

Abstract
Cyclotides are plant-derived proteins that naturally exhibit various biological activities and whose unique cyclic structure makes them remarkably stable and resistant to denaturation or degradation. These attributes, among others, make them ideally suited for use as drug development tools. This study investigated the cellular uptake of cyclotide, MCoTI-I in live HeLa cells. Using real time confocal fluorescence microscopy imaging, we show that MCoTI-I is readily internalized in live HeLa cells and that its endocytosis is temperature-dependent. Endocytosis of MCoTI-I in HeLa cells is achieved primarily through fluid-phase endocytosis, as evidenced by its significant colocalization with 10K-dextran, but also through other pathways as well, as evidenced by its colocalization with markers for cholesterol-dependent and clathrin-mediated endocytosis, cholera toxin B and EGF respectively. Uptake does not appear to occur only via macropinocytosis as inhibition of this pathway by Latrunculin B-induced disassembly of actin filaments did not affect MCoTI-I uptake and treatment with EIPA which also seemed to inhibit other pathways collectively inhibited approximately 80% of cellular uptake. As well, a significant amount of MCoTI-I accumulates in late endosomal and lysosomal compartments and MCoTI-I-containing vesicles continue to exhibit directed movements. These findings demonstrate internalization of MCoTI-I through multiple endocytic pathways that are dominant in the cell type investigated, suggesting that this cyclotide has ready access to general endosomal/lysosomal pathways but could readily be re-targeted to specific receptors through addition of targeting ligands.
Introduction
Cyclotides are fascinating micro-proteins ranging from 28 to 37 amino acid residues that are naturally expressed in plants and exhibit various biological activities such as anti-microbial, insecticidal, cytotoxic, antiviral (against HIV), and protease inhibitory activity, as well as exert hormone-like effects [3–6]. They share a unique head-to-tail circular knotted topology of three disulfide bridges, with one disulfide penetrating through a macrocycle formed by the other two disulfides and inter-connecting peptide backbones, forming what is called a cystine knot topology (Fig. 1). This cyclic cystine knot (CCK) framework gives the cyclotides exceptional resistance to thermal and chemical denaturation, and enzymatic degradation [6, 7]. In fact, the use of cyclotide-containing plants in indigenous medicine first highlighted the fact that the peptides are resistant to boiling and are apparently orally bioavailable [8].
Figure 1. Primary and tertiary structures of MCoTI and kalata cyclotides.

The structures of MCoTI-II (pdb ID: 1IB9 [1] and kalata B1 (pdb ID: 1NB1 [2]) are shown. Conserved cysteine residues and disulfide bonds are shown in yellow. An arrow marks residue Lys4 located at loop 1 in MCoTI-cyclotides. This residue in MCoTI-I was used for the site-specific conjugation of AlexaFluor488 N-hydroxysuccinimide ester (AF488-OSu) through a stable amide bond.
Cyclotides have been isolated from plants in the Rubiaceae, Violaceae, Cucurbitacea [6, 9] and most recently Fabaceae families [10]. Around 160 different cyclotide sequences have been reported in the literature [11, 12], although it has been estimated that ≈ 50,000 cyclotides might exist [13, 14]. Despite the sequence diversity, all cyclotides share the same CCK motif (Fig.1). Hence, these micro-proteins can be considered natural combinatorial peptide libraries structurally constrained by the cystine-knot scaffold [4] and head-to-tail cyclization, but in which hypermutation of essentially all residues is permitted with the exception of the strictly conserved cysteines that comprise the knot.
Cyclotides are ribosomally produced in plants from precursors that comprise between one and three cyclotide domains. However, the mechanism of excision of the cyclotide domains and ligation of the free N- and C-termini to produce the circular peptides has not been completely elucidated yet. It is suspected, however, that specific aspariginyl endopeptidases are involved in the proteolytic processing and cyclization of the precursor proteins [15–17]. Cyclotides can also be produced chemically using solid-phase peptide synthesis in combination with native chemical ligation [18–21], or recombinantly in bacteria by using modified protein splicing units or inteins [22, 23]. The latter method can generate folded cyclotides either in vivo or in vitro using standard bacterial expression systems [22–24] and opens the possibility of producing large libraries of genetically encoded cyclotides which can be analyzed by high throughput cell-based screening for selection of specific sequences able to bind to particular biomolecular targets [23, 25].
Cyclotides have been classified into three main subfamilies. The Möbius and bracelet cyclotide subfamilies differ in the presence or absence of a cis-Pro residue, which introduces a twist in the circular backbone topology [26]. A third subfamily comprises the cyclic trypsin inhibitors MCoTI-I/II (Fig. 1), which have been recently isolated from the dormant seeds of Momordica cochinchinensis, a plant member of the cucurbitaceae family, and are powerful trypsin inhibitors (Ki ≈ 20 – 30 pM) [27]. These cyclotides do not share significant sequence homology with other cyclotides beyond the presence of the three-cystine bridges, but structural analysis by NMR has shown that they adopt a similar backbone-cyclic cystine-knot topology [1, 28]. MCoTI cyclotides, however, show high sequence homology with related linear cystine-knot squash trypsin inhibitors [27], and therefore represent interesting molecular scaffolds for drug design [21, 29–31]. Indeed, acyclic squash inhibitors have already been used as scaffolds for the incorporation of novel bioactive peptides to render de-novo engineered knottins with novel biological activities [32, 33].
All these features make cyclotides ideal drug development tools [21, 29–31]. They are remarkably stable due to the cyclic cystine knot [34]. They are relatively small, making them readily accessible to chemical synthesis [18]. They can also be encoded within standard cloning vectors, and expressed in cells [22–24], and are amenable to substantial sequence variation [35], making them ideal substrates for molecular grafting of biological peptide epitopes [6]. They are also amenable to molecular evolution strategies to enable generation and selection of compounds with optimal binding and inhibitory characteristics [24, 35]. Even more importantly, MCoTI-cyclotides have been recently shown to be able to enter human macrophages and breast cancer cell lines [36]. Internalization into macrophages was shown to be mediated mainly through macropinocytosis, a form of endocytosis that is actin-mediated and results in formation of large vesicles termed macropinosomes [37, 38]. It should be noted, however, that in this study the visualization of MCoTI-II uptake was done in fixed, not live, cells. Analysis of live cells provides the ability to visualize events in real time without the possible complication of fixation artifacts that have confounded interpretations of the uptake of Tat and other related peptides for instance [39, 40]. As well, macropinocytosis is a dominant mechanism for endocytic uptake in macrophages [41–43], unlike other cells that are not specialized for large-scale sampling of extracellular fluid, and which use multiple alternative endocytic mechanisms. These mechanisms can include clathrin-mediated endocytosis, caveolar endocytosis, macropinocytosis, phagocytosis, flotillin-dependent endocytosis, as well as multiple other as yet under-characterized mechanisms [44, 45]. Intrigued by these results, we explored the cellular uptake of site-specific fluorescently-labeled MCoTI-I cyclotides and studied the cellular uptake mechanisms in HeLa cells using live cell imaging by confocal fluorescence microscopy.
In this work we report for the first time the cellular uptake of MCoTI-cyclotides monitored by real time confocal fluorescence microscopy imaging in live HeLa cells. Our results clearly show that HeLa cells readily internalize fluorescently-labeled-MCoTI-I. We found that this process is temperature-dependent and can be reversibly inhibited at 4°C, which indicates an active mechanism of internalization. The internalized cyclotide also seems to colocalize in live cells with multiple endocytic markers including, to the greatest extent, the fluid-phase endocytic marker dextran (10 KDa dextran,10K-Dex). Internalized MCoTI-I was colocalized to a lesser extent with the cholesterol/lipid dependent endocytic marker cholera toxin B (CTX-B) and the clathrin-mediated endocytic marker, EGF. Internalized MCoTI-I was localized within a fairly rapid time course within late endosomal and lysosomal compartments which engaged in rapid and directed movements suggestive of cytoskeletal involvement. MCoTI-I uptake in HeLa cells was not impaired by Latrunculin B (Lat B) although its uptake was approximately 80% inhibited by the less specific macropinocytosis inhibitor EIPA. Altogether, these data seem to indicate that MCoTI-I cyclotide is capable of internalization in live cells through multiple endocytic pathways that may be dominant in the particular cell type under study and may also include macropinocytosis. The lack of strong preference for MCoTI-I internalization via a specific cellular internalization pathway is of significant value since the lack of endogenous affinity for a particular pathway can enable the ready re-targeting by introduction of targeting peptides, within the scaffold, that may enable specific and targeted endocytic uptake to a particular target cell. At the same time, the ready uptake of MCoTI-I by multiple pathways suggests accessibility, in the untargeted form, to essentially all cells.
Materials and Methods
Analytical characterization of cyclotides
Analytical HPLC was performed on a HP1100 series instrument with 220 and 280 nm detection using a Vydac C18 column (5 micron, 4.6 × 150 mm) at a flow rate of 1 mL/min. Preparative and semi-preparative HPLC were performed on a Waters Delta Prep system fitted with a Waters 2487 UV-visible detector using a Vydac C18 (15–20 μm, 10 × 250 mm) at a flow rate of 5 mL/min. All runs used linear gradients of 0.1% aqueous trifluoroacetic acid (TFA, solvent A) vs. 0.1% TFA, 90% acetonitrile in H2O (solvent B). Ultraviolet-visible (UV-vis) spectroscopy was carried out on an Agilent 8453 diode array spectrophotometer. Electrospray mass spectrometry (ES-MS) analysis was routinely applied to all compounds and components of reaction mixtures. ES-MS was performed on an Applied Biosystems API 3000 triple quadrupole electrospray mass spectrometer using Analyst 1.4.2. Calculated masses were obtained using Analyst 1.4.2. All chemicals involved in synthesis or analysis were obtained from Aldrich (Milwaukee, WI) or Novabiochem (San Diego, CA) unless otherwise indicated.
Preparation of Fmoc-Tyr(tBu)-F
Fmoc-Tyr(tBu)-F was prepared using diethylaminosulfur trifluoride DAST [46] and quickly used afterwards. Briefly, to a stirred solution of Fmoc-Tyr(tBu)-OH (459.6 mg, 1 mmol) in 10 mL of dry dichloromethane (DCM), containing dry pyridine (800 μL, 1 mmol) and (1.1 mL, 1.2 mmol) of DAST was added dropwise at 25° C under nitrogen current. After 20 minutes, the mixture was washed with ice-cold water (3 × 20 mL). The organic layer was separated and dried over anhydrous MgSO4. The solvent was removed under reduced pressure to give the corresponding Fmoc-amino acyl fluoride as white solid that was used immediately. Amino acid fluorides should be used immediately as they are extremely unstable and prone to hydrolysis.
Loading of 4-sulfamylbutyryl AM resin with Fmoc-Tyr(tBu)-F
Loading of the first residue was accomplished using Fmoc-Tyr(tBu)-F according to standard protocols [47]. Briefly, 4-Sulfamylbutyryl AM resin (420mg, 0.33 mmol) (Novabiochem) was swollen for 20 minutes with dry DCM and then drained. A solution of Fmoc-Tyr(tBu)-F (≈461 mg, 1 mmol) in dry DCM (2 mL) and di-isopropylethylamine (DIEA) (180 μL, 1 mmol) was added to the drained resin and reacted at 25° C for 1 h. The resin was washed with dry DCM (5 × 5 mL), dried and kept at −20°C until use.
Chemical synthesis of MCoTI-I
Solid-phase synthesis was carried out on an automatic peptide synthesizer ABI433A (Applied Biosystems) using the Fast-Fmoc chemistry with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) activation protocol at 0.1 mmole scale on a Fmoc-Tyr(tBu)-sulfamylbutyryl AM resin. Side-chain protection was employed as previously described for the synthesis of peptide α-thiesters by the Fmoc-protocol [48], except for the N-terminal Cys residue, which was introduced as Boc-Cys(Trt)-OH. After chain assembly, the alkylation, thiolytic cleavage and deprotection were performed as previously described [49, 50]. Briefly, ≈100 mg of protected peptide resin were first alkylated two times with ICH2CN (174 μL, 2.4 mmol; previously filtered through basic silica) and DIEA (82 μL, 0.46 mmol) in N-methylpyrrolidone (NMP) (2.2 mL) for 12 h. The resin was then washed with NMP (3 × 5 mL) and DCM (3 × 5 mL). The alkylated peptide resin was cleaved with HSCH2CH2CO2Et (200 μL, 1.8 mmol) in the presence of a catalytic amount of sodium thiophenolate (NaSPh, 3 mg, 22 μmol) in dimethylformamide (DMF):DCM (3:4 v/v, 1.4 mL) for 24 h. The resin was then dried at reduced pressure. The side-chain protecting groups were removed by treating the dried resin with trifluoroacetic acid (TFA):H2O:tri-isopropylsilane (TIS) (95:3:2 v/v, 5 mL) for 3–4 h at room temperature. The resin was filtered and the linear peptide thioester was precipitated in cold Et2O. The crude material was dissolved in the minimal amount of H2O:MeCN (4:1) containing 0.1% TFA and characterized by HPLC and ES-MS as the desired MCoTI-I linear precursor α-thioester [Expected mass (average isotopic composition) = 3608.2 Da; measured = 3608.8 ± 0.3 Da]. Cyclization and folding was accomplished by flash dilution of the MCoTI-I linear α-thioester TFA crude to a final concentration of ≈50 μM into freshly degassed 2 mM reduced glutathione (GSH), 50 mM sodium phosphate buffer at pH 7.5 for 18 h. Folded MCoTI-I was purified by semi-preparative HPLC using a linear gradient of 10–35% solvent B over 30 min. Pure MCoTI-I was characterized by HPLC and ES-MS [Expected mass (average isotopic composition) = 3480.9 Da; measured = 3481.0 ± 0.4 Da].
Recombinant Expression of MCoTI-I
Bacterial expression and purification of MCoTI-I was carried out previously described [24].
Chemical labeling of MCoTI with AlexaFluor488 succinimide ester (AF488-NHS)
MCoTI-I was site-specifically labeled through the ε-amino of residue Lys4 (Fig.1). MCoTI-I only has one Lys residue in its sequence (Fig. 1). Briefly, MCoTI-I (5 mg, 1.4 μmol) was conjugated with two-fold molar excess of AF488-MHS in 0.2 M sodium phosphate buffer (2.5 mL) at pH 7.5 for 2 h. The reaction was quenched with 6 mM NH2-OH solution at pH 4. AF488-labeled MCoTI-I was purified by semi-preparative HPLC using a linear gradient of 15–35% solvent B over 30 min. Pure labeled MCoTI-I was characterized by HPLC and ES-MS [Expected mass (average isotopic composition) = 3997.9 Da; measured = 3997.4 ± 0.3 Da].
Purification of synthetic MCoTI-I using trypsin-Sepharose beads
Preparation of trypsin-Sepharose beads was done as previously described [23–25]. Pull down experiments with synthetic MCoTI-I were performed as follows: Synthetic MCoTI-I cyclization/folding crude reactions were typically incubated with 0.2 mL of trypsin-Sepharose for one hour at room temperature with gentle rocking, and centrifuged at 3000 rpm for 1 min. The beads were washed with 50 volumes of PBS containing 0.1% Triton X-100, then rinsed with 50 volumes of PBS, and drained of excess PBS. Bound MCoTI-I was eluted with 0.4 mL of 8 M GdmCl and fractions were analyzed by RP-HPLC and ES-MS.
Endocytosis Experiments
For studies of endocytic uptake mechanisms, methyl-β-cyclodextrin (MBCD) and 5-(N-ethyl-N-isopropyl)amiloride (EIPA) were purchased from Sigma-Aldrich. Latrunculin B (Lat B) was purchased from Calbiochem. Texas Red-EGF (TR-EGF), LysoTracker™ Red DN-99 (LysoRed), AF594 cholera toxin B (AF594 CTX-B), Texas Red 10,000 MW dextran (TR-10K Dex) and CellLight™ Lysosomes-RFP (RFP-Lamp1), rhodamine-phalloidin, and DAPI were all purchased from Invitrogen (Carlsbad, CA).
Cell Culture
HeLa cells were obtained from the American Type Culture Collection (ATCC) and were cultured in a humidified incubator at 37°C in 95% air/5% CO2 in phenol red-free Dulbecco’s modified essential medium (DMEM)(4.5 g/L glucose with 10% FBS, 1% glutamine, and 1% non-essential amino acids) and split with trypsin/EDTA as recommended by the manufacturer.
Confocal Fluorescence Microscopy
For MCoTI-I uptake studies, HeLa cells were seeded on 35 mm glass-bottom culture dishes (MatTek, Ashland, MA) at a density of 8.5 × 104 cells/dish. On day 2 of culture, the cells were rinsed with PBS and the media replaced with incubation buffer (phenol red-free, serum-free DMEM with 1% P/S and 20 mM HEPES) prior to addition of AF488-MCoTI-I (25 μM) and incubation at 37°C for 1 hr. Following this time, excess MCoTI-I was rinsed off with a gentle PBS wash and the media replaced prior to imaging. Intracellular distribution was analyzed at 1 hr and again at regular intervals for up to 10 hours. For assessment of distribution and colocalization of AF488-MCoTI-I and LysoTracker™ Red, RFP-Lamp1, AF594-Cholera toxin B, TR-10K Dex, or TR-EGF in live cells, we utilized a Zeiss LSM 510 Meta NLO imaging system equipped with Argon and HeNe lasers and mounted on a vibration-free table for confocal fluorescence microscopy. For analysis of the effects of Lat B or EIPA pre-treatment, 2 μM Lat B or 50 μM EIPA was added for 30 min at 37°C prior to addition of AF488-MCoTI-I. For colocalization studies, LysoTracker™ Red (50 nM), AF594-Cholera toxin B (10 μg/mL), TR-10K Dex (1 mg/mL), or TR-EGF (400 ng/mL) was added to cells simultaneously with MCoTI-I prior to incubation at 37°C. Analysis of the extent of colocalization was done at 1 hr of uptake. For colocalization with RFP-Lamp1, cells were treated with RFP-Lamp1-expressing BacMam (2 × 107 particles/plate) on the previous day. For temperature-dependent uptake studies, cells were cooled on ice for 30 min prior to the addition of AF488-MCoTI-I in incubation buffer. After incubation at 4°C for 30 min, the cells were imaged and subsequently incubated at 37°C for 1 hour before imaging again. For fixation and visualization of actin filaments following treatment with or without 2 μM Lat B, the cells were fixed with 4% paraformaldehyde prior to the addition of rhodamine-phalloidin and DAPI. For analysis of fluorescent pixel colocalization, cells from at least 3 different experiments were analyzed individually. Using the Zeiss LSM 510 software colocalization tool, regions of interest (ROI) were selected and marked with an overlay to encompass all pixels, following the Zeiss manual protocol. The threshold was automatically set from these ROIs. For time-lapse imaging, cells were incubated with 25 μM AF488-MCoTI-I for 1 h at 37°C. Following this time, excess MCoTI-I was rinsed off with a gentle PBS wash and the media replaced prior to imaging. The time series image capture was set to a 2.5 second delay between scans.
Results and Discussion
In order to study the cellular uptake of MCoTI-cyclotides, we decided to use MCoTI-I. MCoTI-I contains only one Lys residue located in loop 1 versus MCoTI-II, which contains three Lys residues in the same loop (Fig. 1). The presence of only one Lys residue facilitates the site-specific introduction of a unique fluorophore on the sequence, thus minimizing any affect that the introduction of this group may have on the cellular uptake properties of the cyclotide.
Folded MCoTI-I cyclotide was produced either by recombinant or synthetic methods. In both cases the backbone cyclization was performed by an intramolecular native chemical ligation (NCL) [51–54] using the native Cys located on the beginning of loop 6 to facilitate the cyclization. This ligation site has been shown to give very good cyclization yields [23, 24]. Intramolecular NCL requires the presence of an N-terminal Cys residue and C-terminal α-thioester group in the same linear precursor [53, 55]. In the biosynthetic approach, the MCoTI-I linear precursor was fused in frame at the C- and N-terminus to a modified Mxe Gyrase A intein and a Met residue, respectively, and expressed in Escherichia coli [25]. This allows the generation of the required C-terminal thioester and N-terminal Cys residue after in vivo processing by endogenous Met aminopeptidase (MAP) [22, 56]. Cyclization and folding can be accomplished very efficiently in vitro by incubating the MCoTI-I intein fusion construct in sodium phosphate buffer at pH 7.4 in the presence of reduced glutathione (GSH). Biosynthetic MCoTI-cyclotides generated this way have been shown to adopt a native folded structure by NMR and trypsin inhibitory assays [22, 24, 34].
Natively folded MCoTI-II has been already successfully produced using Fmoc-based solid-phase peptide synthesis [20, 21]. Encouraged by these results we also explored the production of MCoTI-I by chemical synthesis (Fig. 2). For this purpose the MCoTI-I linear precursor α-thioester was assembled by Fmoc-based solid-phase peptide synthesis on a sulfonamide resin [49, 50] (Fig. 2A). Activation of the sulfonamide linker with iodoacetonitrile, followed by cleavage with ethyl mercaptoacetate and acidolytic deprotection with TFA, provided the fully protected linear peptide α-thioester (Fig. 2B). The synthetic linear precursor thioester was then efficiently cyclized and folded in a one-pot reaction using sodium phosphate buffer at pH 7.5 in the presence of 2 mM GSH. The reaction was complete in 18 h and the folded product was purified by reverse-phase HPLC and characterized by ES-MS. The expected mass for folded MCoTI-I was in agreement with a folded structure (Expected mass = 3480.9 Da; measured = 3481.0 ± 0.4 Da). Synthetic folded MCoTI-II was also shown to co-elute by HPLC with recombinant natively folded MCoTI-I (data not shown). The biological activity of synthetic MCoTI-I was assayed by using a trypsin pull-down experiment [24, 25]. As shown in Figure 2B, synthetic folded MCoTI-I was specifically captured from a cyclization/folding crude reaction by trypsin-immobilized Sepharose beads [23–25], thus indicating that it adopted a native cyclotide fold.
Figure 2. Chemical synthesis of MCoTI-I.

(A) Synthetic scheme used for the chemical synthesis of cyclotide MCoTI-I by Fmoc-based solid-phase peptide synthesis (B) Analytical reverse-phase HPLC traces of MCoTI-I linear precursor α-thioester, cyclization/folding crude and purified MCoTI-I by either affinity chromatograpgy using trypsin-immobilized Sepharose beads or semipreparative reverse-phase HPLC. HPLC analysis was performed in all the cases using a linear gradient of 0% to 70% buffer B over 30 min. Detection was carried out at 220 nm. An arrow indicates the desired product in each case.
Purified MCoTI-I was site-specifically labeled with AlexaFluor 488 (AF488) for live confocal imaging. The ε-amino group of Lys4 residue located in loop 1 was conjugated to AF488-NHS in sodium phosphate buffer at pH 7.5 for 2 h (Fig. 3A). Under these conditions, the main product of the reaction was mono-labeled AF488-MCoTI as characterized by HPLC and ES-MS (expected average mass = 3997.9 Da; measured = 3997.4 ± 0.3 Da) (Fig. 3C). AF488-labeled MCoTI-I was then purified by reverse-phase HPLC to remove any trace of unreacted materials (Fig. 3B).
Figure 3. Site-specific labeling of MCoTI-I with AlexaFluor-488 N-hydroxysuccinimide ester (AF488-OSu).

(A) Scheme depicting the bioconjugation process and localization of the fluorescent probe at residue Lys4 in loop 1. (B) Analytical reverse-phase HPLC trace of pure AF488-MCoTI-I. HPLC analysis was performed using a linear gradient of 0% to 70% buffer B over 30 min. Detection was carried at 220 nm (C) ES-MS spectra of pure AF488-MCoTI-I.
In order to infer the correct conclusions regarding data obtained on the cellular uptake of native MCoTI-I when using modified cyclotides, like AF488-MCoTI-I for example, it is critical to be sure that they still adopt structures similar to that of the native form. MCoTI-cyclotides are extremely stable to chemical and thermal denaturation, and they have been shown to be able to withstand procedures like reverse-phase chromatography in the presence of organic solvents under acidic conditions without affecting their tertiary structure [18, 20–23, 34]. It is also unlikely that the acylation of the ε-amino group of Lys4 in MCoTI-I may disrupt the tertiary structure of this cyclotide. Craik and co-workers have previously shown that biotinylation of the three Lys residues located in loop 1 in MCoTI-II, including Lys4 (Fig. 1) does not disrupt the native cyclotide fold of this cyclotide as determined by 1H-NMR [36]. We have also recently shown that mutation of residue Lys4 by Ala does not seem to affect the ability of this mutant to adopt a native cyclotide fold, thus indicating that the presence of positive charge residue in this position is not critical for the tertiary structure of MCoTI-I [24]. Similar findings have also been shown by Leatherbarrow and coworkers, where mutation of this residue by Phe or Val, rendered MCoTI-cyclotides able to fold correctly and have inhibitory activity against chymotrypsin and human elastase, respectively [21]. Altogether these facts suggest that residue Lys4 is not critical for adopting the native cyclotide fold or disturbing the tertiary structure of MCoTI-cyclotides.
To study the cellular uptake of AF488-MCoTI-I we used HeLa cells. The internalization studies were all carried out with 25 μM AF488-MCoTI-I. This concentration provided a good signal/noise ratio for live cell confocal fluorescence microscopy studies and did not show any cytotoxic effect. This is in agreement with the cellular tolerance of wild-type and biotinylated MCoTI-II reported for other types of human cell lines [36]. First, we analyzed the time course of changes in cellular distribution following uptake of 25 μM AF488-MCoTI-I by incubating with the cyclotide for 1 hr and then analyzing its distribution after 1, 2, 4, 8 and 10 h. As shown in Figure 4, the internalized cyclotide was clearly visible within perinuclear punctate spots inside the cells after 1 h incubation. Observation of cells pulsed with AF488-MCoTI-I for one hour and then incubated for longer periods of time in the absence of cyclotide did not show any evidence for decreased intracellular fluorescence, while the largely perinuclear distribution of internalized MCoTI-I appeared comparable at all time points. Similar results have been also been recently reported on the internalization of biotinylated MCoTI-II in macrophage and breast cancer cell lines [36], these studies however, used fixed cells to visualize the internalized cyclotide.
Figure 4. MCoTI-I distribution in HeLa cells.

HeLa cells were incubated with 25 μM MCoTI-I for 1 hour, followed by removal with gentle rinsing in PBS, and then monitored for distribution of intracellular fluorescence at intervals from 1–10 hours using confocal fluorescence microscopy. Bar = 10μm.
In order to study the mechanism of internalization of AF488-MCoTI-I in live HeLa cells, we first explored the effect of temperature on the uptake process. Active and energy-dependent endocytic mechanisms of internalization are inhibited at 4° C [57]. The internalization of AF488-MCoTI-I was totally inhibited after a 1 h incubation at 4°C (Fig. 5). This inhibition was completely reversible and when the same cells were incubated again at 37°C for 1 h, the punctate intracellular fluorescence labeling pattern was restored. This result confirmed that the uptake of AF488-MCoTI-I in HeLa cells follows a temperature dependent active endocytic internalization pathway. It should be noted that no significant surface binding was detected at 4°C, suggesting that MCoTI-I does not bind a surface receptor, even nonspecifically. This is in agreement with studies on the MCoTI-II in fixed cells, so both the MCoTI-I and MCoTI-II appear to lack specific affinity for proteins or lipids in cell membranes, unlike the kalata B1 cyclotide which shows membrane affinity [36]. This lack of endogenous affinity for a specific surface receptor or membrane constituent makes MCoTI-I ideal for engineering using more specific, receptor-directed, peptide-based, internalization motifs, within the scaffold, that might enable members of this family to have enhanced targeting to a specific cell type.
Figure 5. Endocytosis of MCoTI-I is temperature-dependent.
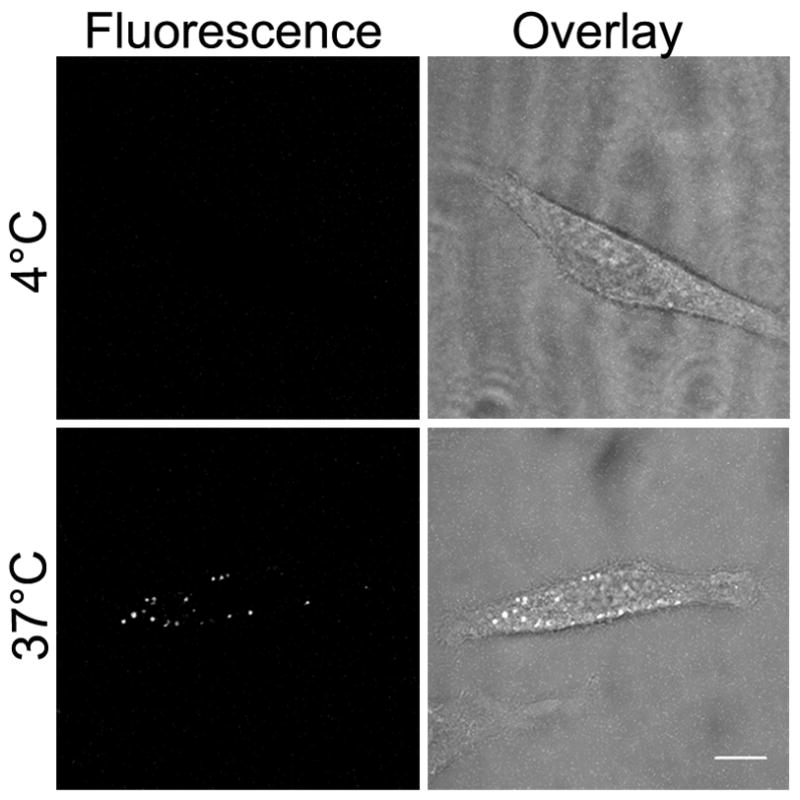
HeLa cells were incubated with 25 μM MCoTI-I for 1 hr at 4°C. After removal of the MCoTI-I-containing media, and a gentle PBS wash, the cells were imaged. Following imaging, the MCoTI-I-containing media was replaced and the cells incubated at 37°C for 1 hr and imaged again. Bar = 10 μm.
Next, we investigated the internalization pathway used by labeled-MCoTI-I to enter HeLa cells. There are several known and well-characterized mechanisms of endocytosis [58]. It is also now well established that almost all cell-penetrating peptides (CPPs) use a combination of different endocytic pathways rather than a single endocytic mechanism [58]. A recent study showed that several CPPs (including Antennapedia/penetratin, nona-Arg and Tat peptides) can be internalized into cells by multiple endocytic pathways including macropinocytosis, clathrin-mediated endocytosis, and caveolae/lipid raft mediated endocytosis [59]. To investigate if that was the case with the internalization of AF488-MCoTI-I in HeLa cells, we decided to look at its colocalization with various endocytic markers (Fig. 6). 10K-Dex has previously been used as a marker of fluid-phase endocytosis [37, 60–62]. CTX-B has been used as a marker for various lipid-dependent endocytic pathways [45, 63], while EGF has traditionally been a marker of clathrin-mediated endocytosis [64–66]. As shown in Figure 6, colocalization studies showed that after 1 h, AF488-MCoTI-I fluorescence was significantly colocalized with the fluorescence associated with 10K-Dex (59 ± 4% of total cyclotide fluorescence pixels were colocalized with 10K-Dex fluorescence pixels). Less colocalization was observed with fluorescent CTX-B (39 ± 4 %) and fluorescent EGF (21 ± 2 %). This data seems to suggest that AF488-MCoTI-I is primarily entering cells through fluid-phase endocytosis. The observed traces of colocalization with CTX-B and EGF also suggest that AF488-MCoTI-I could be using alternative or additional endocytic pathways. The colocalization results could also be attributed, however, to the merging of endosomal uptake vesicles generated by different pathways at the level of an early endosome. To address whether the major uptake and colocalization of AF488-MCoTI-I with 10K-Dex was due to cointernalization by macropinocytosis, we explored the inhibition of AF488-MCoTI-I uptake by Lat B, a potent inhibitor of actin polymerization, which is an essential element of macropinocytlsis [67–70]. As shown in Figure 7, Lat B did not significantly inhibit uptake of AF488-MCoTI-I (Fig. 7A) nor of 10K-Dex (data not shown). Treatment of HeLa cells with this agent resulted in a total disruption of the actin filament network (Fig. 7B). We also explored the inhibition of AF488-MCoTI-I uptake by 5-N-ethyl-N-isopropyl amiloride (EIPA). EIPA is a potent and specific inhibitor of the Na+/H+ exchanger, whose activity is important for macropinosome formation [71]. Treatment of HeLa cells with 50 μM EIPA significantly inhibited the uptake of both AF488-MCoTI-I (≈80%) and 10K-Dex (Supplemental Figure 1). The same treatment had no or little effect on the uptake of EGF (data not shown), but did significantly inhibit uptake of CTX-B (Supplemental Figure 1), suggesting that at the concentration used, EIPA is affecting multiple endocytic mechanisms in these HeLa cells beyond simply macropinocytosis.
Figure 6. Colocalization of MCoTI-I with markers of endocytosis.

(A) HeLa cells were incubated with 25 μM MCoTI-I and either 1 mg/ml 10K-dextran (10K-Dex), 10 μg/ml cholera toxin B (CTX-B), or 400 ng/ml epidermal growth factor (EGF) for 1 hour at 37°C as described in Materials and Methods and then imaged. Bar = 10 μm. (B) Quantification of pixel colocalization was done using the Zeiss LSM software for image analysis and measures the % of total fluorescent AF488 MCoTI-I pixels in the ROI relative to red pixels associated with different endocytic markers. (n = 13 cells for 10K-Dex, n = 11 cells for CTX-B and n = 10 cells for EGF, with cells assessed across 3 different experiments, * p ≤ 0.05 relative to 10K-Dex, # p ≤ 0.05 relative to CTX-B).
Figure 7. Disruption of actin does not inhibit MCoTI-I uptake.
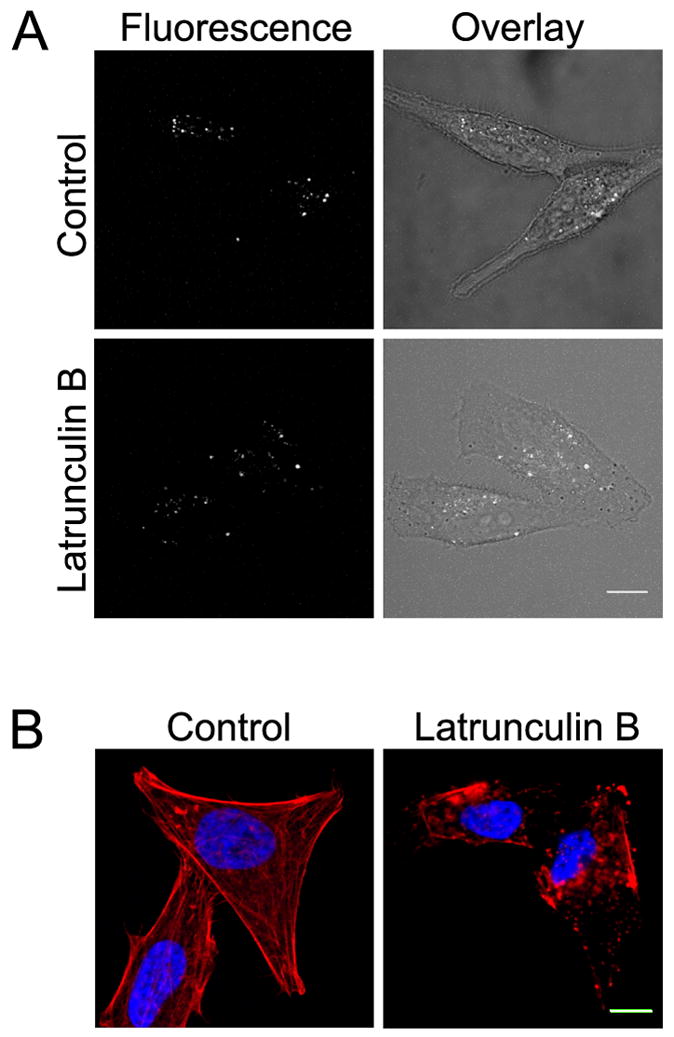
(A) HeLa cells were untreated (control) or treated with Lat B (2 μM) for 30 min at 37°C prior to addition of 25 μM MCoTI-I. Following uptake for 1 hr at 37°C, the cells were imaged using confocal fluorescence microscopy. Bar = 10 μm. (B) HeLa cells without treatment (control) or treated with 2 μM Lat B for 30 min at 37°C were fixed and labeled with rhodamine—phalloidin to label actin (red) and DAPI to label nuclei (blue). Bar = 10 μm.
As an extension of these inhibition studies, cells were also treated with MBCD, a well-established cholesterol-depleting agent employed for studying the involvement of lipid rafts/caveolae in endocytosis [72, 73]. Preliminary studies with MBCD suggested no significant inhibition of AF488-MCoTI-I (data not shown). Since the extent of total colocalization of AF488-MCoTI-I with CTX-B was less than 40%, it is unsurprising that no marked effect was seen by live cell microscopy. Taken together, these results seem to suggest that the uptake of AF488-MCoTI-I in HeLa cells is following multiple endocytic pathways, which is in agreement with what has been recently reported for different CPPs [59].
Next we explored the fate of the endocytic vesicles containing labeled MCoTI-I. There are at least two pathways that involve the cellular trafficking of endosomal vesicles. The degradative pathway includes routing of internalized materials from early endosomes via late endosomes to lysosomes where degradation of internalized materials occurs within the cells. On the other hand, recycling endosomes sort material internalized into early endosomes and are responsible for effluxing internalized material back to the cellular membrane [74]. If labeled-MCoTI-I was localized in recycling endosomes, it would be expected that its concentration in the cell would decrease and/or accumulate on the membrane over time, which was not the case in the time course experiment following the cellular fate of internalized cyclotide (Fig. 4). To explore the potential localization of labeled-MCoTI-I in lysosomes we first used LysoTracker Red (LysoRed). This pH sensitive fluorescent probe is utilized for identifying acidic organelles, such as lysosomes and late endosomes, in live cells. As shown in Figure 8A, significant colocalization (60 ± 4.0% as determined by pixel colocalization analysis) of LysoRed and AF488-MCoTI-I was observed after treating the cells for 1h with both agents. As an extension of these experiments, we also investigated the colocalization of labeled-MCoTI-I and lysosomal-associated membrane protein 1 (Lamp1), an established mature lysosomal marker [75, 76]. For this experiment, live HeLa cells were first infected with a Red Fluorescent Protein (RFP)-Lamp1-expressing BacMam virus. The next day the cells were incubated with AF488-MCoTI-I for 1 h and imaged. As shown in Figure 8B, colocalization was also seen for AF488-MCoTI-I and RFP-Lamp1 (38 ± 5%, as determined by pixel colocalization analysis), suggesting that even after 1 h, significant MCoTI-I has already reached the lysosomal compartments. Our data suggest that after 1 h, a significant amount of MCoTI-I (≈40%) has trafficked through the endosomal pathway to the lysosomes and that ≈20% is already localized in late endosomes or other types of acidic organelles. It has previously been reported that the perinuclear steady-state distribution of lysosomes is a balance between movement on microtubules and actin filaments [77–79]. Likewise, movement from early endosomal compartments to late endosomes to lysosomes has also been shown to rely on the microtubule network [80, 81]. As an extension of these experiments, and to investigate whether MCoTI-I-containing vesicles were actively trafficking inside the cell, we captured time-lapse video of cells after incubation with MCoTI-I for 1 h. Indeed, the time-lapse capture showed active movements of MCoTI-I-containing vesicles (Fig. 9). Directed short- and long-range movements could be seen, characteristic of movement on cytoskeletal filaments. These results suggest that while a large portion of MCoTI-I has reached lysosomal compartments by 1 h, and some of the movements seen may be attributed to the steady-state distribution of lysosomes, the remaining cyclotide may still be trafficking through the cell from other membrane compartments, likely within late endosomes.
Figure 8. MCoTI-I is colocalized with lysosomal compartments.

Untreated (A) or BacMam-RFP-Lamp1 treated (B) HeLa cells were incubated with 25 μM MCoTI-I and LysoTracker Red, or MCoTI-I alone, for 1 hr at 37°C as described in Materials and Methods and then imaged. Bar = 10 μm. (C) Quantification of % of total fluorescent AF488-MCoTI-I pixel colocalization with fluorescent pixels associated with both markers was done using the Zeiss LSM software for image analysis. (n = 14 cells for LysoTracker Red and n = 11 cells for Lamp1 with cells selected from 3 separate experiments).
Figure 9. MCoTI-I-containing vesicles are in motion.

HeLa cells were incubated with 25 μM MCoTI-I for 1 hr at 37°C and then imaged using time-lapse microscopy as described in Materials and Methods. Arrows indicate position of the moving vesicle at 0 min while displacement of the fluorescent vesicle relative to the arrow shows the extent of movement over time. Bar = 2 μm.
Craik and co-workers have recently reported the uptake of biotinylated-MCoTI-II by human macrophages and breast cancer cell lines [36]. This work concluded that the uptake of MCoTI-II in macrophages is mediated by macropinocytosis and that the cyclotide accumulates in macropinosomes without trafficking to the lysosome. MCoTI-II shares high homology with MCoTI-I (≈97% homology, see Fig. 1) and similar biological activity. Despite their similarities, the differences in the cellular uptake and trafficking of MCoTI-cyclotides by macrophages versus HeLa cells could be attributed to the cellular differences in endocytic preferences for these two very different cell types. Macrophage cells are specialized in large scale sampling of extracellular fluid using macropinocytosis as the dominant endocytic pathway. Meanwhile, other types of cells may employ multiple endocytic pathways as has been recently shown for the uptake of different CPPs in HeLa cells [59].
At this point we cannot be certain if some labeled MCoTI-I is able to escape from endosomal/lysosomal compartments into the cytosol. The ability to track the release of fluorescent-labeled molecules from cellular vesicles is limited using live cell imaging of fluorescence signal primarily due to the large dilution effect if the molecule is able to escape the highly confined volume of the vesicle into the larger cytosolic volume. One way to demonstrate the release of peptide into the cytosol, however, would be by using labels with better detection sensitivity or incorporating a biological activity that can be measured in the cellular cytosol. For example, the presence of Tyr residues in both MCoTI-cyclotides should facilitate the incorporation of radioactive iodine into the phenolic ring of Tyr with minimal disruption of the native structure of the cyclotide. The incorporation or grafting of biological peptides into the MCoTI scaffold could also provide proof of endosomal/lysosomal escape if such biological activity could be measured only in the cytosol. This approach has already been used to demonstrate endosomal escape of CPPs such as the TAT peptide [82, 83]. The retention of fluorescence signal in the perinuclear, lysosomal compartments for a period of up to 10 hours suggests that most of the cyclotide remains within these compartments. However, given the flexibility of the cyclotide backbone to accommodate multiple peptide sequences, subsequent studies may explore the ability of targeting and endosomolytic sequences for concomitant targeted entry and endosomal/lysosomal escape into cytosol.
Conclusion
This study reports the first analysis of intracellular uptake of MCoTI-I cyclotide using live cell imaging by confocal fluorescence microscopy. Cyclotides represent a novel platform for drug development. Their stability, conferred by the cyclic cystine knot, their small size, their amenability to both chemical and biological synthesis, and their flexible tolerance to sequence variation make them ideal for grafting of biologically-active therapeutic epitopes. As we show herein, they are also capable, in the unmodified state, of utilizing multiple cellular endocytic pathways for internalization. Their ease of access makes them readily accessible, in their current state, to endosomal/lysosomal compartments of virtually any cell. Without an apparent strong preference for an existing cellular pathway, nor surface-expressed epitope in HeLa cells (nor in other studies with MCoTI-II in macrophages [36]), they appear highly amenable to retargeting to exploit a particular target cell’s dominant internalization pathway and/or unique surface receptor repertoire, along with the targeted introduction of biologically-active therapeutic motifs.
Supplementary Material
Acknowledgments
The authors acknowledge the support of NIH grants EY017293 (to SHA), GM090323 (to JAC), Department of Defense Congressionally Directed Medical Research Program Grant PC09305 (to JAC) and the support of a Ruth Kirchstein NIH minority predoctoral fellowship (F31EY018807) to JC.
Abbreviations
- Boc
tert-butyloxy carbonyl
- CCK
cyclic cystine knot
- CPPs
cell-penetrating peptides
- CTX-B
cholera toxin B
- DAST
diethylaminosulfur trifluoride
- DCM
dichloromethane
- 10K-Dex
10,000MW-dextran
- DIEA
N,N-diisopropylethylamine
- DMF
dimethyl formamide
- EGF
epidermal growth factor
- EIPA
5-(N-ethyl-N-isopropyl)amiloride
- ES-MS
electrospray-mass spectrometry
- Fmoc
9-fluorenyloxy carbonyl
- HBTU
2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HPLC
high performance liquid chromatography
- Lat B
latrunculin B
- MCoTI
Momordica cochinchinensis trypsin inhibitor
- NMP
N-methyl-pyrrolidone
- NMR
nuclear magnetic resonance
- RFP-Lamp1
Red Fluorescent Protein-lysosomal associated protein 1
- RP-HPLC
reverse phase-high performance liquid chromatography
- TFA
trifluoroacetic acid
- TIS
tri-isopropylsilane
- TR
Texas Red
- Trt
trityl
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Felizmenio-Quimio ME, Daly NL, Craik DJ. Circular proteins in plants: solution structure of a novel macrocyclic trypsin inhibitor from Momordica cochinchinensis. J Biol Chem. 2001;276(25):22875–22882. doi: 10.1074/jbc.M101666200. [DOI] [PubMed] [Google Scholar]
- 2.Rosengren KJ, Daly NL, Plan MR, Waine C, Craik DJ. Twists, knots, and rings in proteins. Structural definition of the cyclotide framework. J Biol Chem. 2003;278(10):8606–8616. doi: 10.1074/jbc.M211147200. [DOI] [PubMed] [Google Scholar]
- 3.Craik DJ, Simonsen S, Daly NL. The cyclotides: novel macrocyclic peptides as scaffolds in drug design. Curr Opin Drug Discov Devel. 2002;5(2):251–260. [PubMed] [Google Scholar]
- 4.Craik DJ, Cemazar M, Wang CK, Daly NL. The cyclotide family of circular miniproteins: nature’s combinatorial peptide template. Biopolymers. 2006;84(3):250–266. doi: 10.1002/bip.20451. [DOI] [PubMed] [Google Scholar]
- 5.Jagadish K, Camarero JA. Cyclotides, a promising molecular scaffold for peptide-based therapeutics. Biopolymers. 94(5):611–616. doi: 10.1002/bip.21433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia AE, Camarero JA. Biological activities of natural and engineered cyclotides, a novel molecular scaffold for peptide-based therapeutics. Curr Mol Pharmacol. 2010;3(3):153–163. doi: 10.2174/1874467211003030153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daly NL, Rosengren KJ, Craik DJ. Discovery, structure and biological activities of cyclotides. Adv Drug Deliv Rev. 2009;61(11):918–930. doi: 10.1016/j.addr.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Saether O, Craik DJ, Campbell ID, Sletten K, Juul J, Norman DG. Elucidation of the primary and three-dimensional structure of the uterotonic polypeptide kalata B1. Biochemistry. 1995;34(13):4147–4158. doi: 10.1021/bi00013a002. [DOI] [PubMed] [Google Scholar]
- 9.Cascales L, Craik DJ. Naturally occurring circular proteins: distribution, biosynthesis and evolution. Org Biomol Chem. 2010;8(22):5035–5047. doi: 10.1039/c0ob00139b. [DOI] [PubMed] [Google Scholar]
- 10.Poth AG, Colgrave ML, Lyons RE, Daly NL, Craik DJ. Discovery of an unusual biosynthetic origin for circular proteins in legumes. Proc Natl Acad Sci U S A. 2011;108(25):1027–1032. doi: 10.1073/pnas.1103660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulvenna JP, Wang C, Craik DJ. CyBase: a database of cyclic protein sequence and structure. Nucleic Acids Res. 2006;34(Database issue):D192–194. doi: 10.1093/nar/gkj005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang CK, Kaas Q, Chiche L, Craik DJ. CyBase: a database of cyclic protein sequences and structures, with applications in protein discovery and engineering. Nucleic Acids Res. 2008;36(Database issue):D206–210. doi: 10.1093/nar/gkm953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mulvenna JP, Mylne JS, Bharathi R, Burton RA, Shirley NJ, Fincher GB, Anderson MA, Craik DJ. Discovery of cyclotide-like protein sequences in graminaceous crop plants: ancestral precursors of circular proteins? Plant Cell. 2006;18(9):2134–2144. doi: 10.1105/tpc.106.042812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gruber CW, Elliott AG, Ireland DC, Delprete PG, Dessein S, Goransson U, Trabi M, Wang CK, Kinghorn AB, Robbrecht E, Craik DJ. Distribution and evolution of circular miniproteins in flowering plants. Plant Cell. 2008;20(9):2471–2483. doi: 10.1105/tpc.108.062331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saska I, Gillon AD, Hatsugai N, Dietzgen RG, Hara-Nishimura I, Anderson MA, Craik DJ. An asparaginyl endopeptidase mediates in vivo protein backbone cyclization. The Journal of biological chemistry. 2007;282(40):29721–29728. doi: 10.1074/jbc.M705185200. [DOI] [PubMed] [Google Scholar]
- 16.Gillon AD, Saska I, Jennings CV, Guarino RF, Craik DJ, Anderson MA. Biosynthesis of circular proteins in plants. Plant J. 2008;53(3):505–515. doi: 10.1111/j.1365-313X.2007.03357.x. [DOI] [PubMed] [Google Scholar]
- 17.Conlan BF, Gillon AD, Craik DJ, Anderson MA. Circular proteins and mechanisms of cyclization. Biopolymers. 2010;94(5):573–583. doi: 10.1002/bip.21422. [DOI] [PubMed] [Google Scholar]
- 18.Daly NL, Love S, Alewood PF, Craik DJ. Chemical synthesis and folding pathways of large cyclic polypeptides: studies of the cystine knot polypeptide kalata B1. Biochemistry. 1999;38(32):10606–10614. doi: 10.1021/bi990605b. [DOI] [PubMed] [Google Scholar]
- 19.Tam JP, Lu YA, Yang JL, Chiu KW. An unusual structural motif of antimicrobial peptides containing end-to-end macrocycle and cystine-knot disulfides. Proc Natl Acad Sci U S A. 1999;96(16):8913–8918. doi: 10.1073/pnas.96.16.8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thongyoo P, Tate EW, Leatherbarrow RJ. Total synthesis of the macrocyclic cysteine knot microprotein MCoTI-II. Chem Commun (Camb) 2006;27:2848–2850. doi: 10.1039/b607324g. [DOI] [PubMed] [Google Scholar]
- 21.Thongyoo P, Roque-Rosell N, Leatherbarrow RJ, Tate EW. Chemical and biomimetic total syntheses of natural and engineered MCoTI cyclotides. Org Biomol Chem. 2008;6(8):1462–1470. doi: 10.1039/b801667d. [DOI] [PubMed] [Google Scholar]
- 22.Kimura RH, Tran AT, Camarero JA. Biosynthesis of the cyclotide kalata B1 by using protein splicing. Angew Chem Int Ed. 2006;45(6):973–976. doi: 10.1002/anie.200503882. [DOI] [PubMed] [Google Scholar]
- 23.Camarero JA, Kimura RH, Woo YH, Shekhtman A, Cantor J. Biosynthesis of a fully functional cyclotide inside living bacterial cells. Chembiochem. 2007;8(12):1363–1366. doi: 10.1002/cbic.200700183. [DOI] [PubMed] [Google Scholar]
- 24.Austin J, Wang W, Puttamadappa S, Shekhtman A, Camarero JA. Biosynthesis and biological screening of a genetically encoded library based on the cyclotide MCoTI-I. Chembiochem. 2009;10(16):2663–2670. doi: 10.1002/cbic.200900534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Austin J, Kimura RH, Woo YH, Camarero JA. In vivo biosynthesis of an Ala-scan library based on the cyclic peptide SFTI-1. Amino Acids. 2010;38(5):1313–1322. doi: 10.1007/s00726-009-0338-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daly NL, Gustafson KR, Craik DJ. The role of the cyclic peptide backbone in the anti-HIV activity of the cyclotide kalata B1. FEBS Lett. 2004;574(1–3):69–72. doi: 10.1016/j.febslet.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 27.Hernandez JF, Gagnon J, Chiche L, Nguyen TM, Andrieu JP, Heitz A, Trinh Hong T, Pham TT, Le Nguyen D. Squash trypsin inhibitors from Momordica cochinchinensis exhibit an atypical macrocyclic structure. Biochemistry. 2000;39(19):5722–5730. doi: 10.1021/bi9929756. [DOI] [PubMed] [Google Scholar]
- 28.Heitz A, Hernandez JF, Gagnon J, Hong TT, Pham TT, Nguyen TM, Le-Nguyen D, Chiche L. Solution structure of the squash trypsin inhibitor MCoTI-II. A new family for cyclic knottins. Biochemistry. 2001;40(27):7973–7983. doi: 10.1021/bi0106639. [DOI] [PubMed] [Google Scholar]
- 29.Clark RJ, Daly NL, Craik DJ. Structural plasticity of the cyclic-cystine-knot framework: implications for biological activity and drug design. The Biochemical journal. 2006;394(Pt 1):85–93. doi: 10.1042/BJ20051691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Craik DJ, Cemazar M, Daly NL. The cyclotides and related macrocyclic peptides as scaffolds in drug design. Current opinion in drug discovery & development. 2006;9(2):251–260. [PubMed] [Google Scholar]
- 31.Craik DJ, Daly NL, Mulvenna J, Plan MR, Trabi M. Discovery, structure and biological activities of the cyclotides. Curr Protein Pept Sci. 2004;5(5):297–315. doi: 10.2174/1389203043379512. [DOI] [PubMed] [Google Scholar]
- 32.Reiss S, Sieber M, Oberle V, Wentzel A, Spangenberg P, Claus R, Kolmar H, Losche W. Inhibition of platelet aggregation by grafting RGD and KGD sequences on the structural scaffold of small disulfide-rich proteins. Platelets. 2006;17(3):153–157. doi: 10.1080/09537100500436663. [DOI] [PubMed] [Google Scholar]
- 33.Kolmar H. Biological diversity and therapeutic potential of natural and engineered cystine knot miniproteins. Curr Opin Pharmacol. 2009;9(5):608–614. doi: 10.1016/j.coph.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Puttamadappa SS, Jagadish K, Shekhtman A, Camarero JA. Backbone Dynamics of Cyclotide MCoTI-I Free and Complexed with Trypsin. Angew Chem Int Ed Engl. 2010;49(39):7030–7034. doi: 10.1002/anie.201002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sancheti H, Camarero JA. “Splicing up” drug discovery. Cell-based expression and screening of genetically-encoded libraries of backbone-cyclized polypeptides. Adv Drug Deliv Rev. 2009;61(11):908–917. doi: 10.1016/j.addr.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenwood KP, Daly NL, Brown DL, Stow JL, Craik DJ. The cyclic cystine knot miniprotein MCoTI-II is internalized into cells by macropinocytosis. Int J Biochem Cell Biol. 2007;39(12):2252–2264. doi: 10.1016/j.biocel.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 37.Hewlett LJ, Prescott AR, Watts C. The coated pit and macropinocytic pathways serve distinct endosome populations. J Cell Biol. 1994;124(5):689–703. doi: 10.1083/jcb.124.5.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerr MC, Teasdale RD. Defining macropinocytosis. Traffic. 2009;10(4):364–371. doi: 10.1111/j.1600-0854.2009.00878.x. [DOI] [PubMed] [Google Scholar]
- 39.Lundberg M, Johansson M. Positively charged DNA-binding proteins cause apparent cell membrane translocation. Biochem Biophys Res Commun. 2002;291(2):367–371. doi: 10.1006/bbrc.2002.6450. [DOI] [PubMed] [Google Scholar]
- 40.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278(1):585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 41.Lim JP, Gleeson PA. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol. 2011 doi: 10.1038/icb.2011.20. [DOI] [PubMed] [Google Scholar]
- 42.Norbury CC, Hewlett LJ, Prescott AR, Shastri N, Watts C. Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity. 1995;3(6):783–791. doi: 10.1016/1074-7613(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 43.Steinman RM, Brodie SE, Cohn ZA. Membrane flow during pinocytosis. A stereologic analysis. J Cell Biol. 1976;68(3):665–687. doi: 10.1083/jcb.68.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422(6927):37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 45.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 46.Kaduk C, Wenschuh H, Beyermann M, Forner K, Carpino LA, Bienert M. Synthesis of Fmoc-amino acid fluorides via DAST, an alternative fluorinating agent. Lett Pept Sci. 1997;2(5):285–288. [Google Scholar]
- 47.Ingenito R, Dreznjak D, Guffler S, Wenschuh H. Efficient loading of sulfonamide safety-catch linkers by Fmoc amino acid fluorides. Org Lett. 2002;4(7):1187–1188. doi: 10.1021/ol0256320. [DOI] [PubMed] [Google Scholar]
- 48.Camarero JA, Mitchell AR. Synthesis of proteins by native chemical ligation using Fmoc-based chemistry. Protein Pept Lett. 2005;12(8):723–728. doi: 10.2174/0929866054864166. [DOI] [PubMed] [Google Scholar]
- 49.Ingenito R, Bianchi E, Fattori D, Pessi A. Solid phase synthesis of peptide C-terminal thioesters by Fmoc/t-Bu chemistry Source. J Am Chem Soc. 1999;121(49):11369–11374. [Google Scholar]
- 50.Shin Y, Winans KA, Backes BJ, Kent SBH, Ellman JA, Bertozzi CR. Fmoc-Based Synthesis of Peptide. J Am Chem Soc. 1999;121 :11684–11689. [Google Scholar]
- 51.Camarero JA, Thioesters TW-A. Application to the Total Chemical Synthesis of a Glycoprotein by Native Chemical Ligation. Muir, Chemoselective backbone cyclization of unprotected peptides. Chem Comm. 1997;1997:1369–1370. [Google Scholar]
- 52.Zhang L, Tam JP. Synthesis and application of unprotected cyclic peptides as building blocks for peptide dendrimers. J Am Chem Soc. 1997;119:2363–2370. [Google Scholar]
- 53.Camarero JA, Pavel J, Muir TW. Chemical Synthesis of a Circular Protein Domain: Evidence for Folding-Assisted Cyclization. Angew Chem Int Ed. 1998;37(3):347–349. doi: 10.1002/(SICI)1521-3773(19980216)37:3<347::AID-ANIE347>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 54.Shao Y, Lu WY, Kent SBH. A novel method to synthesize cyclic peptides. Tetrahedron Lett. 1998;39(23):3911–3914. [Google Scholar]
- 55.Camarero JA, Muir TW. Biosynthesis of a Head-to-Tail Cyclized Protein with Improved Biological Activity. J Am Chem Soc. 1999;121:5597–5598. [Google Scholar]
- 56.Camarero JA, Fushman D, Cowburn D, Muir TW. Peptide chemical ligation inside living cells: in vivo generation of a circular protein domain. Bioorg Med Chem. 2001;9(9):2479–2484. doi: 10.1016/s0968-0896(01)00217-6. [DOI] [PubMed] [Google Scholar]
- 57.Silverstein SC, Steinman RM, Cohn ZA. Endocytosis. Annu Rev Biochem. 1977;46:669–722. doi: 10.1146/annurev.bi.46.070177.003321. [DOI] [PubMed] [Google Scholar]
- 58.Fonseca SB, Pereira MP, Kelley SO. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv Drug Deliv Rev. 2009;61(11):953–964. doi: 10.1016/j.addr.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 59.Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R, Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic. 2007;8(7):848–866. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 60.Brown D, Sabolic I. Endosomal pathways for water channel and proton pump recycling in kidney epithelial cells. J Cell Sci Suppl. 1993;17:49–59. doi: 10.1242/jcs.1993.supplement_17.8. [DOI] [PubMed] [Google Scholar]
- 61.Shurety W, Stewart NL, Stow JL. Fluid-phase markers in the basolateral endocytic pathway accumulate in response to the actin assembly-promoting drug Jasplakinolide. Mol Biol Cell. 1998;9(4):957–975. doi: 10.1091/mbc.9.4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thompson K, Rogers MJ, Coxon FP, Crockett JC. Cytosolic entry of bisphosphonate drugs requires acidification of vesicles after fluid-phase endocytosis. Mol Pharmacol. 2006;69(5):1624–1632. doi: 10.1124/mol.105.020776. [DOI] [PubMed] [Google Scholar]
- 63.Nabi IR, Le PU. Caveolae/raft-dependent endocytosis. J Cell Biol. 2003;161(4):673–677. doi: 10.1083/jcb.200302028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang F, Khvorova A, Marshall W, Sorkin A. Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. J Biol Chem. 2004;279(16):16657–16661. doi: 10.1074/jbc.C400046200. [DOI] [PubMed] [Google Scholar]
- 65.Jiang X, Huang F, Marusyk A, Sorkin A. Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol Biol Cell. 2003;14(3):858–870. doi: 10.1091/mbc.E02-08-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sorkin A, Von Zastrow M. Signal transduction and endocytosis: close encounters of many kinds. Nat Rev Mol Cell Biol. 2002;3(8):600–614. doi: 10.1038/nrm883. [DOI] [PubMed] [Google Scholar]
- 67.Burckhardt CJ, Greber UF. Virus movements on the plasma membrane support infection and transmission between cells. PLoS Pathog. 2009;5(11):e1000621. doi: 10.1371/journal.ppat.1000621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jerdeva GV, Wu K, Yarber FA, Rhodes CJ, Kalman D, Schechter JE, Hamm-Alvarez SF. Actin and non-muscle myosin II facilitate apical exocytosis of tear proteins in rabbit lacrimal acinar epithelial cells. J Cell Sci. 2005;118(Pt 20):4797–4812. doi: 10.1242/jcs.02573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mercer J, Helenius A. Virus entry by macropinocytosis. Nat Cell Biol. 2009;11(5):510–520. doi: 10.1038/ncb0509-510. [DOI] [PubMed] [Google Scholar]
- 70.Peterson JR, Mitchison TJ. Small molecules, big impact: a history of chemical inhibitors and the cytoskeleton. Chem Biol. 2002;9(12):1275–1285. doi: 10.1016/s1074-5521(02)00284-3. [DOI] [PubMed] [Google Scholar]
- 71.Mercer J, Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science. 2008;320(5875):531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- 72.Le PU, Nabi IR. Distinct caveolae-mediated endocytic pathways target the Golgi apparatus and the endoplasmic reticulum. J Cell Sci. 2003;116(Pt 6):1059–1071. doi: 10.1242/jcs.00327. [DOI] [PubMed] [Google Scholar]
- 73.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8(3):185–194. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 74.Spang A. On the fate of early endosomes. Biol Chem. 2009;390(8):753–759. doi: 10.1515/BC.2009.056. [DOI] [PubMed] [Google Scholar]
- 75.Eskelinen EL, Tanaka Y, Saftig P. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003;13(3):137–145. doi: 10.1016/s0962-8924(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 76.Fukuda M. Lysosomal membrane glycoproteins. Structure, biosynthesis, and intracellular trafficking. J Biol Chem. 1991;266(32):21327–21330. [PubMed] [Google Scholar]
- 77.Cordonnier MN, Dauzonne D, Louvard D, Coudrier E. Actin filaments and myosin I alpha cooperate with microtubules for the movement of lysosomes. Mol Biol Cell. 2001;12(12):4013–4029. doi: 10.1091/mbc.12.12.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matteoni R, Kreis TE. Translocation and clustering of endosomes and lysosomes depends on microtubules. J Cell Biol. 1987;105(3):1253–1265. doi: 10.1083/jcb.105.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Taunton J, Rowning BA, Coughlin ML, Wu M, Moon RT, Mitchison TJ, Larabell CA. Actin-dependent propulsion of endosomes and lysosomes by recruitment of N-WASP. J Cell Biol. 2000;148(3):519–530. doi: 10.1083/jcb.148.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aniento F, Emans N, Griffiths G, Gruenberg J. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J Cell Biol. 1993;123(6 Pt 1):1373–1387. doi: 10.1083/jcb.123.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Loubery S, Wilhelm C, Hurbain I, Neveu S, Louvard D, Coudrier E. Different microtubule motors move early and late endocytic compartments. Traffic. 2008;9(4):492–509. doi: 10.1111/j.1600-0854.2008.00704.x. [DOI] [PubMed] [Google Scholar]
- 82.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55(6):1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 83.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11(11):1355–1362. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.