

Abstract
Multiple endocrine neoplasia type 1 (MEN1) results from mutations in the tumor suppressor gene, MEN1, which encodes nuclear protein menin. Menin is important for suppressing tumorigenesis in various endocrine and certain non-endocrine tissues. Although menin suppresses MEN1 through a variety of mechanisms including regulating apoptosis and DNA repair, the role of menin in regulating cell proliferation is one of the best-studied functions. Here, we focus on reviewing various mechanisms underlying menin-mediated inhibition of cell proliferation. Menin inhibits cell proliferation to repress MEN1 through multiple mechanisms. 1) Menin interacts with various histonemodifying enzymes, such as MLL, EZH2 and HDACs, to affect gene transcription, leading to repression of cell proliferation. 2) Menin also interacts with various transcription factors, such as JunD, NF-κB, PPARγ and VDR, to induce or suppress gene transcription. As these various transcription factors are known to regulate cell proliferation, their interaction with menin may be relevant to menin's role in inhibiting cell proliferation. 3) Menin inhibits cell proliferation via TGF-β signaling and Wnt/β-catenin signaling pathways. 4) Menin represses certain pro-proliferative factors involved in endocrine tumors such as IGFBP-2, IGF2 and PTHrP to repress cell proliferation. 5) Menin affects cell cycle progression to inhibit cell proliferation. This review is helpful in our understanding of the comprehensive mechanisms whereby menin represses MEN1 through inhibiting cell proliferation.
Keywords: Menin, cell proliferation, gene transcription, epigenetics, cell cycle
1. Introduction
Multiple endocrine neoplasia type 1 (MEN1) is a dominantly inherited disorder characterized with the development of various combinations of tumors in multiple endocrine glands, including the pituitary, parathyroid or pancreas [1-3], and sometimes in non-endocrine tissues [4]. The human MEN1 gene, whose mutation is responsible for the MEN1 syndrome, was localized to chromosome band 11q13 and was identified by positional cloning [5]. The human MEN1 gene contains 10 exons and encodes a ubiquitously expressed mRNA of 2.8 kb [5]. Menin, the predicted 610 amino acid protein product of the MEN1 gene, is highly conserved from Drosophila to human, however exhibits no apparent similarities to any previously known proteins [5]. Thus, its molecular function cannot be deduced from its structure. Menin contains nuclear localization signals at its carboxyl-terminal region and resides mainly in the nucleus [6]. Several studies have also found that menin is localized in cytoplasm and membrane fractions [7-8], albeit at a lower abundance, suggesting a possible function outside the nucleus. Menin is ubiquitously expressed in tissues examined, although its expression levels are variable among different tissues [9-12].
Heterozygous germline mutations of the MEN1 gene have been identified in approximately 95% of familial MEN1 [13], and about 1336 mutations in MEN1 patients have been identified [13]. Investigating the function of menin in various types of cells is important for us to understand how MEN1 tumors develop, even though these studies remain insufficient to explain the propensity of tumor development in endocrine tissues or the origin of MEN1-specific tumors. Targeted heterozygous Men1 inactivation in mice produces a spectrum of endocrine tumors similar to those observed in human patients exhibiting MEN1 syndrome [14-16]. Studies involving Men1 knockout mice support menin's role as a tumor suppressor. In addition, studies using endocrine cells and certain non-endocrine cells, such as mouse embryonic fibroblasts (MEFs), are also helpful to unravel menin's molecular function in suppressing cell proliferation.
Numerous studies have revealed direct and indirect interactions between menin and diverse proteins with known function, shedding light on its molecular function. These menin-interacting proteins involve chromatin modifying proteins, transcription factors (including nuclear receptors), DNA-repair/replication proteins and cytoskeletal proteins. Menin-interacting chromatin modifying proteins include histone methyltransferase MLL [17-23], EZH2 [24] and histone deacetylases (HDACs) [25-27], and menin affects binding of these histone modifiers to the promoter of its target genes. Menin also interacts with transcription factors, such as JunD [25-26, 28], NF-κB [29] and Pem [30], to regulate gene transcription. Recent studies have shown that menin interacts with nuclear receptors, such as Erα [31], PPARγ [32] and VDR [33], thus regulating nuclear receptor-mediated transcription. In addition, menin interacts with DNA-repair/replication proteins, such as the Fanconi anemia complementation group D2 (FANCD2) [34] and replication protein A2 (RPA2) [35], suggesting that menin plays an important role in maintenance of genomic stability. Moreover, menin also interacts with cytoskeletal proteins including glial fibrillary acid protein (GFAP) [8], vimentin[8] and IQGAP1 [36]. An excellent and detailed review on menin interacting proteins can be found elsewhere [37].
Recent data suggest that interactions between menin and menin-interacting proteins have a role in physiological regulation of embryological development [38], cell differentiation [39], cell proliferation [40], apoptosis [41], DNA-damage repair [34-35], and endocrine/metabolic functions [42]. The role of menin in regulating cellular proliferation is one of its best-studied functions. For instance, menin over expression slows proliferation of RAS-transformed NIH3T3 cells and suppresses tumorigenesis from Ras-transformed cells [40]. Mouse embryonic fibroblasts (MEFs) isolated from homozygous Men1-/-mice proliferate more rapidly than wild-type cells, while re-expression of menin in Men1-/-cells represses proliferation of these cells [43].
Furthermore, Men1 excision in mice enhances cell proliferation in pancreatic islets in vivo, through an accelerated S-phase entry [44]. As a result, we focus on reviewing partners and pathways that have a definitive or likely role for menin-mediated regulation of cell proliferation.
2. Menin interacts with histone modifiers: MLL histone H3K4 methyltransferase, Polycomb group EZH2 and histone deacetylases (HDACs)
Menin is predominantly nuclear and may act as a scaffold protein to regulate gene transcription by coordinating various chromatin-associating proteins [21, 45-47]. Several findings indicate that menin is involved in the regulation of gene transcription through histone modifications. Menin can interact with histone methyltransferase MLL [17-23], EZH2 [24], and histone deacetylases HDACs in regulating transcription of genes involved in cell proliferation [25-27]. However, to date, direct interaction between menin and EZH2 has not been reported.
2.1. Menin inhibits cell proliferation by up regulating cyclin-dependent kinase inhibitors, p27kip1 and p18Ink4c, via interacting with MLL
Several reports have shown that menin directly regulates expression of the cyclin-dependent kinase inhibiting (CDKI) genes Cdkn1b and Cdkn2c, which encode p27Kip1 and p18Ink4c respectively [20, 44, 48]. P27kip1 and p18Ink4c play a crucial role in repressing cell proliferation [49-50]. Menin recruits mixed lineage leukemia (MLL) protein to the Cdkn1b and Cdkn2c promoters and coding regions, where MLL catalyzes histone H3 lysine 4 (H3K4) methylation to up regulate their transcription [20, 44, 48] (Figure 1A). The menin/MLL histone methyltransferase complex also contains other components including Ash2, Rbbp5, and WDR5 in HeLa cells [20].
Figure 1.
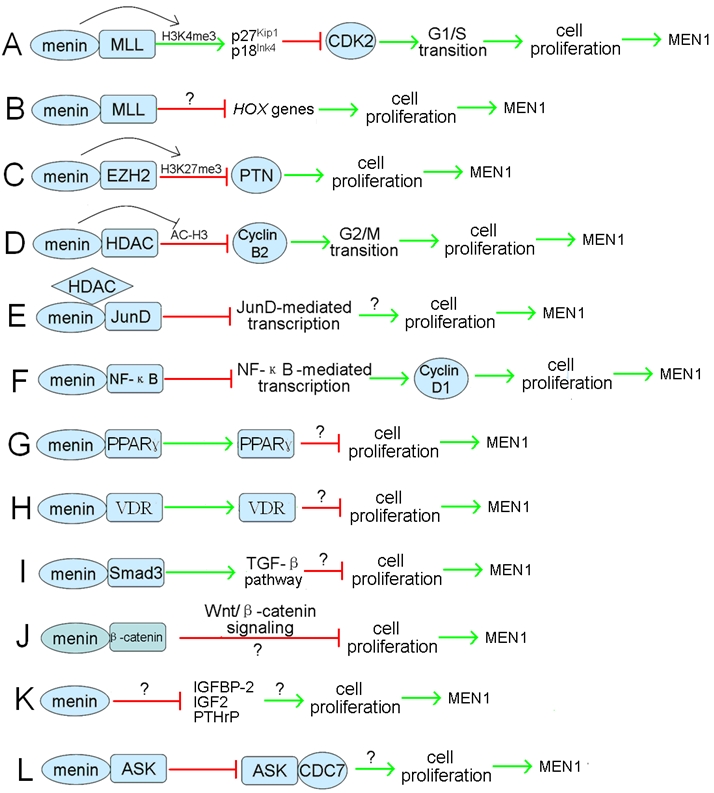
Menin inhibits cell proliferation via various mechanisms. A, Menin interacts with MLL and recruits MLL to the promoter of CDKN1B and CDKN2C (encoding p27Kip1 and p18Ink4c proteins) and enhances H3K4me3 level. Expression of p27Kip1 and p18Ink4c is activated, and the cell cycle G1/S transition is arrested. B, Menin interacts with MLL and represses the expression of Hox genes to inhibit cell proliferation. C, Menin recruits EZH2 to the promoter of PTN and represses PTN transcription through increasing H3K27me3 level, thus inhibiting cell proliferation. D, Menin recruits HDACs to the promoter of cyclin B2 and represses cyclin B2 transcription via decreasing histone H3 acetylation level, leading to arrest at G2/M transition. E, Menin associates with HDACs and interacts with the transcription factor, JunD, to represses JunD-activated transcription leading to inhibition of cell proliferation. F, Menin interacts with NF-κB and represses NF-κB-mediated Cyclin D1 transcription and inhibition of cell proliferation. G, Menin interacts with the nuclear receptor PPARγ to repress cell proliferation. H, Menin interacts with the nuclear receptor VDR to repress cell proliferation. I, Menin interacts with Smad3 and enhance the TGF-β signaling pathway to inhibit cell proliferation. J, Menin interacts with β-catenin and affects the Wnt/β-catenin signaling pathway to repress cell proliferation. K, Menin represses IGFBP-2, IGF2, and PTHrP proliferative factors involved in endocrine tumors to repress cell proliferation. L, Menin interacts with the activator of S-phase kinase (ASK) and represses ASK -induced cell proliferation. The question mark represents the related mechanism needs to be further investigated.
In cell culture, loss of either MLL or menin results in down-regulation of p27Kip1 and p18Ink4c and deregulated cell proliferation of MEFs [20, 48]. In MIN6 cells, a well-established cell line derived from mouse pancreatic β cell tumors [51], ectopic menin expression by transfection increased levels of p27Kip1 and p18Ink4c proteins and repressed cell proliferation [48].
Mice deficient for both p27Kip1 and p18Ink4c develop pituitary tumors much more rapidly than mice with either deficiency alone, suggesting that the two CDK inhibitors collaborate to suppress tumorigenesis [52]. The menin-dependent H3K4 methylation may maintain the in vivo expression of the CDK inhibitors to prevent the development of pancreatic islet tumors. In vivo expression of CDK inhibitors, including p27Kip1 and p18Ink4c, and other cell cycle regulators is reduced in islet tumors of a majority of mice with Men1 disruption [48].
An acute effect of Men1 mutation is accelerated S-phase entry and enhanced cell proliferation in pancreatic islets [44]. As early as 7 days following Men1 excision, pancreatic islet cells display increased proliferation, leading to detectable enlargement of pancreatic islets 14 days after Men1 excision [44]. Excision of the floxed Men1 in MEFs accelerates cell cycle G0/G1 to S phase entry and enhanced cyclin-dependent kinase 2 (CDK2) activity, as well as reduced expression of CDK inhibitors p27Kip1 and p18Ink4c [44]. Consistently, complementation of menin-null cells with wild-type menin represses S-phase entry. Together, these results suggest a molecular mechanism whereby menin suppresses MEN1 tumorigenesis at least partly through repression of G0/G1 to S transition by interaction with menin and MLL, leading to transcriptional activation of Cdkn1b and Cdkn2c by menin and MLL, even though the direct effect of MLL on beta cell proliferation remains to be examined.
2.2. Loss of menin deregulates homeobox (HOX) genes and promotes cell proliferation in MEN1 parathyroid tumors
Studies have suggested that menin is an oncogenic cofactor for transcription of HOX genes in a subset of leukemia via interacting with MLL fusion proteins [22-23]. Menin directly regulates the expression of Hox genes such as Hoxa9 by binding to the loci of various Hox genes during hematopoiesis and myeloid transformation [18]. On the other hand, menin may play a role in repressing tumorigenesis by affecting expression of HOX genes in human parathyroid tissue [53].
A recent study has compared the expression profiles of the 39 HOX genes in human familial MEN1 (fMEN1) parathyroid tumors and sporadic parathyroid adenomas with normal tissues and has identified a large set of 23 HOX genes that are specifically dysregulated in fMEN1 parathyroid tumors, while only 5 HOX genes are dysregulated in sporadic parathyroid tumors [53]. This study also reveals the existence of menin-dependent and menin-independent regulation of HOX genes that could be critical for the development of human parathyroid tumors [53].
In association with MLL, menin positively regulates Hoxc8 in MEFs [19], Hoxa9 in mouse bone marrow cells [18] and HOXA7, HOXA9, and HOXA10 in human HeLa cells or leukemia cells [22-23]. However, the loss of menin leads to up regulation of expression for HOXA9, HOXA10 and HOXC8 in familial parathyroid tumors. These findings suggest that menin might be a positive or negative regulator for HOX gene transcription depending on cell type (Figure 1B). HOX genes are important for cell proliferation, differentiation and morphogenesis, however, the complex in vivo role of menin-regulated HOX genes in proliferation of parathyroid tumor cells and other endocrine cells remains to be determined.
2.3. Menin inhibits cell proliferation by repressing transcription of Pleiotrophin (PTN) gene via interaction with Polycomb group (PcG) proteins
Loss of menin in MEFs increases expression of PTN 11-fold [54]. PTN is a heparin-binding growth factor that is highly expressed in certain solid cancers such as breast and lung cancer [55-56]. It has been proposed that PTN binds to its cell surface receptor, protein tyrosine phosphatase receptor Z1, and inhibits phosphatase activity of the receptor toward another oncogenic kinase, ALK [57-60]. PTN is highly expressed in a number of cancers including lung cancer, and PTN concentrations in serum were over 10-fold higher in lung cancer patients as compared with the control group [55].
It has been reported that menin inhibits human lung cancer cells through repression of PTN and its cell surface receptors including ALK [24]. Ectopic menin expression reduced PTN expression at the mRNA and protein levels of PTN in human A549 lung cancer cells [24]. Notably, Men1 knockdown in lung cancer cells and MEFs also increases the levels of mRNA and the intra-cellular and secreted PTN. Menin represses, but PTN promotes, growth of xenografts of human lung adenocarcinoma cells in mice, highlighting the crucial role of menin and PTN in controlling growth of the tumors in vivo [24]. Thus, menin represses lung cancer proliferation at least partly through inhibiting PTN expression.
Unlike the classic role of menin in recruiting MLL and enhancing MLL-mediated H3K4 methylation to promote transcription of genes, such as Cdkn1b and Cdkn2c [20, 44, 48], menin may repress transcription of Ptn via recruiting PcG proteins to enhance H3K27 methylation at the locus in lung epithelial cells [24] (Figure 1C).
PcG genes encode various proteins including Polycomb Repressive Complex 2 (PRC2), which contains Enhancer of Zeste Homolog 2 (EZH2) and its regulatory protein SUZ12 [61-63]. EZH2 is also a chromatin-associating protein with a conserved SET domain [64]. However, unlike the SET domain in MLL that methylates H3K4 [23], the EZH2 SET domain specifically methylates H3K27. The methylated H3K27 can be recognized by other specific binding proteins to compress chromatin structure leading to repression of gene transcription [63-64]. Menin binds the promoter of PTN and recruits the PcG complex including EZH2 and SUZ12 to the locus, resulting in H3K27 trimethylation, PTN suppression and inhibition of proliferation of lung cancer cells [24]. Loss of menin notably abrogates binding of EZH2 and SUZ12 to the PTN locus and reduced H3K27me3 at the locus. However, it remains unclear whether menin directly interacts with EZH2.
These results suggest that menin recruits the PcG complex, including EZH2 and SUZ12 to the PTN locus, to methylate H3K27 and then silence PTN expression. This mechanism is quite distinct from menin-mediated suppression of endocrine tumors through up regulating a positive histone mark, H3K4 methylation at the loci of Cdkn1b and cdkn2c [48]. These studies expand the role of menin-mediated suppression of endocrine tumors to suppression of human lung cancer. Further studies remain to determine whether menin is also crucial for repression of PTN expression in endocrine cells.
2.4. Menin inhibits cell proliferation by repressing cyclin B2 via interaction with HDACs
It has been reported that menin represses transcriptional factor Jun D in a histone deacetylase (HDAC)-dependent manner, and mSin3A, a general co-repressor, bridges HDACs and menin in the interaction [25-26, 28]. In our recent study, it has been shown that menin represses an endogenous proproliferative gene via recruiting HDAC3 [27].
In MEFs, menin binds to the Ccnb2 promoter in the region containing 3 CCAAT boxes, cell cycle-dependent element (CDE) and cell cycle genes homology region (CHR); menin represses transcription of Ccnb2 [27]. Further, menin recruited HDAC3 to the Ccnb2 locus and reduced the histone H3 acetylation level. Notably, MEN1 disease-related menin point-mutants, L22R and A242V, fail to repress histone H3 acetylation at the Ccnb2 locus as well as transcription of Ccnb2 [27]. Consistent with these findings, insulinomas in Men1-mutated mice express a high level of cyclin B2 [65].
Although it has been reported that menin represses G1/S phase transition [66], more menin-expressing MEFs are blocked at G2/M phase than menin-null cells [27, 44, 54], and fewer menin-expressing cells are at M-phase, as shown by staining for phospho-histone H3 (Ser10) [27]. Therefore, menin also blocks cells at G2/M phase. When menin-null and menin-expressing cells are synchronized at G2/M phase, the menin-null cells exit from G2/M phase more rapidly than menin-expressing cells. These findings indicate that menin-induced repression of cyclin B2 plays an important role in menin-dependent inhibition of G2/M transition and cell proliferation (Figure 1D).
3. Menin interacts with transcription factors
Menin directly interacts with a number of transcription factors including JunD and NF-κB, two transcription factors involved in regulating cell proliferation, and represses JunD-mediated and NF-κB-mediated transcription. Menin may exert its repressive effect on gene transcription partly by recruiting repressors such as histone deacetylases [25]. Further studies are needed to establish the role of menin-JunD and menin-NF-κB interactions in suppressing cell proliferation by regulating the endogenous and downstream genes.
3.1. Menin inhibits cell proliferation by interacting with JunD
JunD is a transcription factor belonging to the AP1 transcription complex family and is involved in negative control of cell proliferation [67]. Menin-JunD interaction has been identified by the yeast two-hybrid method in the human brain cDNA library and has been confirmed in vitro and in vivo [28]. The interaction is mediated via the N-terminal transcription activation domain of JunD and the C-terminal part of menin. Menin represses JunD-activated transcription through a histone deacetylase (HDAC)-dependent mechanism [25] in which mSin3A, a general corepressor, recruits HDAC to menin [26]. Several naturally occurring and clustered MEN1 missense mutations disrupted menin's interaction with JunD. These observations suggest that menin's tumor suppressing function involves direct binding to JunD and inhibition of JunD-activated transcription [28] (Figure 1E).
At first sight, it seems paradoxical that menin, a tumor suppressor, suppresses the activity of another protein, JunD, that inhibits cell proliferation. However, recent studies have suggested that JunD, as a repressor of cell proliferation, might bind directly to menin and exist as a complex, rather than as a ‘free’ form [68]. Association with menin appears to reverse the effect of JunD on cell proliferation, converting JunD to a suppressor of cell proliferation. In contrast, Jun D may act as a proliferation promoter when it is unable to bind menin. Binding of ‘free’ JunD (a proliferation promoter) to menin (a proliferation suppressor) leads to the formation of a proliferation-suppressing complex [69].
Two isoforms of JunD, the full-length isoform of JunD (JunD-FL) and the truncated isoform (ΔJunD), resulting from alternative translation start codons within JunD mRNA, are ubiquitously expressed [68]. Menin suppresses transcriptional activity of JunD-FL, but not of ΔJunD, highlighting JunD-FL as a functional partner of menin. Disruption of JunD-FL-menin interaction by point mutations, followed by examining the impact on cell proliferation, may further reveal the importance of this interaction in repressing cell proliferation [68]. It remains unclear what are the key endogenous target genes of the menin-JunD complex in regulating cell proliferation.
3.2. Menin inhibits cell proliferation by interacting with NF-κB
Interaction of menin and NF-κB has been identified by immunoprecipitation in HEK 293 cells [29]. The NF-κB proteins p50, p52 and p65 are found to interact specifically with menin in vitro and in vivo. Furthermore, in HeLa cells, Cos7 cells and NTERA-2 cells, menin represses p65-mediated transcriptional activation on NF-κB sites in a reporter gene in a dose-dependent manner, and suppresses PMA (phorbol 12-myristate 13-acetate)-stimulated or TNFα-stimulated NF-κB activation [29]. Loss of menin-mediated repression of NF-κB activity may thus contribute to oncogenesis (Figure 1F).
Recent studies have revealed a negative correlation between menin expression and p65 phosphorylation in human parathyroid tumors, as well as a decrease in cyclin D1 expression in response to inhibition of NF-κB [70]. Cyclin D1 is a key regulator of G1/0-S phase transition and promotes cell proliferation. Over expression of cyclin D1, as well as loss of menin, has been demonstrated to induce parathyroid hyperplasia in transgenic mice [16, 71-72]. A blockade of the NF-κB activity caused a significant decrease in cyclin D1 expression in human parathyroid cells, suggesting a proliferative role for NF-κB in parathyroid cells. Nevertheless, it remains to be determined whether menin and NF-κB are colocalized to the loci of their target genes to control cell proliferation.
Cyclin D1 expression is increased in menin-knockdown IEC-17 cells, a non-transformed crypt-like cell line representative of the proliferative compartment of the small intestine, at least partly due to up regulation of NF-κB-mediated transcription [73]. A blockade of the NF-κB activity reduces proliferation, cell cycle, and cyclin D1 expression levels in IEC-17 cells to the levels observed in the presence of menin [73]. These studies indicate a strong correlation between NF-κB activity and cyclin D1 expression. Menin expression in the IEC-17 epithelial cells and its repression of NF-κB/cyclin D1 may be relevant to the physiological function of menin in regulating proliferation of the intestinal epithelial cells. Moreover, the reduction of menin expression in the IEC-17 cell line leads to tumorigenic transformation. It remains unclear whether menin also regulates NF-κB/cyclin D1 in endocrine cells and whether all of them directly bind to the loci of their target genes.
3.3. Menin inhibits cell proliferation by interacting with nuclear receptors
Both immunoprecipitation experiments and yeast two-hybrid studies show that menin interacts with nuclear receptors PPARγ [32] and VDR [33] in a ligand-independent manner. Menin augments PPARγ- and VDR-target gene expression through recruitment of H3K4 methyltransferase activity (Figure 1G and H). Nuclear receptors play an important role in the development of human cancers.
PPARγ is expressed in MEN1-associated tumor types, such as pituitary adenomas and neuroendocrine tumors [74-75]. Moreover, p18Ink4c is found to be up regulated in mouse fibroblasts that ectopically express PPARγ [76]. Menin suppresses adaptive pancreatic β-cell proliferation in obese mice, in which PPARγ is also involved, raising an interesting possibility that the interaction between menin and PPARγ is important for regulating cell proliferation as well [77-78]. In addition, PPARγ is involved in adipocyte differentiation and proliferation and PPARγ plays a role in lipoma development [32].
Several reports suggest that aberrant VDR function can contribute to parathyroid adenoma formation. VDR-null mice develop parathyroid hyperplasia [79]. The VDR gene polymorphisms are associated with primary hyperparathyroidism and alterations of VDR mRNA and protein levels have been reported to occur in parathyroid adenomas [80-82]. In addition, p27Kip1 and p18Ink4c are also regulated by VDR [83-85]. Reduced p27Kip1, p18Ink4c and p21Waf1/Cip1 levels are reported in sporadic parathyroid adenomas [86]. P27Kip1 is significantly reduced in both sporadic and MEN1-related parathyroid adenomas, as compared to normal parathyroid tissue [33].
4. Menin inhibits cell proliferation by interacting with cell signaling pathways
Recent studies have shown that menin can inhibit cell proliferation by collaborating with or regulating cell signaling pathways, such as the transforming growth factor-β (TGF-β) and the Wnt/β-catenin signaling pathways.
4.1. Menin represses cell proliferation via the transforming growth factor-β (TGF-β) signaling pathway
Menin has been shown to interfere with the TGF -β signaling pathway by interacting with Smad3
to suppress proliferation of GH4C1, a rat pituitary cell line [87]. Knockdown of menin interrupts Smad3 binding to DNA, thereby blocking the TGF-β signaling pathway (Figure 1I). The Smad-family proteins are critical components of the TGF-β signaling pathway. TGF-β exerts inhibitory proliferation and transcriptional activities through two receptor-regulated Smads, Smad2 and Smad3 [88]. Receptor-mediated phosphorylation of Smad2 or Smad3 induces their association with the common partner Smad4, followed by translocation of the Smad proteins into the nucleus, in which these complexes activate transcription of specific genes [89]. TGF-β is thought to regulate the proliferation of pituitary cells by inhibiting their growth in an autocrine or paracrine manner [90]. Reduced menin expression leads to loss of TGF-β-mediated inhibition of proliferation of primary parathyroid cells [42]. TGF-β is an important autocrine/paracrine negative regulator of parathyroid cell proliferation and Parathyroid Hormone (PTH) secretion, and loss of TGF-β signaling resulting from menin inactivation may contribute to parathyroid tumorigenesis [42].
In recent studies, Leydig cell tumors (LCT) developed from heterozygous Men1 mutant mice show deregulation of the TGF-β pathway [91]. Both immunostaining and Western Blotting analyses demonstrate markedly reduced nuclear expression of Smad1, 3, 4, and 5 in the tumors. Furthermore, the expression of p18, p27 and cyclin dependant kinase 4 (Cdk4), targets of the TGF-β pathway, is altered in the Leydig cell tumors [91]. Further comparison of the global gene expression profiles of testis and ovary adenomas, along with other endocrine tumors, from control and Men1 heterozygous mice also reveal deregulation of the TGF-β signaling pathway in sex cord stromal tumors [92].
4.2. Menin represses cell proliferation via the Wnt/β-catenin signaling pathway
It has been reported that menin directly interacts with β-catenin and carries β-catenin out of the nucleus through its two functional nuclear export signals [93]. In Men1-null MEFs and insulinomas tissues from β-cell-specific Men1 knockout mice, β-catenin is accumulated in the nucleus [93]. Since over expression of menin reduces nuclear accumulation of β-catenin and its transcriptional activity [93], menin may repress cell proliferation through suppression of the Wnt/β-catenin signaling, which is pro-proliferative [93] (Figure 1J). However, in cultured rodent islet tumor cells, menin may repress cell proliferation partly by activating the Wnt/β-catenin signaling [94]. When menin and activated β-catenin is over expressed, the Wnt/ β-catenin downstream target gene, Axin2, is significantly enhanced correlating with increased H3K4 trimethylation at the promoter of the Axin2 gene [94].
These different results regarding the impact of menin on the Wnt signaling pathway and cell proliferation are unexpected, and may be attributed to the different cell models used, in which menin's primary role could be different in distinct context of cells. Relevant to this possibility, in mice stably expressing β-catenin (β-catactive), expression of β-catenin in early embryonic development disrupts pancreatic development, whereas its late expression leads to an increase of islets mainly by increasing the number of proliferating cells [95]. These findings suggest that β-catenin functions differently in regulating cell proliferation at different stages of pancreatic development. The distinct regulations of β-catenin by menin in insulinomas from β-cell-specific Men1 knockout mice or in cultured rodent islet tumors may be attributed to the distinct functions of β-catenin in a context-dependent manner. Nevertheless, the precise role of beta catenin in regulating pancreatic islet cells is less clear. Further studies are needed to elucidate the detailed mechanisms underlying the functional interaction between menin and the Wnt signaling pathway in regulating beta cell proliferation.
5. Menin represses certain pro-proliferative factors that may promote proliferation of endocrine tumor cells
Some proliferative factors that are considered to be relevant to endocrine tumors, such as insulin-like growth factor binding protein 2 (IGFBP-2), insulin-like growth factor 2 (IGF2) and parathyroid hormone-related protein (PTHrP) are inhibited by menin [54, 65] (Figure 1K).
Targeted disruption of Men1 leads to up regulation of IGFBP-2, and complementation of menin-null MEFs with wild-type menin reduces expression of IGFBP-2 [54]. MEN1-related point mutant menin fails to repress expression of IGFBP-2. Consistent with this, the promoter of IGFBP-2 is repressed by wild-type menin, but not by a MEN1-related point mutant. Menin also alters the chromatin structure surrounding the promoter of the IGFBP-2 gene. Furthermore, nuclear localization signals in menin are crucial for repressing the expression of IGFBP-2 [54].
IGFBP-2 was originally identified for its ability to bind insulin-like growth factors (IGFs) [96]. IGFBP-2 is a member of the IGFBP family and plays a crucial role in regulating cell proliferation. It can stimulate cell proliferation in an IGF-independent manner in certain types of cells, but also inhibits cell proliferation by suppressing the activities of IGFs [96]. For example, IGFBP-2 inhibits proliferation of normal epithelial cells but stimulates proliferation of cancer cells [97-98]. Moreover, it has been reported that IGFBP-2 partly mediates TGF-β-induced inhibition of proliferation of mink lung epithelial cells [99]. It is well known that TGF-β inhibits proliferation of normal epithelial cells but stimulates proliferation of many cancer cells [96, 100]. This positive and negative regulation of cell proliferation may be similar to what is observed for the role of IGFBP-2.
Another study reveals increased expression of IGF2 in mouse Men1 insulinomas [65]. The IGF2 is known to play an important role in β-cell proliferation. Further analysis reveals that high expression of IGF2 in murine Men1 insulinomas is accompanied by the hypermethylation of the intragenic differentially methylated region 2 (DMR2), which is associated with an increased level of transcription through methylation [65]. The up-regulation of IGF2 expression could contribute to tumorigenesis of β-cells, as a result of menin inactivation. In addition, PTHrP is also up-regulated in mouse Men1 insulinomas [65].
However, the precise role of menin in repressing cell proliferation through repressing IGFBP-2, IGF2 and PTHrp remains to be further explored.
6. Menin in cell cycle
Menin plays a crucial role in regulating the cell cycle. Expression of menin has been examined at various cell cycle stages in a rat pituitary cell line, GH4C1 [101], although in different types of cells, this phenomenon may vary. Menin expression fluctuates during cell cycle progression, mimicking expression of cyclins [101]. The expression level of menin is relatively high in quiescent cells during G0/G1, then transiently decreases when the cells enter the cycle in G1, and increases again as the cells enter S phase [101]. During and immediately after cell division, menin resurges in the cytoplasm and the nucleus. However, during S phase, menin must be translocated to the cytoplasm for the cell cycle to proceed [7, 102]. In non-dividing cells synchronized at the G2/M phases of the cell cycle, localization of menin occurs predominantly in the nucleus [101]. Menin has also been shown to increase the number of cells in G2/M, suggesting that menin causes a potential block in the G2/M phases, which is consistent with its nuclear localization during this phase. This fluctuation and nuclear/cytoplasm shuttling are consistent with the function of menin in regulating cell cycle progression. However, it remains unclear whether fluctuation of menin levels directly affects expression of cell cycle genes and cell cycle progression. Further studies are necessary to address this question.
Studies have demonstrated that menin represses the cell cycle G0/G1 to S phase transition and G2/M phase transition. The function of cell cycle control by menin is partly overlapped with the function of menin in interacting with histone modifiers, which alter expression of cell cycle-regulators. Moreover, menin has been shown to interact with the activator of S-phase kinase (ASK), a component of the Cdc7/ASK kinase complex, and represses ASK-induced cell proliferation [66] (Figure 1L). In addition, a recent study has shown that acute Men1 excision promotes the proliferation of pancreatic β cells in mice via increasing the expression of several genes involved in the cell cycle, such as Cyclin A and Pbk, a PDZ-binding kinase regulating mitosis [103]. Collectively, these findings suggest that menin exerts its function of repressing proliferation through coordinately inhibiting cell cycle progression. How menin is regulated at either transcriptional or post-transcriptional levels during cell cycling remains to be investigated .
7. Summary and Perspective
Menin likely exerts an important function in suppressing tumorigenesis via repressing cell proliferation, and menin inhibits cell proliferation to repress MEN1 through various mechanisms (Figure 1). Menin may interact with histone modifying enzymes, transcription factors including nuclear receptors to suppress cell proliferation. In addition, menin also affects signaling pathways, expression of endocrine tumor-related proliferative factors and cell cycle progression to inhibit cell proliferation. Further study of these regulations will be helpful for us to understand menin's tumor-suppressing function thoroughly.
Most of the mechanisms for menin-mediated suppression of cell proliferation are closely related to menin's role in regulation of gene transcription. The mechanisms whereby menin regulates transcription are diverse and complex. Menin not only promotes but also represses gene transcription. Menin can directly or indirectly associate with the promoters of various genes and recruit histone-modifying enzymes, such as MLL, EZH2 and HDACs, to modify histones and thus determine the status of gene transcription. Also, menin interacts with and affects activity of various transcriptional factors, such as JunD and NF-κB, to regulate gene transcription. Despite distinct or even opposite roles of menin in either activating or inactivating gene transcription, these opposite roles of menin in gene transcription may be all important for suppressing tumorigenesis. How does menin play a positive or negative role in regulating gene expression? Is that determined by a tissue-specific or gene-specific manner, or both? Answers to these questions will likely further our understanding of how menin functions in a tissue-specific manner.
Another interesting question is how menin itself is regulated. Considering its expression fluctuates with cell cycle progression, it could be regulated both at a transcriptional level and post-transcriptional level. Protein translation or protein stability could be involved in post-transcriptional regulation. Protein modifications, such as phosphorylation, ubiquitination, and sumoylation should be considered.
Interestingly, accumulating evidence in recent years suggests that menin is not only a tumor suppressor gene in multiple endocrine organs, but also an oncogenic cofactor promoting proliferation of leukemia cells and development of mixed lineage leukemia. The dual role of menin in suppressing or promoting cell proliferation seems to be related to the menin-MLL dual function in suppressing or promoting cell proliferation. Elucidating mechanisms for menin's tissue-specific and gene-specific activities will likely deepen our understanding of the role of menin in regulating cell proliferation and suppressing MEN1 tumorigenesis.
Acknowledgments
This work was supported, in part, by a National Institute of Health Grant (R01-CA-113962 to XH) and by an American Diabetes Association Grant (7-07-RA-60 to XH.). We thank Shivani Sethi for reading and editing the manuscript. We apologize for not being able to cite all the relevant references due to space limitation.
Conflicts of interests
The authors have no conflicts of interests to declare.
References
- 1.Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW, Tomassetti P, Tonelli F, Wells SA, Jr, Marx SJ. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86:5658–5671. doi: 10.1210/jcem.86.12.8070. [DOI] [PubMed] [Google Scholar]
- 2.Gardner DG. Recent advances in multiple endocrine neoplasia syndromes. Adv Intern Med. 1997;42:597–627. [PubMed] [Google Scholar]
- 3.Trump D, Farren B, Wooding C, Pang JT, Besser GM, Buchanan KD, Edwards CR, Heath DA, Jackson CE, Jansen S, Lips K, Monson JP, O'Halloran D, Sampson J, Shalet SM, Wheeler MH, Zink A, Thakker RV. Clinical studies of multiple endocrine neoplasia type 1 (MEN1) Qjm. 1996;89:653–669. doi: 10.1093/qjmed/89.9.653. [DOI] [PubMed] [Google Scholar]
- 4.Tsukada T, Nagamura Y, Ohkura N. MEN1 gene and its mutations: Basic and clinical implications. Cancer Sci. 2008 doi: 10.1111/j.1349-7006.2008.01034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 6.Guru SC, Goldsmith PK, Burns AL, Marx SJ, Spiegel AM, Collins FS, Chandrasekharappa SC. Menin, the product of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci U S A. 1998;95:1630–1634. doi: 10.1073/pnas.95.4.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang SC, Zhuang Z, Weil RJ, Pack S, Wang C, Krutzsch HC, Pham TA, Lubensky IA. Nuclear/cytoplasmic localization of the multiple endocrine neoplasia type 1 gene product, menin. Lab Invest. 1999;79:301–310. [PubMed] [Google Scholar]
- 8.Lopez-Egido J, Cunningham J, Berg M, Oberg K, Bongcam-Rudloff E, Gobl A. Menin's interaction with glial fibrillary acidic protein and vimentin suggests a role for the intermediate filament network in regulating menin activity. Exp Cell Res. 2002;278:175–183. doi: 10.1006/excr.2002.5575. [DOI] [PubMed] [Google Scholar]
- 9.Ikeo Y, Sakurai A, Suzuki R, Zhang MX, Koizumi S, Takeuchi Y, Yumita W, Nakayama J, Hashizume K. Proliferation-associated expression of the MEN1 gene as revealed by in situ hybridization: possible role of the menin as a negative regulator of cell proliferation under DNA damage. Lab Invest. 2000;80:797–804. doi: 10.1038/labinvest.3780084. [DOI] [PubMed] [Google Scholar]
- 10.Maruyama K, Tsukada T, Hosono T, Ohkura N, Kishi M, Honda M, Nara-Ashizawa N, Nagasaki K, Yamaguchi K. Structure and distribution of rat menin mRNA. Mol Cell Endocrinol. 1999;156:25–33. doi: 10.1016/s0303-7207(99)00150-1. [DOI] [PubMed] [Google Scholar]
- 11.Stewart C, Parente F, Piehl F, Farnebo F, Quincey D, Silins G, Bergman L, Carle GF, Lemmens I, Grimmond S, Xian CZ, Khodei S, Teh BT, Lagercrantz J, Siggers P, Calender A, Van de Vem V, Kas K, Weber G, Hayward N, Gaudray P, Larsson C. Characterization of the mouse Men1 gene and its expression during development. Oncogene. 1998;17:2485–2493. doi: 10.1038/sj.onc.1202164. [DOI] [PubMed] [Google Scholar]
- 12.Wautot V, Khodaei S, Frappart L, Buisson N, Baro E, Lenoir GM, Calender A, Zhang CX, Weber G. Expression analysis of endogenous menin, the product of the multiple endocrine neoplasia type 1 gene, in cell lines and human tissues. Int J Cancer. 2000;85:877–881. doi: 10.1002/(sici)1097-0215(20000315)85:6<877::aid-ijc23>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 13.Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2008;29:22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- 14.Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, Lorang D, Libutti SK, Chandrasekharappa SC, Marx SJ, Spiegel AM, Collins FS. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A. 2001;98:1118–1123. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biondi C, Gartside M, Tonks I, Paterson C, Hayward NK, Kay GF. Targeting and conditional inactivation of the murine Men1 locus using the Cre recombinase: loxP system. Genesis. 2002;32:150–151. doi: 10.1002/gene.10061. [DOI] [PubMed] [Google Scholar]
- 16.Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol Endocrinol. 2003;17:1880–1892. doi: 10.1210/me.2003-0154. [DOI] [PubMed] [Google Scholar]
- 17.Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res. 2007;67:7275–7283. doi: 10.1158/0008-5472.CAN-06-2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen YX, Yan J, Keeshan K, Tubbs AT, Wang H, Silva A, Brown EJ, Hess JL, Pear WS, Hua X. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci U S A. 2006;103:1018–1023. doi: 10.1073/pnas.0510347103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, Kay GF, Hayward NK, Hess JL, Meyerson M. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 20.Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, Hua X, Roeder RG, Meyerson M, Hess JL. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancerassociated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLLassociated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 23.Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao SB, Feng ZJ, Xu B, Wu Y, Yin P, Yang Y, Hua X, Jin GH. Suppression of lung adenocarcinoma through menin and polycomb genemediated repression of growth factor pleiotrophin. Oncogene. 2009;28:4095–4104. doi: 10.1038/onc.2009.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gobl AE, Berg M, Lopez-Egido JR, Oberg K, Skogseid B, Westin G. Menin represses JunD-activated transcription by a histone deacetylase-dependent mechanism. Biochim Biophys Acta. 1999;1447:51–56. doi: 10.1016/s0167-4781(99)00132-3. [DOI] [PubMed] [Google Scholar]
- 26.Kim H, Lee JE, Cho EJ, Liu JO, Youn HD. Menin, a tumor suppressor, represses JunDmediated transcriptional activity by association with an mSin3A-histone deacetylase complex. Cancer Res. 2003;63:6135–6139. [PubMed] [Google Scholar]
- 27.Wu T, Zhang X, Huang X, Yang Y, Hua X. Regulation of cyclin B2 expression and cell cycle G2/m transition by menin. J Biol Chem. 2010;285:18291–18300. doi: 10.1074/jbc.M110.106575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, Burns AL. Menin interacts with the AP1 transcription factor JunD and represses JunDactivated transcription. Cell. 1999;96:143–152. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- 29.Heppner C, Bilimoria KY, Agarwal SK, Kester M, Whitty LJ, Guru SC, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, Burns AL. The tumor suppressor protein menin interacts with NF-kappaB proteins and inhibits NFkappaB-mediated transactivation. Oncogene. 2001;20:4917–4925. doi: 10.1038/sj.onc.1204529. [DOI] [PubMed] [Google Scholar]
- 30.Lemmens IH, Forsberg L, Pannett AA, Meyen E, Piehl F, Turner JJ, Van de Ven WJ, Thakker RV, Larsson C, Kas K. Menin interacts directly with the homeobox-containing protein Pem. Biochem Biophys Res Commun. 2001;286:426–431. doi: 10.1006/bbrc.2001.5405. [DOI] [PubMed] [Google Scholar]
- 31.Dreijerink KM, Mulder KW, Winkler GS, Hoppener JW, Lips CJ, Timmers HT. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res. 2006;66:4929–4935. doi: 10.1158/0008-5472.CAN-05-4461. [DOI] [PubMed] [Google Scholar]
- 32.Dreijerink KM, Varier RA, van Beekum O, Jeninga EH, Hoppener JW, Lips CJ, Kummer JA, Kalkhoven E, Timmers HT. The multiple endocrine neoplasia type 1 (MEN1) tumor suppressor regulates peroxisome proliferatoractivated receptor gamma-dependent adipocyte differentiation. Mol Cell Biol. 2009;29:5060–5069. doi: 10.1128/MCB.01001-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dreijerink KM, Varier RA, van Nuland R, Broekhuizen R, Valk GD, van der Wal JE, Lips CJ, Kummer JA, Timmers HT. Regulation of vitamin D receptor function in MEN1-related parathyroid adenomas. Mol Cell Endocrinol. 2009;313:1–8. doi: 10.1016/j.mce.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 34.Jin S, Mao H, Schnepp RW, Sykes SM, Silva AC, D'Andrea AD, Hua X. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 2003;63:4204–4210. [PubMed] [Google Scholar]
- 35.Sukhodolets KE, Hickman AB, Agarwal SK, Sukhodolets MV, Obungu VH, Novotny EA, Crabtree JS, Chandrasekharappa SC, Collins FS, Spiegel AM, Burns AL, Marx SJ. The 32-kilodalton subunit of replication protein A interacts with menin, the product of the MEN1 tumor suppressor gene. Mol Cell Biol. 2003;23:493–509. doi: 10.1128/MCB.23.2.493-509.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan J, Yang Y, Zhang H, King C, Kan HM, Cai Y, Yuan CX, Bloom GS, Hua X. Menin interacts with IQGAP1 to enhance intercellular adhesion of beta-cells. Oncogene. 2009;28:973–982. doi: 10.1038/onc.2008.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balogh K, Racz K, Patocs A, Hunyady L. Menin and its interacting proteins: elucidation of menin function. Trends Endocrinol Metab. 2006;17:357–364. doi: 10.1016/j.tem.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 38.Bertolino P, Radovanovic I, Casse H, Aguzzi A, Wang ZQ, Zhang CX. Genetic ablation of the tumor suppressor menin causes lethality at mid-gestation with defects in multiple organs. Mech Dev. 2003;120:549–560. doi: 10.1016/s0925-4773(03)00039-x. [DOI] [PubMed] [Google Scholar]
- 39.Sowa H, Kaji H, Hendy GN, Canaff L, Komori T, Sugimoto T, Chihara K. Menin is required for bone morphogenetic protein 2- and transforming growth factor beta-regulated osteoblastic differentiation through interaction with Smads and Runx2. J Biol Chem. 2004;279:40267–40275. doi: 10.1074/jbc.M401312200. [DOI] [PubMed] [Google Scholar]
- 40.Kim YS, Burns AL, Goldsmith PK, Heppner C, Park SY, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ. Stable overexpression of MEN1 suppresses tumorigenicity of RAS. Oncogene. 1999;18:5936–5942. doi: 10.1038/sj.onc.1203005. [DOI] [PubMed] [Google Scholar]
- 41.La P, Yang Y, Karnik SK, Silva AC, Schnepp RW, Kim SK, Hua X. Menin-mediated caspase 8 expression in suppressing multiple endocrine neoplasia type 1. J Biol Chem. 2007;282:31332–31340. doi: 10.1074/jbc.M609555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sowa H, Kaji H, Kitazawa R, Kitazawa S, Tsukamoto T, Yano S, Tsukada T, Canaff L, Hendy GN, Sugimoto T, Chihara K. Menin inactivation leads to loss of transforming growth factor beta inhibition of parathyroid cell proliferation and parathyroid hormone secretion. Cancer Res. 2004;64:2222–2228. doi: 10.1158/0008-5472.can-03-3334. [DOI] [PubMed] [Google Scholar]
- 43.Schnepp RW, Mao H, Sykes SM, Zong WX, Silva A, La P, Hua X. Menin induces apoptosis in murine embryonic fibroblasts. J Biol Chem. 2004;279:10685–10691. doi: 10.1074/jbc.M308073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schnepp RW, Chen YX, Wang H, Cash T, Silva A, Diehl JA, Brown E, Hua X. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006;66:5707–5715. doi: 10.1158/0008-5472.CAN-05-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Y, Hua X. In search of tumor suppressing functions of menin. Mol Cell Endocrinol. 2007;265-266:34–41. doi: 10.1016/j.mce.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.La P, Desmond A, Hou Z, Silva AC, Schnepp RW, Hua X. Tumor suppressor menin: the essential role of nuclear localization signal domains in coordinating gene expression. Oncogene. 2006;25:3537–3546. doi: 10.1038/sj.onc.1209400. [DOI] [PubMed] [Google Scholar]
- 47.Jin S, Zhao H, Yi Y, Nakata Y, Kalota A, Gewirtz AM. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J Clin Invest. 2010;120:593–606. doi: 10.1172/JCI38030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, Kim SK. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zindy F, Cunningham JJ, Sherr CJ, Jogal S, Smeyne RJ, Roussel MF. Postnatal neuronal proliferation in mice lacking Ink4d and Kip1 inhibitors of cyclin-dependent kinases. Proc Natl Acad Sci U S A. 1999;96:13462–13467. doi: 10.1073/pnas.96.23.13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zindy F, Nilsson LM, Nguyen L, Meunier C, Smeyne RJ, Rehg JE, Eberhart C, Sherr CJ, Roussel MF. Hemangiosarcomas, medulloblastomas, and other tumors in Ink4c/p53-null mice. Cancer Res. 2003;63:5420–5427. [PubMed] [Google Scholar]
- 51.Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 52.Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, Su L, Xiong Y. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998;12:2899–2911. doi: 10.1101/gad.12.18.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen HC, Rosen JE, Yang LM, Savage SA, Burns AL, Mateo CM, Agarwal SK, Chandrasekharappa SC, Spiegel AM, Collins FS, Marx SJ, Libutti SK. Parathyroid tumor development involves deregulation of homeobox genes. Endocr Relat Cancer. 2008;15:267–275. doi: 10.1677/ERC-07-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.La P, Schnepp RW, C DP, A CS, Hua X. Tumor suppressor menin regulates expression of insulin-like growth factor binding protein 2. Endocrinology. 2004;145:3443–3450. doi: 10.1210/en.2004-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jager R, List B, Knabbe C, Souttou B, Raulais D, Zeiler T, Wellstein A, Aigner A, Neubauer A, Zugmaier G. Serum levels of the angiogenic factor pleiotrophin in relation to disease stage in lung cancer patients. Br J Cancer. 2002;86:858–863. doi: 10.1038/sj.bjc.6600202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perez-Pinera P, Chang Y, Deuel TF. Pleiotrophin, a multifunctional tumor promoter through induction of tumor angiogenesis, remodeling of the tumor microenvironment, and activation of stromal fibroblasts. Cell Cycle. 2007;6:2877–2883. doi: 10.4161/cc.6.23.5090. [DOI] [PubMed] [Google Scholar]
- 57.Lu KV, Jong KA, Kim GY, Singh J, Dia EQ, Yoshimoto K, Wang MY, Cloughesy TF, Nelson SF, Mischel PS. Differential induction of glioblastoma migration and growth by two forms of pleiotrophin. J Biol Chem. 2005;280:26953–26964. doi: 10.1074/jbc.M502614200. [DOI] [PubMed] [Google Scholar]
- 58.Meng K, Rodriguez-Pena A, Dimitrov T, Chen W, Yamin M, Noda M, Deuel TF. Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor -type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci U S A. 2000;97:2603–2608. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Powers C, Aigner A, Stoica GE, McDonnell K, Wellstein A. Pleiotrophin signaling through anaplastic lymphoma kinase is rate-limiting for glioblastoma growth. J Biol Chem. 2002;277:14153–14158. doi: 10.1074/jbc.M112354200. [DOI] [PubMed] [Google Scholar]
- 60.Stoica GE, Kuo A, Aigner A, Sunitha I, Souttou B, Malerczyk C, Caughey DJ, Wen D, Karavanov A, Riegel AT, Wellstein A. Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J Biol Chem. 2001;276:16772–16779. doi: 10.1074/jbc.M010660200. [DOI] [PubMed] [Google Scholar]
- 61.Ringrose L, Paro R. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development. 2007;134:223–232. doi: 10.1242/dev.02723. [DOI] [PubMed] [Google Scholar]
- 62.Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 63.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 64.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 65.Fontaniere S, Tost J, Wierinckx A, Lachuer J, Lu J, Hussein N, Busato F, Gut I, Wang ZQ, Zhang CX. Gene expression profiling in insulinomas of Men1 beta-cell mutant mice reveals early genetic and epigenetic events involved in pancreatic beta-cell tumorigenesis. Endocr Relat Cancer. 2006;13:1223–1236. doi: 10.1677/erc.1.01294. [DOI] [PubMed] [Google Scholar]
- 66.Schnepp RW, Hou Z, Wang H, Petersen C, Silva A, Masai H, Hua X. Functional interaction between tumor suppressor menin and activator of S-phase kinase. Cancer Res. 2004;64:6791–6796. doi: 10.1158/0008-5472.CAN-04-0724. [DOI] [PubMed] [Google Scholar]
- 67.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 68.Yazgan O, Pfarr CM. Differential binding of the Menin tumor suppressor protein to JunD isoforms. Cancer Res. 2001;61:916–920. [PubMed] [Google Scholar]
- 69.Agarwal SK, Novotny EA, Crabtree JS, Weitzman JB, Yaniv M, Burns AL, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ. Transcription factor JunD, deprived of menin, switches from growth suppressor to growth promoter. Proc Natl Acad Sci U S A. 2003;100:10770–10775. doi: 10.1073/pnas.1834524100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Corbetta S, Vicentini L, Ferrero S, Lania A, Mantovani G, Cordella D, Beck-Peccoz P, Spada A. Activity and function of the nuclear factor kappaB pathway in human parathyroid tumors. Endocr Relat Cancer. 2005;12:929–937. doi: 10.1677/erc.1.00970. [DOI] [PubMed] [Google Scholar]
- 71.Imanishi Y, Hosokawa Y, Yoshimoto K, Schipani E, Mallya S, Papanikolaou A, Kifor O, Tokura T, Sablosky M, Ledgard F, Gronowicz G, Wang TC, Schmidt EV, Hall C, Brown EM, Bronson R, Arnold A. Primary hyperparathyroidism caused by parathyroid-targeted overexpression of cyclin D1 in transgenic mice. J Clin Invest. 2001;107:1093–1102. doi: 10.1172/JCI10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Libutti SK, Crabtree JS, Lorang D, Burns AL, Mazzanti C, Hewitt SM, O'Connor S, Ward JM, Emmert-Buck MR, Remaley A, Miller M, Turner E, Alexander HR, Arnold A, Marx SJ, Collins FS, Spiegel AM. Parathyroid gland-specific deletion of the mouse Men1 gene results in parathyroid neoplasia and hypercalcemic hyperparathyroidism. Cancer Res. 2003;63:8022–8028. [PubMed] [Google Scholar]
- 73.Theillaumas A, Blanc M, Couderc C, Poncet G, Bazzi W, Bernard C, Cordier-Bussat M, Scoazec JY, Roche C. Relation between menin expression and NF-kappaB activity in an intestinal cell line. Mol Cell Endocrinol. 2008;291:109–115. doi: 10.1016/j.mce.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 74.Heaney AP, Fernando M, Melmed S. PPARgamma receptor ligands: novel therapy for pituitary adenomas. J Clin Invest. 2003;111:1381–1388. doi: 10.1172/JCI16575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Messager M, Carriere C, Bertagna X, de Keyzer Y. RT-PCR analysis of corticotrophassociated genes expression in carcinoid tumours in the ectopic-ACTH syndrome. Eur J Endocrinol. 2006;154:159–166. doi: 10.1530/eje.1.02077. [DOI] [PubMed] [Google Scholar]
- 76.Morrison RF, Farmer SR. Role of PPARgamma in regulating a cascade expression of cyclin-dependent kinase inhibitors, p18(INK4c) and p21(Waf1/Cip1), during adipogenesis. J Biol Chem. 1999;274:17088–17097. doi: 10.1074/jbc.274.24.17088. [DOI] [PubMed] [Google Scholar]
- 77.Karnik SK, Chen H, McLean GW, Heit JJ, Gu X, Zhang AY, Fontaine M, Yen MH, Kim SK. Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science. 2007;318:806–809. doi: 10.1126/science.1146812. [DOI] [PubMed] [Google Scholar]
- 78.Rosen ED, Kulkarni RN, Sarraf P, Ozcan U, Okada T, Hsu CH, Eisenman D, Magnuson MA, Gonzalez FJ, Kahn CR, Spiegelman BM. Targeted elimination of peroxisome proliferator-activated receptor gamma in beta cells leads to abnormalities in islet mass without compromising glucose homeostasis. Mol Cell Biol. 2003;23:7222–7229. doi: 10.1128/MCB.23.20.7222-7229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bouillon R, Carmeliet G, Verlinden L, van Etten E, Verstuyf A, Luderer HF, Lieben L, Mathieu C, Demay M. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev. 2008;29:726–776. doi: 10.1210/er.2008-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carling T, Kindmark A, Hellman P, Lundgren E, Ljunghall S, Rastad J, Akerstrom G, Melhus H. Vitamin D receptor genotypes in primary hyperparathyroidism. Nat Med. 1995;1:1309–1311. doi: 10.1038/nm1295-1309. [DOI] [PubMed] [Google Scholar]
- 81.Carling T, Rastad J, Szabo E, Westin G, Akerstrom G. Reduced parathyroid vitamin D receptor messenger ribonucleic acid levels in primary and secondary hyperparathyroidism. J Clin Endocrinol Metab. 2000;85:2000–2003. doi: 10.1210/jcem.85.5.6607. [DOI] [PubMed] [Google Scholar]
- 82.Sudhaker Rao D, Han ZH, Phillips ER, Palnitkar S, Parfitt AM. Reduced vitamin D receptor expression in parathyroid adenomas: implications for pathogenesis. Clin Endocrinol (Oxf) 2000;53:373–381. doi: 10.1046/j.1365-2265.2000.01081.x. [DOI] [PubMed] [Google Scholar]
- 83.Gedlicka C, Hager G, Weissenbock M, Gedlicka W, Knerer B, Kornfehl J, Formanek M. 1,25(OH)2Vitamin D3 induces elevated expression of the cell cycle inhibitor p18 in a squamous cell carcinoma cell line of the head and neck. J Oral Pathol Med. 2006;35:472–478. doi: 10.1111/j.1600-0714.2006.00407.x. [DOI] [PubMed] [Google Scholar]
- 84.Cheng HT, Chen JY, Huang YC, Chang HC, Hung WC. Functional role of VDR in the activation of p27Kip1 by the VDR/Sp1 complex. J Cell Biochem. 2006;98:1450–1456. doi: 10.1002/jcb.20780. [DOI] [PubMed] [Google Scholar]
- 85.Saramaki A, Banwell CM, Campbell MJ, Carlberg C. Regulation of the human p21 (waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006;34:543–554. doi: 10.1093/nar/gkj460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Buchwald PC, Akerstrom G, Westin G. Reduced p18INK4c, p21CIP1/WAF1 and p27KIP1 mRNA levels in tumours of primary and secondary hyperparathyroidism. Clin Endocrinol (Oxf) 2004;60:389–393. doi: 10.1111/j.1365-2265.2004.01995.x. [DOI] [PubMed] [Google Scholar]
- 87.Kaji H, Canaff L, Lebrun JJ, Goltzman D, Hendy GN. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc Natl Acad Sci U S A. 2001;98:3837–3842. doi: 10.1073/pnas.061358098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.ten Dijke P, Miyazono K, Heldin CH. Signaling inputs converge on nuclear effectors in TGF-beta signaling. Trends Biochem Sci. 2000;25:64–70. doi: 10.1016/s0968-0004(99)01519-4. [DOI] [PubMed] [Google Scholar]
- 89.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 90.Asa SL, Ezzat S. The cytogenesis and pathogenesis of pituitary adenomas. Endocr Rev. 1998;19:798–827. doi: 10.1210/edrv.19.6.0350. [DOI] [PubMed] [Google Scholar]
- 91.Hussein N, Lu J, Casse H, Fontaniere S, Morera AM, Guittot SM, Calender A, Di Clemente N, Zhang CX. Deregulation of anti-Mullerian hormone/BMP and transforming growth factor-beta pathways in Leydig cell lesions developed in male heterozygous multiple endocrine neoplasia type 1 mutant mice. Endocr Relat Cancer. 2008;15:217–227. doi: 10.1677/ERC-06-0046. [DOI] [PubMed] [Google Scholar]
- 92.Mould AW, Duncan R, Serewko-Auret M, Loffler KA, Biondi C, Gartside M, Kay GF, Hayward NK. Global expression profiling of sex cord stromal tumors from Men1 heterozygous mice identifies altered TGF-beta signaling, decreased Gata6 and increased Csf1r expression. Int J Cancer. 2009;124:1122–1132. doi: 10.1002/ijc.24057. [DOI] [PubMed] [Google Scholar]
- 93.Cao Y, Liu R, Jiang X, Lu J, Jiang J, Zhang C, Li X, Ning G. Nuclear-cytoplasmic shuttling of menin regulates nuclear translocation of {beta}-catenin. Mol Cell Biol. 2009;29:5477–5487. doi: 10.1128/MCB.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen G, A J, Wang M, Farley S, Lee LY, Lee LC, Sawicki MP. Menin promotes the Wnt signaling pathway in pancreatic endocrine cells. Mol Cancer Res. 2008;6:1894–1907. doi: 10.1158/1541-7786.MCR-07-2206. [DOI] [PubMed] [Google Scholar]
- 95.Heiser PW, Lau J, Taketo MM, Herrera PL, Hebrok M. Stabilization of beta-catenin impacts pancreas growth. Development. 2006;133:2023–2032. doi: 10.1242/dev.02366. [DOI] [PubMed] [Google Scholar]
- 96.Hoeflich A, Reisinger R, Lahm H, Kiess W, Blum WF, Kolb HJ, Weber MM, Wolf E. Insulin-like growth factor-binding protein 2 in tumorigenesis: protector or promoter? Cancer Res. 2001;61:8601–8610. [PubMed] [Google Scholar]
- 97.Hoeflich A, Fettscher O, Lahm H, Blum WF, Kolb HJ, Engelhardt D, Wolf E, Weber MM. Overexpression of insulin-like growth factorbinding protein-2 results in increased tumorigenic potential in Y-1 adrenocortical tumor cells. Cancer Res. 2000;60:834–838. [PubMed] [Google Scholar]
- 98.Moore MG, Wetterau LA, Francis MJ, Peehl DM, Cohen P. Novel stimulatory role for insulin-like growth factor binding protein-2 in prostate cancer cells. Int J Cancer. 2003;105:14–19. doi: 10.1002/ijc.11015. [DOI] [PubMed] [Google Scholar]
- 99.Dong F, Wu HB, Hong J, Rechler MM. Insulin-like growth factor binding protein-2 mediates the inhibition of DNA synthesis by transforming growth factor-beta in mink lung epithelial cells. J Cell Physiol. 2002;190:63–73. doi: 10.1002/jcp.10034. [DOI] [PubMed] [Google Scholar]
- 100.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 101.Kaji H, Canaff L, Goltzman D, Hendy GN. Cell cycle regulation of menin expression. Cancer Res. 1999;59:5097–5101. [PubMed] [Google Scholar]
- 102.Suphapeetiporn K, Greally JM, Walpita D, Ashley T, Bale AE. MEN1 tumor-suppressor protein localizes to telomeres during meiosis. Genes Chromosomes Cancer. 2002;35:81–85. doi: 10.1002/gcc.10113. [DOI] [PubMed] [Google Scholar]
- 103.Yang Y, Gurung B, Wu T, Wang H, Stoffers DA, Hua X. Reversal of preexisting hyperglycemia in diabetic mice by acute deletion of the Men1 gene. Proc Natl Acad Sci U S A. 2010;107:20358–20363. doi: 10.1073/pnas.1012257107. [DOI] [PMC free article] [PubMed] [Google Scholar]