

Abstract
A cis-acting region, Δ4, located in the 3′-untranslated region of N-methyl-D-aspartate R (NR) mRNA interacts with several trans-acting proteins present in polysomes purified from fetal cortical neurons. Chronic ethanol exposure of fetal cortical neurons increases Δ4 RNA–protein interactions. This increased interaction is due to an increase in one of the Δ4-binding trans-acting proteins identified as beta subunit of alpha glucosidase II (GIIβ). In this study, we examined whether ethanol-mediated regulation of NR1 mRNA in vivo is similar to that in vitro and whether Δ4–trans interactions are important for ethanol-mediated NR1 mRNA stability. Our data show that polysomal proteins from adult mouse cerebral cortex (CC) formed a complex with Δ4 RNA, suggesting the presence of NR1 mRNA-binding trans-acting proteins in CC polysomes. The intensity of the Δ4 RNA–protein complex was increased with polysomes from chronic ethanol-exposed CC. The Δ4 RNA–protein complex harbored GIIβ and a second trans-acting protein identified as annexin A2 (AnxA2). Ethanol-sensitive GIIβ was upregulated by 70% in ethanol-exposed CC. Heparin, a known binding partner of AnxA2, inhibited Δ4 RNA–protein complex formation. Transient transfection studies using chimeric constructs with and without the Δ4 region revealed that cis–trans interactions are important for ethanol-mediated stability of NR1 mRNA. Furthermore, our data highlight, for the first time, the presence of a binding site on the 3′-untranslated region of NR1 mRNA for AnxA2 and demonstrate the regulation of NR1 mRNA by AnxA2, GIIβ and a third NR1 mRNA-binding protein, which is yet to be identified.
Keywords: cerebral cortex, cis-acting regions, fetal cortical neurons, mouse, mRNA stability, trans-acting proteins
Introduction
Excitatory N-methyl-D-aspartate (NMDA) receptors play a vital role in neurodevelopment (Endele et al., 2010), synaptic plasticity including learning and memory (McDonald et al., 2006; Dingeldine et al., 1999), and neurotoxicity (Malenka & Nicoll, 1999). Functional heteromeric NMDA receptors consist of NMDA R (NR)1 subunit and members of the NR2 and NR3 family (Dingledine et al., 1999). The NR1 subunit, the functional subunit of the NMDA receptor, has eight full-length splice variants (Hollmann et al., 1993). NMDA receptors are a target of alcohol (ethanol) and are involved in the development of alcohol tolerance, dependence and withdrawal syndrome (Kumari & Ticku, 2000; Hoffman, 2003; Krystal et al., 2003). Chronic ethanol exposure, both in vivo and in vitro, increases NMDA receptor function and number (Iorio et al., 1992; Lovinger, 1993; Michaelis et al., 1993; Freund & Anderson, 1996) with a concomitant increase in NR1 and NR2B polypeptide levels (Kumari & Ticku, 2000; Hoffman, 2003). Chronic ethanol exposure in vivo and in vitro does not affect total NR1 mRNA levels (Follesa & Ticku, 1995; Hu et al., 1996; Snell et al., 1996) but selectively increases exon 5 lacking splice variants (Hardy et al., 1999; Winkler et al., 1999; Kumari, 2001).
Chronic ethanol exposure increases the half-life of NR1 mRNA in fetal cortical neurons (FCNs) (Kumari & Ticku, 1998) and this half-life returns to control values when new protein synthesis is inhibited in ethanol-exposed FCNs (Kumari et al., 2003), highlighting the importance of proteins in ethanol-mediated stabilization of NR1 mRNA. A cis-acting region, Δ4, identified in the 3′-untranslated region (UTR) of NR1 mRNA interacts with proteins present in FCN polysomes. The intensity of the Δ4 RNA–protein complex was higher in ethanol-exposed FCN polysomes as compared with control polysomes and this difference is attributed to the ethanol-sensitive NR1 mRNA-binding protein, beta subunit of alpha glucosidase II (GIIβ) (Anji & Kumari, 2006). Although in-vitro studies are unfolding the molecular mechanism(s) regulating NR1 gene expression, similar in-vivo studies have not been performed to date. Therefore, we initiated studies to understand the ethanol-mediated regulation of NR1 mRNA in vivo using C57 BL/6 mice. Specifically, we examined the expression of NR1 mRNA-binding trans-acting proteins in the cerebral cortex (CC) and the effect of chronic ethanol on these proteins. We purified and identified a second NR1 mRNA-binding protein. Using transient transfection assays we tested the importance of the Δ4 region in ethanol-mediated NR1 mRNA stability.
Our data demonstrated that the CC expressed NR1 mRNA-binding proteins and that the intensity of the Δ4 RNA–protein complex is enhanced with chronic ethanol-exposed CC polysomes, suggesting that NR1 regulation in vivo and in vitro is very similar. We show, for the first time, that the Δ4 RNA–CC protein complex harbors two binding sites, one for GIIβ and a second for annexin A2 (AnxA2). The GIIβ polypeptide was upregulated by 70% in ethanol-exposed CC. Heparin, a binding partner of AnxA2, prevented Δ4 RNA–protein complex formation. We also show, for the first time, that cis–trans interactions are an important regulator of the ethanol-mediated stability of NR1 mRNA.
Materials and Methods
Materials
The liquid diet was purchased from Bioserv (Frenchtown, NJ, USA). [32P]-α-CTP (specific activity 3000 Ci/mmol) was purchased from Perkin Elmer Life and Analytical Sciences (Waltham, MA, USA), Fugene HD and Sephadex G50 columns from Roche Applied Science (Indianapolis, IN, USA), Hybond polyvinylidenedifluoride membrane from GE Healthcare Life Sciences (Piscataway, NJ, USA), Pfu polymerase and QuikChange XL site-directed mutagenesis kit from Agilent Technologies (formerly Stratagene) (La Jolla, CA, USA), dual-luciferase reporter assay system from Promega (Madison, WI, USA), T4 DNA ligase from New England Biolabs Inc. (Beverly, MA, USA), and PureLink™ Quick Gel Extraction Kit and PureLink™ HiPure Plasmid Filter Maxiprep Kit from InVitrogen (Carlsbad, CA, USA). The GIIβ antibody was obtained from Dr Hanne Ostergaard (Department of Medical Microbiology & Immunology, University of Alberta, Edmonton, AB, Canada), the AnxA2 antibody was a kind gift from Dr Tony Hunter (Molecular and Cell Biology Laboratory, Salk Institute Cancer Center, San Diego, CA, USA) and the recombinant AnxA2 protein was obtained from Dr David Waisman (Department of Biochemistry and Molecular Biology, Dalhouse University, Halifax, NS, Canada). All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) and all restriction endonucleases were purchased from either New England Biolabs (Ipswich, MA, USA) or Promega.
Animals
Experiments were performed with 6-8-week-old male mice (strain C57 BL/6) bred at our animal resource facility. Animals were housed individually under a 12 h light/12 h dark illumination cycle (lights on at 09:00 h). Animals were used in accordance with institutional guidelines and procedures were approved by the Institutional Animal Care and Use Committee of Kansas State University.
Chronic ethanol treatment of mice
The chronic ethanol (alcohol) treatment used in this study has been described previously (Anji & Kumari, 2008). Briefly, after a 1 week acclimation period, animals were divided into control and ethanol-exposed groups. Ethanol-exposed animals were introduced to the ethanol-containing liquid diet with a step-wise increase in ethanol: days 1 and 2, 0% ethanol; days 3 and 4, 2.3% ethanol; days 5 and 6, 4.7% ethanol and days 7-16, 7% ethanol. Both control and ethanol-exposed mice also received a measured amount of Purina rat chow (∼1.0 g/day) from day 9 to day 16. The amount of ethanol-containing liquid diet consumed was measured every morning (09:00 h). Control animals were pair-fed and received the same volume of control liquid diet as the ethanol-exposed mice had consumed the previous day. Maltose-dextrin was substituted isocalorically for ethanol. Body weights of ethanol-exposed and control mice were monitored daily. The level of intoxication of mice was determined by measuring blood ethanol levels on the day of killing.
Determination of blood ethanol levels
Blood ethanol levels were determined enzymatically as described previously (Draski et al., 2001). Briefly, a 40 μL venous blood sample obtained from the retro-orbital sinus or trunk was mixed with 2 mL of 3.0% (v/v) perchloric acid. Samples were centrifuged and a known volume of the supernatant (200 μL) from each sample was mixed with 2 mL of the reaction mixture (0.5 M Tris.HCl buffer, pH 8.8, containing alcohol dehydrogenase and NAD+) and incubated for 15 min at 30 °C. The ethanol content in the sample was determined by measuring the amount of NADH(-) produced from NAD+. All of the samples were read in a Beckman DU 640 spectrophotometer at 340 nm and blood ethanol levels were calculated from a standard curve (0-500 mg% alcohol), analyzed simultaneously with the unknown samples.
Cell culture
Fetal cortical neurons isolated from 14- to 15-day-old mouse fetuses were cultured as described previously (Kumari, 2001). Mice (strain C57 BL/6) purchased from Harlan (Indianapolis, IN, USA) were bred at our animal resource facility and used in accordance with institutional guidelines, and procedures were approved by the animal welfare committee.
Preparation of polysomes
At 24 h after the last ethanol treatment, animals were killed and brains were dissected. CCs from each mouse were processed independently to prepare polysomal fractions according to the procedure described (Brewer & Ross, 1990). Briefly, tissue was minced and homogenized in a glass homogenizer with a tight pestle (10 strokes) in 1× Buffer A (10 mM Tris.HCl, pH 7.4, 1 mM potassium acetate, 1.5 mM magnesium acetate, 0.32 M sucrose, supplemented with 1× protease inhibitor cocktail). Homogenates were centrifuged at 10 000 g at 4 °C for 10 min and pellets containing broken cells, nuclei, mitochondria and synaptosomes were discarded. Supernatants (S10 fraction) were centrifuged at 100 000 g at 4 °C for 2.5 h and pellets containing microsomes (polysomal fraction) were resuspended in Buffer A. The supernatant (S100 fraction) was also recovered. Aliquots of polysomes were stored at -80 °C until further use. The protein concentration in polysome fractions was determined by the Bradford method (Bradford, 1976). To verify the consistency of polysomal preparations we routinely confirmed, by western blotting, the ethanol-mediated upregulation of NR1 protein in the polysomal fractions (Kumari, 2001). We also processed the polysomal preparations for examination under the electron microscope.
In-vitro transcription
A 156-nucleotide-long cis-acting region termed Δ4 (Supporting Information Fig. S1A) was previously mapped within the 3′-UTR of NR1 mRNA (Anji & Kumari, 2006). Sense RNA spanning the Δ4 region was generated by in-vitro transcription of Nhe I linearized plasmid in the presence of [32P]-α-CTP and T3 RNA polymerase, and purified using a Sephadex G50 column. Purified [32P]-labeled Δ4 sense RNA was used as a probe for RNA gel mobility shift assays, northwestern analysis and RNA gel mobility supershift assays.
RNA gel mobility shift assay
These assays were performed according to the method described by Anji & Kumari (2006) using polysomes (5 μg protein) from control and ethanol-treated mouse CC or purified recombinant protein(s). When appropriate, heparin was included in the RNA gel shift incubation mix.
Purification of N-methyl-D-aspartate R1 mRNA-binding proteins
To purify the mRNA-binding proteins from the shifted RNA in the gel (Stenger et al., 2004), an appropriate amount of [32P]-labeled Δ4 RNA was incubated with 20 μg polysomal protein for 30 min at 30 °C. The incubation mix was separated at 200 V for 3 h on a 2-mm-thick 5% non-denaturing cooled acrylamide gel with a half-inch well size. After completion of electrophoresis, the gel was wrapped in saran wrap and exposed to X-ray film for 2-3 h at room temperature. The image on the developed film was aligned with the gel. The gel area containing the shifted Δ4 RNA was excised with a new scalpel blade and used as a source of NR1 mRNA-binding trans-acting proteins. Proteins were eluted from the excised gel pieces containing the RNA–protein complex. Proteins were concentrated and separated by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis. The protein bands were stained with colloidal blue and the 47 kDa protein band was excised for mass spectroscopy (see below).
Mass spectroscopy and bioinformatics analysis
The excised protein band in gel was sent to the Harvard Microchemistry Facility for protein identification. After in-gel digestion with trypsin, multiple peptide sequences were determined in a single run by microcapillary reverse-phase HPLC nano-electrospray tandem mass spectrometry on a Finnigan LCQ DECA XP Plus quadruple ion trap mass spectrometer. MS/MS spectra were correlated with known sequences using the algorithm Sequest (Yates et al., 1995) and the program ‘FuzzyIons’ (Chittum et al., 1998). MS/MS sequences were confirmed for consensus with known proteins and the results were manually confirmed for fidelity.
RNA gel mobility supershift analysis
Binding reactions were performed as for RNA gel mobility shift assays. Following incubation of polysomal proteins with radiolabeled Δ4 RNA, varying concentrations of GIIβ antibody, AnxA2 antibody or normal rabbit serum were added and reaction mixes were incubated for an additional 20 min at room temperature. At the end of the incubation, RNA–protein complexes were processed as in RNA gel mobility shift assays.
Northwestern analysis
Polysomes (10 μg) from control and ethanol-exposed CC and control FCNs were separated on 7.5% SDS–polyacrylamide gel. Following electrophoresis, proteins were electroblotted onto a nitrocellulose membrane. Proteins were then allowed to refold in binding buffer (Tian et al., 1998). The membrane was hybridized at room temperature with radiolabeled Δ4 RNA (2 × 106 cpm) in binding buffer for 90 min. Following hybridization, unbound RNA was removed by washing in binding buffer (3 × 2 min and 1 × 7 min) and the membrane was exposed to PhosphorImager screen and analyzed using ImageQuant software.
Western blot analysis
This was performed as described previously (Kumari, 2001). To determine the effect of chronic ethanol exposure on GIIβ expression, polysomal proteins (10 μg) from the CC of control and ethanol-exposed mice were separated on 8% SDS–polyacrylamide gel electrophoresis, blotted onto PVDF membrane, and processed to detect the GIIβ-immunoreactive band using GIIβ antibody. To normalize results, membranes were stripped and reprobed with β-actin antibody.
Generation of chimeric constructs
Reporter gene construct with full-length N-methyl-D-aspartate R1 3′-untranslated region (pGLNR1-3UTR)
The pGL3 control (pGL3C) vector (Promega) containing luciferase reporter gene driven by SV40 promoter was used to test the effect of the NR1 3′-UTR on mRNA stability because the SV40 promoter is known to provide higher expression of the reporter gene in primary cultures of cortical neurons (Anji et al., 2003) and luciferase enzyme activity can be easily measured using a luminometer. A restriction endonuclease EcoRV site was created at the end of the luciferase coding region in pGL3C vector by mutating three nucleotides 1918AAG1920 to GAT (pGL3CRV) using a QuikChange XL site-directed mutagenesis kit. The NR1 3′-UTR sequence (accession no. AY616022) and the NR1 sequence encoding the last 17 amino acids (= DITGPLNLSDPSVSTVV) of the C2′ cassette were excised out of pKSNR1-3UTR by digestion with EcoRV and Xba I and subcloned, in-frame, into EcoRV/Xba I-digested pGL3CRV to generate pGLNR1-3UTR. Correct subcloning of the full-length NR1 3′-UTR into pGL3CRV was confirmed initially by restriction digestion and subsequently by DNA sequencing. Because expression of pGLNR1-3UTR will generate luciferase enzyme with 17 amino acids of the NR1 C2′ cassette at the C-terminal domain, we initially assessed the ability of pGLNR1-3UTR to generate an active luciferase enzyme by transfecting P19 cells. Expression of 17 amino acids of the C2′ cassette at the C-terminal domain of luciferase enzyme did not interfere with its enzyme activity (data not shown).
Deletion mutant lacking the Δ4 region of the N-methyl-D-aspartate R1 3′-untranslated region (pGLNR1-Δ4)
A 156-nucleotide-long cis-acting region, Δ4 (Anji & Kumari, 2006), can be excised out of pKSNR1-3UTR by digestion with Apa I (at the 5′ end) and Nhe I (at the 3′ end) (Supporting Information Fig. S1A); however, these two ends are not compatible. Therefore, to delete the Δ4 region in the NR1 3′-UTR, we employed a novel polymerase chain reaction-based method. A known amount of plasmid, pKSNR1-3UTR, was amplified by polymerase chain reaction using 200 ng each of upstream oligonucleotide (5′-CGACCTTTCCCAGGGCTAGCCTTC-3′) spanning the Nhe I site present at the 3′ end of the Δ4 region and downstream oligonucleotide (5′-CAAGCTACAGTGGG CTAGCAAAAGCAAC-3′) with a Nhe I site at the 3′ end of the oligonucleotide in a 100 μL reaction containing 1 × polymerase chain reaction buffer, 200 μM each dNTP, 0.2% dimethyl sulfoxide (v/v) and 1.2 μL of high fidelity Pfu polymerase. Optimized temperature cycling for polymerase chain reaction was as follows: step 1, 2.5 min at 95 °C × 1 cycle; step 2, 50 s at 95 °C, 35 s at 54 °C, 8 min at 72 °C × 25 cycles; step 3, 10 min at 72 °C × 1 cycle, and soak at 4 °C. Amplified DNA was precipitated with ethanol and recovered DNA was digested to completion with Nhe I at 37 °C. Digested DNA was separated on agarose gel and the correct size DNA fragment was excised to extract DNA using a PureLink™ Quick Gel Extraction Kit. Purified DNA (50 ng) was allowed to self-ligate overnight at 16 °C using T4 DNA ligase. Ligated DNA was processed for transformation of DH5α bacteria, and plasmid with the desired deletion of the Δ4 region in pKSNR1-3UTR (pKSNR13UTR-Δ4) was generated. Deletion of the Δ4 region was confirmed initially by restriction digestion (Supporting Information Fig. S1B) and subsequently by DNA sequencing. The NR1 3′-UTR sequence lacking the Δ4 region and the NR1 sequence encoding the last 17 amino acids of the C2′ cassette were excised out of pKSNR13UTR-Δ4 by digestion with EcoRV and Xba I and subcloned, in-frame, into EcoRV/Xba I-digested pGL3CRV to generate pGLNR1-Δ4. All plasmid DNA was generated in large quantities by Maxiprep using a PureLink™ HiPure Plasmid Filter Maxiprep Kit.
Transient transfection and reporter gene assay
Transient transfection of FCNs cultured in the absence or presence of 50 mM ethanol was performed as described previously (Anji et al., 2003) except that Fugene HD was used instead of lipofectamine. Cells were transfected independently with 1 pmol DNA of control vectors or test plasmids containing firefly luciferase reporter gene and co-transfected with 0.5 pmol DNA of pRL-TK, Renilla luciferase reporter plasmid, as an internal control, to normalize the transfection results. The culture medium was changed every 24 h. At 48 h post-transfection, cells were washed with phosphate-buffered saline and lysed in 1× passive lysis buffer. Cell lysates were centrifuged at 16 100 g for 30 s. Supernatants were recovered and an aliquot was used for protein estimation by the Bradford method (Bradford, 1976). A known amount of protein from each supernatant was utilized to measure firefly luciferase and Renilla luciferase activities using the dual-luciferase reporter assay system with a TD 20/20 luminometer. Relative luciferase activity was obtained by normalizing firefly luciferase activity against the internal control Renilla luciferase activity.
Statistical analysis
Images scanned on PhosphorImager Storm 840 were analyzed using ImageQuant TL software. Statistical analysis was performed using ANOVA and Scheffe's test.
Results
Polysomes of adult mouse cerebral cortex contain the N-methyl-D-aspartate R1 3′-untranslated region-binding trans-acting proteins
Our deletion analyses of the 3′-UTR of NR1 mRNA using FCN polysomes identified a 156-nucleotide-long cis-acting region (Δ4) that interacts with several trans-acting proteins in FCNs (Anji & Kumari, 2006). To test our hypothesis that such cis–trans interactions also occur in adult brain (in vivo) and that these interactions are altered by chronic ethanol exposure in vivo, we exposed young adult male mice to chronic ethanol and control mice were pair-fed. Blood ethanol levels were assessed at the time of killing, at 24 h after the last ethanol treatment. The average blood ethanol level was 362.65 mg%. Polysomal fractions were purified from the CC of individual control and ethanol-exposed mice. A known amount of polysomal protein from control and ethanol-exposed CC was incubated with [32P]-labeled Δ4 RNA and samples were processed for RNA gel mobility shift assays to assess the ability of Δ4 RNA to bind trans-acting proteins. As a positive control, FCN polysomes were included in the RNA gel mobility shift assay. The autoradiogram shown in Fig. 1A shows a shift of the Δ4 RNA when CC polysomes from control mice were utilized (Fig. 1A, lane Adult C). The intensity of the Δ4 RNA–protein complex was significantly enhanced when CC polysomes from ethanol-exposed mice were employed for the RNA gel mobility shift assay (Fig. 1A, lane Adult E and Fig. 1B). These data are similar to control and ethanol-exposed FCN polysomes that were included as a positive control in this analysis. These results suggest that NR1 mRNA-binding trans-acting proteins are expressed in the CC of adult mice, and that some of the trans-acting proteins may be ethanol sensitive.
Fig. 1.
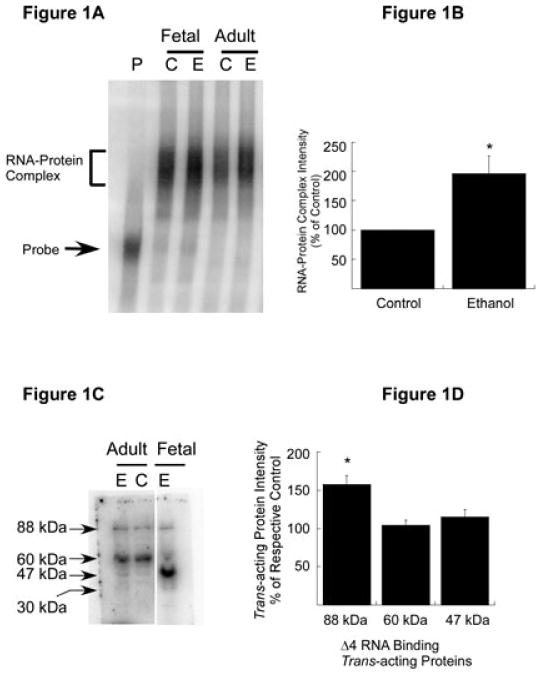
Expression of trans-acting proteins in the CC of adult mouse and effect of chronic ethanol treatment. (A) RNA gel shift analysis. [32P]-labeled Δ4 sense RNA was incubated with polysomes purified from the CC of control (C) and chronic ethanol (E)-exposed adult mice (Adult). Polysomes from control (C) and ethanol-exposed (E) FCNs (Fetal) were included as positive controls. Binding of trans-acting proteins to Δ4 RNA was detected by RNA gel mobility shift assays. A representative autoradiogram is shown here. The Δ4 RNA–protein complex (bracket) and free probe (solid arrow) are indicated on the left. (B) Effect of chronic ethanol on RNA–protein interactions. The intensity of the protein–RNA complex in A was quantitated using ImageQuantTL software. Data are expressed as a percentage of control and plotted values represent the mean + SEM of three independent experiments. Statistical analysis was performed by ANOVA and Scheffe's test (*p < 0.01). (C) Nature of Δ4 RNA-binding trans-acting protein(s) in the CC of adult mice. Polysomal proteins from the CC of control (C) and chronic ethanol-exposed (E) adult mice and ethanol-exposed (E) (50 mM, 5 days) FCNs (Fetal) were separated on SDS–polyacrylamide gel electrophoresis, allowed to refold, blotted onto nitrocellulose membrane and probed with [32P]-labeled Δ4 sense RNA. Three trans-acting proteins that interact with Δ4 RNA were detected in the CC of control and ethanol-exposed mice and four in ethanol-exposed FCNs. Solid arrows on the left indicate the apparent molecular mass of Δ4 RNA-binding trans-acting proteins. Note that gel lanes between Adult and Fetal samples have been omitted. (D) Effect of chronic ethanol on NR1 mRNA-binding trans-acting proteins. Intensities of 88, 60 and 47 kDa protein–Δ4 RNA complexes in C were quantitated using ImageQuantTL software. Data are expressed as a percentage of their respective controls and plotted values represent the mean + SEM of three independent experiments. Statistical analysis was performed by ANOVA and Scheffe's test (*p < 0.01).
Nature of the N-methyl-D-aspartate R1 mRNA-binding proteins
We performed northwestern analysis to determine the number of trans-acting proteins in the Δ4 RNA–CC protein complex and their apparent molecular mass. Polysomal proteins from control and ethanol-exposed adult CC were separated on SDS–polyacrylamide gel electrophoresis along with protein molecular weight markers, blotted onto nitrocellulose membrane and probed with radiolabeled Δ4 RNA. FCN polysomes that are known to express NR1 mRNA-binding trans-acting proteins were included as a positive control. Upon overnight exposure to a PhosphorImager screen, we detected three protein bands in adult CC irrespective of ethanol treatment (Fig. 1C, lanes Adult C and E) and four protein bands in FCNs (Fig. 1C, lane Fetal E). The calculated apparent molecular mass of these proteins was approximately 88, 60 and 47 kDa, respectively (Fig. 1C). The intensity of the 88 kDa band was increased by 57% in polysomes from ethanol-exposed adult CC as compared with polysomes from control adult CC (Fig. 1C and D), suggesting that this protein is ethanol sensitive. The intensity of the 60 and 45 kDa bands, however, was not significantly different in ethanol-treated polysomes when compared with their respective controls (Fig. 1D).
To ensure that the binding of [32P]-labeled Δ4 RNA to the respective trans-acting proteins in northwestern analysis was specific, we performed competition northwestern analysis where membranes were incubated with [32P]-labeled Δ4 RNA in the presence of a 20-fold molar excess of unlabeled Δ4 RNA. As we have shown previously (Anji & Kumari, 2006), no radioactive bands were detected in the presence of a 20-fold molar excess of unlabeled Δ4 RNA (data not shown here).
Beta subunit of alpha glucosidase II is an N-methyl-D-aspartate R1 mRNA-binding trans-acting protein expressed in adult cerebral cortex
One of the NR1 mRNA-binding proteins identified by northwestern analysis using CC polysomes had a calculated molecular mass of 88 kDa (Fig. 1C). Previously, using FCN polysomes, we identified this 88 kDa trans-acting protein as GIIβ. To confirm whether this RNA-binding protein is indeed GIIβ and a component of the Δ4 RNA–CC polysomal protein complex, we performed RNA gel mobility supershift analysis. Polysomes from ethanol-exposed adult CC were incubated with [32P]-labeled Δ4 RNA for 30 min at 30 °C. Increasing concentrations of anti-GIIβ were added to the incubation mix and then incubated for an additional 20 min at room temperature. Samples were analyzed as for RNA gel shift assays. Addition of anti-GIIβ to the incubation mix produced a supershift of the Δ4 RNA–protein complex (Fig. 2A). The intensity of the supershift of the Δ4 RNA–protein complex increased with increasing concentrations of anti-GIIβ with a corresponding decrease in the intensity of the Δ4 RNA–protein complex in these lanes (Fig. 2A, lanes 2-4). Similar results were obtained for control adult CC (data not shown here). To determine the specificity of the supershift, increasing concentrations of normal rabbit serum were added instead of anti-GIIβ. No supershift of the Δ4 RNA–protein complex was observed with normal rabbit serum (Fig. 2A, lanes 5-7). These data suggested that GIIβ is present in the CC polysomes and binds specifically to Δ4 RNA.
Fig. 2.
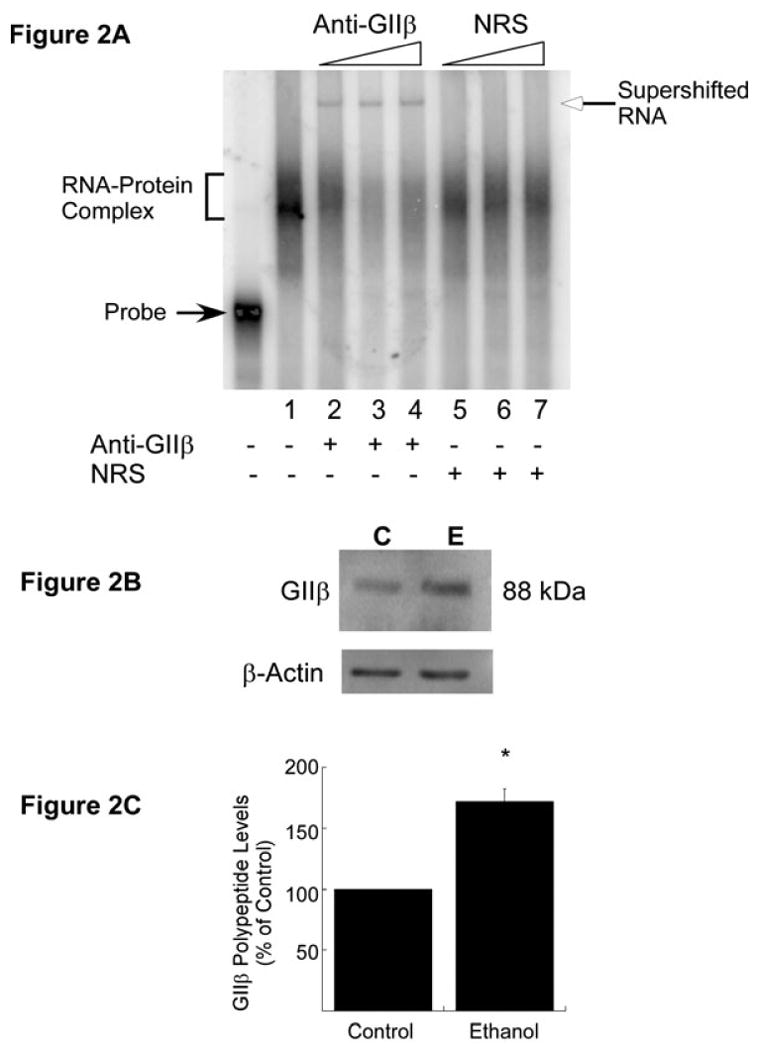
Trans-acting protein GIIβ in the CC of adult mouse. (A) RNA gel mobility supershift analysis to detect the presence of GIIβ in Δ4 RNA–CC polysomal protein complex. The [32P]-labeled Δ4 sense RNA was first incubated with CC polysomes from ethanol-exposed mice as in RNA gel shift assays. After the addition of increasing concentrations of either anti-GIIβ (lane 1, 0.04 μL; lane 2, 0.0625 μL; lane 3, 0.125 μL; lane 4, 0.25 μL of antiserum) or normal rabbit serum (NRS) (lane 5, 0.0625 μL; lane 6, 0.125 μL; lane 7, 0.25 μL), samples were further incubated at room temperature and processed as in RNA gel shift assays. A representative RNA supershift autoradiogram is shown in A. The Δ4 RNA–protein complex (bracket) and free probe (solid arrow) are indicated on the left. The addition (+) of anti-GIIβ or NRS is indicated below the autoradiogram. An increase in anti-GIIβ or NRS concentration in the reaction mix is indicated by triangles above the autoradiogram. Experiments were repeated three times with similar results. (B) In-vivo effects of chronic ethanol exposure on GIIβ polypeptide levels. CC polysomes from control (C) and ethanol-exposed (E) adult mice were separated by SDS–polyacrylamide gel electrophoresis, blotted onto PVDF membrane and probed with anti-GIIβ. Immunoreactive GIIβ bands were visualized using ECL Plus solution as described in Materials and methods. To control for equal loading, membranes were stripped and reprobed with anti-β-actin. (C) Quantitation of GIIβ polypeptide levels was performed using ImageQuantTL software. The signal intensity of the GIIβ-immunoreactive band was divided by the signal intensity of the β-actin band from the same lane and normalized data are expressed as a percentage of control (mean + SEM of three independent experiments). Statistical analysis was performed by ANOVA and Scheffe's test (*p<0.05).
Chronic ethanol exposure increases expression of beta subunit of alpha glucosidase II polypeptide in adult cerebral cortex
Northwestern results exhibited a significant increase in the intensity of the 88 kDa protein band in ethanol-exposed adult CC as compared with control CC polysomes (Fig. 1C and D). To determine whether chronic ethanol treatment has an effect on the polypeptide levels of GIIβ in vivo, western blot analysis was performed using CC polysomes from control and ethanol-treated adult mice. Membranes were probed with anti-GIIβ. To normalize the results, membranes were stripped and reprobed with β-actin antibody. A 88 kDa GIIβ-immunoreactive band was detected in both control and chronic ethanol-exposed adult CC (Fig. 2B). Analysis of the normalized GIIβ-immunoreactive band showed an ∼70% increase in GIIβ polypeptide levels in ethanol-exposed CC as compared with the untreated controls (Fig. 2C).
Identification of second N-methyl-D-aspartate R1 mRNA-binding trans-acting protein
To identify other NR1 mRNA-binding trans-acting proteins, RNA gel mobility shifts were performed using CC polysomes from ethanol-exposed adult animals. The gel areas containing the Δ4 RNA–protein complex were excised and proteins from these gel pieces were recovered. Recovered proteins were separated on SDS–polyacrylamide gel electrophoresis and a colloidal blue-stained 47 kDa band (see Materials and methods for more details) was processed for in-gel digestion with trypsin. Peptide fragments were analyzed by microcapillary reverse-phase HPLC nano-electrospray tandem mass spectrometry. The correlation of MS/MS spectra with known proteins identified eight peptides (Fig. 3A) comprising 27% sequence coverage that matched 100% with the sequence of the protein, AnxA2 (Fig. 3B).
Fig. 3.

AnxA2 – an NR1 3′-UTR-binding trans-acting protein. A 47 kDa protein band in gel obtained using polysomes from ethanol-exposed CC was processed for in-gel trypsin digestion. Tryptic fragments were analyzed by microcapillary reverse-phase HPLC nano-electrospray tandem mass spectrometry and MS spectra were correlated with known proteins using two different programs. (A) Amino acid sequence of peptides that matched 100% to AnxA2 (gi|18645167). (B) Amino acid sequence of murine AnxA2 (339 residues). Shaded boxes with bold letters indicate the amino acid sequence that matched with eight peptides. Two peptides that spanned the same region of AnxA2 are highlighted by a double underline. Comparison of amino acid sequence of eight matched peptides with AnxA2 demonstrated a 27% sequence coverage.
Annexin A2 is an N-methyl-D-aspartate R1 mRNA-binding trans-acting protein
To verify our mass spectrometric results, we performed RNA gel mobility supershift analysis using a polyclonal antibody against AnxA2 (kindly provided by Tony Hunter). Polysomes from ethanol-exposed adult CC were incubated with radiolabeled Δ4 sense RNA as in the RNA gel shift assay. After adding an increasing concentration of anti-AnxA2, samples were further incubated for 20 min at room temperature and then analyzed as for the RNA gel mobility shift assays. A supershift of the Δ4 RNA–protein complex was observed following the addition of anti-AnxA2 to the reaction mix (Fig. 4A). The intensity of the supershift of the Δ4 RNA–protein complex increased with increasing concentrations of anti-AnxA2 with a corresponding decrease in the intensity of the Δ4 RNA–protein complex (Fig. 4A, lanes 2-6). The RNA-binding complex was completely abolished with the maximal concentration of anti-AnxA2 (Fig. 4, lane 6). Similar observations were made for control CC and FCN polysomes (data not shown here). The specificity of the supershift was confirmed by adding normal rabbit serum instead of anti-AnxA2 to the incubation mix. No supershift of the Δ4 RNA–protein complex was observed when increasing concentrations of normal rabbit serum were added to the incubation mix (Fig. 4A, lanes 7-9). These data demonstrated that AnxA2 is indeed a component of the Δ4 RNA–protein complex.
Fig. 4.
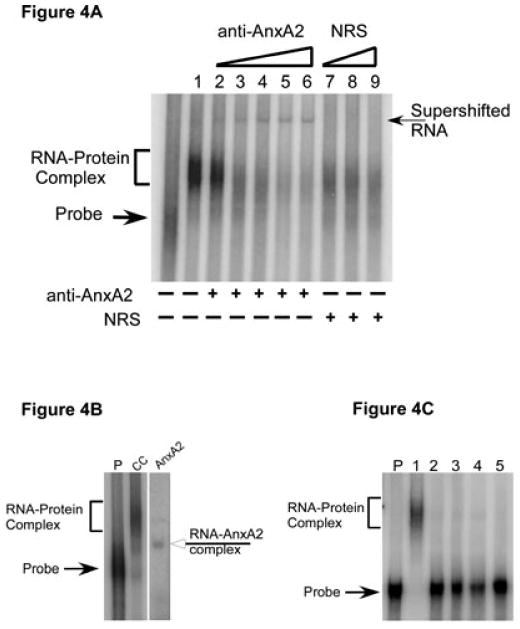
Confirmation of the presence of AnxA2 in Δ4 RNA–protein complex. (A) RNA gel mobility supershift analysis. CC polysomes from ethanol-exposed mice were incubated with 32P-labeled Δ4 sense RNA and then increasing concentrations of either anti-AnxA2 (lane 2, 0.1 μL; lane 3, 0.2 μL; lane 4, 0.3 μL; lane 5, 0.4 μL; lane 6, 0.5 μL) or normal rabbit serum (NRS) (lane 7, 0.3 μL; lane 8, 0.4 μL; lane 9, 0.5 μL) were added. Samples were incubated for an additional 20 min and processed as for RNA gel shift analysis. A representative autoradiogram is shown here. A supershift (arrow on the right) was observed upon addition of anti-AnxA2 (lanes 2-6) but not with normal rabbit serum (lanes 7-9). Free probe and Δ4 RNA–protein complex are indicated on the left. Addition of anti-AnxA2 or normal rabbit serum to the incubation mix is indicated by (+) below the lanes, whereas an increase in the concentration of antibody or normal rabbit serum is indicated by the open triangles above the gel. Experiments were repeated three times with similar results. (B) RNA gel mobility assays. Purified recombinant AnxA2 protein or CC polysomes from control mice were incubated with [32P]-labeled Δ4 sense RNA and samples were processed as detailed in Materials and methods. A shift of Δ4 RNA was observed upon incubation of RNA with CC polysomes (bracket on the left) or full-length recombinant AnxA2 protein (open arrow on the right). Arrow on the left indicates the position of free probe. Note that gel lanes between lane CC and lane AnxA2 have been omitted. Experiments were repeated three times with similar results. (C) Specificity of AnxA2 binding to Δ4 RNA. CC polysomes from control mice (lane 1), increasing concentration of purified recombinant thyroglobulin (lane 2, 0.25 μg protein; lane 3, 0.5 μg protein; lane 4, 1.0 μg protein) and bovine serum albumin (lane 5, 1.0 μg protein) were independently incubated with [32P]-labeled Δ4 sense RNA and samples were processed as detailed in Materials and methods. A shift of Δ4 RNA was observed upon incubation of RNA with CC polysomes (bracket on the left) but not with thyroglobulin or bovine serum albumin. Arrow on the left indicates the position of free probe. Experiments were repeated three times with similar results.
To further confirm the Δ4 RNA–AnxA2 interaction, RNA gel mobility shift assays were performed using purified recombinant AnxA2 protein (kindly provided by David Waisman) and [32P]-labeled Δ4 RNA. CC polysomes from normal adult mouse were included as a positive control for the RNA–protein shift. Two non-specific proteins, thyroglobulin and bovine serum albumin, that do not bind any mRNA were used as negative controls. A shift of Δ4 RNA was observed upon incubation with CC polysomes (Fig. 4B, lane CC) and recombinant AnxA2 (Fig. 4B, lane AnxA2). However, the Δ4 RNA–AnxA2 complex migrated more rapidly as compared with the Δ4 RNA–polysomal protein complex (Fig. 4B). Inclusion of bovine serum albumin or an increasing concentration of thyroglobulin did not produce any shift of Δ4 RNA (Fig. 4C), suggesting that binding of AnxA2 to Δ4 RNA is highly specific.
Heparin disrupts N-methyl-D-aspartate R1 mRNA–protein interaction
Annexin A2 binds to heparin (Kassam et al., 1997) as well as certain mRNAs (Mickleburgh et al., 2005; Hollas et al., 2006). Both the RNA- and heparin-binding domains lie in domain IV of AnxA2 (Kassam et al., 1997; Aukrust et al., 2007). Therefore, we reasoned that heparin may interfere with binding of AnxA2 to Δ4 RNA. This was tested by RNA gel mobility shift assays. Polysomes were incubated with [32P]-labeled Δ4 RNA in the absence or presence of heparin and processed for analysis by RNA gel mobility shift assays. As expected, a shift of Δ4 RNA was observed when polysomes from control (Fig. 5A, lane C) and ethanol-exposed (Fig. 5A, lane E) FCNs were incubated with [32P]-labeled Δ4 RNA in the absence of heparin. In contrast, addition of heparin to the incubation mix prevented the binding of ethanol-exposed CC polysomal proteins to [32P]-labeled Δ4 RNA. No shift of Δ4 RNA was observed. Instead, [32P]-labeled Δ4 RNA degraded into multiple smaller fragments (Fig. 5A, lane H, open arrows on the right). That the lack of Δ4 RNA–polysomal protein complex formation in the presence of heparin was due to binding of heparin to AnxA2 was substantiated by performing additional RNA gel shift assays using purified recombinant AnxA2 protein. Recombinant AnxA2 protein formed a complex with [32P]-labeled Δ4 RNA in the absence (Fig. 5B, lanes 1, 2 and 4) but not in the presence (Fig. 5B, lane 3) of heparin. Interestingly, Δ4 RNA did not degrade upon incubation with purified AnxA2 in the presence of heparin (Fig. 5B, lane 3).
Fig. 5.
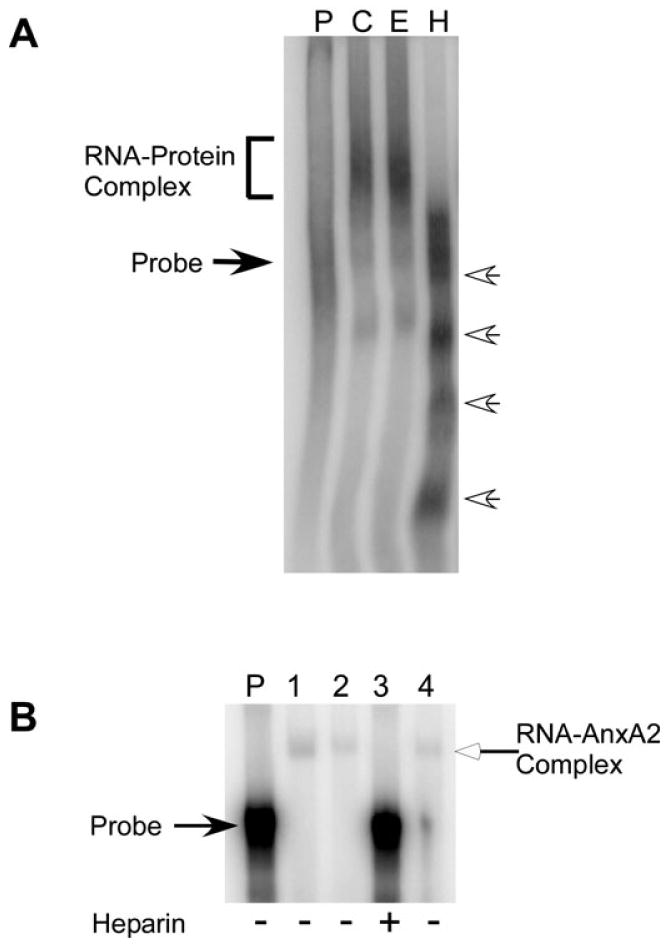
Effect of heparin on Δ4 RNA–protein interactions. (A) Polysomes from control (lane C) and ethanol-exposed FCNs (lane E) were incubated with [32P]-labeled Δ4 sense RNA in the absence of heparin and ethanol-exposed CC polysomes were incubated with [32P]-labeled Δ4 sense RNA in the presence of heparin (H). At the end of the incubation, samples were processed for RNA gel shift analysis and results were analyzed on PhosphorImager. A shift of Δ4 RNA is seen in the absence of heparin (bracket on the left). Degradation of Δ4 RNA in the presence of heparin into smaller fragments is indicated by open arrows on the right. Free probe (lane P) is indicated by solid arrow on the left. Experiments were repeated three times with similar results. (B) Purified recombinant AnxA2 protein was incubated with [32P]-labeled Δ4 sense RNA in the absence (lanes 1, 2 and 4) or presence (lane 3) of heparin. Following incubation, samples were processed for RNA gel mobility shift assays. A shift of Δ4 RNA (indicated by open arrow on the right) is observed in the absence (lanes 1, 2 and 4) but not in the presence (lane 3) of heparin. Location of free probe (lane P) is indicated on the left and the addition of heparin to the incubation mix is indicated by (+) below the lanes. Experiments were repeated twice with similar results.
The cis-acting region, Δ4, within the N-methyl-D-aspartate R1 3′-untranslated region contains mRNA stability element
The RNA–protein interactions play an important role in regulating mRNA stability. Several trans-acting proteins interact with Δ4 sense RNA. As the Δ4 region is the only cis-acting region identified in 3′-UTR to date we tested whether this cis-acting region is important in the ethanol-mediated stability of NR1 mRNA in cultured neurons. Several chimeric constructs were generated using the pGL3 control vector backbone (Fig. 6). The NR1 3′-UTR along with the last 51 nucleotides of the NR1-4 coding region were subcloned in-frame 3′ to the luciferase ORF (pGLNR1-3UTR), whereas the 5′-UTR and first few NR1 exons were subcloned in-frame 5′ to the luciferase ORF (pGL-5UTRNR1). To determine whether NR1 UTRs allow the expression of enzymatically active luciferase, we transiently transfected control FCNs with pGL-5UTRNR1 and pGLNR1-3UTR. The pGL3 basic vector lacking the SV40 promoter served as a negative control and the pGL3 control vector containing the SV40 promoter was the positive control (Fig. 6A). To normalize the transfection results, neurons were co-transfected with pRL-TK vector expressing Renilla luciferase. Transfected cells lysed in passive lysis buffer were cleared by centrifugation to remove the insoluble particulate fraction. A known amount of supernatant protein from each sample was used to measure firefly and Renilla luciferase activities. Interestingly, when FCNs were transfected with pGL-5UTRNR1, firefly luciferase activity was always found in the insoluble fraction and not in the soluble fraction. Therefore, cell lysates from pGL-5UTRNR1-transfected FCNs were not centrifuged before measuring enzyme activities. Normalized data revealed minimal luciferase activity when FCNs were transfected with pGL3 basic vector and the pGL-5UTRNR1 construct (Fig. 6A). The pGL3 basic vector is expected to produce little or no luciferase activity as it lacks the SV40 promoter. However, the negative effect of NR1 5′-UTR on luciferase expression was unexpected. It is possible that the reduced activity occurred as a result of improper folding of the luciferase protein and/or retention in the endoplasmic reticulum. However, the pGL3 control vector containing the SV40 promoter showed maximal activity (Fig. 6A). The chimeric construct containing the NR1 3′-UTR, pGLNR1-3UTR, showed more activity than the pGL-5UTRNR1 construct but less than the positive control, pGL3 control vector (Fig. 6A). These data indicated that the NR1 3′-UTR regulates expression of luciferase-NR1 3′-UTR chimeric mRNA without affecting the enzymatic activity of luciferase. Based upon these data, we utilized pGLNR1-3UTR to test the role of the cis-acting region, Δ4, on ethanol-mediated NR1 mRNA stability. Another chimeric construct, pGLNR1-Δ4, was generated. The pGLNR1-Δ4 is similar to pGLNR1-3UTR except that it lacked the cis-acting region, Δ4.
Fig. 6.
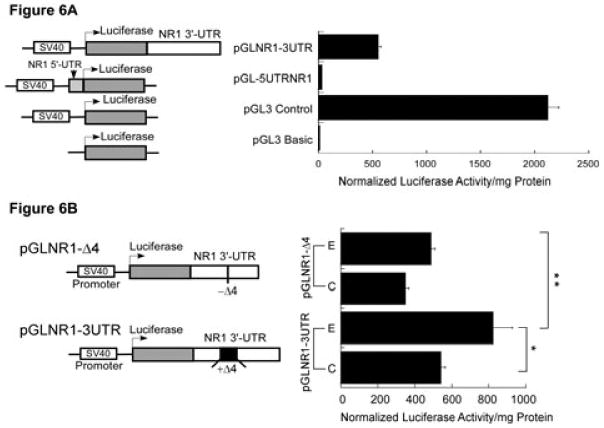
Functional analysis of the Δ4 region in the regulation of NR1 mRNA stability. (A) Effect of NR1 UTRs on luciferase enzyme activity. A schematic representation of the structure of chimeric constructs is shown on the left. Transcription (bent arrow) of the luciferase gene is driven by the constitutive SV40 promoter located upstream of the luciferase gene. The 3′-UTR of NR1 (white box) was subcloned in-frame downstream of the luciferase gene, whereas the 5′-UTR of NR1 (gray box) was subcloned in-frame upstream of the luciferase gene. These chimeric plasmids were independently transfected into FCNs and co-transfected with plasmid pRL-TK. Firefly and Renilla luciferase activities were determined by the dual-luciferase reporter assay system. Firefly luciferase activity was normalized and expressed as normalized luciferase enzyme activity per mg protein (histogram on the right). (B) Effect of the Δ4 region on expression of luciferase activity. A schematic representation of the structure of two chimeric constructs, pGLNR1-3UTR (containing the full-length NR1 3′-UTR including the Δ4 region) and pGLNR1-Δ4 (excluding the Δ4 region in the NR1 3′-UTR), is shown on the left. Control and ethanol-exposed FCNs were independently transfected with pGLNR1-3UTR and pGLNR1-Δ4, and co-transfected with pRL-TK. At 2 days after transfection, cell lysates from transfected FCNs were used to determine firefly and Renilla luciferase activities. Firefly luciferase activity was normalized as above. The histogram on the right shows the normalized firefly luciferase activity per mg soluble cellular protein. Data points represent the mean + SEM of three or more experiments. Statistical analysis was performed by ANOVA and Scheffe's test (*p<0.05, **p<0.01).
The FCNs cultured in the absence or presence of 50 mM ethanol were independently transfected with pGLNR1-3UTR or pGLNR1-Δ4 and co-transfected with pRL-TK vector. At 2 days following transfection, cells were lysed to measure firefly luciferase and Renilla luciferase activities as above and firefly luciferase activity was normalized. Analysis of data showed that, upon transfection with pGLNR1-3UTR, firefly luciferase activity per mg protein was significantly higher in chronic ethanol-exposed FCNs as compared with control FCNs (Fig. 6B). In contrast, deletion of the Δ4 region from the NR1 3′-UTR (pGLNR1-Δ4) significantly reduced the firefly luciferase activity per mg protein in ethanol-exposed FCNs such that the luciferase activity in control FCNs was similar to ethanol-exposed FCNs (Fig. 6B). A comparison of luciferase activity between two ethanol-exposed FCNs (Fig. 6B) showed a significant reduction of luciferase activity in ethanol-exposed FCNs transfected with pGLNR1-Δ4 as compared with ethanol-exposed FCNs transfected with pGLNR1-3UTR. These results indicate that the Δ4 region is an important regulator of ethanol-mediated NR1 mRNA stability.
Discussion
The present study was undertaken to determine whether chronic ethanol-mediated post- transcriptional regulation of NR1 mRNA in adult CC (in vivo) is similar to cultured FCNs and whether the cis-acting region, Δ4, in the 3′-UTR of NR1 mRNA confers stability to NR1 mRNA following chronic ethanol exposure in vitro. Our data demonstrate that NR1 mRNA-binding trans-acting proteins are indeed expressed in the CC. Chronic ethanol exposure in vivo enhanced the binding of trans-acting proteins to the Δ4 RNA and this effect was due to an increase in expression of GIIβ, one of the trans-acting proteins. AnxA2 was identified as a second NR1 mRNA-binding trans-acting protein in vivo and in vitro. Our in-vitro studies implicate interactions between the cis-acting region, Δ4, and trans-acting proteins as an important determinant of the ethanol-mediated regulation of NR1 subunit gene expression.
The NR1 subunit is the functional subunit of the NMDA receptor (Forrest et al., 1994) and the half-life of its mRNA is increased in response to chronic ethanol exposure in vitro (Kumari & Ticku, 1998). Proteins are crucial for the ethanol-mediated stability of NR1 mRNA as inhibition of de-novo protein synthesis in ethanol-exposed FCNs returns the NR1 mRNA half-life to control levels (Kumari et al., 2003). Our previous studies described the presence of a cis-acting region, Δ4, in the 3′-UTR of NR1 mRNA that interacts with three trans-acting proteins expressed in FCNs (Anji & Kumari, 2006). To determine whether these cis–trans interactions observed using FCN polysomes are a phenomenon of neuronal development or are a general regulatory process for NR1 mRNA throughout the life-span of neurons, we conducted experiments using polysomes from adult mouse CC. In the present study, we selected adult mouse CC as the source of cultured fetal neurons was mouse fetal cortices. Incubation of CC polysomes from control mice with [32P]-labeled Δ4 RNA produced a shift of Δ4 RNA, suggesting the presence of NR1 mRNA-binding trans-acting proteins in adult mouse cortex. The Δ4 RNA–CC polysomal protein complex consisted of three trans-acting proteins with an apparent molecular mass of 88, 60 and 47 kDa, respectively. Binding of multiple proteins to a cis-acting region is not unique to NR1 mRNA. For example, three trans-acting proteins, namely hnRNP D, hnRNP I and KSRP, interact with AU-rich elements in the 3′-UTR of human LDL receptor mRNA (Li et al., 2009) and PTH mRNA (Nechama et al., 2009).
Both intrinsic and extrinsic stimuli can increase cis–trans interactions resulting in stabilization of specific mRNAs. For example, epidermal growth factor-mediated binding of trans-acting proteins including poly(C)-binding protein 1 and perhaps HuR to cis-acting elements in the 3′-UTR of p21WAF1 mRNA increase its mRNA stability (Giles et al., 2003). Likewise, binding of trans-acting proteins, HuR and NF90, to U-rich motifs in the 3′-UTR of MAP kinase phosphatase 1 mRNA increases its stability in response to oxidative stress (Kuwano et al., 2008). We examined whether chronic ethanol exposure in vivo enhances the cis–trans interactions for NR1 mRNA. Incubation of CC polysomes from chronic ethanol-exposed mice with [32P]-labeled Δ4 RNA produced a shift of Δ4 RNA similar to control CC polysomes. However, the intensity of the Δ4 RNA–polysomal protein complex in ethanol-exposed CC polysomes was enhanced as compared with control CC polysomes. Although the number of proteins in the Δ4 RNA–CC polysomes complex were identical in the CC of control and chronic ethanol-exposed mice, the intensity of the 88 kDa protein was significantly higher in ethanol-exposed CC as compared with control CC. This result indicated that the 88 kDa protein is perhaps an ethanol-sensitive protein in the adult brain. Previously, we identified an 88 kDa trans-acting protein as GIIβ (Anji & Kumari, 2006). To confirm whether the 88 kDa protein in adult CC is indeed GIIβ, RNA gel mobility supershift assays were performed using anti-GIIβ. A supershift of Δ4 RNA observed upon addition of anti-GIIβ to the incubation mix but not normal rabbit serum confirmed the identity of the 88 kDa protein as GIIβ. These data also demonstrated that the interaction of Δ4 RNA and GIIβ is highly specific. The ethanol sensitivity of GIIβ in adult CC was analyzed by western blotting. A significant increase (70%) in polypeptide levels of GIIβ in the CC of ethanol-exposed mice was detected as compared with the CC from control mice. The GIIβ amino acid sequence analysis does not harbor any previously identified RNA-binding domain(s), suggesting that GIIβ may not belong to any known class of RNA-binding proteins and can be classified as a novel RNA-binding protein. Interestingly, a number of enzymes are known to bind RNA and have been categorized as trans-acting proteins (Hollams et al., 2002).
In the present study, we purified and identified a second NR1 mRNA-binding trans-acting protein by mass spectrometry. Eight peptide fragments obtained by in-gel trypsin digestion showed 27% coverage of the human AnxA2 sequence. Sequence comparison by ‘blastp’ (Altschul et al., 1997; Altschul et al., 2005) showed a 100% match with human (accession no. AAH23990.1) and mouse (accession no. NP_031611.1) AnxA2. The presence of AnxA2 in the Δ4 RNA–CC protein complex was established by RNA gel mobility supershift analysis using rabbit polyclonal AnxA2 antibody. A supershift of Δ4 RNA was observed with anti-AnxA2 but not with normal rabbit serum. These data demonstrated that AnxA2 is specifically bound to Δ4 RNA. The specificity of AnxA2 binding to Δ4 RNA was further confirmed by replacing polysomal proteins with purified recombinant AnxA2 protein or other purified proteins such as recombinant thyroglobulin and bovine serum albumin in the RNA gel mobility shift assay. A shift of Δ4 RNA was seen only with AnxA2 but not with bovine serum albumin or thyroglobulin. The Δ4 RNA–AnxA2 complex migrated more rapidly as compared with the Δ4 RNA–polysomal protein complex. This difference in migration is probably due to the presence of multiple proteins in the Δ4 RNA–polysomal protein complex.
Annexin A2, a known mRNA-binding protein (Filipenko et al., 2004; Mickleburgh et al., 2005; Hollas et al., 2006), is expressed in adult rat brain as well as in cultured cortical neurons (Zhao et al., 2004; Zhao & Lu, 2007). It can exist as a monomer, a heterodimer (one subunit of AnxA2 bound to one subunit of 3-phosphoglycerate kinase) or a heterotetramer (two subunits of AnxA2 linked to two subunits of S100A10) (AIIt) (Waisman, 1995). Both the monomer AnxA2 and the heterotetramer AIIt can interact with c-myc RNA (Filipenko et al., 2004). However, in the case of NR1 mRNA, we found that only the AnxA2 monomer binds to Δ4 RNA. Vedeler & Hollas (2000) isolated AnxA2 from the cellular fraction containing cytoskeleton-bound polysomes. In the same study, they showed that AnxA2 directly binds to mRNAs associated with the cytoskeleton. In this regard, it is important to note that NR1 mRNA in cultured mouse FCNs is always associated with membrane-bound polysomes. Polysomes isolated with our cell fractionation procedure also contain cytoskeletal elements (Kumari et al., 2003).
Annexin A2 protein interacts with a putative RNA-binding motif consisting of five nucleotides, 5′-AA(C/G)(A/U)G (Hollas et al., 2006). This consensus motif is present in the 3′-UTR of AnxA2 mRNA and c-myc mRNA (Hollas et al., 2006). The cis-acting region, Δ4, in the 3′-UTR of NR1 mRNA also contains this consensus motif (present study). Binding of recombinant AnxA2 protein to Δ4 RNA provided evidence that the AnxA2 protein indeed interacts directly with NR1 mRNA. To understand the functional implications of the AnxA2–Δ4 RNA interaction, we included heparin in the RNA gel shift incubation mix. Heparin is a known AnxA2 binding partner (Kassam et al., 1997; Aukrust et al., 2007). The inclusion of heparin not only prevented the formation of the Δ4 RNA–protein complex but also degraded the Δ4 RNA into discrete smaller fragments. As the heparin- and RNA-binding sites lie within domain IV of AnxA2 (Kassam et al., 1997; Aukrust et al., 2007), it is conceivable that heparin inhibited binding of AnxA2 to Δ4 RNA. It is possible that, as a result, the Δ4 RNA was unprotected and became a target of endoribonucleases present in the polysomes. Support for this notion stems from our observation that degradation of Δ4 RNA is not seen when purified AnxA2 protein is utilized for the RNA gel shift assay instead of polysomes. If binding of trans-acting proteins to the cis-acting region of NR1 mRNA is important to prevent its degradation, then deletion of the cis-acting region should affect the half-life of NR1 mRNA. To preclude the detection of endogenous NR1 mRNA, we generated chimeric constructs containing luciferase cDNA and NR1 3′-UTR with and without the Δ4 region and tested them by transient transfection of FCNs. A significant reduction of luciferase activity was observed when the cis-acting region, Δ4, was absent in the chimeric construct. This decrease in luciferase enzyme activity cannot be attributed to altered efficiency of translation of chimeric mRNA as the only difference between the two chimeric mRNAs is the presence or absence of the Δ4 region in the 3′-UTR. Thus, these data strongly suggest that the Δ4 region of NR1 3′-UTR harbors an mRNA stabilizing element as its deletion resulted in decreased reporter gene activity. However, deletion of the Δ4 region did not completely abolish the reporter gene activity, suggesting that the NR1 3′-UTR may contain additional cis-acting elements that are necessary for ethanol-mediated stabilization of the NR1 mRNA. Indeed, we have identified three additional cis-acting regions within the NR1 3′-UTR (unpublished observation). Given that RNA molecules are able to form secondary and tertiary structures, it is possible for cis-acting regions at some distance to interact with each other and alter the mRNA half-life. In fact, several mRNAs are known to contain multiple regulatory elements in their respective 3′-UTRs (Cok & Morrison, 2001; Mawji et al., 2004; Moncini et al., 2007). A well-known example is the α-globin mRNA. Cooperative binding between trans-acting proteins and three cis-acting regions in the 3′-UTR is essential for the stability of α-globin mRNA (Conne et al., 2000). Despite the fact that the Δ4 region appears to require additional regulatory domains to stabilize the NR1 mRNA after ethanol exposure, an important point to note is that, when the Δ4 region is deleted, reporter gene activities in control and ethanol-exposed neurons were not significantly different from each other, lending support to our hypothesis that this cis-acting region within the 3′-UTR of the NR1 mRNA is ethanol-responsive.
In summary, we presented evidence for the expression of NR1 mRNA-binding trans-acting proteins in adult CC. Interaction between the cis-acting region, Δ4, in the 3′-UTR of NR1 mRNA and CC trans-acting proteins is enhanced following chronic ethanol exposure of mice. One of the trans-acting proteins present in CC polysomes is an ethanol-sensitive protein, GIIβ. A second NR1 mRNA-binding trans-acting protein, identified for the first time in the present study, is AnxA2. Binding of these two trans-acting proteins to Δ4 RNA is highly specific. Finally, our studies are the first to present evidence that the Δ4 cis-acting region is an important regulator of NR1 mRNA stability after chronic ethanol exposure.
Supplementary Material
Acknowledgments
We are grateful to Dr Hanne Ostergaard (University of Alberta) and Dr Tony Hunter (Salk Institute) for providing us with the antibodies to GIIβ and AnxA2, respectively. We are also indebted to Dr David Waisman (Dalhouse University) for providing us with recombinant AnxA2 protein. We also thank Dr Waisman and Dr P. Madureira (Dalhouse University) for critical and helpful comments. We thank Mahjabeen Raza, Sarah Kingsley and Barbara Breazeale for excellent laboratory assistance. Support for the present work was provided by NIH-NIAAA 12070 and the Alcoholic Beverage Medical Research Foundation.
Abbreviations
- AnxA2
annexin A2
- CC
cerebral cortex
- FCN
fetal cortical neuron
- GIIβ
beta subunit of alpha glucosidase II
- NMDA
N-methyl-D-aspartate
- NR
N-methyl-D-aspartate R
- SDS
sodium dodecyl sulfate
- UTR
untranslated region
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schaffer AA, Yu YK. Protein database searches using compositionally adjusted substitution matrices. Febs J. 2005;272:5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anji A, Kumari M. A novel RNA binding protein that interacts with NMDA R1 mRNA: regulation by ethanol. Eur J Neurosci. 2006;23:2339–2350. doi: 10.1111/j.1460-9568.2006.04776.x. [DOI] [PubMed] [Google Scholar]
- Anji A, Kumari M. Supplementing the liquid alcohol diet with chow enhances alcohol intake in C57 BL/6 mice. Drug Alcohol Depend. 2008;97:86–93. doi: 10.1016/j.drugalcdep.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anji A, Shaik KA, Kumari M. Effect of ethanol on lipid-mediated transfection of primary cortical neurons. Ann NY Acad Sci. 2003;993:95–102. doi: 10.1111/j.1749-6632.2003.tb07516.x. discussion 123-104. [DOI] [PubMed] [Google Scholar]
- Aukrust I, Hollas H, Strand E, Evensen L, Trave G, Flatmark T, Vedeler A. The mRNA-binding site of annexin A2 resides in helices C-D of its domain IV. J Mol Biol. 2007;368:1367–1378. doi: 10.1016/j.jmb.2007.02.094. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Brewer G, Ross J. Messenger RNA turnover in cell-free extracts. Methods Enzymol. 1990;181:202–209. doi: 10.1016/0076-6879(90)81122-b. [DOI] [PubMed] [Google Scholar]
- Chittum HS, Lane WS, Carlson BA, Roller PP, Lung FD, Lee BJ, Hatfield DL. Rabbit beta-globin is extended beyond its UGA stop codon by multiple suppressions and translational reading gaps. Biochemistry. 1998;37:10866–10870. doi: 10.1021/bi981042r. [DOI] [PubMed] [Google Scholar]
- Cok SJ, Morrison AR. The 3′-untranslated region of murine cyclooxygenase-2 contains multiple regulatory elements that alter message stability and translational efficiency. J Biol Chem. 2001;276:23179–23185. doi: 10.1074/jbc.M008461200. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Draski LJ, Bice PJ, Deitrich RA. Developmental alterations of ethanol sensitivity in selectively bred high and low alcohol sensitive rats. Pharmacology, biochemistry, and behavior. 2001;70:387–396. doi: 10.1016/s0091-3057(01)00621-9. [DOI] [PubMed] [Google Scholar]
- Filipenko NR, MacLeod TJ, Yoon CS, Waisman DM. Annexin A2 is a novel RNA-binding protein. J Biol Chem. 2004;279:8723–8731. doi: 10.1074/jbc.M311951200. [DOI] [PubMed] [Google Scholar]
- Follesa P, Ticku MK. Chronic ethanol treatment differentially regulates NMDA receptor subunit mRNA expression in rat brain. Brain Res Mol Brain Res. 1995;29:99–106. doi: 10.1016/0169-328x(94)00235-7. [DOI] [PubMed] [Google Scholar]
- Giles KM, Daly JM, Beveridge DJ, Thomson AM, Voon DC, Furneaux HM, Jazayeri JA, Leedman PJ. The 3′-untranslated region of p21WAF1 mRNA is a composite cis-acting sequence bound by RNA-binding proteins from breast cancer cells, including HuR and poly(C)-binding protein. J Biol Chem. 2003;278:2937–2946. doi: 10.1074/jbc.M208439200. [DOI] [PubMed] [Google Scholar]
- Hollams EM, Giles KM, Thomson AM, Leedman PJ. MRNA stability and the control of gene expression: implications for human disease. Neurochem Res. 2002;27:957–980. doi: 10.1023/a:1020992418511. [DOI] [PubMed] [Google Scholar]
- Hollas H, Aukrust I, Grimmer S, Strand E, Flatmark T, Vedeler A. Annexin A2 recognises a specific region in the 3′-UTR of its cognate messenger RNA. Biochim Biophys Acta. 2006;1763:1325–1334. doi: 10.1016/j.bbamcr.2006.08.043. [DOI] [PubMed] [Google Scholar]
- Hu XJ, Follesa P, Ticku MK. Chronic ethanol treatment produces a selective upregulation of the NMDA receptor subunit gene expression in mammalian cultured cortical neurons. Brain Res Mol Brain Res. 1996;36:211–218. doi: 10.1016/0169-328x(95)00223-f. [DOI] [PubMed] [Google Scholar]
- Kassam G, Manro A, Braat CE, Louie P, Fitzpatrick SL, Waisman DM. Characterization of the heparin binding properties of annexin II tetramer. J Biol Chem. 1997;272:15093–15100. doi: 10.1074/jbc.272.24.15093. [DOI] [PubMed] [Google Scholar]
- Kumari M. Differential effects of chronic ethanol treatment on N-methyl-D-aspartate R1 splice variants in fetal cortical neurons. J Biol Chem. 2001;276:29764–29771. doi: 10.1074/jbc.M100317200. [DOI] [PubMed] [Google Scholar]
- Kumari M, Anji A, Woods H, Jr, Ticku MK. The molecular effects of alcohol: clues to the enigmatic action of alcohol. Ann NY Acad Sci. 2003;993:82–94. doi: 10.1111/j.1749-6632.2003.tb07515.x. discussion 123-124. [DOI] [PubMed] [Google Scholar]
- Kumari M, Ticku MK. Ethanol and regulation of the NMDA receptor subunits in fetal cortical neurons. J Neurochem. 1998;70:1467–1473. doi: 10.1046/j.1471-4159.1998.70041467.x. [DOI] [PubMed] [Google Scholar]
- Kuwano Y, Kim HH, Abdelmohsen K, Pullmann R, Jr, Martindale JL, Yang X, Gorospe M. MKP-1 mRNA stabilization and translational control by RNA-binding proteins HuR and NF90. Mol Cell Biol. 2008;28:4562–4575. doi: 10.1128/MCB.00165-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chen W, Zhou Y, Abidi P, Sharpe O, Robinson WH, Kraemer FB, Liu J. Identification of mRNA binding proteins that regulate the stability of LDL receptor mRNA through AU-rich elements. J Lipid Res. 2009;50:820–831. doi: 10.1194/jlr.M800375-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawji IA, Robb GB, Tai SC, Marsden PA. Role of the 3′-untranslated region of human endothelin-1 in vascular endothelial cells. Contribution to transcript lability and the cellular heat shock response. J Biol Chem. 2004;279:8655–8667. doi: 10.1074/jbc.M312190200. [DOI] [PubMed] [Google Scholar]
- Mickleburgh I, Burtle B, Hollas H, Campbell G, Chrzanowska-Lightowlers Z, Vedeler A, Hesketh J. Annexin A2 binds to the localization signal in the 3′ untranslated region of c-myc mRNA. Febs J. 2005;272:413–421. doi: 10.1111/j.1742-4658.2004.04481.x. [DOI] [PubMed] [Google Scholar]
- Moncini S, Bevilacqua A, Venturin M, Fallini C, Ratti A, Nicolin A, Riva P. The 3′ untranslated region of human Cyclin-Dependent Kinase 5 Regulatory subunit 1 contains regulatory elements affecting transcript stability. BMC Mol Biol. 2007;8:111. doi: 10.1186/1471-2199-8-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechama M, Ben-Dov IZ, Silver J, Naveh-Many T. Regulation of PTH mRNA stability by the calcimimetic R568 and the phosphorus binder lanthanum carbonate in CKD. Am J Physiol Renal Physiol. 2009;296:F795–800. doi: 10.1152/ajprenal.90625.2008. [DOI] [PubMed] [Google Scholar]
- Snell LD, Nunley KR, Lickteig RL, Browning MD, Tabakoff B, Hoffman PL. Regional and subunit specific changes in NMDA receptor mRNA and immunoreactivity in mouse brain following chronic ethanol ingestion. Brain Res Mol Brain Res. 1996;40:71–78. doi: 10.1016/0169-328x(96)00038-1. [DOI] [PubMed] [Google Scholar]
- Stenger D, Gruissem W, Baginsky S. Mass spectrometric identification of RNA binding proteins from dried EMSA gels. J Proteome Res. 2004;3:662–664. doi: 10.1021/pr049966q. [DOI] [PubMed] [Google Scholar]
- Tian D, Huang D, Brown RC, Jungmann RA. Protein Kinase A Stimulates Binding of Multiple Proteins to a U-Rich Domain in the 3â€2-Untranslated Region of Lactate Dehydrogenase A mRNA That Is Required for the Regulation of mRNA Stability. Journal of Biological Chemistry. 1998;273:28454–28460. doi: 10.1074/jbc.273.43.28454. [DOI] [PubMed] [Google Scholar]
- Vedeler A, Hollas H. Annexin II is associated with mRNAs which may constitute a distinct subpopulation. Biochem J. 2000;348(Pt 3):565–572. [PMC free article] [PubMed] [Google Scholar]
- Waisman DM. Annexin II tetramer: structure and function. Mol Cell Biochem. 1995;149-150:301–322. doi: 10.1007/BF01076592. [DOI] [PubMed] [Google Scholar]
- Yates JR, 3rd, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67:1426–1436. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, Lu B. Expression of annexin A2 in GABAergic interneurons in the normal rat brain. J Neurochem. 2007;100:1211–1223. doi: 10.1111/j.1471-4159.2006.04311.x. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, Waisman DM, Grimaldi M. Specific localization of the annexin II heterotetramer in brain lipid raft fractions and its changes in spatial learning. J Neurochem. 2004;90:609–620. doi: 10.1111/j.1471-4159.2004.02509.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.