreatment with kaempferol notably inhibited the LPS-induced MMP-3 expression, but did not change the level of MMP-9 (Figure 6).
Figure 6.
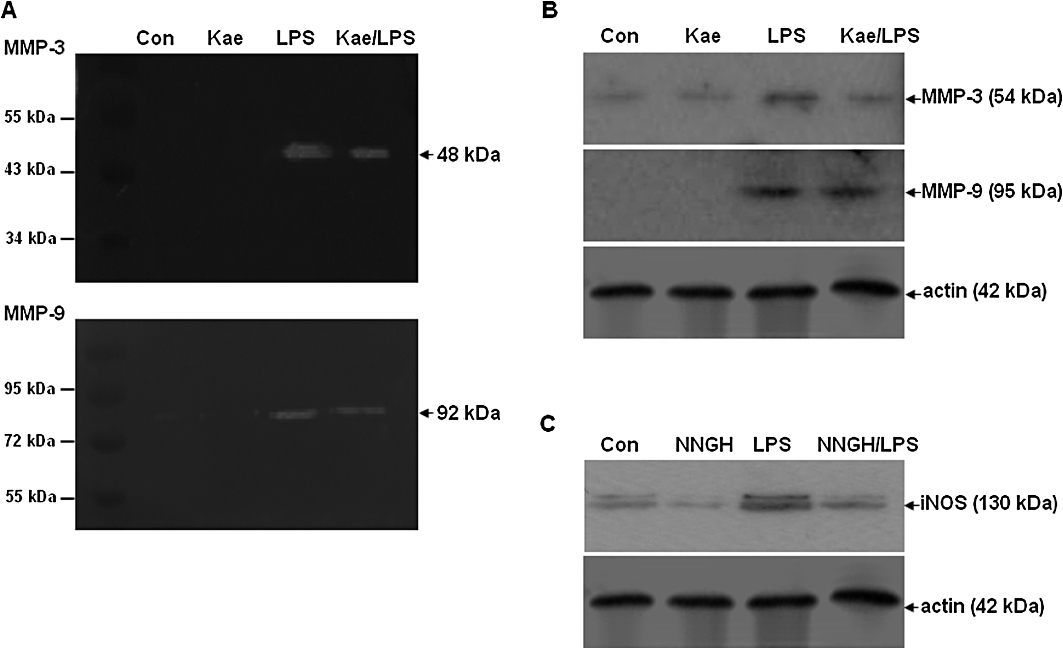
Effects of kaempferol on the LPS-induced expression of MMP-3 and MMP-9, and involvement of MMP-3 in LPS-induced iNOS expression in BV2 cells. Cells were pretreated with 100 µM kaempferol for 1 h, followed by 1.0 µg·mL−1 LPS for 24 h. (A) Zymographic analysis of MMP-3 and MMP-9 activity. The caseinolytic activity of MMP-3 (top), and the gelatinolytic activity of MMP-9 (bottom). (B) Western blot analysis of expression of MMP-3, and MMP-9. Actin was used as an internal loading control. (C) BV2 cells were pretreated with NNGH, a specific inhibitor of MMP-3 for 1 h, followed by LPS for 24 h. Expression of the iNOS was detected by immunoblot assay. Con, control; Kae, kaempferol.
Whether the inhibition of MMP-3 expression is accompanied by down-regulation of proinflammatory mediator was then tested. To investigate this, BV2 cells were treated with NNGH – a specific MMP-3 inhibitor before stimulation with LPS. As shown in Figure 6C, inhibition of MMP-3 significantly repressed the LPS-induced protein level expression of iNOS.
Kaempferol inhibits LPS-induced TLR4 expression
To examine whether kaempferol is able to modulate TLR4 expression in response to LPS exposure, immunoblot assay was performed. As shown in Figure 7, LPS exposure significantly increased TLR4 protein levels compared to those in untreated control BV2 cells. This LPS-induced TLR4 activation was dramatically reversed by kaempferol, indicating that it blocks the activation of this sensor molecule of the neuroinflammatory pathway.
Figure 7.
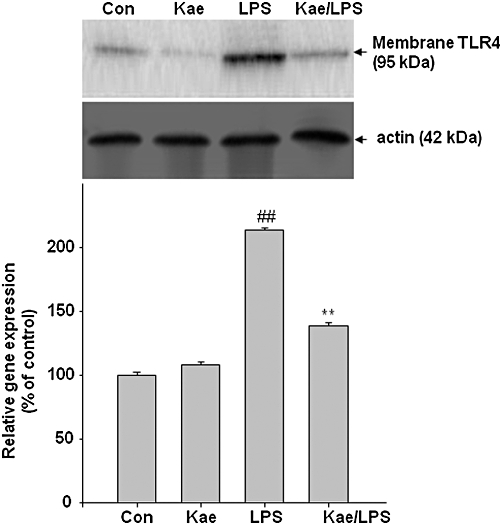
Effect of kaempferol on the LPS-induced activation of toll-like receptor-4 (TLR4) in BV2 cells. Cells were pretreated with 100 µM kaempferol for 1 h, followed by 1.0 µg·mL−1 LPS for 24 h. (A) Membrane protein was extracted from lysed cells and TLR4 protein expression was determined by immunoblot assay. Actin was used as an internal loading control. Results are expressed as mean ± SD of three independent experiments. ##P < 0.01, compared with control group. **P < 0.01, compared with LPS treated alone. Con, control; Kae, kaempferol.
Kaempferol attenuates LPS-induced NF-κB activation in BV2 cells
NF-κB activation involves the translocation of the p65 subunit of NF-κB into nucleus. Thus, we investigated whether kaempferol inhibits the p65 nuclear translocation. Immunocytochemistry analysis showed that the p65 protein was primarily located in the cytosol during the untreated condition. When BV2 cells were exposed to LPS, the p65 protein appeared in nuclei within 1 h and BV2 cells changed their morphology (Figure 8A). Kaempferol pretreatment reduced the p65 nuclear immunoreactivity, as well as the morphological change induced by LPS. Western blot analysis also revealed that p65 protein levels were increased in the nuclei of the cells whereas the levels of cytosolic IκBα and p65 protein were decreased when BV2 cells were exposed to LPS (Figure 8B). Interestingly, these effects were attenuated by kaempferol treatment.
Figure 8.
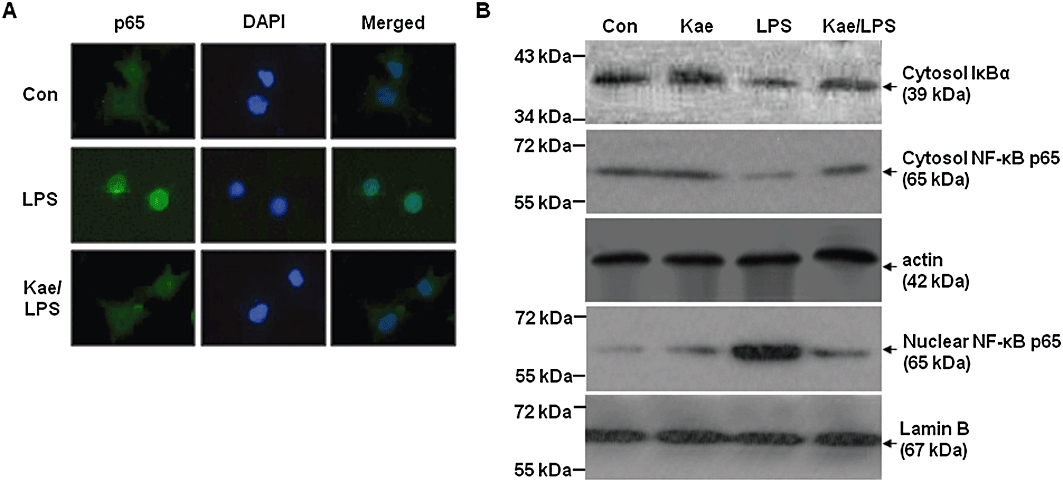
Inhibition of LPS-induced NF-κB activation by kaempferol in BV2 cells. Cells were activated with LPS (1.0 µg·mL−1) for 1 h with or without kaempferol (100 µM). (A) The translocation of p65 subunit of NF-κB was determined by immunocytochemistry. Representative pictures from three independent experiments are shown. (B) Cytoplasmic and nuclear fractions of treated cells were used to detect IκBα and NF-κB expression. Actin and Lamin B were used as an internal loading control for cytosolic and nuclear fractions respectively. Con, control; DAPI, 4′, 6-diamidino-2-phenylindole; Kae, kaempferol.
In order to further examine whether NF-κB activation is involved in the signal transduction pathway caused by LPS that leads to iNOS expression, the NF-κB inhibitor PDTC was used. Figure 9A and B show that PDTC (50 µM) antagonized the LPS-induced enhancement of NO production and iNOS expression in BV2 cells.
Figure 9.
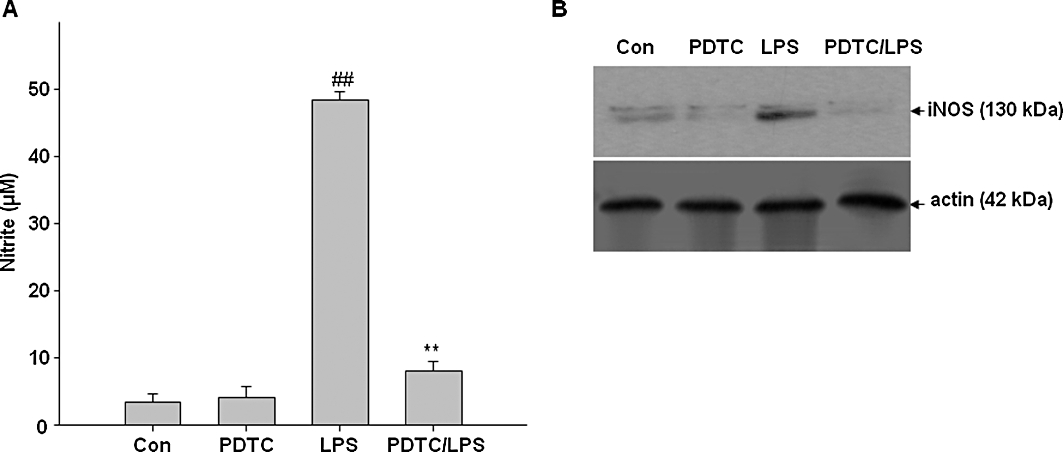
Involvement of NF-κB in LPS-induced iNOS expression and NO production in BV2 cells. BV2 cells were pretreated with pyrrolidine dithiocarbamate (PDTC), a specific inhibitor of NF-κB for 1 h, and then the cells were exposed to LPS for 24 h. (A) The concentration of nitrite in culture medium was monitored as described in methods. (B) Expression of iNOS was determined by Western blot analysis. Actin was used as an internal loading control. Results are expressed as mean ± SD of three independent experiments. ##P < 0.01, compared with control group. **P < 0.01, compared with LPS treated alone. Con, control; Kae, kaempferol.
Effects of kaempferol on LPS-induced NF-κB binding in BV2 cells
As NF-κB has been implicated in the transcriptional activation of various inflammatory cytokines (Baeuerle and Henkel, 1994; Guha and Mackman, 2001), the effects of kaempferol on this signalling pathway were investigated by EMSA. BV2 cells were pretreated with kaempferol for 1 h and then stimulated with LPS for 24 h. Nuclear extracts prepared from the cells were then subjected to EMSA using an oligonucleotide probe corresponding to the consensus binding sites for NF-κB. As shown in Figure 10, NF-κB activity was increased by LPS in BV2 cells, and this increase was attenuated by pretreatment with kaempferol.
Figure 10.
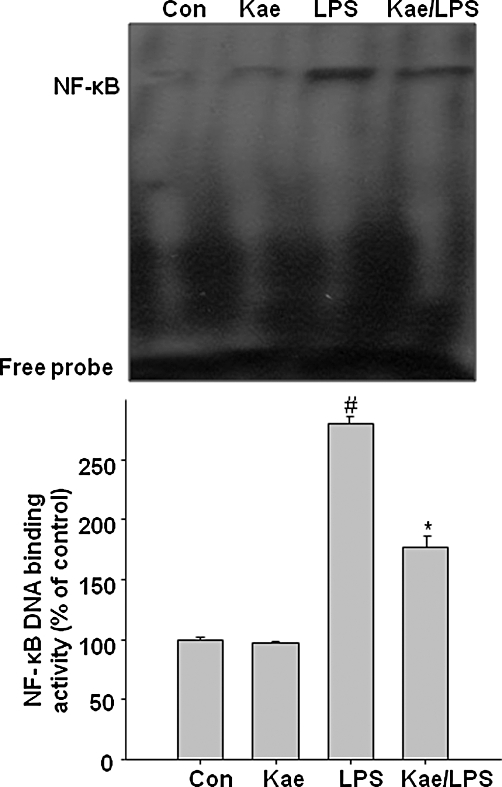
Inhibitory effects of kaempferol on LPS-induced DNA binding of NF-κB in BV2 cells. Cells were pretreated with 100 µM kaempferol for 1 h before being incubated with LPS (1.0 µg·mL−1) for 24 h. Nuclear extracts were prepared and DNA binding activity of NF-κB was determined by EMSA using a NF-κB-specific oligonucleotide probe. Densitographs represent means ± SD of three independent experiments. #P < 0.05, compared with control group. *P < 0.05, compared with LPS treated alone. Con, control; Kae, kaempferol.
Kaempferol inhibits LPS-induced JNK, p38 and AKT phosphorylation
We investigated the effects of kaempferol on the activation of ERK1/2, JNK, p38 MAPK and AKT in LPS-stimulated BV2 cells. As shown in Figure 11, LPS strongly activated ERK2, p38 MAPK and AKT (their phosphorylated forms, pERK2, pp38 and pAKT, are shown) whereas ERK1 and JNK were weakly activated. Kaempferol had significant inhibitory effects on LPS-stimulated JNK, p38 MAPK and AKT activation. However, phosphorylation of ERK1/2 was not affected by kaempferol pretreatment.
Figure 11.
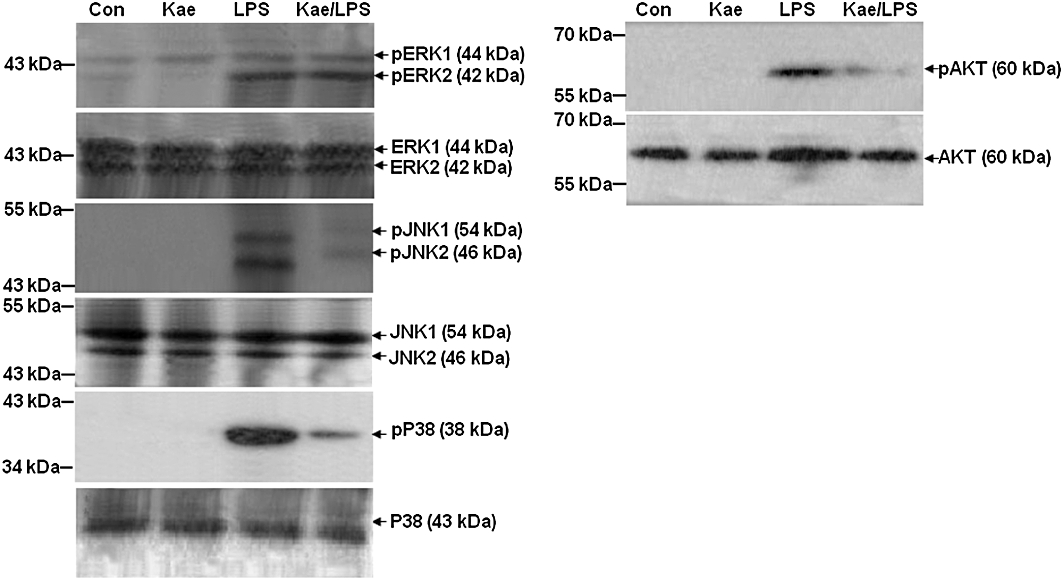
Inhibitory effects of kaempferol on LPS-induced JNK, p38 MAPK and AKT activation in BV2 cells. Cells were pretreated with 100 µM kaempferol for 1 h, followed by stimulation with 1.0 µg·mL−1 of LPS for 1 h. Whole cell extracts were prepared and analysed by immunoblotting using MAPKs and AKT antibodies. Con, untreated control; Kae, kaempferol.
Involvement of MAPKs, AKT and kaempferol in LPS-induced NO, PGE2 production and iNOS expression in BV2 cells
In order to determine the involvement of JNK, p38 MAPK and AKT in the LPS-induced increase in NO, PGE2 production and iNOS expression, SB203580 (a selective p38 inhibitor) and SP600125 (a selective JNK inhibitor) and LY294002 (PI3K/AKT inhibitor) were administered to BV2 cultures 1 h before LPS exposure, while LY294002, a PI3K/AKT specific inhibitor was administered 30 min before. As shown in Figure 12, LPS significantly increased NO and PGE2 production as well as iNOS expression. However, inhibition of p38 MAPK, JNK and AKT activities by pretreatment with the inhibitors blocked the LPS-induced NO, PGE2 production and iNOS expression. As expected, supplementation of the culture medium with kaempferol significantly attenuated NO and PGE2 production and decreased iNOS protein levels in BV2 cells. These results indicate that JNK, p38 MAPK and AKT pathways are involved in the inhibitory effects of kaempferol on LPS-stimulated iNOS and COX-2 expression.
Figure 12.
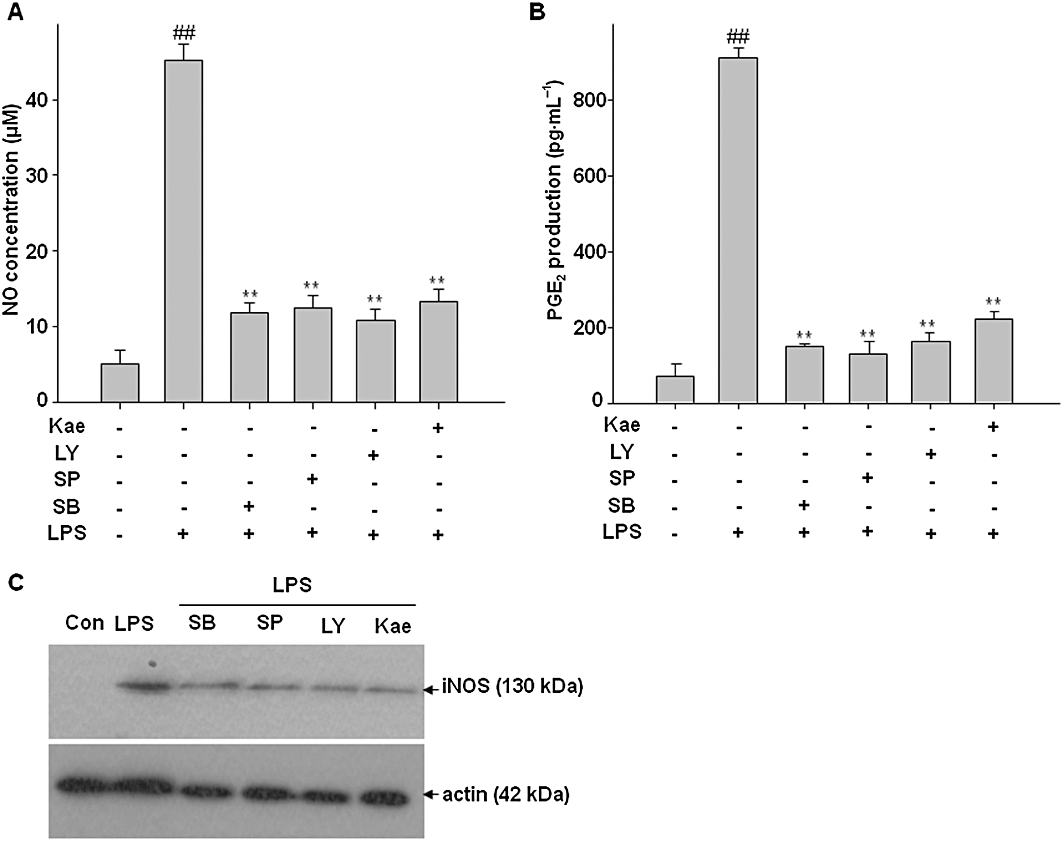
Involvement of kaempferol, MAPKs and AKT in LPS-induced NO, PGE2 production and expression of iNOS in BV2 cells. Cells were pre-incubated with kaempferol, SB203580 (a selective p38 inhibitor), SP600125 (a selective JNK inhibitor) for 1 h and LY294002 (PI3K/AKT inhibitor) for 30 min, then cells were exposed to LPS for 24 h. (A) The concentration of nitrite in culture medium was monitored by Griess reaction. (B) Levels of PGE2 in the culture medium were determined by EIA analysis. (C) Expression of iNOS was determined by Western blot analysis. Actin was used as an internal loading control. Results are expressed as mean ± SD of three independent experiments. ##P < 0.01, compared with control group. **P < 0.01, compared with LPS treated alone. Con, control; Kae, kaempferol.
Kaempferol suppresses the microglia-mediated neurotoxicity
Several studies have demonstrated that activated microglia exert neurotoxic effects by releasing inflammatory mediators (Block et al., 2007). We investigated whether the inhibition of microglial activation by kaempferol can protect dopaminergic neurons. As shown in Figure 13, when conditioned media from LPS-stimulated microglia was added to cultured SH-SY5Y cells, neuronal cell death, as measured by the MTT assay was significantly increased after 24 h. However, pretreatment of BV2 cells with kaempferol prior to LPS stimulation and addition of conditioned media to dopaminergic neurons significantly reduced neuronal cell death in a concentration-dependent manner, demonstrating the neuroprotective effects of kaempferol.
Figure 13.
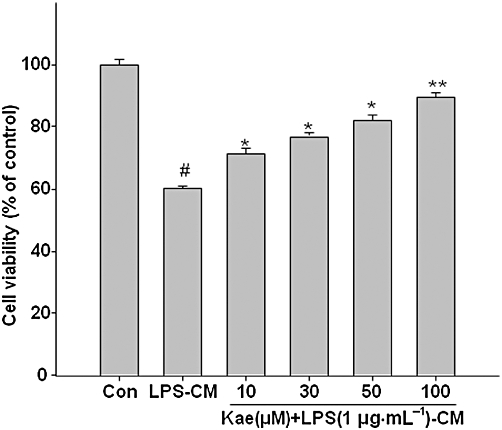
Kaempferol protects against microglial-mediated neurotoxicity. SH-SY5Y cells were treated with conditioned media (CM) from BV2 cells exposed toLPS (1.0 µg·mL−1) with or without kaempferol (10–100 µM) pretreatment for 1 h. After 24 h incubation, the viability of SH-SY5Y cells was assessed by MTT assay. Results are expressed as mean ± SD of three independent experiments. #P < 0.05, compared with control group. *P < 0.05, **P < 0.01, compared with LPS treated alone. Con, control; Kae, kaempferol.
Discussion
Activation of microglia is one of the universal components of neuroinflammation. Activated microglia secrete a variety of pro-inflammatory mediators that are believed to induce neurodegeneration. Therefore, inhibiting neuroinflammatory mediators could be an effective method of treating neurodegenerative diseases. In this study, we examined the effect of kaempferol on LPS-activated BV2 microglial cells. We found that kaempferol exhibited an inhibitory effect on LPS-activated TLR4 and the corresponding downstream NF-κB, JNK, p38 MAPK and AKT activation, which subsequently reduced LPS-induced MMP-3 up-regulation, and TNF-α, IL-1β, NO, iNOS, PGE2, COX-2 and ROS production.
It is generally accepted that excessive production and accumulation of NO by activated microglial cells can contribute to neurodegeneration. Several lines of evidence support the neurotoxic role of NO in neuronal cell death both in vivo and in vitro. Studies using selective and non-selective inhibitors of NOS have afforded significant neuroprotection (Bal-Price and Brown, 2001; Munch et al., 2003). PGE2 is a major eicosanoid found in many inflammatory conditions including CNS inflammatory diseases (McGeer and McGeer, 1995). Chronic treatment with a non-steroidal anti-inflammatory drug, which inhibits prostaglandins synthesis, is associated with decreased risk of developing AD (McGeer and McGeer, 1995; Breitner, 1996). Interestingly, in the present study, kaempferol, a non-steroidal phyto-oestrogen, strongly suppressed not only NO production but also PGE2 generation in LPS-activated BV2 cells.
In order to evaluate whether the inhibitory effect of kaempferol on NO and PGE2 production was related to a modulation of iNOS and COX-2 production, we measured iNOS and COX-2 expression in BV2 cells. We found that kaempferol inhibited iNOS and COX-2 protein expression in LPS-treated BV2 cells. COX-2 and iNOS are part of a family of primary inflammatory response genes, whereby COX-2 and iNOS expression are co-ordinately modulated by LPS and their sustained up-regulation in microglia contributes to the progressive damage in neurodegenerative diseases. It was found that iNOS and COX-2 are expressed in glial cells of substantia nigra of post-mortem PD patients (Knott et al., 2000) and in rodent brain after LPS treatment (Boje and Arora, 1992; Minghetti et al., 1999). Inhibitors of iNOS and COX-2 expression has been found to exert neuroprotective effects in several models of neurodegeneration and inflammation (Liang et al., 1999; Egger et al., 2003). Epidemiological studies suggest that COX inhibitors reduce the risk of PD and AD (Choi et al., 2009). These findings suggest that kaempferol may be a promising candidate to inhibit the primary steps in the neuroinflammatory pathway.
Inflammatory cytokines such as TNF-α and IL-1β have been shown to promote neuronal injury (Lim et al., 2000; Tan et al., 2002). TNF-α is a key downstream mediator in inflammatory responses, and IL-1β is known to be the major microglial signal that promotes the cascade of glial cell reactions. Previous studies have demonstrated that kaempferol represses the gene expression of LPS-induced TNF-α and IL-1β in J7774.2 macrophages (Kowalski et al., 2005). These observations combined with our results, that showed kaempferol inhibits TNF-α and IL-1β production in LPS-stimulated BV2 microglial cells, suggest that kaempferol may possess a potent anti-inflammatory property.
Accumulating evidence suggests that MMPs are instrumental in the production and maintenance of a pro-inflammatory microenvironment. In recent years, MMP-3, and -9 have received much attention for their apparent role in the pathogenesis of acute brain injury and neuroinflammation. Studies have shown that LPS is a potent stimulus for MMP-3 and -9 in microglia (Woo et al., 2008; Candelario-Jalil et al., 2009). Pharmacological intervention of MMPs expression was found to be beneficial for the treatment of inflammatory diseases in the CNS caused by over activation of microglial cells (Woo et al., 2008; Candelario-Jalil et al., 2009). Experiments with the MMP-3 inhibitor, NNGH, indicated that MMP-3 is positively associated with iNOS regulation in LPS-stimulated BV2 cells. Interestingly, kaempferol suppresses the LPS-induced increase in MMP-3 expression, but no changes were observed in the expression of MMP-9. Why MMP-9 is not suppressed by kaempferol is still not clear. As MMP-3 gene is known to have an activator protein-1 (AP-1) element on its promoter (Westermarck and Kahari,1999), it is possible that JNK acts as a transcription factor that binds to the AP-1 element of the MMP-3 gene. Moreover, it has been suggested that the ERK/MAPK signalling pathway is essential for MMP-9 up-regulation (Arai et al., 2003), whereas the JNK/MAPK pathway is associated with MMP-3 activation (Ahmed et al., 2005; Choi et al., 2008). Consistent with these findings, the JNK/MAPK signalling pathway was shown to be involved in our kaempferol-mediated inhibitory effect on neuroinflammation in LPS-activated BV2 cells.
Toll-like receptors are intimately involved in several neurodegenerative disorders. TLR4 has been identified in the innate immunity and inflammation response to LPS. Excessive activation of TLR4 on microglia may lead to the accumulation of cytotoxic compounds such as ROS and pro-inflammatory cytokines causing damage and eventual neuronal loss (Akiyama et al., 2000). Therefore, modulation of the TLR4 signalling may be a potential therapeutic target in neurodegenerative diseases. Importantly, in this study, kaempferol markedly inhibited the LPS-induced increase in TLR4 expression in BV2 cells. Following TLR4 activation, a MyD88-independent pathway can be activated. This culminates in MAPK signalling and activation of the transcription factor NF-κB (Okun et al., 2009). Microglia have been shown to produce different ROS in response to LPS. Accumulation of intracellular ROS triggers the release of inflammatory mediators in microglia through the activation of downstream signalling molecules such as MPAKs and NF-κB (Rosenberger et al., 2001; Lal et al., 2002). It has been reported that antioxidants inhibit NF-κB activation and block the production of inflammatory cytokines by inhibiting the generation of ROS (Chen et al., 2007; Mendis et al., 2008). Being a potent antioxidant, we found that kaempferol effectively inhibited intracellular ROS generation and attenuated LPS-induced IκBα degradation as well as nuclear translocation of p65 in BV2 microglia. These results suggest that kaempferol may suppress NF-κB signalling at least in part by inhibiting the generation of ROS in LPS-activated microglia. Moreover, NF-κB was found to be positively related to LPS-signalling in BV2 cells because PDTC, a specific inhibitor of NF-κB reduced LPS-induced iNOS protein and NO production. It has been suggested that activation of NF-κB is involved in LPS-induced iNOS expression (Nomura, 2001). This indicates that kaempferol-mediated inhibition of NF-κB activation is one of the possible mechanisms underlying its inhibitory actions on iNOS expression and NO production.
Phagocytosis is one of the innate functions of microglia to remove a pathogen and cellular debris from the brain during neuropathological conditions. Microglial activation occurs in response to inflammogens such as LPS and inflammatory cells show phagocytotic activity (Vallières et al., 2006). In the present study we found that kaempferol had a protective effect against LPS-induced phagocytotic activity in BV2 cells, suggesting that kaempferol could be used as a therapeutic compound to regulate microglial activation.
In this study, we further examined the involvement of MAPKs and AKT signalling pathways in the anti-inflammatory effects of kaempferol. We found that kaempferol inhibited LPS-induced activation of p38 MAPK, JNK and AKT in BV2 cells. P38 MAPK, JNK and AKT have been reported to be involved in inflammatory mediator production in response to a wide variety of stimuli including LPS. The p38 MAPK has been implicated in a signal transduction pathway responsible for the increased in iNOS gene expression in glial cells (Park et al., 2007). It has been reported that JNK is an essential mediator of several pro-inflammatory stimuli in microglia (Waetzig et al., 2005) and intervention of this pathway may be a therapeutic approach for treating inflammatory neurological diseases (Borsello and Forloni, 2007; Jang et al., 2008). Moreover, inhibition of AKT phosphorylation is found to be involved in inhibition of iNOS and COX-2 in microglia (Nam et al., 2008). In this study, it was also found that the specific p38 MAPK inhibitor SB203580, JNK inhibitor SP600125 and AKT inhibitor LY294002 markedly reduced the NO, PGE2 production and iNOS expression in the LPS-stimulated BV2 cells. Moreover, studies have suggested that signalling through p38 MAPK and JNK results in increased AP-1 activity, while signalling through AKT results in increased NF-κB activity. Activation of AP-1 and NF-κB results in the expression and production of inflammatory mediators. Thus, it appears that reduced p38 MAPK, JNK and AKT phosphorylation by kaempferol in LPS-stimulated microglia is sufficient to induce the down-regulation of inflammatory mediators. Although, studies have shown that ERK1/2 is also involved in the production of inflammatory mediators, kaempferol did not reduce its phosphorylation in LPS-activated BV2 cells. These observations suggest that JNK-, p38 MAPK- and AKT-dependent pathways are involved in the inhibitory effect of kaempferol on the production of inflammatory mediators. The proposed signalling mechanism involved in the anti-neuroinflammatory effects of kaempferol is shown in Figure 14.
Figure 14.
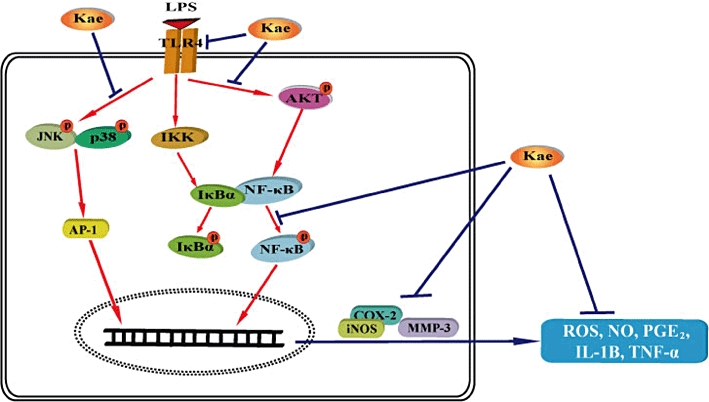
Proposed signalling mechanism for the effects of kaempferol on LPS-induced neuroinflammation in BV2 cells. Activation of toll-like receptor-4 (TLR4) with LPS leads to activation of IKK complex, and MAPKs and AKT pathways. The IKK complex phosphorylates IκB, which leads to the degradation of IκBα and the subsequent nuclear translocation of NF-κB. Similarly, p38 MAPK and JNK phosphorylate and activate AP-1. Phosphorylation of AKT also leads to activation of NF-κB. Activation of NF-κB and AP-1 results in the expression and production of pro-inflammatory mediators. Kaempferol disrupts the neurotoxic pathway and protects against LPS-induced toxicity through inhibition of NF-κB, MAPKs and AKT signalling. IKK, inhibitor of nuclear factor-κB (IκB)-kinase; Kae, kaempferol.
The present results apply to BV2 cells of a certain passage range, maintained under particular conditions as mentioned above, and microglial cells in situ might or might not respond similarly. Our microglia conditioned media results indicate that kaempferol can be neuroprotective by suppressing microglial activation and subsequent neurotoxicity. Moreover, in vivo studies have shown that kaempferol can protect against brain oxidative damages induced by different stimuli (Lopez-Sanchez et al., 2007; Lagoa et al., 2009). Recently, Filomeni et al. (2010) have demonstrated that kaempferol protects human dopaminergic cells and primary neurons from rotenone toxicity. These results indicate that kaempferol may have a multipotential neuroprotective action.
In conclusion, the present observations identify a potential anti-inflammatory role of kaempferol against neuroinflammatory toxicity induced by LPS-activated microglia through the inhibition of TLR4, NF-κB, p38 MAPK, JNK and AKT activation. Although, the neuroprotective actions of kaempferol need to be explored further, our results indicate that kaempferol is a novel compound with potential for the treatment of deurodegenerative diseases associated with neuroinflammation.
Acknowledgments
This research was supported by Technology Development Program for fisheries, Ministry for Food, Agriculture, Forestry and Fisheries, Korea.
Glossary
Abbreviations
- AKT
PKB
- iNOS
inducible NOS
- ROS
reactive oxygen species
- TLR-4
toll-like receptor-4 (TLR4)
Conflicts of interest
None.
References
- Agrawal SM, Lau L, Yong VW. MMPs in the central nervous system: where the good guys go bad. Semin Cell Dev Biol. 2007;19:42–51. doi: 10.1016/j.semcdb.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Wang N, Hafeez BB, Cheruvu VK, Haqqi TM. Punica granatum L. extract inhibits IL-1beta-induced expression of matrix metalloproteinases by inhibiting the activation of MAP kinases and NF-kappaB in human chondrocytes in vitro. J Nutr. 2005;135:2096–2102. doi: 10.1093/jn/135.9.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Lee SR, Lo EH. Essential role for ERK mitogen-activated protein kinase in matrix metalloproteinase-9 regulation in rat cortical astrocytes. Glia. 2003;43:254–264. doi: 10.1002/glia.10255. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T. Function and activation of NF-k B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21:6480–6491. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhathena SJ, Velasquez MT. Beneficial role of dietary phytoestrogens in obesity and diabetes. Am J Clin Nutr. 2002;76:1191–1201. doi: 10.1093/ajcn/76.6.1191. [DOI] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- Borsello T, Forloni G. JNK signalling: a possible target to prevent neurodegeneration. Curr Pharm Des. 2007;13:1875–1886. doi: 10.2174/138161207780858384. [DOI] [PubMed] [Google Scholar]
- Breitner JCS. Inflammatory processes and antiinflammatory drugs in Alzheimers disease-a current appraisal. Neurobiol Aging. 1996;17:789–794. doi: 10.1016/0197-4580(96)00109-1. [DOI] [PubMed] [Google Scholar]
- Byrnes KR, Loane DJ, Faden AI. Metabotropic glutamate receptors as targets for multipotential treatment of neurological disorders. Neurotherapeutics. 2009;6:94–107. doi: 10.1016/j.nurt.2008.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelario-Jalil E, Yang Y, Rosenberg GA. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 2009;158:983–994. doi: 10.1016/j.neuroscience.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S. Tumor necrosis factor-alpha potentiates glutamate neurotoxicity in human fetal brain cell cultures. Dev Neurosci. 1994;16:172–179. doi: 10.1159/000112104. [DOI] [PubMed] [Google Scholar]
- Chen CY, Peng WH, Tsai KD, Hsu SL. Luteolin suppresses inflammation-associated gene expression by blocking NF-κB and AP-1 activation pathway in mouse alveolar macrophages. Life Sci. 2007;81:1602–1614. doi: 10.1016/j.lfs.2007.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DH, Kim EM, Son HJ, Joh TH, Kim YS, Kim D, et al. A novel intracellular role of matrix metalloproteinase- 3 during apoptosis of dopaminergic cells. J Neurochem. 2008;106:405–415. doi: 10.1111/j.1471-4159.2008.05399.x. [DOI] [PubMed] [Google Scholar]
- Choi SH, Aid S, Bosetti F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends Pharmacol Sci. 2009;30:174–181. doi: 10.1016/j.tips.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71:479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- Egger T, Schuligoi R, Wintersperger A, Amann R, Malle E, Sattler W. Vitamin E (alpha-tocopherol) attenuates cyclo-oxygenase 2 transcription and synthesis in immortalized murine BV-2 microglia. Biochem J. 2003;370:459–467. doi: 10.1042/BJ20021358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filomeni G, Graziani I, De Zio D, Dini L, Centonze D, Rotili G, et al. Neuroprotection of kaempferol by autophagy in models of rotenone-mediated acute toxicity: possible implications for Parkinson's disease. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.05.021. doi: 10.1016/j.neurobiolaging. 2010.05.021. [DOI] [PubMed] [Google Scholar]
- García-Mediavilla V, Crespo I, Collado PS, Esteller A, Sánchez-Campos S, Tuñón MJ, et al. The anti-inflammatory flavones quercetin and kaempferol cause inhibition of inducible nitric oxide synthase, cyclooxygenase-2 and reactive C-protein, and down-regulation of the nuclear factor kappaB pathway in Chang Liver cells. Eur J Pharmacol. 2007;557:221–229. doi: 10.1016/j.ejphar.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and (15N) nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32132. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- Hämäläinen M, Nieminen R, Vuorela P, Heinonen M, Moilanen E. Anti-inflammatory effects of flavonoids: genistein, kaempferol, quercetin, and daidzein inhibit STAT-1 and NF-kappaB activations, whereas flavone, isorhamnetin, naringenin, and pelargonidin inhibit only NF-kappaB activation along with their inhibitory effect on iNOS expression and NO production in activated macrophages. Mediators Inflamm. 2007;2007:45673. doi: 10.1155/2007/45673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, Kelley KW, Johnson RW. Luteolin reduces IL-6 production in microglia by inhibiting JNK phosphorylation and activation of AP-1. Proc Natl Acad Sci U S A. 2008;105:7534–7539. doi: 10.1073/pnas.0802865105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, Park HR, Lee JS, Chung TS, Ching HY, Chung J. Down-regulation of iNOS and TNF-alpha expression by kaempferol via NF-kappaB inactivation in aged rat gingival tissues. Biogerontology. 2007;8:399–408. doi: 10.1007/s10522-007-9083-9. [DOI] [PubMed] [Google Scholar]
- Kim SK, Kim HJ, Choi SE, Park KH, Choi HK, Lee MW. Anti-oxidative and inhibitory activities on nitric oxide (NO) and prostaglandin E2 (COX-2) production of flavonoids from seeds of Prunus tomentosa Thunberg. Arch Pharm Res. 2008;31:424–428. doi: 10.1007/s12272-001-1174-9. [DOI] [PubMed] [Google Scholar]
- Knott C, Stern G, Wilkin GP. Inflammatory regulators in Parkinson's disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol Cell Neurosci. 2000;16:724–739. doi: 10.1006/mcne.2000.0914. [DOI] [PubMed] [Google Scholar]
- Kowalski J, Samojedny A, Paul M, Pietsz G, Wilczok T. Effect of kaempferol on the production and gene expression of monocyte chemoattractant protein-1 in J774.2 macrophages. Pharmacol Rep. 2005;57:107–112. [PubMed] [Google Scholar]
- Lagoa R, Lopez-Sanchez C, Samhan-Arias AK, Ganan CM, Garcia-Martinez V, Gutierrez-Merino C. Kaempferol protects against rat striatal degeneration induced by 3-nitropropionic acid. J Neurochem. 2009;111:473–487. doi: 10.1111/j.1471-4159.2009.06331.x. [DOI] [PubMed] [Google Scholar]
- Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84:49–59. doi: 10.1139/Y05-143. [DOI] [PubMed] [Google Scholar]
- Lal MA, Brismar H, Eklof AC, Aperia A. Role of oxidative stress in advanced glycation end product-induced mesangial cell activation. Kidney Int. 2002;61:2006–2014. doi: 10.1046/j.1523-1755.2002.00367.x. [DOI] [PubMed] [Google Scholar]
- Le W, Rowe D, Xie W, Ortiz I, He Y, Appel SH. Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson's disease. J Neurosci. 2001;21:8447–8455. doi: 10.1523/JNEUROSCI.21-21-08447.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KM, Lee KW, Jung SK, Lee EJ, Heo YS, Bode AM, et al. Kaempferol inhibits UVB-induced COX-2 expression by suppressing Src kinase activity. Biochem Pharmacol. 2010;80:2042–2049. doi: 10.1016/j.bcp.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YC, Huang YT, Tsai SH, Lin-Shiau SY, Chen CF, Lin JK. Suppression of inducible cyclooxygenase and inducible nitric oxide synthase by apigenin and related flavonoids in mouse macrophages. Carcinogenesis. 1999;20:1945–1952. doi: 10.1093/carcin/20.10.1945. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Wang K, Gao HM, Mandavilli B, Wang JY, Hong JS. Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J Neurochem. 2001;77:182–189. doi: 10.1046/j.1471-4159.2001.t01-1-00216.x. [DOI] [PubMed] [Google Scholar]
- Lopez-Sanchez C, Martin-Romero FJ, Sun F, Luis L, Samhan-Arias AK, Garcia-Martinez V, et al. Blood micromolar concentrations of kaempferol afford protection against ischemia/ reperfusion-induced damage in rat brain. Biochem Pharmacol. 2007;1182:123–137. doi: 10.1016/j.brainres.2007.08.087. [DOI] [PubMed] [Google Scholar]
- Lorenzl S, Calingasan N, Yang L, Albers DS, Shugama S, Gregorio J, et al. Matrix metalloproteinase-9 is elevated in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridineinduced parkinsonism in mice. Neuromuscular Med. 2004;5:119–132. doi: 10.1385/NMM:5:2:119. [DOI] [PubMed] [Google Scholar]
- Mahat MY, Kulkarni NM, Vishwakarma SL, Khan FR, Thippeswamy BS, Hebballi V, et al. Modulation of the cyclooxygenase pathway via inhibition of nitric oxide production contributes to the anti-inflammatory activity of kaempferol. Eur J Pharmacol. 2010;642:169–176. doi: 10.1016/j.ejphar.2010.05.062. [DOI] [PubMed] [Google Scholar]
- Manicone AM, McGuire JK. Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol. 2008;19:34–41. doi: 10.1016/j.semcdb.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Mendis E, Kim MM, Rajapakse N, Kim SK. Sulfated glucosamine inhibits oxidation of biomolecules in cells via a mechanism involving intracellular free radical scavenging. Eur J Pharmacol. 2008;579:74–85. doi: 10.1016/j.ejphar.2007.10.027. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Walsh DT, Levi G, Perry VH. In vivo expression of cyclooxygenase-2 in rat brain following intraparenchymal injection of bacterial endotoxin and inflammatory cytokines. J Neuropathol Exp Neurol. 1999;58:1184–1191. doi: 10.1097/00005072-199911000-00008. [DOI] [PubMed] [Google Scholar]
- Munch G, Gasic-Milenkovic J, Dukic-Stefanovic S, Kuhla B, Heinrich K, Riederer P, et al. Microglial activation induces cell death, inhibits neurite outgrowth and causes neurite retraction of differentiated neuroblastoma cells. Exp Brain Res. 2003;150:1–8. doi: 10.1007/s00221-003-1389-5. [DOI] [PubMed] [Google Scholar]
- Nam KN, Son MS, Park JH, Lee EH. Shikonins attenuate microglial inflammatory responses by inhibition of ERK, Akt, and NF-κB: neuroprotective implications. Neuropharmacology. 2008;55:819–825. doi: 10.1016/j.neuropharm.2008.06.065. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci. 2002;3:216–227. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and IκBa degradation in a stimulus-specific manner in microglia. J Neurochem. 2006;96:314–323. doi: 10.1111/j.1471-4159.2005.03520.x. [DOI] [PubMed] [Google Scholar]
- Nomura Y. NF-kappaB activation and IkappaB alpha dynamism involved in iNOS and chemokine induction in astroglial cells. Life Sci. 2001;68:1695–1701. doi: 10.1016/s0024-3205(01)00967-5. [DOI] [PubMed] [Google Scholar]
- Nuttall RK, Silva C, Hader W, Bar-Or A, Patel KD, Edwards DR, et al. Metalloproteinases are enriched in microglia compared with leukocytes and they regulate cytokine levels in activated microglia. Glia. 2007;55:516–526. doi: 10.1002/glia.20478. [DOI] [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Rev. 2009;59:278–292. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewska M. Separation of quercetin, sexangularetin, kaempferol and isorhamnetin for simultaneous HPLC determination of flavonoid aglycones in inflorescences, leaves and fruits of three Sorbus species. J Pharm Biomed Ana. 2008;48:629–635. doi: 10.1016/j.jpba.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Park JS, Woo MS, Kim DH, Hyun JW, Kim WK, Lee JC, et al. Antiinflammatory mechanisms of isoflavone metabolites in lipopolysaccharide-stimulated microglial cells. J Pharmacol Exp Ther. 2007;320:1237–1245. doi: 10.1124/jpet.106.114322. [DOI] [PubMed] [Google Scholar]
- Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- Rao KM. MAP kinase activation in macrophages. J Leukoc Biol. 2001;69:3–10. [PubMed] [Google Scholar]
- Richter-Landsberg C, Besser A. Effects of organotins on rat brain astrocytes in culture. J Neurochem. 1994;63:2202–2209. doi: 10.1046/j.1471-4159.1994.63062202.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Dencoff JE, Correa JN, Reiners M, Ford CC. Effects of steroids on CSF matrix metalloproteinases in multiple sclerosis: relation to blood-brain barrier injury. Neurology. 1996;46:1626–1632. doi: 10.1212/wnl.46.6.1626. [DOI] [PubMed] [Google Scholar]
- Rosenberger J, Petrovics G, Buzas B. Oxidative stress induces proorphanin FQ and proenkephalin gene expression in astrocytes through p38- and ERK-MAP kinases and NF-kappaB. J Neurochem. 2001;79:35–44. doi: 10.1046/j.1471-4159.2001.00520.x. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, et al. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's disease. Nat Neurosci. 2002;5:1288–1293. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T. Pharmaceutical prospects of phytoestrogens. Endocr J. 2006;53:7–20. doi: 10.1507/endocrj.53.7. [DOI] [PubMed] [Google Scholar]
- Vallières N, Berard JL, David S, Lacroix S. Systemic injections of lipopolysaccharide accelerates myelin phagocytosis during Wallerian degeneration in the injured mouse spinal cord. Glia. 2006;53:103–113. doi: 10.1002/glia.20266. [DOI] [PubMed] [Google Scholar]
- Waetzig V, Czeloth K, Hidding U, Mielke K, Kanzow M, Brecht S, et al. c-Jun N-Terminal Kinases (JNKs) mediate proinflammatory actions of microglia. Glia. 2005;50:235–246. doi: 10.1002/glia.20173. [DOI] [PubMed] [Google Scholar]
- Walker DG, Lue LF. Investigations with cultured human microglia on pathogenic mechanisms of Alzheimer's disease and other neurodegenerative diseases. J Neurosci Res. 2005;81:412–425. doi: 10.1002/jnr.20484. [DOI] [PubMed] [Google Scholar]
- Westermarck J, Kahari VM. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999;13:781–792. [PubMed] [Google Scholar]
- Woo MS, Park JS, Choi IY, Kim WK, Kim HS. Inhibition of MMP-3 or -9 suppresses lipopolysaccharide-induced expression of proinflammatory cytokines and iNOS in microglia. J Neurochem. 2008;106:770–780. doi: 10.1111/j.1471-4159.2008.05430.x. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-β peptide catabolism. J Neurosci. 2006;26:10939–10948. doi: 10.1523/JNEUROSCI.2085-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR. Matrix metalloproteinases and diseases of the CNS. Trends Neurosci. 1998;21:75–80. doi: 10.1016/s0166-2236(97)01169-7. [DOI] [PubMed] [Google Scholar]
- Yong VW, Power C, Forsyth P, Edwards DR. Matrix metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001;2:502–511. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]