

Abstract
Doxorubicin (DOX) is one of most common anti-cancer chemotherapeutic drugs, but its clinical use is associated with dose-limiting cardiotoxicity. We have recently developed a series of PEG-oligocholic acid based telodendrimers, which can efficiently encapsulate hydrophobic drugs and self-assemble to form stable micelles in aqueous condition. In the present study, two representative telodendrimers (PEG5k-CA8 and PEG2k-CA4) have been applied to prepare DOX micellar formulations for the targeted delivery of DOX to lymphoma. PEG2k-CA4 micelles, compared to PEG5k-CA8 micelles, were found to have higher DOX loading capacity (14.8% vs. 8.2%, w/w), superior stability in physiological condition, and more sustained release profile. Both of these DOX-loaded micelles can be efficiently internalized and release the drug in Raji lymphoma cells. DOX-loaded micelles were found to exhibit similar in vitro cytotoxic activities against both T- and B- lymphoma cells as the free DOX. The maximum tolerated dose (MTD) of DOX-loaded PEG2k-CA4 micelles in mice was approximately 15 mg/kg, which was 1.5-fold higher of the MTD of free DOX. Pharmacokinetics and biodistribution studies demonstrated both DOX-loaded micelles were able to prolong the blood retention time, preferentially accumulate and penetrate in B-cell lymphomas via the enhanced permeability and retention (EPR) effect. Finally, DOX-PEG2k-CA4 micelles achieved enhanced anti-cancer efficacy and prolonged survival in Raji lymphoma bearing mice, compared to free DOX and PEGylated liposomal DOX (Doxil®) at the equivalent dose. In addition, the analysis of creatine kinase (CK) and lactate dehydrogenase (LDH) serum enzymes level indicated that DOX micellar formulations significantly reduced the cardiotoxicity associated with free DOX.
Keywords: doxorubicin, polymeric micelles, drug delivery, biodistribution, cardiotoxicity, cancer therapy
1. Introduction
Doxorubicin (DOX) is a commonly used anti-cancer chemotherapeutic agent that is effective for the treatment of a wide range of cancers, including hematological malignancies, many types of carcinoma and soft tissue sarcomas. It works through the interaction with DNA by intercalation and the inhibition of the topoisomerase II enzyme activity. However, the long-term clinical use of DOX is often compromised by its severe side effects such as cardiotoxicity, which may result in irreversible cardiomyopathy and subsequent congestive heart failure [1]. One of the promising approaches for limiting DOX associated cardiotoxicity is to selectively deliver the drug to tumor tissue while decreasing its exposure to normal tissues including the heart.
Nanomedicine is an emerging field that has shown great promise for the development of novel diagnostic, imaging, and therapeutic agents for a variety of diseases, including cancer. To limit some of the side effects associated with many chemotherapeutics agents such as DOX, several nanocarrier systems have been developed. These nanomaterials include solid nanoparticles (NPs) [2], liposomes [3], liposome-nanogel [4], dendrimers [5], polymeric micelles [6], water soluble polymers, and protein aggregates. It is believed that such nanoformulation will decrease the toxicity to normal organs, prolong the circulation time, slow down the metabolism of the drug, and facilitate the delivery of the drug to the tumor sites via the enhanced permeability and retention (EPR) effect. PEGylated liposomal DOX (Doxil®) is the first nanoformulations of DOX approved by FDA for the treatment of AIDS-related Kaposi's sarcoma, breast cancer, and ovarian cancer. Although the encapsulation of DOX into liposome does result in decreased DOX-induced cardiomyopathy, its clinical anti-cancer efficacy is only marginally better than the parent drug. This could in part be explained by its relatively large size (ca. 140 nm), thus limiting the tumor tissue penetration and obviating the EPR effects [7]. In addition, one of dose limiting toxicities of Doxil® is hand-foot syndrome (HFS) [8], which may be due to the trapping of the larger nanoaggregates and therefore drug leakage at the capillaries of palms and soles.
Polymeric micelles are a large family of nano-sized particles with unique core-shell structure, and they may offer some therapeutic advantages over liposomes as their size could be significantly smaller (20–100 nm). Compared to liposomes, the smaller polymeric micelles have been shown to have an enhanced tumor-penetrating capability [9, 10]. We have recently developed a series of micelle nanocarriers, which were formed by the self-assembly of novel linear-dendritic block copolymers (named as telodendrimer) with engineerable and well defined structures, comprising polyethylene glycol (PEG) and dendritic oligo-cholic acids (CA) [11–15]. By varying the length of PEG chain and the number of cholic acids in the dendritic core of the telodendrimers, the particle size of the resulting micelles could be easily tuned from 17 nm to 150 nm after drug loading. It has been demonstrated that the biodistribution of these telodendrimer micelles depends greatly on the particle size [13]. The smaller micellar NPs (17–64 nm) could preferentially accumulate in the tumor site via EPR effects, whereas most of the larger NPs (154 nm) were trapped in liver and lung where macrophages were located. The representative PEG5k-CA8 micelles (superscript “5k” represents the molecular weight of PEG (Dalton), while subscript “8” indicates the number of CA subunits in the telodendrimer) have been shown to have high loading capacity for some hydrophobic drugs such as paclitaxel (PTX), outstanding stability (over 6 months at 4 °C), and sustainable drug release profiles. PTX-loaded PEG5k-CA8 micelles were found to exhibit superior anti-cancer efficacy and toxicity profile in SKOV-3 ovarian cancer xenograft mouse models when compared to free PTX formulation (Taxol®) and the albumin-bound PTX formulation (Abraxane®) at equivalent PTX dose [11]. Furthermore, we have recently completed a phase I clinical trial of PTX-loaded PEG5k-CA8 micelles in companion dogs with lymphoma, and demonstrated that it was well tolerated and exhibited significant anti-cancer efficacy.
Although PEG5k-CA8 micellar NPs have been successfully applied in the delivery of PTX, unfortunately, the delivery of DOX using this nanocarrier is limited by the relatively low drug loading capacity and poor stability. However, another telodendrimer with the similar structure, PEG2k-CA4, was found to be able to encapsulate DOX to form stable micelles with higher drug loading capacity, and prolonged drug release rate, when compared to PEG5k-CA8 micelle formulation. In the present study, in an attempt to overcome the cardiotoxicity of DOX, and to prevent the problems associated with Doxil® (HFS and insufficient tumor penetration), two micelle-based nanoformulations of DOX have been prepared and systematically evaluated for their efficacies in the treatment of cancers such as lymphoma. DOX is one of the four drugs (doxorubicin, vincristine, cytoxan, and prednisone) in the standard CHOP regimen commonly used for the treatment of lymphoma. The morphology, particle size distribution, stability, and drug release profiles of DOX-loaded micelles were characterized by transmission electron microscopy (TEM), dynamic light scattering (DLS), and dialysis method, respectively. The in vitro cytotoxicity of different DOX formulations (free DOX, Doxil® and DOX-loaded micelles) were examined in both T and B lymphoma cell lines using MTS assay. The pharmacokinetics, biodistribution, and tumor accumulation of DOX micellar formulations were investigated in normal or lymphoma bearing mice via the DOX fluorescence measurement and near-infrared fluorescence (NIRF) optical imaging approach, respectively. Finally, the in vivo anti-cancer efficacy and toxicity profiles including cardiotoxicity of DOX-loaded micelles were evaluated in the subcutaneous Raji lymphoma mouse model, when compared with clinical formulations (free DOX and Doxil®, the liposomal formulation).
2. Experimental Section
2.1. Materials
Doxorubicin hydrochloride (DOX·HCl) (Novaplus) and Doxil® (Ben Venue Laboratories, Inc., Bedford, OH) were obtained from the UC Davis Cancer Center Pharmacy. Monomethyl-terminated poly (ethylene glycol) monoamine (MeO-PEG-NH2, Mw = 2 kDa and 5 kDa, respectively) were purchased from Rapp Polymere (Tübingen, Germany). (Fmoc)lys(Boc)-OH, (Fmoc)Lys(Dde)-OH, (Fmoc)Lys(Fmoc)-OH were obtained from AnaSpec Inc. (San Jose, CA). NIRF dye DiD (1,1'-dioctadecyl-3,3,3',3'-tetramethylindodicarbocyanine perchlorate, D-307), BODIPY 650/665 (6-(((4,4-difluoro-5-(2-pyrrolyl)-4-bora-3a,4a-diaza-s-indacene-3-yl)styryloxy) acetyl) aminohexanoic acid, succinimidyl ester), and 4', 6-diamidino-2-phenylindole (DAPI) were purchased from Invitrogen. Tetrazolium compound [3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, MTS] and phenazine methosulfate (PMS) were purchased from Promega (Madison, Wisconsin). Cholic acid, triethylamine (TEA), and all other chemicals were purchased from Sigma-Aldrich (St. Louis).
2.2 Synthesis of PEG5k-CA8 and PEG2k-CA4 telodendrimers
The telodendrimers were synthesized via solution-phase condensation reactions using Meo-PEG-NH2, lysine and cholic acid as building blocks as described previously [13]. BODIPY650/665 (NIRF dye) labeled telodendrimers were synthesized by coupling BODIPY NHS ester to the amino group of the proximal lysine between PEG and cholic acid after the removal of 1-(4,4-dimethyl-2,6-dioxocyclohex-1-yldine)ethyl (Dde) protecting group by 2% (v/v) hydrazine in DMF.
2.3 Preparation and characterization of DOX-loaded PEG5k-CA8 and PEG2k-CA4 micelles
DOX-loaded PEG5k-CA8 (DOX-PEG5k-CA8) and DOX-loaded PEG2k-CA4 (DOX-PEG2k-CA4) micelles were prepared, respectively, via a dry-down (evaporation) method as described previously [11]. Before the encapsulation of DOX into the polymeric micelles, DOX·HCl was stirred with 3 molar equivalent of triethylamine in chloroform (CHCl3)/methanol (MeOH) (1:1, v/v) overnight to remove HCl from DOX·HCl. 20 mg PEG5k-CA8 or PEG2k-CA4 telodendrimer along with different amount of neutralized DOX were first dissolved in CHCl3/MeOH, mixed, and evaporated on rotavapor to obtain a homogeneous dry polymer film. The film was reconstituted in 1 mL phosphate buffered solution (PBS), followed by sonication for 30 min, allowing the sample film to disperse into micelle solution. To track their in vivo fates, hydrophobic NIRF dye DiD was encapsulated into the micelles using the same method as described above. Finally, the micelle formulation was filtered with 0.22 μm filter to sterilize the sample. To determine the amount of DOX, DOX-loaded micelles were diluted with DMSO (micelle solution/DMSO: 1:9, v/v) to dissociate micelle nanoparticles and the fluorescence was measured by NanoDrop 2000 spectrophotometer (Thermo Scientific), wherein calibration curve was obtained using a series of DOX/DMSO standard solutions with different concentrations. The drug loading content (DL) and encapsulation efficiency (EE) were calculated according to the following formula:
The morphology and particle size distribution of DOX-PEG5k-CA8 and DOX-PEG2k-CA4 micelles were characterized by Philips CM-120 transmission electron microscopy (TEM) and dynamic light scattering (DLS, Microtrac), respectively. The stability of DOX-loaded micelles in the physiological condition was evaluated by monitoring the particle sizes of micelles in 50% fetal bovine serum (FBS) over time using DLS.
The in vitro drug release profiles from DOX-loaded micelles were measured by the dialysis method. The DOX loading levels of both micelles used in the drug release study were 2 mg/ml DOX in 20 mg/ml telodendrimer. Aliquots of free DOX or DOX-loaded micelles solution were injected into dialysis cartridges with the MWCO of 3.5 kDa (Thermo Scientific, Rochford, IL), respectively. The cartridge was dialyzed against 1 L PBS and gently shaken at 37 °C at 100 rpm with activated charcoal to create a sink condition. The concentration of DOX remained in the dialysis cartridge at different time points were measured using a Molecular Devices SpectraMax M2 (Sunnyvale, CA) at excitation 470 nm/emission 590 nm. Data were reported as the average percentage of DOX accumulative release for each triplicate samples.
2.4 Cell culture and animals
T-cell lymphoma cell lines (Jurkat and MOLT-4) and B-cell lymphoma cell lines (Raji and Ramos) were purchased from American Type Culture Collection (ATCC; Manassas,VA, USA). All these cells were cultured in ATCC-formulated RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin G, and 100 μg/mL streptomycin at 37 °C using a humidified 5% CO2 incubator.
Female SPF BALB/c mice, 8–10 weeks age, were purchased from Charles River (Hollister, CA); female athymic nude mice (Nu/Nu strain), 6–8 weeks age, were purchased from Harlan (Livermore, CA). All animals were kept under pathogen-free conditions according to AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) guidelines and were allowed to acclimatize for at least 4 days prior to any experiments. All animal experiments were performed in compliance with institutional guidelines and according to protocol No. 07-13119 and No. 09-15584 approved by the Animal Use and Care Administrative Advisory Committee at the University of California, Davis. Lymphoma xenograft mouse models were established by subcutaneously injecting 1×107 Raji, Ramos or MOLT-4 lymphoma cells in a 100 μL of mixture of PBS and Matrigel (1:1 v/v) at the right flank in female nude mice.
2.5 Cellular uptake study
2.5.1. Confocal fluorescence microscopy
Raji lymphoma cells were incubated with DOX·HCl and DOX-loaded micelles at the final DOX concentration of 10 μM for 30 min or 2 h at 37 °C, respectively. The cells were washed three times with cold PBS, spinned down onto glass slides using CytoSpin III Cytocentrifuge (Shandon), and fixed with 4% paraformaldehyde for 10 min. The nuclei were counterstained by DAPI. The slides were mounted with coverslips and cells were imaged with an Olympus FV1000 laser scanning confocal fluorescence microscopy.
2.5.2. Flow cytometric analysis
The quantitative cellular uptake of various DOX formulations by Raji cells was analyzed by flow cytometry. Briefly, 3×105 Raji cells were incubated with DOX·HCl, DOX-PEG5k-CA8 or DOX-PEG2k-CA4 at different DOX concentrations (1, 3 and 9 μM) for 30 min or 2 h at 37 °C, respectively. Then, the cells were washed with cold PBS three times and resuspended in PBS for the flow cytometry analysis using the FACScan (Becton Dickinson, San Jose, CA). Cell-associated DOX was excited with an argon laser (488 nm), and fluorescence was detected at 560 nm. 10, 000 events were collected for each sample.
2.6 In vitro cytotoxicity assay
The MTS assay was used to evaluate the effects of DOX-loaded micellar NPs on the cell viability against both T and B lymphoma cell lines. MOLT-4, Jurkat, Raji and Ramos cells were seeded in 96-well plate at the cell densities of 4 × 103 cells/well, respectively. After overnight incubation, the cells were treated with different concentrations of DOX·HCl, Doxil®, and DOX-loaded micelles, as well as the equivalent doses of blank micelles. After 72 h incubation, CellTiter 96® Aqueous Cell Proliferation Reagent, which is composed of MTS and an electron coupling reagent PMS, was added to each well according to the manufacturer's instructions. The cell viability was determined by measuring the absorbance at 490 nm using a microplate reader (SpectraMax M2, Molecular Devices, USA). Untreated cells served as a control. Results were shown as the average cell viability [(ODtreat−ODblank)/(ODcontrol−ODblank)×100%] of triplicate wells.
2.7 Maximum tolerated dose (MTD) studies
Healthy female SPF BALB/c mice were administered intravenously with DOX·HCl or DOX-PEG2k-CA4 (3 mg/ml DOX in 20 mg/ml PEG2k-CA4 telodendrimer) at the dose of 5, 10, 15, 20 and 30 mg DOX/kg body weight, respectively (n = 4). Mice survival and body weight change were monitored daily for two weeks. The MTD was defined as the allowance of a median body weight loss of 15% and causes neither death due to toxic effects nor remarkable changes in the general signs within two weeks after administration.
2.8 Pharmacokinetics and biodistribution studies
Various DOX formulations (DOX·HCl, DOX-PEG5k-CA8 and DOX-PEG2k-CA4) were injected into female BALB/c mice via tail vein at a dose of 10 mg/kg body weight, respectively (n = 4 for each group). At different time points (3 min, 8 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, and 24 h) post-injection, blood was collected in a heparinized tube. The blood was separated by centrifugation and plasma was isolated for the analysis. Twenty microliters of plasma were added to 180 μL of extraction buffer (10% Triton X-100, deionized water, and acidified isopropanol (0.75 N HCl) with 1:2:15 volumetric ratio), and DOX was extracted overnight at −20 °C. Standard curves for DOX drug in blood were generated by the addition of free DOX with different concentrations to whole blood followed by extraction and quantification. The fluorescence of the supernatant was determined at excitation/emission of 470/590 nm to calculate DOX concentration. Non-compartmental pharmacokinetic analysis was done using Microsoft Excel 2008 version 12.2.9 (Seattle, WA). The area under plasma concentration (AUC) versus time curve from time zero to the time of the last measurable drug concentration (AUC0-t) was calculated using the linear trapezoidal rule. The extrapolated AUC from last measurable timepoint to infinity (AUCinf), was calculated by dividing the last measurable plasma concentration by elimination rate constant of the terminal phase (ke). The half-life of the terminal phase (t1/2) was calculated by ln(2)/ke. Total body clearance (Cl) was calculated as dose divided by AUC0-inf and adjusted for body weight.
In another set of experiment, DOX·HCl, DOX-PEG5k-CA8 and DOX-PEG2k-CA4 micelles were intravenously injected into Raji lymphoma bearing mice at the dose of 10 mg DOX/kg, respectively (n = 4). At 16 h post-injection, major organs (heart, liver, spleen, lung and kidney) and tumor tissue were harvested from the mice. 100 mg tissues were mixed with 900 μL extraction buffer, and homogenized using POLYTRON® PT 10–35 homogenizer (Kinematica, AG, Littau/Lucerne, Switzerland). DOX was extracted overnight at −20 °C using the same method mentioned above. The samples were centrifuged at 3000 rpm for 5 min after sufficient mixing, and the supernatant was used for the fluorescence measurement. The recovery standard curve for each tissue was separately generated by mixing different concentration of free DOX with different tissues followed by extraction and quantification. The biodistribution data was expressed as the % injected dose per gram tissue.
2.9 NIRF optical imaging
NIRF dyes (DiD or BODIPY650/665) fluorescently labeled micelles and free DiD in 100 μL PBS were injected into lymphomas bearing nude mice via the tail vein, respectively (n = 3). At different time points (1 h, 2 h, 4 h, 8 h, 24 h and 48 h) post-injection, mice were scanned using a Kodak multimodal imaging system IS2000MM with the excitation at 625 nm and the emission at 700 nm. The mice were anaesthetized by intraperitoneal injection of pentobarbital (60 mg/kg) before each imaging. All animals were euthanized by CO2 overdose at 24 h or 48 h after injection. Tumors and major organs were excised and imaged with the Kodak imaging station. For the histological evaluation, excised tumors were frozen in O.C.T. (cryo-embedding medium) at 80 °C. The corresponding slices (10 μm) were then prepared with a Zeiss Microm HM500 microtome cryostat (Zeiss, Walldorf, Germany), air-dried for 30 min and fixed with 4% paraformaldehyde for 10 min. The nuclei were stained with DAPI, and the slides were mounted with coverslips and imaged with an Olympus FV1000 confocal fluorescence microscopy.
2.10 In vivo anti-cancer efficacy study
Subcutaneous Raji lymphoma xenograft mouse model was used to evaluate the therapeutic efficacy of different DOX formulations. When tumor volume reached 150–300 mm3, mice were intravenously administrated with PBS, DOX·HCl, Doxil®, DOX-PEG5k-CA8 and DOX-PEG2k-CA4 at the dose of 10 mg/kg DOX equivalent (MTD of free DOX), respectively (n = 5–8). The treatment was given every four day on days 0, 4 and 8 for total three doses. Tumor sizes were measured with a digital caliper twice per week. Tumor volume was calculated by the formula (L×W2)/2, where L is the longest, and W is the shortest in tumor diameters (mm). To compare between groups, relative tumor volume (RTV) was calculated at each measurement time point (where RTV equals the tumor volume at given time point divided by the tumor volume prior to initial treatment). For humane reasons, animals were sacrificed when the implanted tumor volume reached 2000 mm3, which was considered as the end point of survival data. At day 7 after the last dosage, blood samples were obtained from all the mice for the measurement of blood cell counts, serum chemistry including alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and markers of cardiotoxicity such as creatine kinase (CK) and lactate dehydrogenase (LDH). One mouse from each group was also sacrificed, and its heart was submitted for histopathology evaluation.
2.11 Statistical analysis
The level of significance in all statistical analyses was set at a probability of P < 0.05. Data are presented as means ± standard error (SEM). Statistical analysis was performed by Student's t-test for comparison of two groups, and one-way analysis of variance (ANOVA) for multiple groups, followed by Newman-Keuls test if overall P < 0.05.
3. Results and discussion
3.1 Preparation and characterization of DOX-loaded PEG5k-CA8 and PEG2k-CA4 micelles
The chemical structure of PEG2k-CA4 telodendrimer, with four dendritic cholic acid molecules attached to one terminus of linear PEG chain (2k), is shown in Fig. 1A. PEG2kCA4 and PEG5k-CA8 telodendrimers were synthesized via stepwise solution-phase condensation reactions using MeO-PEG-NH2, lysine and cholic acid as building blocks, respectively. As previously reported [13], the chemical structures of both telodendrimers were determined by 1H-NMR spectrometry, and their molecular weights were analyzed using MALDI-TOF mass spectrometry. The monodispersed mass traces were detected for both telodendrimers and their molecular weights obtained from both NMR and MALDI-TOF measurements were very similar to the theoretical values, indicating their well-defined structures.
Fig.1.

(A) the chemical structure of PEG2k-CA4 telodendrimer; Particle size distribution of blank (B) and DOX-loaded PEG2k-CA micelles (C) obtained by DLS; (D) TEM image of DOX-loaded PEG2k-CA4 micelles. DOX loading level was 3 mg/ml DOX in 20 mg/ml PEG2k-CA4 telodendrimer.
Both PEG5k-CA8 and PEG2k-CA4 telodendrimers are able to self-assemble in aqueous solution to form core/shell micellar NPs with the particle size of around 16 nm and 10 nm, respectively (Fig. 1B). Using the dry-down method, both telodendrimers can efficiently encapsulate some hydrophobic drugs such as PTX into the core of micelles with relatively high loading capacity (up to 7.3 mg/ml PTX in 20 mg/ml telodendrimer) [11]. However, in the present study, PEG2k-CA4 telodendrimer has been shown to have relatively higher drug loading content and encapsulation efficiency (EE) for DOX, compared to PEG5k-CA8 telodendrimer. As summarized in Table 1, the EE of DOX in PEG5k-CA8 micelles was 91% when the feeding amount of DOX/telodendrimer (wt/wt) was < 5%, whereas it decreased to 78% and 55%, when the feeding amount increased to 10% and 15%, respectively. In contrast, the EE of DOX into PEG2k-CA4 micelles was almost 100% when DOX feeding amount was < 15%, and gradually decreased as the feeding amount was increased. It should be noted that DOX loading capacity of PEG2k-CA4 micelles is superior to Doxil® (2 mg/ml) and other reported micelle formulations such as PEGylated PLGA NPs (< 5% DOX/polymer, w/w) [1]. After DOX was loaded into micelles solution, the 1H NMR signals of drug and hydrophobic oligocholane of the telodendrimers were significantly suppressed in D2O, which indicated the formation of solid core of the aggregates for both PEG5k-CA8 and PEG2k-CA4 micelles (Supplementary, Fig. S-1). In addition, no free DOX signals were observed in the proton NMR spectra of the DOX-loaded micelles, indicating the negligible concentration of free DOX existing in the micelles solutions. Subsequent 1H NMR spectra obtained for the DOX-loaded micelle samples after one month's storage at 4 °C were identical to those obtained previously, with the absence of free DOX signals, which further suggested the overall stability of the micelles, particularly in their capability of retaining DOX. The differential loading capacity of PEG5k-CA8 telodendrimer micelles for PTX and DOX may be explained by the miscibility and interactions between cargo and nanocarriers, including hydrogen bonding, electrostatic interaction and dipole-dipole interaction [16]. However, given that PEG5k-CA8 and PEG2k-CA4 micelles have the same chemical components, the differential DOX loading capacity and stability may be caused by the difference in the thermo-dynamic character of the drug-loaded system, including the hydrophilic-lipophilic balance (HLB) [17] and the molecular weight of the telodendrimers. To further understand this micellar system, a combination of experimental and theoretical studies such as molecular dynamics analysis will be needed.
Table 1.
Physicochemical characterization of DOX-loaded PEG5k -CAs8 and PEG2k -CA4 micelles
| Telodendrimers | DOX/polymer (w/w, %) | Particle size (nm)a | DL (%) | EE (%) | DOX concentration in micellesb(mg/ml) |
|---|---|---|---|---|---|
| PEG5k-CA8 | 0 | 16 ± 3 | N/A | N/A | N/A |
| 5 | 15 ± 5 | 4.6 | 91 | 0.91 | |
| 10 | 17 ± 7* | 7.8 | 78 | 1.56 | |
| 15 | 20 ± 9* | 8.2 | 55 | 1.65 | |
| PEG2k-CA4 | 0 | 10 ± 2 | N/A | N/A | N/A |
| 5 | 10 ± 3 | 4.8 | 99 | 0.99 | |
| 10 | 10 ± 4 | 9.7 | 97 | 1.94 | |
| 15 | 12 ± 5 | 14.8 | 98 | 2.94 | |
| 20 | 18 ± 6* | 13.2 | 66 | 2.64 | |
| 25 | 22 ± 8* | 7.3 | 30 | 1.5 |
The sample was filtered through 0.22 μM filter due to the precipitation.
Data represent the mean ± standard deviation (n ± 3).
The telodendrimer concentration was kept at 20 mg/ml.
The morphology and particle size distribution of DOX-loaded micelles were characterized by TEM and DLS, respectively. The particle sizes of DOX-loaded PEG5k-CA8 and PEG2k-CA4 micelles were in the range of 10–20 nm in diameter, when their DOX loading content is lower than 5% and 15%, respectively. TEM image (Fig. 1D) showed that DOX-loaded PEG2k-CA4 micelles (3 mg/ml DOX in 20 mg/ml PEG2k-CA4 telodendrimer) were spherical, with an average diameter of around 15 nm, which was consistent with the results obtained from the DLS particle sizer (Fig. 1C).
The release profiles of DOX from DOX-loaded micelles were determined by dialysis against PBS at 37 °C. As shown in Fig. 2A, 90% of free DOX diffused out of the dialysis cartridge within the first 4 hours. In contrast, the rate of DOX release was considerably slowed down by the micelle encapsulation. The DOX release patterns from both DOX-loaded micelles were biphasic, with the initial fast release of DOX (approximately 50% from DOX-PEG5k-CA8, and 35% from DOX-PEG2k-CA4) during the first 6 hours, followed by the slow linear release in the next 7 days. However, the DOX release rate from DOX-PEG2k-CA4 micelles was significantly slower than that from DOX-PEG5k-CA8 micelles. Even after one week, DOX-PEG2k-CA4 micelle solution still had deep red color indicative of DOX remained in the dialysis cartridge (Supplementary, Fig. S-2), suggesting more sustained drug release feature of DOX-PEG2k-CA4 micelles. The slower DOX release rate from DOX-PEG2k-CA4 micelles also reflects their better stability and stronger interaction between the nanocarrier and DOX drug [18].
Fig.2.

(A) Cumulative DOX release profiles from free DOX and DOX-loaded micelles measured by dialysis against PBS with activated charcoal at 37 °C. (B) DLS measurement of particle size change of DOX-loaded micelles, and Doxil® in 50% FBS over time at 37 °C. DOX loading level was 2 mg/ml DOX in 20 mg/ml telodendrimer, respectively.
The stability of DOX-loaded micelles was monitored by the DLS particle sizer. Both DOX-PEG5k-CA8 and DOX-PEG2k-CA4 micelles (2 mg/ml DOX in 20 mg telodendrimer) were found to be very stable at 4 °C for over one month, with no significant change in particle sizes. To further test their stability in the physiological condition, both DOX-loaded micelles and Doxil® were incubated with 50% FBS for up to 72 h at 37 °C. DOX-PEG2k-CA4 micelles as well as Doxil® were able to maintain their initial particle sizes over 72 h incubation in the presence of 50% FBS. However, DOX-PEG5k-CA8 micelles started to form bigger aggregates (around 300 nm) after a 6-hour of incubation with 50% FBS (Fig. 2B). The above results suggest that the DOX-PEG2k-CA4 formulation may exhibit higher in vivo stability than the DOX-PEG5k-CA8 formulation.
3.2 Cellular uptake and intracellular distribution of DOX-loaded micelles
The intracellular uptake and localization of free DOX and DOX-loaded micelles in Raji cells were first investigated using confocal fluorescence microscopy. At 30 min after incubation, DOX fluorescence from both free DOX and DOX-loaded micelles were mainly distributed in the cytoplasm with slight fluorescence in the nuclear region (Fig. 3A). Interestingly, some dot-shape fluorescent foci were observed at the perinuclear region of the cytoplasm in the cells treated with DOX-loaded micelles, but not in the cells treated with free DOX. Similar observation was also reported by Shuai and coworkers in the MCF-7 cells incubated with DOX-loaded PEG-PCL (poly (ε-caprolactone)) micelles [19]. These fluorescent foci are probably due to the micelles trapped in the endocytic vesicles (i.e. endosomes and lysosomes) upon cellular uptake. This suggests that the endocytosis pathway is likely to be involved in the internalization process of DOX-loaded micelles in this study, which is different from the diffusion pathway of free DOX. At 2 hrs after incubation, the majority of DOX were transported into the nuclei as indicated by the weak DOX fluorescence in the cytoplasm, although some perinuclear cytoplasmic fluorescent foci did remain in the DOX-PEG2k-CA4 treated sample, as shown in Fig. 3B. This indicated that DOX was released from micelles in the endocytic compartments with a much faster rate than the in vitro release study. The mechanism of faster intracellular drug release from those micelles is still unclear. The complexity of intracellular environment, e.g. lower pH, and the presence of many enzymes in the endosomes and lysosomes likely plays an important role.
Fig.3.

Confocal images of Raji cells incubated with 10 μM of free DOX and DOX-loaded micelles for 30 min (A) and 2 h (B); Quantitative analysis of cellular uptake of various DOX formulations by Raji cells after 30 min (C) and 2 h (D) incubation via flow cytometry.
The uptake efficiencies of various DOX formulations by Raji cells were further quantified by the flow cytometric analysis. The cellular uptake of each DOX formulation was in concentration- and time- dependent manners (Fig. 3C and D). However, the micellar formulations were demonstrated to have higher cellular uptake than free DOX. For examples, when Raji cells were incubated with 1 μM of various DOX formulations for 2 h, the median fluorescence intensity (MFI) of cells treated with DOX-PEG5k-CA8 and DOX-PEG2k-CA4 micelles were 1.8-fold and 1.9-fold higher than that of free DOX, respectively. Anthracycline drugs such as DOX have been proven to be P-glycoprotein (P-gp) substrates [20]. P-gp could efflux drugs out of tumor cells, contributing to multiple drug resistance (MDR) in tumor cells [21]. Several types of NPs drug delivery systems including polymeric micelles have demonstrated to be able to overcome MDR by preventing the drug efflux mediated by P-gp [22–24]. Therefore, the enhanced cellular uptake of DOX-loaded micelles demonstrated in this study suggests that our micellar formulations might prevent the extracellular efflux of DOX and overcome the DOX-associated MDR.
3.3 In vitro cytotoxicities
Cytotoxicities of blank and DOX-loaded micelles, free DOX and Doxil® against T-cell lymphoma cell lines (Jurkat and MOLT-4) and B-cell lymphoma cell lines (Raji and Ramos) after 72 h continuous exposure were determined by MTS assay. As shown in Fig. 4 and supplementary Fig. S-3, no obvious cytotoxic effects were observed with blank PEG5k-CA8 and PEG2k-CA4 micelles at the tested concentrations. However, both DOX-loaded micelles showed similar cytotoxicities against both T and B lymphoma cells compared with free DOX and Doxil®, with the IC50 values ranging from 20 to 50 ng/ml DOX.
Fig.4.
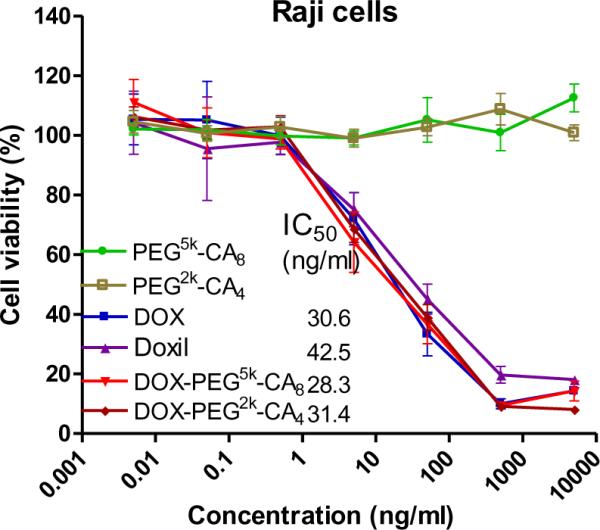
The cell viability of Raji lymphoma cells after 72 h of incubation with blank and DOX-loaded micelles, compared with free DOX and Doxil®.
3.4 MTD study
Dose-finding studies were conducted to establish the MTD for DOX-loaded micelles in non-tumor bearing female BALB/c mice. Four mice per group were injected intravenously with a single dose of either free DOX or DOX-loaded PEG2k-CA4 micelles at a dose range of 5–30 mg/kg. There were no obvious body weight loss and other toxicity signs in the mice treated with DOX-PEG2k-CA4 micelles at the doses of 5–10 mg DOX/kg throughout the experiment (Supplementary, Fig. S-4). Only moderate body weight loss (< 10%) was found in mice treated with 15 mg/kg of DOX-PEG2k-CA4 micelles during the first week of treatment, which was completely recovered at the end of the second week. In contrast, 15 mg/kg of free DOX caused a sharp decrease (> 15%) in the body weight, and resulted in the death of all the animals in this group (Supplementary, Table S-1). Although the mice treated with lower than 10 mg/kg of free DOX survived with moderate body weight loss. Therefore, the MTD of DOX-PEG2k-CA4 micelles and free DOX in BLAB/c mice are approximately 15 mg/kg and 10 mg/kg, respectively. The MTD of free DOX reported here is also consistent with previous results (8–12 mg/kg) from several independent studies [9, 25, 26]. Whereas, the MTDs of Doxil®, a very stable nanoformulation of DOX, were reported to be only comparable or even lower than that of free DOX, respectively in dogs and mice [27, 28]. In this study, the 1.5-fold higher of MTD for DOX-PEG2k-CA4 micelles compared to free DOX is likely related to the prolonged circulation time and the controlled drug release property, as well as the improved biodistribution profile of the micellar formulation.
3.5 Pharmacokinetics and biodistribution
Free DOX, DOX-PEG5k-CA8 and DOX-PEG2k-CA4 at the dose of 10 mg/kg were administered intravenously into tumor-free BALB/c mice, respectively. The blood were collected and extracted for the DOX fluorescence measurement at different time points [1]. A standard curve of DOX in blood was generated by the extraction and measurement of predetermined concentrations of DOX in the blood, and there was a good linear correlation (R2 = 0.9994) between the fluorescence intensity and the DOX concentration in the range of 0.39–100 μg/ml (Supplementary, Fig. S-5A). As shown in Fig. 5A, when compared to free DOX, both DOX micellar formulations significantly increased the retention time of DOX in the blood. The pharmacokinetic parameters were summarized in supplementary Table S-2, and the results revealed that the administration of the micellar formulations DOX-PEG5k-CA8 and DOX-PEG2k-CA4 resulted in substantially higher Cmax and AUC compared to free DOX. The AUC0-inf of DOX-PEG5k-CA8 and DOX-PEG2k-CA4 were 10-fold and 14-fold, respectively higher than that of free DOX, whereas the Vd and CL were significantly lower than that of free DOX, suggesting the prolonged blood circulation property of the micellar DOX formulations.
Fig.5.

Pharmacokinetics (A) and biodistribution data (B) of free DOX and DOX-loaded micelles given intravenously at a dose of 10 mg/kg in mice.
In another set of experiment, various DOX formulations at the dose of 10 mg/kg body weight were injected intravenously into Raji lymphoma bearing mice, respectively. The recovery standard curve for each tissue is present in supplementary Fig. S-5 (B–G). A strong linear correlation between DOX concentration and fluorescence intensity was found in each tissue, with the correlation coefficient (R2) in tumor, heart, liver, spleen, lung, and kidney being 0.9983, 0.9978, 0.9827, 0.9992, 0.9999 and 0.9957, respectively. At 16 h post-injection, major organs and tumor tissue were excised for the measurement of DOX amount. Data in Fig. 5B demonstrated that both micellar formulations significantly enhanced the accumulation of DOX into the tumor tissue, compared to free drug (P < 0.05). The DOX uptake in the tumor tissue of mice treated with DOX-PEG5k-CA8 and DOX-PEG2k-CA4 micelles were 2.0-fold, and 2.2-fold higher than that in the free DOX group, respectively. The preferential tumor uptake of both DOX micellar formulations is likely due to the prolonged circulation and the EPR effect. It was also noted that both micellar formulations reduced significant amounts of drug distribution into the heart compared with free DOX, which may lower the DOX-associated cardiotoxicity. It is not surprising that both micellar formulations have relatively higher uptake in the liver and spleen compared with free DOX, which could be explained by the nonspecific elimination of micellar NPs via the reticuloendothelial system (RES) such as macrophage in the liver and spleen. However, liver function tests were found to be normal in the following in vivo therapeutic studies (Supplementary, Table S-4).
3.6 NIRF optical imaging
NIRF dyes with high penetration, low tissue absorption and scattering enable the deep tissue imaging [29]. To track the in vivo fate of the payload or the nanocarriers, NIRF dye DiD or BODIPY 650/665 was either physically encapsulated into the micelles or chemically conjugated to the telodendrimer, respectively [30]. The in vivo optical images in Fig. 6A demonstrated that DiD-loaded PEG2k-CA4 micelles were able to gradually accumulate at the Raji tumor starting at 1 h after intravenous administration, and the micellar NPs were retained in the tumor throughout the 48 h period. In contrast, no obvious tumor accumulation was observed all the time in the mice injected with free DiD dye. At 48 h post-injection, all tumors and other major organs were excised for ex vivo NIRF imaging to determine the tissue distribution of NPs. As shown in Fig. 6B, DiD-loaded PEG2k-CA4 micelles mainly accumulated in the tumor tissue, with significantly higher fluorescence signal than other normal organs, whereas in the mice treated with the free DiD, DiD was mainly taken up by spleen and lung, instead of the tumor. The preferential tumor uptake of DiD-loaded PEG2k-CA4 micelles in the NIRF optical imaging study correlates very well with the above biodistribution results of DOX-PEG2k-CA4 micelles.
Fig.6.

In vivo (A) and ex vivo (B) NIRF optical images of Raji lymphoma bearing mice injected intravenously with free DiD dye and DiD-loaded PEG2k-CA4 micelles, respectively. Tumors and major organs were excised for ex vivo imaging at 48 h post-injection.
To simultaneously monitor the in vivo fates of both DOX drug payload and the telodendrimer carrier, BODIPY650/665 dye chemically conjugated PEG5k-CA8 micelles loaded with DOX were intravenously injected into the nude mice bearing B-cell lymphoma (Raji and Ramos) and T-cell lymphoma (MOLT-4), respectively. The in vivo and ex vivo NIRF optical images at 24 h post-injection demonstrated that DOX-loaded BODIPY650/665 labeled PEG5k-CA8 micelles could predominantly accumulate in B-cell lymphomas such as Raji and Ramos (Supplementary, Fig. S-6A and B). However, the uptake of PEG5k-CA8 micelles in the MOLT-4 T-cell lymphoma was relatively low (Supplementary, Fig. S-6C). This could be attributed to the insufficient “leaky” vasculature network in the MOLT-4 tumor as demonstrated in our previous study [30], which is essential for the EPR effects of NPs in tumors. The microscopic analysis further demonstrated that the majority of micellar NPs were able to penetrate deep into the tumor, and efficiently release the DOX payload from the micelles after their permeation from the blood vessels. This is evidenced by the strong BODIPY650/665 fluorescence from the micellar carrier especially in the perivascular region, and the even distribution of DOX fluorescence throughout the tumor tissue in Raji lymphoma (Supplementary, Fig. S-6D).
3.7 In vivo therapeutic study
The anti-tumor activities of DOX micellar formulations were evaluated in the subcutaneous Raji lymphoma mouse model in comparison with the clinical formulations of DOX. Free DOX, Doxil®, DOX-PEG5k-CA8 and DOX-PEG2k-CA4 micelles at the equivalent DOX dose of 10 mg/kg (MTD of free DOX), as well as PBS control were administered intravenously into Raji lymphoma bearing mice every four days on days 0, 4, and 8, respectively (n = 5–8). The tumor growth inhibition and survival rate of mice in different groups were compared and the results are shown in Fig 7. Compared with the control group, mice in all the DOX treatment groups showed significant inhibition of tumor growth (P < 0.05). However, the tumor growth rates of mice treated with both liposomal and micellar DOX formulations were significantly lower (P < 0.05), compared to those in the free DOX treatment group. It can be attributed to the higher amount of DOX that reached the tumor site via the EPR effects for both liposomal and micellar NPs. More importantly, DOX-PEG2k-CA4 micelles exhibited even better tumor growth inhibition (P <0.05) than both DOX-PEG5k-CA8 micelles and Doxil®. For example, by day 28, the median RTV was 9.9 for mice treated with free DOX, while the RTVs for mice treated with Doxil®, DOX-PEG5k-CA8 and DOX-PEG2k-CA4 micelles were 7.6, 7.7 and 6.8, respectively. Compared to Doxil®, the superior tumor inhibition of DOX-PEG2k-CA4 micelles could be partially explained by the deeper penetration capability throughout the tumor tissue due to their significantly smaller particle sizes (12 nm VS 140 nm) when reaching the tumor site via efficient EPR effect. Compared to DOX-PEG5k-CA8 micelles, DOX-PEG2k-CA4 micelles have relatively longer retention time and slower drug release rate as demonstrated earlier, which may facilitate the delivery of more DOX drugs to the tumor site, resulting in the improved anti-tumor efficacy. The survival rate of mice in each group is presented by the Kaplan-Meier survival curve, respectively (Fig. 7B). In general, compared to PBS control, all the DOX formulations significantly prolonged the survival rates of tumor bearing mice. However, mice treated with DOX-PEG2k-CA4 micelles achieved the longest survival time among all the DOX formulations. The median survival time of mice in the group of PBS control, free DOX, Doxil®, DOX-PEG5k-CA8 and DOX-PEG2k-CA4 micelles was 20, 32, 36.5, 36 and 41 days, respectively. Such observations are also consistent with the above tumor growth inhibition result, owing to more efficient in vivo delivery of DOX to cancer cells via PEG2k-CA4 micelles, compared to free DOX and other nanoformulations in this study.
Fig.7.

In vivo anti-tumor efficacy (A), Kaplan-Meier survival curve (B) and body weight changes (C) after intravenous treatment of different DOX formulations (10 mg/kg, three doses) in Raji lymphoma bearing mice (n = 5–8).
The toxicities of all the treated mice were monitored by the body weight change, blood cell counts and serum chemistry including hepatic and renal function panels as well as cardiac enzymes. Compared to PBS control group, mice given all the DOX formulations exhibited initial body weight loss to varying extent, followed by the recovery of body weight one week after the end of treatment (Fig. 7C). However, the body weight loss of mice in the free DOX group was significantly higher than other DOX nanoformulation groups (P < 0.05), leading to one death on day 16. One week after the last dose of DOX formulations, blood samples were collected on day 15 for blood cell counts and serum chemistry analysis. Compared to PBS control group, the WBC count in free DOX group significantly decreased (P < 0.05), whereas the WBC counts in all the DOX nanoformulations groups were within the normal range (Supplementary, Table S-3). The hepatic and renal function tests including ALT, AST and BUN were within the normal ranges for all the groups (Supplementary, Table S-4). Importantly, encapsulation of DOX in the micellar NPs was found to decrease the cardiotoxicity compared with free drug. Serum CK and LDH levels are two of well-characterized markers for cellular damage in a variety of cardiac disease models. The induction of CK and LDH enzymes in the serum of mice treated with free DOX was significantly increased (P < 0.05), compared with untreated mice (Fig. 8). However, when mice were treated with DOX-loaded micelles, both serum CK and LDH levels were not significantly different from the PBS control group (P > 0.05), but significantly lower than those in the group treated with free DOX (P < 0.05). The decreased cardiotoxicity of DOX micellar formulations can be attributed to the reduced uptake in the heart, as demonstrated in the above in vivo biodistribution study. Although there were no significant findings in the histological examination of the heart at one week after completion of treatment, it is likely that the short follow-up time was not sufficient to discern major histological changes.
Fig.8.

Serum creatine kinase (CK) (A) and lactate dehydrogenase (LDH) (B) on day 7 after the last dosage of different DOX formulations in Raji tumor bearing mice. Each data point is represented as mean ± SEM. * P < 0.05.
A high-affinity and high-specificity peptidomimetic ligand (LLP2A; IC50 = 2 pM) against both T- and B- cell lymphomas has been identified through screening the one-bead one-compound (OBOC) combinatorial libraries in our laboratory [29]. It is expected that the decoration of our telodendrimer micelle system with LLP2A targeting ligand will further enhance the tumor accumulation and anti-cancer efficacy of delivered drugs, while reducing the side effects. The physicochemical characterization, in vitro and in vivo therapeutic efficacy of LLP2A-targted micelle system for the treatment of lymphoma are currently being investigated in our laboratory.
4. Conclusion
PEG-oligocholic acid telodendrimer micellar system has been successfully applied for the efficient delivery of DOX in the treatment of B-cell lymphoma. Compared with the conventional PEG5k-CA8 micelles reported previously, PEG2k-CA4 micelles exhibited higher drug loading capacity for DOX, better stability in physiological condition, and much slower drug release profiles. Both DOX-loaded micellar formulations were able to be efficiently taken up by Raji lymphoma cells via the endocytosis pathway, which may overcome the MDR. Both DOX-loaded micelles exhibited similar in vitro cytotoxicity with free DOX. Compare to free DOX, DOX-loaded micelles (especially DOX-PEG2k-CA4) had prolonged blood residence time, and preferential uptake in B-cell lymphoma via the EPR effect. Furthermore, the MTD of DOX-PEG2k-CA4 was found to be 1.5-fold higher than that of free DOX, suggesting that the micellar formulation is more tolerated. Finally, DOX-PEG2k-CA4 micelles were demonstrated to be more efficacious than the equivalent dose of free DOX and liposomal DOX formulation (Doxil®) in Raji lymphoma bearing mice, and significantly reduced the DOX-associated cardiotoxicity.
Supplementary Material
Acknowledgments
The authors thank the financial support from NIH/NCI R01CA140449 (Luo), R01CA115483 (Lam), and US Department of Defense Postdoctoral Training Award (W81XWH-10-1-0817 Xiao).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Park J, Fong PM, Lu J, Russell KS, Booth CJ, Saltzman WM, Fahmy TM. PEGylated PLGA nanoparticles for the improved delivery of doxorubicin. Nanomedicine. 2009;5(4):410–418. doi: 10.1016/j.nano.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fukumori Y, Ichikawa H. Nanoparticles for cancer therapy and diagnosis. Advanced Powder Technology. 2006;17(1):1–28. [Google Scholar]
- [3].Sharma G, Anabousi S, Ehrhardt C, Kumar MNVR. Liposomes as targeted drug delivery systems in the treatment of breast cancer. Journal of Drug Targeting. 2006;14(5):301–310. doi: 10.1080/10611860600809112. [DOI] [PubMed] [Google Scholar]
- [4].Kazakov S, Levon K. Liposome-nanogel structures for future pharmaceutical applications. Current Pharmaceutical Design. 2006;12(36):4713–4728. doi: 10.2174/138161206779026281. [DOI] [PubMed] [Google Scholar]
- [5].Gillies ER, Frechet JMJ. Dendrimers and dendritic polymers in drug delivery. Drug Discovery Today. 2005;10(1):35–43. doi: 10.1016/S1359-6446(04)03276-3. [DOI] [PubMed] [Google Scholar]
- [6].Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Advanced Drug Delivery Reviews. 2004;56(9):1273–1289. doi: 10.1016/j.addr.2003.12.004. [DOI] [PubMed] [Google Scholar]
- [7].Andresen TL, Jensen SS, Jorgensen K. Advanced strategies in liposomal cancer therapy: problems and prospects of active and tumor specific drug release. Prog Lipid Res. 2005;44(1):68–97. doi: 10.1016/j.plipres.2004.12.001. [DOI] [PubMed] [Google Scholar]
- [8].El-Rayes BF, Ibrahim D, Shields AF, LoRusso PM, Zalupski MM, Philip PA. Phase I study of liposomal doxorubicin (Doxil) and cyclophosphamide in solid tumors. Invest New Drugs. 2005;23(1):57–62. doi: 10.1023/B:DRUG.0000047106.20104.a7. [DOI] [PubMed] [Google Scholar]
- [9].Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjug Chem. 2005;16(1):122–130. doi: 10.1021/bc0498166. [DOI] [PubMed] [Google Scholar]
- [10].Tsukioka Y, Matsumura Y, Hamaguchi T, Koike H, Moriyasu F, Kakizoe T. Pharmaceutical and biomedical differences between micellar doxorubicin (NK911) and liposomal doxorubicin (Doxil) Japanese Journal of Cancer Research. 2002;93(10):1145–1153. doi: 10.1111/j.1349-7006.2002.tb01217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xiao K, Luo J, Fowler WL, Li Y, Lee JS, Xing L, Cheng RH, Wang L, Lam KS. A self-assembling nanoparticle for paclitaxel delivery in ovarian cancer. Biomaterials. 2009;30(30):6006–6016. doi: 10.1016/j.biomaterials.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li Y, Xiao K, Luo J, Lee J, Pan S, Lam KS. A novel size-tunable nanocarrier system for targeted anticancer drug delivery. J Control Release. 2010;144(3):314–323. doi: 10.1016/j.jconrel.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Luo J, Xiao K, Li Y, Lee JS, Shi L, Tan YH, Xing L, Holland Cheng R, Liu GY, Lam KS. Well-defined, size-tunable, multifunctional micelles for efficient paclitaxel delivery for cancer treatment. Bioconjug Chem. 2010;21(7):1216–1224. doi: 10.1021/bc1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32(13):3435–3446. doi: 10.1016/j.biomaterials.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li Y, Xiao K, Luo J, Xiao W, Lee JS, Gonik AM, Kato J, Dong TA, Lam KS. Well-defined, reversible disulfide cross-linked micelles for on-demand paclitaxel delivery. Biomaterials. 2011 doi: 10.1016/j.biomaterials.2011.05.050. In Press, Corrected Proof, DOI: 10.1016/j.biomaterials.2011.1005.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kim S, Shi Y, Kim JY, Park K, Cheng JX. Overcoming the barriers in micellar drug delivery: loading efficiency, in vivo stability, and micelle-cell interaction. Expert Opin Drug Deliv. 7(1):49–62. doi: 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
- [17].Letchford K, Liggins R, Burt H. Solubilization of hydrophobic drugs by methoxy poly(ethylene glycol)-block-polycaprolactone diblock copolymer micelles: theoretical and experimental data and correlations. J Pharm Sci. 2008;97(3):1179–1190. doi: 10.1002/jps.21037. [DOI] [PubMed] [Google Scholar]
- [18].Du YZ, Weng Q, Yuan H, Hu FQ. Synthesis and antitumor activity of stearate-g-dextran micelles for intracellular doxorubicin delivery. ACS Nano. 4(11):6894–6902. doi: 10.1021/nn100927t. [DOI] [PubMed] [Google Scholar]
- [19].Shuai X, Ai H, Nasongkla N, Kim S, Gao J. Micellar carriers based on block copolymers of poly(epsilon-caprolactone) and poly(ethylene glycol) for doxorubicin delivery. J Control Release. 2004;98(3):415–426. doi: 10.1016/j.jconrel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- [20].Lum BL, Gosland MP, Kaubisch S, Sikic BI. Molecular targets in oncology: implications of the multidrug resistance gene. Pharmacotherapy. 1993;13(2):88–109. [PubMed] [Google Scholar]
- [21].Wang F, Wang YC, Dou S, Xiong MH, Sun TM, Wang J. Doxorubicin-Tethered Responsive Gold Nanoparticles Facilitate Intracellular Drug Delivery for Overcoming Multidrug Resistance in Cancer Cells. ACS Nano. 2011;5(5):3679–3692. doi: 10.1021/nn200007z. [DOI] [PubMed] [Google Scholar]
- [22].Hu FQ, Liu LN, Du YZ, Yuan H. Synthesis and antitumor activity of doxorubicin conjugated stearic acid-g-chitosan oligosaccharide polymeric micelles. Biomaterials. 2009;30(36):6955–6963. doi: 10.1016/j.biomaterials.2009.09.008. [DOI] [PubMed] [Google Scholar]
- [23].Shieh MJ, Hsu CY, Huang LY, Chen HY, Huang FH, Lai PS. Reversal of doxorubicin-resistance by multifunctional nanoparticles in MCF-7/ADR cells. J Control Release. 2011;152(3):418–425. doi: 10.1016/j.jconrel.2011.03.017. [DOI] [PubMed] [Google Scholar]
- [24].Kievit FM, Wang FY, Fang C, Mok H, Wang K, Silber JR, Ellenbogen RG, Zhang M. Doxorubicin loaded iron oxide nanoparticles overcome multidrug resistance in cancer in vitro. J Control Release. 2011;152(1):76–83. doi: 10.1016/j.jconrel.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Breistol K, Hendriks HR, Fodstad O. Superior therapeutic efficacy of N-L-leucyl-doxorubicin versus doxorubicin in human melanoma xenografts correlates with higher tumour concentrations of free drug. Eur J Cancer. 1999;35(7):1143–1149. doi: 10.1016/s0959-8049(99)00074-x. [DOI] [PubMed] [Google Scholar]
- [26].Boven E, Schluper HM, Erkelens CA, Pinedo HM. Doxorubicin compared with related compounds in a nude mouse model for human ovarian cancer. Eur J Cancer. 1990;26(9):983–986. doi: 10.1016/0277-5379(90)90626-5. [DOI] [PubMed] [Google Scholar]
- [27].Gabizon AA. Pegylated liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy. Cancer Invest. 2001;19(4):424–436. doi: 10.1081/cnv-100103136. [DOI] [PubMed] [Google Scholar]
- [28].Vail DM, Kravis LD, Cooley AJ, Chun R, MacEwen EG. Preclinical trial of doxorubicin entrapped in sterically stabilized liposomes in dogs with spontaneously arising malignant tumors. Cancer Chemother Pharmacol. 1997;39(5):410–416. doi: 10.1007/s002800050591. [DOI] [PubMed] [Google Scholar]
- [29].Peng L, Liu R, Marik J, Wang X, Takada Y, Lam KS. Combinatorial chemistry identifies high-affinity peptidomimetics against alpha4beta1 integrin for in vivo tumor imaging. Nat Chem Biol. 2006;2(7):381–389. doi: 10.1038/nchembio798. [DOI] [PubMed] [Google Scholar]
- [30].Xiao K, Luo J, Li Y, Xiao W, Lee JS, Gonik AM, Lam KS. The passive targeting of polymeric micelles in various types and sizes of tumor models. Nanosci Nanotechnol Lett. 2010;2(2):79–85. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.