

Abstract
The neuropathological hallmark of the majority of amyotrophic lateral sclerosis (ALS) and a class of frontotemporal lobar degeneration is ubiquitinated cytoplasmic aggregates composed of transactive response DNA binding protein 43 kDa (TDP-43). Genetic manipulation of TDP-43 in animal models has been used to study the protein's role in pathogenesis. Transgenic rodents for TDP-43 have recapitulated key aspects of ALS such as paralysis, loss of spinal motor neurons and muscle atrophy. Viral vectors are an alternate approach to express pathological proteins in animals. Use of the recombinant adeno-associated virus vector serotype 9 has permitted widespread transgene expression throughout the central nervous system after intravenous administration. Expressing TDP-43 in rats with this method produced a phenotype that was consistent with and similar to TDP-43 transgenic lines. Increased levels of TDP-43 in the nucleus are toxic to neurons and sufficient to produce ALS-like symptoms. Animal models based on TDP-43 will address the relationships between TDP-43 expression levels, pathology, neuronal loss, muscle atrophy, motor function and causative mechanisms of disease. New targets that modify TDP-43 function, or targets from previous ALS models and other models of spinal cord diseases, could be tested for efficacy in the recent rodent models of ALS based on TDP-43. The vector approach could be an important therapeutic channel because the entire spinal cord can be affected from a one-time peripheral administration.
Keywords: adeno-associated virus, amyotrophic lateral sclerosis, frontotemporal lobar degeneration, gene therapy, gene transfer
Introduction
Advances in the development of animal models expressing neuropathological proteins have led to increased understanding of neurodegenerative disease mechanisms. Rodents are a gateway for drug development to man because of their similar anatomy and well-characterized models. Rodent models are also faster than larger animals such as cats, dogs, pigs and monkeys. Transgenic and knockout manipulation of the germ-line is a gold-standard approach to study disease hallmark pathologies because of the permanent and consistent effect on gene expression levels. Complementary expression systems can offer higher throughput and more rapid and thus cost-effective means to address key questions about pathogenesis. Research on amyotrophic lateral sclerosis (ALS) has historically been focused on familial forms of the disease involving mutations in superoxide dismutase 1 (SOD1). Following the discovery of the role of transactive response DNA binding protein 43 kDa (TDP-43) in ALS and other neurodegenerative diseases, a number of model systems have been used, which were developed during the course of studying other hallmark neuropathological proteins such as amyloid precursor protein, microtubule-associated protein tau, alpha-synuclein, huntingtin and SOD1. Based on the knowledge gained from the earlier research on these proteins, advances on animal models of TDP-43 and the underlying disease mechanisms have evolved quickly. As most neurodegenerative diseases have limited therapeutic options, it is naturally hoped that the rapid advancements in model development will drive towards clinical translation.
TDP-43 is a predominantly nuclear protein with functions related to transcription and RNA processing and transport (Buratti & Baralle, 2008). In 2006, it was discovered that TDP-43 is a major component of the cytoplasmic ubiquitinated inclusions in the neurodegenerative diseases frontotemporal lobar degeneration with ubiquitin-positive inclusions and ALS (Arai et al., 2006; Neumann et al., 2006). The branch of frontotemporal lobar degeneration with TDP-43 pathology has since been renamed FTLD–TDP (Mackenzie et al., 2010). The neuropathological pattern in these diseases involves TDP-43 mislocalization to the cytoplasm, a clearing of nuclear TDP-43, and ubiquitinated and hyperphosphorylated inclusions found throughout the central nervous system (CNS) (Neumann et al., 2006; Geser et al., 2008), although pathological markers may vary depending on the CNS region affected (Igaz et al., 2008; Neumann, 2009; Mackenzie et al., 2010). A recent study implicates deficient nuclear import of TDP-43 leading to its cytoplasmic deposition (Nishimura et al., 2010). TDP-43 can form inclusions within neuronal nuclei, dystrophic neurites and glia in FTLD–TDP subtypes (Neumann et al., 2007; Mackenzie et al., 2010). FTLD–TDP and ALS may share similar mechanisms of pathogenesis involving TDP-43, and both diseases lack efficacious medicine to slow disease progression. ALS is a neuromuscular disease involving the loss of motor neurons and atrophy of the muscles that they innervate, with differential onset of symptoms that can be tied to early degeneration of either brainstem or spinal motor neurons (Eisen, 2009). Familial mutations account for 10% of the ALS population (Van Damme & Robberecht, 2009). The vast majority of ALS research is focused on SOD1 (Rosen et al., 1993; Boillée et al., 2006), although it has been estimated that only 2% of ALS involves mutations in SOD1 (Mackenzie et al., 2007). However, TDP-43 neuropathology is found in both sporadic and familial ALS, estimated to be as much as 98% of ALS (Mackenzie et al., 2007). An enormous wealth of resources and knowledge has accrued from studying familial SOD1 mutations (Boillée et al., 2006), although there remains much frustration in terms of translational value. Because the vast majority of ALS is now known to involve some degree of TDP-43 pathology, research on this protein may be more broadly relevant to ALS.
Familial mutations in TDP-43 associated with ALS (Gitcho et al., 2008; Sreedharan et al., 2008) suggest either loss of function or toxic gain of function as possible pathogenic mechansims. Yeast (Johnson et al., 2008), Caenorhabditis elegans (Ash et al., 2010; Liachko et al., 2010), Drosophila (Li et al., 2010), zebrafish (Kabashi et al., 2010) and chick embryo (Sreedharan et al., 2008) models have recapitulated the molecular and cellular aspects of FTLD–TDP and ALS in order to address the key question of TDP-43 function/dysfunction in disease (Gendron et al., 2010). These animal models, complemented by cell culture models (e.g. Caccamo et al., 2009; Nonaka et al., 2009; Zhang et al., 2009; Barmada et al., 2010), offer rapid screening of drugs and gene targets to address the mechanisms. The logical next step from any of these high-throughput systems is translation to a mammalian system, usually in a rodent or a non-human primate model. Due to their relevant neuroanatomy and myoanatomy, and well-characterized behavioral and toxicological paradigms, mice and rats are essential for proof of concept discoveries on TDP-43 function as they relate to disease.
TDP-43 overexpression in rodents via germ-line manipulation
The breakthrough study by Neumann et al. (2006) established TDP-43 as a neuropathological substrate protein in FTLD–TDP and ALS, and sparked efforts to generate transgenic mice based on this protein (Wegorzewska et al., 2009; Shan et al., 2010; Stallings et al., 2010; Tsai et al., 2010; Wils et al., 2010; Xu et al., 2010; Zhou et al., 2010; Igaz et al., 2011). Different promoter strategies have been used to drive expression, e.g. ubiquitous expression with the prion promoter (Wegorzewska et al., 2009; Stallings et al., 2010; Xu et al., 2010), specific neuronal expression with the thymus cell antigen 1 promoter (Shan et al., 2010; Wils et al., 2010) or conditional expression with the forebrain specific calcium–calmodulin-dependent kinase II (CaMKII) promoter (Tsai et al., 2010; Igaz et al., 2011). A consensus of motor effects and morbidity and mortality has generally resulted from TDP-43 overexpression despite the different promoter strategies, form of TDP-43 used and the degree of TDP-43 pathology, underscoring great sensitivity to changes in TDP-43 levels and functionality (Table 1). ALS and FTLD–TDP are separated in Table 1, which is an oversimplification because there are both spinal cord and brain effects in the animal models and in disease (Geser et al., 2008).
Table 1.
Rodent models of ALS and FTLD–TDP based on TDP-43
| ALS | Wegorzewska et al. (2009) | Wils et al. (2010) | Shan et al. (2010) | Xu et al. (2010) | Zhou et al. (2010) | Stallings et al. (2010) | Wang et al. (2010) | FTLD–TDP | Tsai et al. (2010) | Igaz et al. (2011) |
|---|---|---|---|---|---|---|---|---|---|---|
| Promoter | PrP | Thy-1 | Thy-1.2 | PrP | miniCMV/tetO | PrP | CBA | Promoter | CaMKII | CaMKII/tetO |
| TDP-43 form | A315T | WT | WT | WT | M337V | A315T | WT | TDP-43 form | WT | WT, ΔNLS |
| Onset of symptoms | 3 months | 0.5–14 months | 0.5–3 months | 3 weeks | 3 weeks | 1 month | 2 weeks | Onset of symptoms | 2–6 months | 1–4 weeks |
| TDP-43 pathological modifications | No | Yes | No | Yes | Yes | Yes | No | TDP-43 pathological modifications | Yes | Yes |
| TDP-43-positive inclusions | no | yes | no | yes | Rare | Rare | no | TDP-43 positive inclusions | yes | Rare |
| TDP-43 fragments | Yes | Yes | no | Yes | Yes | Yes | ND | TDP-43 fragments | Yes | no |
| Motor neurodegeneration | Yes | Yes | no | Yes | ND | Yes | Yes | Forebrain neurodegeneration | Yes | Yes |
| Muscle atrophy | Yes | ND | Yes | No | Yes | Yes | Yes | Cognitive impairment | Yes | ND |
| Motor impairment | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Motor impairment | Yes | Yes |
| Mortality | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Mortality | Yes | No |
Models of ALS have transgene expression in the spinal cord, which results in paralysis, motor neurodegeneration (either spinal motor neuron loss or corticospinal tract degeneration) or muscle atrophy. FTLD–TDP models have TDP-43 expression predominantly in the forebrain, resulting in cognitive dysfunction and neurodegeneration in the cortex or hippocampus. TDP-43 pathological modifications refer to either ubiquitination or phosphorylation, both of which were found in Zhou et al. (2010). PrP, prion protein; Thy, thymus cell antigen; CMV, cytomegalovirus; tetO, tetracycline operator; CBA, cytomegalovirus/chicken beta-actin; ND, not determined; NLS, nuclear localization signal; WT, wild-type.
The first transgenic TDP-43 mice reported in the literature utilized the A315T mutant form of human TDP-43 linked to familial ALS, whereas wild-type TDP-43 mice were embryonic lethal in that study (Wegorzewska et al., 2009). The mutant TDP-43 mice displayed several salient features of ALS, such as limb paralysis and muscle wasting over 3–9 months, and electromyographic traces from the hindlimb consistent with denervation. There was selective ubiquitination in layer V of the cortex and in the spinal cord, which could have included upper and lower motor neurons, although disease-like cytoplasmic hyperphosphorylated aggregates of TDP-43 were not detected. However, there was evidence of disease-relevant TDP-43 protein fragments. The mice used by Wegorzewska et al. (2009) are a milestone in the progress on TDP-43′s role in ALS, and many of their features have been reproduced in other TDP-43 mice and rats. Wils et al. (2010) were able to generate wild-type TDP-43 mice, with several lines with different levels of overexpression. This yielded time- and gene-dosage-dependent effects of TDP-43 to induce functional motor disease states, with more rapid, similar or slower time courses relative to the A315T mice (Wegorzewska et al., 2009) depending on the TDP-43 level. The two studies shared many similar outcomes of paralysis, morbidity, mortality, motor neuron loss, gliosis and, notably, similar ubiquitination in the cortex and spinal cord. The study of Wils et al. (2010) showed evidence of apparent proteolytic cleavage of TDP-43 that corresponded with the severity of the disease state, and disease-relevant cytoplasmic and nuclear inclusions of hyperphosphorylated TDP-43 (Arai et al., 2006, 2010; Neumann et al., 2006), which were not found in the mice used by Wegorzewska et al. (2009). TDP-43 phosphorylation was determined with phospho-specific antibody, and cytoplasmic aggregates have been detected in several studies by this method (Stallings et al., 2010; Xu et al., 2010; Zhou et al., 2010; Igaz et al., 2011). Regarding the relevant recapitulation of TDP-43 proteinopathy in glial cells (Nishihira et al., 2008; Zhang et al., 2008), several transgenic models have used neuron-specific promoters, such as thymus cell antigen 1 and CaMKII (Tsai et al., 2010; Wils et al., 2010; Igaz et al., 2011), whereas one study using the ubiquitous prion protein promoter reported an increase in nuclear, but not cytoplasmic, TDP-43 within glia (Wegorzewska et al., 2009).
Two studies modeled FTLD–TDP by overexpressing TDP-43 with the CaMKII promoter, which is active in the hippocampus, striatum and cortex but not in other brain regions such as the cerebellum and spinal cord (Tsai et al., 2010; Igaz et al., 2011). The CaMKII promoter is also developmentally regulated, which is advantageous to avoid embryonic lethality. In the study of Tsai et al. (2010), TDP-43 mice were deficient in multiple memory tasks by 2 months, concomitant with electrophysiological changes in long-term potentiation and miniature excitatory postsynaptic currents, and neuronal loss in the hippocampus. Surprisingly, motor impairments developed in these mice by 6 months of age, similar to other TDP-43 transgenic mice using different promoters, suggesting that damage to upper motor neurons and the corticospinal tract contributes to the motor pheno-types, as no spinal motor neuron loss or spinal TDP-43 expression was observed. The study of cognitive dysfunction is relevant to frontotemporal lobar degeneration (Chen-Plotkin et al., 2010; Geser et al., 2010) as well as ALS (Abe et al., 1997). However, in TDP-43 transgenic mice with spinal expression, damage to the motor system renders memory-related tasks unfeasible. Furthermore, the sensitivity of the assays of cognitive function may not be able to detect subtle changes, although memory-related performance was impaired when TDP-43 expression was limited to the forebrain (Tsai et al., 2010). Another way of studying the effects of TDP-43 on memory-related behaviors would be to target expression to specific brain regions such as the hippocampus with vector gene transfer, which we have been attempting.
The study of Igaz et al. (2011) utilized a CaMKII/tetracycline operator promoter system, whereby forebrain expression of TDP-43 was induced when mice were withdrawn from the drug doxycycline. The authors used this to compare the effects of human TDP-43 with a defective nuclear localization signal (resulting in cytoplasmic expression) or wild-type TDP-43 (nuclear expression) without developmental effects. Both forms of TDP-43 induced relevant FTLD–TDP neuropathology and degeneration. However, the mutated TDP-43 that forced cytoplasmic expression was surprisingly unable to potentiate disease-relevant pathology in terms of increased cytoplasmic inclusions. Furthermore, the cytoplasmic TDP-43 expression exacerbated the depletion of endogenous mouse TDP-43, which correlated with neuron loss and motor dysfunction. Simultaneous and consistent with the loss of endogenous mouse TDP-43, there were gene expression changes in chromatin assembly pathways. The mice used by Igaz et al. (2011) are unique for the conditional TDP-43 expression pattern, and offer two distinct mechanisms of TDP-43 toxicity, in either the nucleus or cytoplasm, that can be studied.
Mitochondrial dysfunction has been implicated in the neurodegenerative mechanism of TDP-43 (Shan et al., 2010; Xu et al., 2010), based on research on ALS and transgenic SOD1 mice (Boillée et al., 2006). The TDP-43 mice in Xu et al. (2010) showed abnormal clustering of degenerating mitochondria that formed aggregates in the spinal motor neurons, concomitant with changes in expression of specific mitochondrial gene products. These mice also had an overall motor phenotype similar to the TDP-43 mice used by Wegorzewska et al. (2009) and Wils et al. (2010). Utilizing a double transgenic strategy to visualize mitochondria, TDP-43 mice in Shan et al. (2010) also demonstrated mitochondrial clustering in neuron cell bodies coinciding with depletion of mitochondria in neuromuscular junction nerve terminals, which could have been related to their motor dysfunction. This study reported an increase in and altered localization of Gemini coiled bodies, which are nuclear structures involved in premessenger RNA splicing. There were also specific changes in gene expression, such as decreases in neurofilament proteins that could have been related to the axonopathy that was found in the ventral roots of the spinal cord. It is of note that knockdown of TDP-43 expression within this study blocked the formation of Gemini bodies in the nucleus. The onset and severity of motor deficits were more rapid and pronounced in the males of this TDP-43 transgenic line, coinciding with increased TDP-43 accumulation in their spinal cords.
Another emerging theme is the loss of endogenous mouse TDP-43 expression when human TDP-43 is overexpressed. The TDP-43 mice from Xu et al. (2010) and those from Igaz et al. (2011) showed loss of endogenous mouse TDP-43 mRNA and protein, respectively, which could have relevance in terms of the loss of TDP-43 staining in the nucleus in disease samples (Neumann et al., 2006, 2007) and transgenic models (Wegorzewska et al., 2009; Wils et al., 2010). The mice may be downregulating their own TDP-43 levels (Ayala et al., 2011) in the face of overexpression, or cytoplasmic TDP-43 aggregates could be recruiting nuclear TDP-43 to the cytoplasm (Caccamo et al., 2009; Nonaka et al., 2009).
The functional impact of inherited familial mutations in TDP-43 has been studied using transgenic mice. Results from yeast (Johnson et al., 2008), C. elegans (Liachko et al., 2010), zebrafish (Kabashi et al., 2010), chick embryos (Sreedharan et al., 2008) and cell culture (Barmada et al., 2010) have implicated the specific pathogenicity of familial mutations such as the M337V and A315T TDP-43 mutations. Similar conclusions have been reached in TDP-43 transgenic mice with either the A315T or M337V TDP-43 mutations (Stallings et al., 2010) and in TDP-43 transgenic rats using the M337V TDP-43 mutation (Zhou et al., 2010). However, both wild-type (Wils et al., 2010; Xu et al., 2010) and mutant TDP-43 (Wegorzewska et al., 2009; Zhou et al., 2010) have led to comparably robust ALS-relevant phenotypes.
Transgenic rats for mutant TDP-43 manifest an ALS-relevant motor phenotype with paralysis, motor neuron loss and muscle atrophy (Zhou et al., 2010), so the consequence of overexpressing TDP-43 is shared in mice and rats. A `tet-off' promoter system was used to avoid overexpression during embryogenesis. This model yielded unique evidence for the direct ubiquitination of TDP-43 by co-immunoprecipitation, not reported in TDP-43 mice. Rat models are particularly advantageous for fine anatomical manipulations, and specific behavioral and toxicological assays designed for rats, such as food-reward paradigms.
A variety of germ-line strategies have been employed to express mutant and wild-type TDP-43 in rodents involving different regulatable or conditional promoters, achieving TDP-43 expression in select tissues and cell types and at certain ages (Table 1). Despite the different strategies, increasing TDP-43 levels in the nucleus results in consistent functional outcomes such as paresis/paralysis and morbidity (Wegorzewska et al., 2009; Shan et al., 2010; Stallings et al., 2010; Tsai et al., 2010; Wils et al., 2010; Xu et al., 2010; Zhou et al., 2010; Igaz et al., 2011), with several studies showing modest loss of spinal motor neurons (Wegorzewska et al., 2009; Wils et al., 2010; Zhou et al., 2010) and muscle wasting (Wegorzewska et al., 2009; Stallings et al., 2010; Zhou et al., 2010). Despite similar behavioral outcomes, some of the main differences among the lines involve TDP-43 inclusion formation and apparent proteolytic processing (Wegorzewska et al., 2009; Shan et al., 2010; Igaz et al., 2011), which suggest that these features are non-essential for the phenotype of motor paralysis.
TDP-43 knockout mice
Loss of TDP-43 function may play a role in disease pathogenesis due to the clearing of nuclear TDP-43 that occurs concomitantly with cytoplasmic inclusions. TDP-43 knockout flies in the study of Feiguin et al. (2009) had motor impairment and defects in motor neuron synapses, which could be rescued by the expression of human TDP-43. An antisense oligonucleotide strategy in zebrafish reduced TDP-43 levels and induced a swimming impairment, similar to that seen when TDP-43 was overexpressed (Kabashi et al., 2010). That a similar motor phenotype could be caused in zebrafish by either TDP-43 knockdown or TDP-43 overexpression suggests that loss of TDP-43 function could underlie both disease and, paradoxically, the phenotype when TDP-43 is overexpressed in animals. Studying the loss of TDP-43 function in mice has been challenging due to embryonic lethality, and has underscored the great sensitivity to altering levels of TDP-43 in rodents. Homozygous knockouts are consistently embryonic lethal (Kraemer et al., 2010; Sephton et al., 2010; Wu et al., 2010), which suggests a critical function during embryogenesis that lacks redundancy with other ribonucleoproteins. However, heterozygous mice do not show any level of TDP-43 knockdown, consistent with TDP-43 being able to regulate its own levels (Ayala et al., 2011). The TDP-43 heterozygous knockouts from Kraemer et al. (2010) had muscle weakness after 1 year, although there was no evidence of neuronal loss or muscle atrophy. To permit the study of homozygous knockout in mice, Chiang et al. (2010) employed a loxP/Cre recombinase system to create conditional knockout mice when the drug tamoxifen was given. Homozygotes had a drastic decrease in TDP-43 expression that led to weight loss and death, and concomitant dysregulation of genes involved with obesity. Modeling the loss of TDP-43 function in rodents is limited by the embryonic lethality and tight regulation of TDP-43 levels, and has yet to produce ALS-related sequelae such as loss of motor neurons or muscle atrophy.
TDP-43 overexpression in rats via somatic cell gene transfer strategy
The viral vector overexpression of neuropathological proteins is also used to mimic salient features of neurodegenerative diseases in rodents or non-human primates (e.g. Senut et al., 2000; Kirik et al., 2002, 2003; Klein et al., 2002, 2004). The injection of a gene vector can target disease-relevant areas, which was advantageous for overexpression of the Parkinson's disease-related protein alpha-synuclein (Kirik et al., 2002; Klein et al., 2002; Lo Bianco et al., 2002) because, overall, the transgenic mice do not exhibit disease-relevant loss of dopamine neurons in the substantia nigra (Chesselet, 2008). Adeno-associated virus (AAV) is a single-stranded DNA virus that is not attributed to a specific known disease or pathology (Daya & Berns, 2008). The recombinant vectors based on AAV express no viral genes and are therefore considered to be relatively safe. AAV vectors have been administered to humans in clinical research for Parkinson's disease using several different gene targets (Kaplitt et al., 2007; Christine et al., 2009; Marks et al., 2010). Because AAV vectors are efficient for expressing specific genes in neurons without causing nonspecific side effects due to the vector, they have become a key tool in basic neuroscience research.
Compared with germ-line mice, vector-based models do not produce the same level of expression and are more subject to variability due to gene transfer efficiency in each individual subject. However, stable long-term expression levels and reproducible results have demonstrated sufficient consistency to address hypotheses that would take longer and cost more in germ-line transgenics to answer similar questions. Gene variants could be quickly screened and compared in a rodent without having to generate, maintain and genotype transgenic lines. Improved targeting, facility for gene combinations and the presence of within-subject non-transduced internal controls are some of the unique advantages of the vector approach. The ability to turn on expression in specific CNS regions gives the experimenter control of optimizing the expression and disease relevance, as well as avoiding embryonic lethality. If the vector technique can be sufficiently consistent in producing ALS-like symptoms, it could be more cost-effective than transgenic lines, given the advent of intravenous gene delivery with the AAV type 9 vector, which widely transduces the spinal cord and brain (Foust et al., 2009).
Tatom et al. (2009) studied focal injections of an AAV9 human TDP-43 vector to the substantia nigra of rats, a brain region with TDP-43 pathology and/or neuron loss in several neurodegenerative diseases with parkinsonian symptomatology (Leverenz et al., 2007; Geser et al., 2008; Zhang et al., 2008; Wider et al., 2009). The AAV9 TDP-43 produced potent toxicity to the dopamine neurons that was vector dose-dependent, along with a progressive effect on rotational behavior over time. The focal vector injections thus produced a highly potent and consistent assay of TDP-43-induced neurodegeneration and behavioral deficit with disease relevance in the rat. The human TDP-43 expression was mainly nuclear, and estimated to be threefold higher than endogenous rat TDP-43 levels, so as in other TDP-43 models, small alterations in TDP-43 levels in the nucleus were toxic to neurons. There were examples of cytoplasmic TDP-43 deposition and cytoplasmic ubiquitination in the TDP-43 vector group, but they were infrequent and marginal compared with the massive gliosis and neuronal loss that occurred in the TDP-43 transduced substantia nigra. Although TDP-43 is relevant in regard to the substantia nigra, the focal vector approach seemed limited for the transduction of upper and lower motor neurons in relation to ALS. The widespread TDP-43 pathology (Geser et al., 2008) in ALS could be mimicked with the widespread expression in germ-line transgenics (e.g. Wegorzewska et al., 2009), but results with vectors until now could not generate similar widespread expression (e.g. Foust et al., 2008b).
That changed when Foust et al. (2009) demonstrated green fluorescent protein expression throughout the mouse CNS with intravenous AAV9 vector administration. By injecting neonatal mice, a still-developing blood–brain barrier allows vector entry to the CNS that is more difficult to achieve in older subjects. Wang et al. (2010) translated this technique to rats to overexpress TDP-43, which produced paresis and paralysis, an overall functional phenotype common to the TDP-43 transgenic mice and rats. The spread of green fluorescent protein expression in the CNS of rats was unprecedented, visualized by biophotonic imaging in Fig. 1A. The TDP-43 rats had progressive weight loss, morbidity and mortality relative to green fluorescent protein rats, with modest loss of spinal motor neurons, but remarkably severe muscle atrophy (Fig. 1B) and hindlimb paralysis. As cytoplasmic TDP-43 neuropathology was not found in these rats, the data again implicate the toxicity of nuclear TDP-43 overexpression. The hindlimb paralysis was quantified by rotarod and analysis of rearing behavior, and both readouts were highly consistent, demonstrating that the behavioral phenotype for TDP-43-induced paralysis is reproducible and comparable to data from transgenic mice. Although consistent, the onset of the disease state was rapid within a few weeks, so controlling expression to the adult onset of symptoms will be more relevant to ALS. Other refinements will be more selective expression in the CNS and motor neurons, as the intravenous AAV9 injections also caused peripheral expression, which also occurs in TDP-43 transgenic mice using the prion promoter (e.g. Xu et al., 2010). The corticospinal tract of rodents and primates varies because, in rodents, it is located in the dorsomedial spinal cord, whereas in primates it is in the lateral spinal cord (Watson & Harvey, 2008). Functionally, it is also more of a pure motor pathway in primates compared with rodents (Watson & Harvey, 2008). A non-human primate model might be essential for greater relevance, which can be achieved via vector gene transfer. Apart from the utility in modeling neurodegenerative diseases through the overexpression of neurotoxic proteins, viral vectors also have therapeutic potential. Vector gene transfer has been the best method to deliver therapeutic proteins in a stable manner to the CNS, and recent advancements of widespread CNS transduction using a one-time peripheral injection have broadened the scope for affecting large portions of the spinal cord, and thus spinal cord diseases.
Fig. 1.
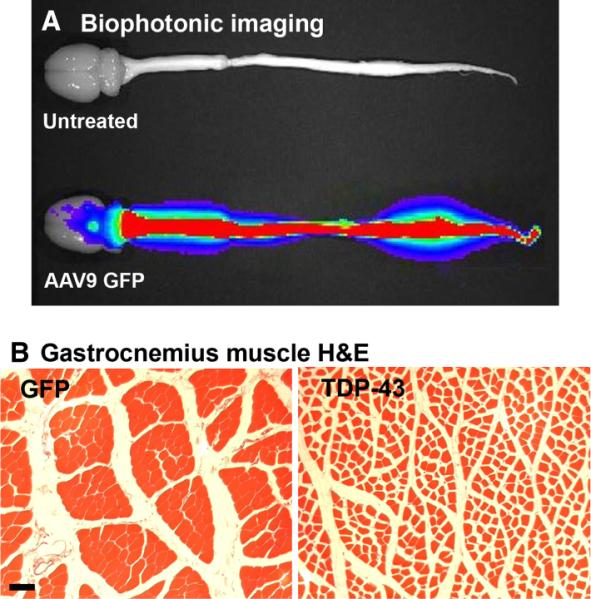
Peripheral intravenous injections of an AAV9 vector to neonatal rats led to widespread CNS expression of green fluorescent protein (GFP) or TDP-43. (A) GFP expression viewed by biophotonic imaging at 12 weeks of age. There is robust expression in the spinal cord of the GFP rat, but not in an age-matched uninjected control. The level sets of photonic emission are reflected by the color scheme, i.e. red denotes greater levels of GFP epifluorescence, and blue denotes lower levels. (B) Hematoxylin and eosin stain from the gastrocnemius muscle of a GFP rat at 4 weeks (left panel). AAV9 TDP-43 caused uniform shrinkage of myofibers indicative of widespread denervation (right panel). Bar: 67 μm for both panels in B. Reprinted from Wang et al. (2010).
Preclinical gene therapy in the spinal cord
Amyotrophic lateral sclerosis is a candidate for gene therapy because it is life threatening and without cure, and because there are forms of the disease with genetic etiology. To counteract the neurodegeneration in ALS, the whole spinal cord may need to be treated on a long-term basis. Several strategies have been used for AAV gene delivery to the spinal cord (Fig. 2), with recent efforts succeeding for widespread and long-term spinal transduction. The discussion is centered on data with AAV, although similar strategies have been used with lentivirus vectors (Azzouz et al., 2004; Ralph et al., 2005; Raoul et al., 2005) or adenovirus vectors (Bordet et al., 2001; Acsadi et al., 2002; Manabe et al., 2002; Yamashita et al., 2002) for gene transfer in mutant SOD1 mice. Peel et al. (1997) used an AAV2 vector to achieve green fluorescent protein expression in the spinal cord via direct injections into the cervical spinal cord. Azzouz et al. (2000) used this route of administration and demonstrated neuroprotection in mutant SOD1 mice using an AAV vector for the antiapoptotic gene B-cell lymphoma 2. Other studies have shown neuroprotection using the growth factors insulin-like growth factor 1 (Lepore et al., 2007; Franz et al., 2009) and granulocyte-colony-stimulating factor (Henriques et al., 2011) via spinal injections. Dodge et al. (2008) injected an insulin-like growth factor 1 vector into the deep cerebellar nuclei, a brain region with extensive spinal projections, resulting in insulin-like growth factor 1 expression throughout the spinal cord. Secretable peptidergic factors can spread beyond the area of the transduced cells to exert more widespread effects.
Fig. 2.

Routes of AAV vector administration for spinal cord transduction. CNS administrations target injections directly to CNS tissues or the cerebrospinal fluid (red). Intraparenchymal injections can be into spinal cord (PSC) tissue, or in brain tissues that have axonal projections into the spinal cord (PB). Injections can be into the intrathecal cavity (T) or the cerebral ventricles (CV), which can enhance vector spread in cerebrospinal fluid. Peripheral routes of administration lead to transduction of motor neurons: intramuscular (M) or intravenous (V) injections (black). Intravenous injections efficiently transduce neurons throughout the brain and spinal cord, although the approach is limited by having to use very young subjects. Modified vectors that can cross the mature blood–brain barrier (Gray et al., 2010) or the use of mannitol, which relaxes the blood–brain barrier (McCarty et al., 2009), could make this route more applicable in adults. Certainly some of these approaches transduce brain neurons in addition to spinal cord neurons (e.g. Foust et al., 2008a, 2009; Passini et al., 2010), although the figure highlights strategies to affect spinal cord diseases.
Different routes of administration that avoid direct injections to the spinal cord parenchyma have been developed (Fig. 2), because it would be beneficial to affect the entire spinal cord without having to inject it. Kaspar et al. (2003) successfully delivered a vector for insulin-like growth factor 1 to muscle, relying on vector uptake in the axons of innervating spinal neurons, to treat SOD1 mice. The spread of spinal cord neuronal transduction was limited, but the secretable growth factors could have spread further. The efficiency of transduction of motor neurons after intramuscular administration has improved with newer types of the AAV vector (Towne et al., 2010). Intracerebroventricular vector administration leads to more widespread spinal transduction (Dodge et al., 2010; Passini et al., 2010), and this route of gene delivery was successful in extending lifespan in a mouse model of spinal muscular atrophy by expressing and thereby rescuing the deficiency in the survival motor neuron protein (Passini et al., 2010). An intravenous route of administration could be preferable because it allows for widespread gene transfer derived from a peripheral administration. An early study administering AAV intravenously to neonatal mice was effective in correcting a model of lysosomal storage disease in the brain (Daly et al., 1999), although transgene expression in the spinal cord was not discussed. Intravenous AAV8 vector delivery to mice led to limited spinal cord and brain expression previously (Foust et al., 2008b). However, the AAV9 vector resulted in more widespread spinal cord expression after intravenous delivery to neonatal mice (Foust et al., 2009), rats (Wang et al., 2010) and macaques (Foust et al., 2010), and adult cats and mice (Duque et al., 2009; Foust et al., 2009). The recent consistent success of intravenous gene delivery of survival motor neuron protein to spinal muscular atrophy mice (Bevan et al., 2010; Foust et al., 2010; Valori et al., 2010; Dominguez et al., 2011) has provided key preclinical data necessary for human translation. A neonatal approach would be limited in order to treat a patient with symptoms. However, some of the data suggest that this approach can efficiently affect the adult spinal cord (Duque et al., 2009), and transduction in adults can be bolstered by temporary disruption of the blood–brain barrier with mannitol (McCarty et al., 2009). Further research will determine if AAV9 is unique in its ability to widely transduce the CNS compared with other natural vector serotypes. However, engineered versions of the vector could improve the gene transfer to adults (Gray et al., 2010).
Several transgene targets have been efficacious in the SOD1 model of ALS using a variety of AAV administration routes in terms of extension of lifespan, phenotypic delay, preservation of spinal motor neurons and reduction of neuropathology (Table 2). AAV strategies have also been used for other spinal cord diseases, such as spinal muscular atrophy, spinal cord injury and neuropathic pain (Table 2). For example, gene delivery of survival motor neuron protein increased lifespan and motor function, and decreased muscle atrophy in an spinal muscular atrophy model (Passini et al., 2010), and gene delivery of neurotrophic factors increased motor function and motor neuron survival in spinal cord injury models (Blits et al., 2003, 2004). It is possible that a secreted growth factor that was efficacious in a mutant SOD1 model could also work in a TDP-43 model if there are shared mechanisms of spinal neurodegeneration. There could also be a mechanistic overlap of deficiency of survival motor neuron protein with ALS (Piao et al., 2011), mutant SOD1 models of ALS (Turner et al., 2009) and TDP-43 models of ALS. As the field of research on TDP-43 develops, the emergence of proteins that can degrade or detoxify pathological TDP-43 will provide new therapeutic targets. The ability to affect the entire spinal cord from a peripheral route of administration will enhance the development of therapeutic strategies in ALS rodent models based on TDP-43, as well as in a variety of other spinal cord disease paradigms.
Table 2.
Preclinical strategies in rodent models of ALS and other spinal cord diseases using AAV vector gene transfer
| Disease | Transgene | Route of administration | Vector | References |
|---|---|---|---|---|
| ALS (SOD1) | Bcl2 | Spinal parenchyma | AAV | Azzouz et al. (2000) |
| ALS (SOD1) | GDNF | Forelimb and hindlimb muscles | AAV2 | Wang et al. (2002) |
| ALS (SOD1) | IGF-1 | Hindlimb and intercostal muscles | AAV2 | Kaspar et al. (2003, 2005) |
| ALS (SOD1) | IGF-1 | Spinal parenchyma | AAV2 | Franz et al. (2009), Lepore et al. (2007) |
| ALS (SOD1) | IGF-1 | Deep cerebellar nuclei | AAV1,2 | Dodge et al. (2008) |
| ALS (SOD1) | IGF-1, VEGF | Ventricles | AAV4 | Dodge et al. (2010) |
| ALS (SOD1) | G-CSF | Spinal parenchyma | AAV1/2 | Henriques et al. (2011) |
| SMA | SMN1 | Intravenous | AAV9 | Bevan et al. (2010), Dominguez et al. (2011), Foust et al. (2010), Valori et al. (2010) |
| SMA | SMN1 | Ventricles and spinal parenchyma | AAV8 | Passini et al. (2010) |
| SCI | BDNF, NT-3, GDNF | Spinal parenchyma | AAV2 | Blits et al. (2003, 2004) |
| SCI | L1 | Spinal parenchyma | AAV5 | Chen et al. (2007) |
| SCI | VEGF, Ang-1 | Spinal parenchyma | AAV8 | Herrera et al. (2010) |
| SCI | NT-3 | Forelimb muscles | AAV5 | Fortun et al. (2009) |
| Neuropathic pain | BDNF | Spinal parenchyma | AAV | Eaton et al. (2002) |
| Neuropathic pain | IL-10 | Intrathecal | AAV2 | Milligan et al. (2005) |
| Neuropathic pain | ppbetaEP, IL-10 | Intrathecal | AAV8 | Storek et al. (2008) |
Various transgenes, delivery methods and AAV serotypes have been used. There are rational targets that are worth testing in the new TDP-43 mouse and rat models of ALS, using either a secretable growth factor or an intraneuronal protein that could directly interact with TDP-43. SMA, spinal muscular atrophy; SCI, spinal cord injury; Bcl2, B-cell lymphoma 2; GDNF, glial cell-line-derived neurotrophic factor; IGF-1, insulin-like growth factor 1; VEGF, vascular endothelial growth factor; G-CSF, granulocyte-colony-stimulating factor; SMN1, survival motor neuron 1; BDNF, brain-derived neurotrophic factor; NT-3, neurotrophin-3; L1, cell adhesion molecule L1; Ang-1, angiopoeitin-1; IL-10, interleukin 10; ppbetaEP, prepro-beta-endorphin. The AAVs used in Azzouz et al. (2000) and Eaton et al. (2002) were probably AAV2, but could not be determined.
Rodent model caveats and outlook
Rodent models for TDP-43 have definitively demonstrated the specific toxicity of this protein to cause motor neuron symptoms. It will be important to determine how well the disease states accurately mimic specific human disease subtypes, and to improve upon the clinical relevance. The diseases now known as TDP-43 proteinopathies include clinicopathological syndromes ranging from dementia to motor neuron disease, subclassified as a spectrum of subtypes based on specific types of TDP-43 pathology and genetic etiologies (recently reviewed in Chen-Plotkin et al., 2010; Geser et al., 2010). The majority of the TDP-43 transgenic mice showed expression and pathology in both the brain and spinal cord, which could represent disease forms with overlapping brain and spinal cord pathology (Chen-Plotkin et al., 2010; Geser et al., 2010). The two studies using the CaMKII promoter successfully limited TDP-43 expression to the forebrain, which is thus more relevant to frontotemporal lobar degeneration subtypes and not ALS (Tsai et al., 2010; Igaz et al., 2011). However, the probability of cytoplasmic TDP-43 inclusions forming in rodent models may be different to that in human disease as most of the models showed predominantly nuclear expression, which underscores a potential nuclear mechanism in disease, at least in these models. The TDP-43 mice used by Wils et al. (2010) may have demonstrated the most comprehensive relevance, with specific loss of motor neurons, specific ubiquitination of motor cortex and spinal cord neurons, nuclear and cytoplasmic TDP-43 aggregates, and other relevant markers such as TDP-43 hyperphosphorylation and proteolytic cleavage (Arai et al., 2006, 2010). Although relevant, the disease states generated in rodents based on overexpression cannot mirror human diseases with normal levels of TDP-43, and thus the disease mechanism may be different. More ideal models would more closely mimic specific subtypes in terms of targeting expression to the neuronal populations that are vulnerable in human disease, with more frequent TDP-43 cytoplasmic inclusions, and with disease state pathogenesis and progression similar to the human time course, e.g. adult onset of symptoms.
Overexpression is an indispensable way of addressing disease mechanisms, but there is overall frustration about the extent of clinical translation that has directly developed from transgenic mouse models. Clinical research derived from beta-amyloid mice regarding immunotherapy or secretase inhibition has so far not translated successfully (Kokjohn & Roher, 2009; Extance, 2010). There have been a number of preclinical therapeutic strategies tested in mutant SOD1 mice for ALS that have also been attempted in clinical trials, such as lithium (Fornai et al., 2008) and antisense oligonucleotides against mutant SOD1 (Smith et al., 2006), although they have not yet yielded new approved drugs for ALS (Siciliano et al., 2010). Although disease models should platform for testing new drugs, some of the biggest gains from rodent models are their contributions to understanding disease mechanisms and determining the most appropriate proteins that could be targeted with a drug (e.g. Ittner et al., 2010; Oddo et al., 2003; Roberson et al., 2007).
Although AAV vector strategies are promising for gene therapy as well as studying disease mechanisms, gene therapy clinical trials for hemophilia, for example, have had difficulty in translation, despite clear-cut efficacy in animal models (Mingozzi & High, 2011). However, current gene therapy clinical trials with AAV for a type of blindness are more promising (Simonelli et al., 2010).
Conclusions
Diverse strategies have been used to generate rodents that overexpress TDP-43 and produce a consensus ALS-like syndrome (Wegorzewska et al., 2009; Wils et al., 2010; Xu et al., 2010). Improved vector gene transfer efficiency of the spinal cord has permitted a consistent disease state with paralysis induced by TDP-43 (Wang et al., 2010). The peripheral to central administration is also an advance that could be used for new gene therapy strategies in ALS animal models based on TDP-43, and for other spinal cord diseases, like ALS, that are orphan diseases with respect to efficacious medication.
Acknowledgements
David Knight and R. Waleed Naseem critiqued the manuscript. This work was supported by Department of Veterans Affairs Merit Review Grant 1147891 (B.C.K.) and NIH/NINDS R01 NS048450 (R.L.K.).
Abbreviations
- AAV
adeno-associated virus
- ALS
amyotrophic lateral sclerosis
- CaMKII
calcium–calmodulin-dependent kinase II
- CNS
central nervous system
- FTLD–TDP
frontotemporal lobar degeneration with TDP-43
- SOD1
superoxide dismutase 1
- TDP-43
transactive response DNA binding protein 43 kDa
References
- Abe K, Fujimura H, Toyooka K, Sakoda S, Yorifuji S, Yanagihara T. Cognitive function in amyotrophic lateral sclerosis. J. Neurol. Sci. 1997;148:95–100. doi: 10.1016/s0022-510x(96)05338-5. [DOI] [PubMed] [Google Scholar]
- Acsadi G, Anguelov RA, Yang H, Toth G, Thomas R, Jani A, Wang Y, Ianakova E, Mohammad S, Lewis RA, Shy ME. Increased survival and function of SOD1 mice after glial cell-derived neurotrophic factor gene therapy. Hum. Gene Ther. 2002;13:1047–1059. doi: 10.1089/104303402753812458. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Nonoka T, Kametani F, Yamashita M, Hosokawa M, Niizato K, Tsuchiya K, Kobayashi Z, Ikeda K, Yoshida M, Onaya M, Fujishiro H, Akiyama H. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology. 2010;30:170–181. doi: 10.1111/j.1440-1789.2009.01089.x. [DOI] [PubMed] [Google Scholar]
- Ash PE, Zhang YJ, Roberts CM, Saldi T, Hutter H, Buratti E, Petrucelli L, Link CD. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 2010;19:3206–3218. doi: 10.1093/hmg/ddq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala YM, De Conti L, Avendaño-Vázquez SE, Dhir A, Romano M, D'Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, Baralle FE. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30:277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzouz M, Hottinger A, Paterna JC, Zurn AD, Aebischer P, Büeler H. Increased motoneuron survival and improved neuromuscular function in transgenic ALS mice after intraspinal injection of an adeno-associated virus encoding Bcl-2. Hum. Mol. Genet. 2000;9:803–811. doi: 10.1093/hmg/9.5.803. [DOI] [PubMed] [Google Scholar]
- Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, Carmeliet P, Mazarakis ND. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429:413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 2010;30:639–649. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan AK, Hutchinson KR, Foust KD, Braun L, McGovern VL, Schmelzer L, Ward JG, Petruska JC, Lucchesi PA, Burghes AH, Kaspar BK. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet. 2010;19:3895–3905. doi: 10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blits B, Oudega M, Boer GJ, Bartlett Bunge M, Verhaagen J. Adeno-associated viral vector-mediated neurotrophin gene transfer in the injured adult rat spinal cord improves hind-limb function. Neuroscience. 2003;118:271–281. doi: 10.1016/s0306-4522(02)00970-3. [DOI] [PubMed] [Google Scholar]
- Blits B, Carlstedt TP, Ruitenberg MJ, de Winter F, Hermens WT, Dijkhuizen PA, Claasens JW, Eggers R, van der Sluis R, Tenenbaum L, Boer GJ, Verhaagen J. Rescue and sprouting of motoneurons following ventral root avulsion and reimplantation combined with intraspinal adeno-associated viral vector-mediated expression of glial cell line-derived neurotrophic factor or brain-derived neurotrophic factor. Exp. Neurol. 2004;189:303–316. doi: 10.1016/j.expneurol.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Bordet T, Lesbordes JC, Rouhani S, Castelnau-Ptakhine L, Schmalbruch H, Haase G, Kahn A. Protective effects of cardiotrophin-1 adenoviral gene transfer on neuromuscular degeneration in transgenic ALS mice. Hum. Mol. Genet. 2001;10:1925–1933. doi: 10.1093/hmg/10.18.1925. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–878. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Deng JJ, Bai Y, Thornton FB, Oddo S. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J. Biol. Chem. 2009;284:27416–27424. doi: 10.1074/jbc.M109.031278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Wu J, Apostolova I, Skup M, Irintchev A, Kügler S, Schachner M. Adeno-associated virus-mediated L1 expression promotes functional recovery after spinal cord injury. Brain. 2007;130:954–969. doi: 10.1093/brain/awm049. [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 2010;6:211–220. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesselet MF. In vivo alpha-synuclein overexpression in rodents: a useful model of Parkinson's disease? Exp. Neurol. 2008;209:22–27. doi: 10.1016/j.expneurol.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PM, Ling J, Jeong YH, Price DL, Aja SM, Wong PC. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc. Natl. Acad. Sci. USA. 2010;107:16320–16324. doi: 10.1073/pnas.1002176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christine CW, Starr PA, Larson PS, Eberling JL, Jagust WJ, Hawkins RA, Van Brocklin HF, Wright JF, Bankiewicz KS, Aminoff MJ. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology. 2009;73:1662–1669. doi: 10.1212/WNL.0b013e3181c29356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly TM, Vogler C, Levy B, Haskins ME, Sands MS. Neonatal gene transfer leads to widespread correction of pathology in a murine model of lysosomal storage disease. Proc. Natl. Acad. Sci. USA. 1999;96:2296–2300. doi: 10.1073/pnas.96.5.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008;21:583–593. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge JC, Haidet AM, Yang W, Passini MA, Hester M, Clarke J, Roskelley EM, Treleaven CM, Rizo L, Martin H, Kim SH, Kaspar R, Taksir TV, Griffiths DA, Cheng SH, Shihabuddin LS, Kaspar BK. Delivery of AAV-IGF-1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol. Ther. 2008;16:1056–1064. doi: 10.1038/mt.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge JC, Treleaven CM, Fidler JA, Hester M, Haidet A, Handy C, Rao M, Eagle A, Matthews JC, Taksir TV, Cheng SH, Shihabuddin LS, Kaspar BK. AAV4-mediated expression of IGF-1 and VEGF within cellular components of the ventricular system improves survival outcome in familial ALS mice. Mol. Ther. 2010;18:2075–2084. doi: 10.1038/mt.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez E, Marais T, Chatauret N, Benkhelifa-Ziyyat S, Duque S, Ravassard P, Carcenac R, Astord S, Pereira de Moura A, Voit T, Barkats M. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2011;20:681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- Duque S, Joussemet B, Riviere C, Marais T, Dubreil L, Douar AM, Fyfe J, Moullier P, Colle MA, Barkats M. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton MJ, Blits B, Ruitenberg MJ, Verhaagen J, Oudega M. Amelioration of chronic neuropathic pain after partial nerve injury by adeno-associated viral (AAV) vector-mediated over-expression of BDNF in the rat spinal cord. Gene Ther. 2002;9:1387–1395. doi: 10.1038/sj.gt.3301814. [DOI] [PubMed] [Google Scholar]
- Eisen A. Amyotrophic lateral sclerosis: a 40-year personal perspective. J. Clin. Neurosci. 2009;16:505–512. doi: 10.1016/j.jocn.2008.07.072. [DOI] [PubMed] [Google Scholar]
- Extance A. Alzheimer's failure raises questions about disease-modifying strategies. Nat. Rev. Drug Discov. 2010;9:749–751. doi: 10.1038/nrd3288. [DOI] [PubMed] [Google Scholar]
- Feiguin F, Godena VK, Romano G, D'ambrogio A, Klima R, Baralle FE. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009;583:1586–1592. doi: 10.1016/j.febslet.2009.04.019. [DOI] [PubMed] [Google Scholar]
- Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, Lazzeri G, Spalloni A, Bellio N, Lenzi P, Modugno N, Siciliano G, Isidoro C, Murri L, Ruggieri S, Paparelli A. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA. 2008;105:2052–2057. doi: 10.1073/pnas.0708022105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortun J, Puzis R, Pearse DD, Gage FH, Bunge MB. Muscle injection of AAV-NT3 promotes anatomical reorganization of CST axons and improves behavioral outcome following SCI. J. Neurotrauma. 2009;26:941–953. doi: 10.1089/neu.2008.0807. [DOI] [PubMed] [Google Scholar]
- Foust KD, Flotte TR, Reier PJ, Mandel RJ. Recombinant adeno-associated virus-mediated global anterograde delivery of glial cell line-derived neurotrophic factor to the spinal cord: comparison of rubrospinal and corticospinal tracts in the rat. Hum. Gene Ther. 2008a;19:71–82. doi: 10.1089/hum.2007.104. [DOI] [PubMed] [Google Scholar]
- Foust KD, Poirier A, Pacak CA, Mandel RJ, Flotte TR. Neonatal intraperitoneal or intravenous injections of recombinant adeno-associated virus type 8 transduce dorsal root ganglia and lower motor neurons. Hum. Gene Ther. 2008b;19:61–70. doi: 10.1089/hum.2007.093. [DOI] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, Le TT, Morales PR, Rich MM, Burghes AH, Kaspar BK. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Franz CK, Federici T, Yang J, Backus C, Oh SS, Teng Q, Carlton E, Bishop KM, Gasmi M, Bartus RT, Feldman EL, Boulis NM. Intraspinal cord delivery of IGF-I mediated by adeno-associated virus 2 is neuroprotective in a rat model of familial ALS. Neurobiol. Dis. 2009;33:473–481. doi: 10.1016/j.nbd.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Josephs KA, Petrucelli L. Review: transactive response DNA-binding protein 43 (TDP-43): mechanisms of neurodegeneration. Neuropathol. Appl. Neurobiol. 2010;36:97–112. doi: 10.1111/j.1365-2990.2010.01060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geser F, Brandmeir NJ, Kwong LK, Martinez-Lage M, Elman L, McCluskey L, Xie SX, Lee VM, Trojanowski JQ. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch. Neurol. 2008;65:636–641. doi: 10.1001/archneur.65.5.636. [DOI] [PubMed] [Google Scholar]
- Geser F, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology. 2010;30:103–112. doi: 10.1111/j.1440-1789.2009.01091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, 3rd, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SJ, Blake BL, Criswell HE, Nicolson SC, Samulski RJ, McCown TJ, Li W. Directed evolution of a novel adeno-associated virus (AAV) vector that crosses the seizure-compromised blood–brain barrier (BBB) Mol. Ther. 2010;18:570–578. doi: 10.1038/mt.2009.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques A, Pitzer C, Dittgen T, Klugmann M, Dupuis L, Schneider A. CNS-targeted viral delivery of G-CSF in an animal model for ALS: improved efficacy and preservation of the neuromuscular unit. Mol. Ther. 2011;19:284–292. doi: 10.1038/mt.2010.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera JJ, Sundberg LM, Zentilin L, Giacca M, Narayana PA. Sustained expression of vascular endothelial growth factor and angiopoietin-1 improves blood-spinal cord barrier integrity and functional recovery after spinal cord injury. J. Neurotrauma. 2010;27:2067–2076. doi: 10.1089/neu.2010.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, McCluskey LF, Trojanowski JQ, Lee VM. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol. 2008;173:182–194. doi: 10.2353/ajpath.2008.080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T, Malunda J, Xu Y, Winton MJ, Trojanowski JQ, Lee VM. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Invest. 2011;121:726–738. doi: 10.1172/JCI44867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Johnson BS, Mccaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA. 2008;105:6439–6444. doi: 10.1073/pnas.0802082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, Rochefort D, Bel Hadj S, Durham HD, Vande Velde C, Rouleau GA, Drapeau P. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 2010;19:671–683. doi: 10.1093/hmg/ddp534. [DOI] [PubMed] [Google Scholar]
- Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, Bland RJ, Young D, Strybing K, Eidelberg D, During MJ. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet. 2007;369:2097–2105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- Kaspar BK, Lladó J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- Kaspar BK, Frost LM, Christian L, Umapathi P, Gage FH. Synergy of insulin-like growth factor-1 and exercise in amyotrophic lateral sclerosis. Ann. Neurol. 2005;57:649–655. doi: 10.1002/ana.20451. [DOI] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Björklund A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J. Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Björklund A. Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson's disease. Proc. Natl. Acad. Sci. USA. 2003;100:2884–2889. doi: 10.1073/pnas.0536383100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, King MA, Hamby ME, Meyer EM. Dopaminergic cell loss induced by human A30P alpha-synuclein gene transfer to the rat substantia nigra. Hum. Gene Ther. 2002;13:605–612. doi: 10.1089/10430340252837206. [DOI] [PubMed] [Google Scholar]
- Klein RL, Lin WL, Dickson DW, Lewis J, Hutton M, Duff K, Meyer EM, King MA. Rapid neurofibrillary tangle formation after localized gene transfer of mutated tau. Am. J. Pathol. 2004;164:347–353. doi: 10.1016/S0002-9440(10)63124-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokjohn TA, Roher AE. Amyloid precursor protein transgenic mouse models and Alzheimer's disease: understanding the paradigms, limitations, and contributions. Alzheimers Dement. 2009;5:340–347. doi: 10.1016/j.jalz.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VM, Schellenberg GD. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119:409–419. doi: 10.1007/s00401-010-0659-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepore AC, Haenggeli C, Gasmi M, Bishop KM, Bartus RT, Maragakis NJ, Rothstein JD. Intraparenchymal spinal cord delivery of adeno-associated virus IGF-1 is protective in the SOD1G93A model of ALS. Brain Res. 2007;1185:256–265. doi: 10.1016/j.brainres.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverenz JB, Yu CE, Montine TJ, Steinbart E, Bekris LM, Zabetian C, Kwong LK, Lee VM, Schellenberg GD, Bird TD. A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain. 2007;130:1360–1374. doi: 10.1093/brain/awm069. [DOI] [PubMed] [Google Scholar]
- Li Y, Ray P, Rao EJ, Shi C, Guo W, Chen X, Woodruff EA, 3rd, Fushimi K, Wu JY. A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA. 2010;107:3169–3174. doi: 10.1073/pnas.0913602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko NF, Guthrie CR, Kraemer BC. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci. 2010;30:16208–16219. doi: 10.1523/JNEUROSCI.2911-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. alpha-Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc. Natl. Acad. Sci. USA. 2002;99:10813–10818. doi: 10.1073/pnas.152339799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM, Trojanowski JQ. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe Y, Nagano I, Gazi MS, Murakami T, Shiote M, Shoji M, Kitagawa H, Setoguchi Y, Abe K. Adenovirus-mediated gene transfer of glial cell line-derived neurotrophic factor prevents motor neuron loss of transgenic model mice for amyotrophic lateral sclerosis. Apoptosis. 2002;7:329–334. doi: 10.1023/a:1016123413038. [DOI] [PubMed] [Google Scholar]
- Marks WJ, Jr, Bartus RT, Siffert J, Davis CS, Lozano A, Boulis N, Vitek J, Stacy M, Turner D, Verhagen L, Bakay R, Watts R, Guthrie B, Jankovic J, Simpson R, Tagliati M, Alterman R, Stern M, Baltuch G, Starr PA, Larson PS, Ostrem JL, Nutt J, Kieburtz K, Kordower JH, Olanow CW. Gene delivery of AAV2-neurturin for Parkinson's disease: a double-blind, randomised, controlled trial. Lancet Neurol. 2010;9:1164–1172. doi: 10.1016/S1474-4422(10)70254-4. [DOI] [PubMed] [Google Scholar]
- McCarty DM, DiRosario J, Gulaid K, Muenzer J, Fu H. Mannitol-facilitated CNS entry of rAAV2 vector significantly delayed the neurological disease progression in MPS IIIB mice. Gene Ther. 2009;16:1340–1352. doi: 10.1038/gt.2009.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Sloane EM, Langer SJ, Cruz PE, Chacur M, Spataro L, Wieseler-Frank J, Hammack SE, Maier SF, Flotte TR, Forsayeth JR, Leinwand LA, Chavez R, Watkins LR. Controlling neuropathic pain by adeno-associated virus driven production of the anti-inflammatory cytokine, interleukin-10. Mol. Pain. 2005;1:9. doi: 10.1186/1744-8069-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- Neumann M. Molecular neuropathology of TDP-43 proteinopathies. Int. J. Mol. Sci. 2009;10:232–246. doi: 10.3390/ijms10010232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J. Neuropathol. Exp. Neurol. 2007;66:152–157. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- Nishihira Y, Tan CF, Onodera O, Toyoshima Y, Yamada M, Morita T, Nishizawa M, Kakita A, Takahashi H. Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol. 2008;116:169–182. doi: 10.1007/s00401-008-0385-z. [DOI] [PubMed] [Google Scholar]
- Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, Gallo JM, Hortobágyi T, Shaw CE, Rogelj B. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain. 2010;133:1763–1771. doi: 10.1093/brain/awq111. [DOI] [PubMed] [Google Scholar]
- Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 2009;18:3353–3364. doi: 10.1093/hmg/ddp275. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol. Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Passini MA, Bu J, Roskelley EM, Richards AM, Sardi SP, O'Riordan CR, Klinger KW, Shihabuddin LS, Cheng SH. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2010;120:1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel AL, Zolotukhin S, Schrimsher GW, Muzyczka N, Reier PJ. Efficient transduction of green fluorescent protein in spinal cord neurons using adeno-associated virus vectors containing cell type-specific promoters. Gene Ther. 1997;4:16–24. doi: 10.1038/sj.gt.3300358. [DOI] [PubMed] [Google Scholar]
- Piao Y, Hashimoto T, Takahama S, Kakita A, Komori T, Morita T, Takahashi H, Mizutani T, Oyanagi K. Survival motor neuron (SMN) protein in the spinal anterior horn cells of patients with sporadic amyotrophic lateral sclerosis. Brain Res. 2011;1372:152–159. doi: 10.1016/j.brainres.2010.11.070. [DOI] [PubMed] [Google Scholar]
- Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA, Lee DC, Wong LF, Bilsland LG, Greensmith L, Kingsman SM, Mitrophanous KA, Mazarakis ND, Azzouz M. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 2005;11:429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- Raoul C, Abbas-Terki T, Bensadoun JC, Guillot S, Haase G, Szulc J, Henderson CE, Aebischer P. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat. Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahman Z, Krizus A, Mckenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van Den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Liang NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Senut MC, Suhr ST, Kaspar B, Gage FH. Intraneuronal aggregate formation and cell death after viral expression of expanded polyglutamine tracts in the adult rat brain. J. Neurosci. 2000;20:219–229. doi: 10.1523/JNEUROSCI.20-01-00219.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Good SK, Atkin S, Dewey CM, Mayer P, 3rd, Herz J, Yu G. TDP-43 is a developmentally regulated protein essential for early embryonic development. J. Biol. Chem. 2010;285:6826–6834. doi: 10.1074/jbc.M109.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan X, Chiang PM, Price DL, Wong PC. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. USA. 2010;107:16325–16330. doi: 10.1073/pnas.1003459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano G, Carlesi C, Pasquali L, Piazza S, Pietracupa S, Fornai F, Ruggieri S, Murri L. Clinical trials for neuroprotection in ALS. CNS Neurol. Disord. Drug Targets. 2010;9:305–313. doi: 10.2174/187152710791292648. [DOI] [PubMed] [Google Scholar]
- Simonelli F, Maguire AM, Testa F, Pierce EA, Mingozzi F, Bennicelli JL, Rossi S, Marshall K, Banfi S, Surace EM, Sun J, Redmond TM, Zhu X, Shindler KS, Ying GS, Ziviello C, Acerra C, Wright JF, McDonnell JW, High KA, Bennett J, Auricchio A. Gene therapy for Leber's congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol. Ther. 2010;18:643–650. doi: 10.1038/mt.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, Lobsiger CS, Ward CM, McAlonis-Downes M, Wei H, Wancewicz EV, Bennett CF, Cleveland DW. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 2006;116:2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings NR, Puttaparthi K, Luther CM, Burns DK, Elliott JL. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol. Dis. 2010;40:404–414. doi: 10.1016/j.nbd.2010.06.017. [DOI] [PubMed] [Google Scholar]
- Storek B, Reinhardt M, Wang C, Janssen WG, Harder NM, Banck MS, Morrison JH, Beutler AS. Sensory neuron targeting by self-complementary AAV8 via lumbar puncture for chronic pain. Proc. Natl. Acad. Sci. USA. 2008;105:1055–1060. doi: 10.1073/pnas.0708003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatom JB, Wang DB, Dayton RD, Skalli O, Hutton ML, Dickson DW, Klein RL. Mimicking aspects of frontotemporal lobar degeneration and Lou Gehrig's disease in rats via TDP-43 overexpression. Mol. Ther. 2009;17:607–613. doi: 10.1038/mt.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towne C, Schneider BL, Kieran D, Redmond DE, Jr, Aebischer P. Efficient transduction of non-human primate motor neurons after intramuscular delivery of recombinant AAV serotype 6. Gene Ther. 2010;17:141–146. doi: 10.1038/gt.2009.119. [DOI] [PubMed] [Google Scholar]
- Tsai KJ, Yang CH, Fang YH, Cho KH, Chien WL, Wang WT, Wu TW, Lin CP, Fu WM, Shen CK. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J. Exp. Med. 2010;207:1661–1673. doi: 10.1084/jem.20092164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BJ, Parkinson NJ, Davies KE, Talbot K. Survival motor neuron deficiency enhances progression in an amyotrophic lateral sclerosis mouse model. Neurobiol. Dis. 2009;34:511–517. doi: 10.1016/j.nbd.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Valori CF, Ning K, Wyles M, Mead RJ, Grierson AJ, Shaw PJ, Azzouz M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2010;2:35–42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Robberecht W. Recent advances in motor neuron disease. Curr. Opin. Neurol. 2009;22:486–492. doi: 10.1097/WCO.0b013e32832ffbe3. [DOI] [PubMed] [Google Scholar]
- Wang LJ, Lu YY, Muramatsu S, Ikeguchi K, Fujimoto K, Okada T, Mizukami H, Matsushita T, Hanazono Y, Kume A, Nagatsu T, Ozawa K, Nakano I. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J. Neurosci. 2002;22:6920–6928. doi: 10.1523/JNEUROSCI.22-16-06920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DB, Dayton RD, Henning PP, Cain CD, Zhao LR, Schrott LM, Orchard EA, Knight DS, Klein RL. Expansive gene transfer in the rat CNS rapidly produces amyotrophic lateral sclerosis relevant sequelae when TDP-43 is overexpressed. Mol. Ther. 2010;18:2064–2074. doi: 10.1038/mt.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson C, Harvey AR. Projections from the brain to the spinal cord. In: Watson C, Paxinos G, Kayalioglu G, editors. The Spinal Cord: A Christopher and Dana Reeve Text and Atlas. Academic Press; Sydney: 2008. pp. 168–179. [Google Scholar]
- Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA. 2009;106:18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wider C, Dickson DW, Stoessl AJ, Tsuboi Y, Chapon F, Gutmann L, Lechevalier B, Calne DB, Personett DA, Hulihan M, Kachergus J, Rademakers R, Baker MC, Grantier LL, Sujith OK, Brown L, Calne S, Farrer MJ, Wszolek ZK. Pallidonigral TDP-43 pathology in Perry syndrome. Parkinsonism Relat. Disord. 2009;15:281–286. doi: 10.1016/j.parkreldis.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA. 2010;107:3858–3863. doi: 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LS, Cheng WC, Hou SC, Yan YT, Jiang ST, Shen CK. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2010;48:56–62. doi: 10.1002/dvg.20584. [DOI] [PubMed] [Google Scholar]
- Xu YF, Gendron TF, Zhang YJ, Lin WL, D'Alton S, Sheng H, Casey MC, Tong J, Knight J, Yu X, Rademakers R, Boylan K, Hutton M, McGowan E, Dickson DW, Lewis J, Petrucelli L. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010;30:10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, Mita S, Kato S, Okado H, Ohama E, Uchino M. Effect on motor neuron survival in mutant SOD1 (G93A) transgenic mice by Bcl-2 expression using retrograde axonal transport of adenoviral vectors. Neurosci. Lett. 2002;328:289–293. doi: 10.1016/s0304-3940(02)00454-8. [DOI] [PubMed] [Google Scholar]
- Zhang H, Tan CF, Mori F, Tanji K, Kakita A, Takahashi H, Wakabayashi K. TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2008;115:115–122. doi: 10.1007/s00401-007-0285-7. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P, Gass J, Rangachari V, Buratti E, Baralle F, Golde TE, Dickson DW, Petrucelli L. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA. 2009;106:7607–7612. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Huang C, Chen H, Wang D, Landel CP, Xia PY, Bowser R, Liu YJ, Xia XG. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010;6:e1000887. doi: 10.1371/journal.pgen.1000887. [DOI] [PMC free article] [PubMed] [Google Scholar]