

Abstract
Background
Currently low molecular weight heparin (LMWH) is administered as subcutaneous injection. This study sought to investigate the feasibility of LMWH as an inhalable dry powder (DPI) formulation and evaluate the interaction of the drug with lactose when used as a carrier. The study also compares the extent of pulmonary absorption of LMWH administered as a dry powder with that administered as an aerosolized aqueous solution.
Methods
The formulations were prepared by mixing LMWH in an aqueous solution of lactose followed by lyophilization of the resulting solution. The lyophilized preparation was then ground and sieved. Physical characterization of the formulations was performed by Fourier transform infrared spectroscopy (FTIR), X-ray powder diffraction (XRD), scanning electron microscopy (SEM), particle size analysis, and determination of aerodynamic diameter. For in vivo studies, formulations were administered to anesthetized rats, and drug absorption was monitored by measuring plasma antifactor Xa activity.
Results and Conclusions
In the FTIR scan, all characteristic peaks of lactose and LMWH were observed, suggesting that there was no strong interaction between lactose and LMWH. Although the aerodynamic diameter of the formulation (DPI-2) that was sieved through 170- and 230-mesh screens was similar to that of the formulation (DPI-1) sieved through 120- and 170-mesh screens, the particle sizes of the two formulations were significantly different. Dry powder formulations of LMWH were better absorbed compared to an inhalable solution of LMWH. One of the dry powder formulations (DPI-2) produced an almost 1.5-fold increase in the relative bioavailability (41.6%) compared to the liquid formulation of LMWH (32.5%). Overall, the data presented here suggest that lactose does not adversely affect the physical-chemical characteristics of the drug, and that lactose can be used as a carrier for pulmonary delivery of LMWH.
Key words: LMWH, inhalation, dry powder, pulmonary absorption
Introduction
Currently, pulmonary drug delivery systems and/or devices are classified into three major categories: nebulizers, pressurized metered dose inhalers (pMDIs), and dry powder inhalers (DPIs). In nebulizers and pMDIs, drugs are dispersed in water or propellant–solvent mixtures, and administered as aerosolized droplets of the dispersed systems. But in DPIs, drugs are administered as a pure powder or after formulating them with inert carriers, including lactose, glucose, and mannitol. Compared to other pulmonary drug delivery systems, DPIs offer several advantages, including enhanced drug stability, greater accuracy in dosing, elimination of hand-to-mouth coordination, breath-actuated delivery, and consequently, an overall improvement in patient compliance.(1) DPIs are particularly attractive for delivery of protein and peptide drugs that are susceptible to degradation in aqueous solution. Because these relatively new inhalable formulations do not require the use of propellants, many drugs used in the treatment of respiratory diseases, such as asthma, chronic obstructive pulmonary diseases, and cystic fibrosis,(2,3) are currently available as DPIs. DPIs are also a very attractive platform technology for needle-free formulations of drugs that undergo degradation in the gastrointestinal tract, have low oral bioavailability, or are required to be administered by injections.(4)
Low molecular weight heparins (LMWHs) are negatively charged compounds that undergo degradation in the acidic environment of the gastrointestinal tract. Currently, LMWHs are available for administration by subcutaneous injection. However, administration of drugs using needles is not desirable in many patient populations, including young children and adults. Although currently available LMWHs can be self-administered, patients are required to be trained in safe administration of a sterile drug formulation. Unsafe administration may result in infection and hematoma at the site of administration, which often leads to noncompliance. For this reason, many patients prefer that drugs be administered by a health care professional, which involves extra cost. Recently, we and others have proposed that administration of LMWH by noninvasive routes such as oral, nasal, and pulmonary can address many of the limitations associated with injectable LMWH. In a series of articles, we have shown that it is feasible to administer LMWH via the oral,(5) nasal,(6) and pulmonary(7) routes and that the absorption of LMWH via the respiratory route can be increased in the presence of absorption promoters, including alkylmaltosides and cyclodextrins.(5–8) However, there are concerns regarding the use of absorption promoters in drug formulations that are required to be administered via the respiratory route for long-term therapy. One of the safest formulation approaches is to prepare an aqueous solution or dry powder of the drug that can be inhaled as aerosolized droplets or particles. Previous studies in our laboratory suggest that an aerosolized aqueous solution of LMWH administered via the nasal and pulmonary routes does not produce therapeutic levels of antifactor Xa.(8) In a separate study, we showed that insulin administered as an inhalable dry powder is better absorbed compared to an aqueous solution of insulin.(9)
Currently, it is not known if LMWH administered as a respirable dry powder in the presence or absence of inert carriers will produce therapeutic levels of antifactor Xa. The interaction of the drug, a negatively charged polysaccharide, with a widely used carrier, lactose, is also unknown. In fact, formulation characteristics are critically important for the overall performance of dry powder-based inhalable formulations. The physicochemical properties of both drug and carriers have important effects on the fluidization, dispersion, delivery, and deposition of the formulations in the airways. Particle size, shape, surface area, and morphology affect the interparticular forces of interaction and aerodynamic properties of the formulations.(3) Therefore, to develop an efficacious and optimal formulation, the physiochemical properties of the drug and formulation adjuvants and their interactions are important to determine. As indicated above, lactose, glucose, mannitol, and sucrose have traditionally been used as inert diluents in inhalable dry powder formulations. Lactose monohydrate is the common carrier used in DPIs. In fact, most of the current DPIs available in United States contain lactose.(10) Lactose has become an ideal carrier for DPIs because of its well-established safety profiles, ready availability, and low cost.(11,12) Furthermore, crystalline lactose has smooth surfaces, a regular shape, and good flow properties.(13) This study, therefore, sought to evaluate the feasibility of LMWH as an inhalable dry powder formulation as well as the interaction of the drug with lactose. The pulmonary absorption of LMWH administered as a dry powder was also compared with that administered as an aerosolized aqueous solution of the drug.
Materials and Methods
Materials
Enoxaparin (Lovenox®) was obtained from Sanofi-Aventis Inc. (Bridgewater, NJ) as a sterile solution containing 30 mg of enoxaparin sodium (3,000 U of antifactor Xa activity, average MW: 4,500 Da) per 0.3 mL. Alpha-lactose monohydrate was a product of Fisher Scientific (Fair Lawn, NJ).
Preparation of dry powder formulations of LMWH
To prepare dry powder formulations of LMWH, a commercial LMWH injection solution (Lovenox®) was first diluted in saline and then dissolved in a 5, 10, or 15% lactose solution. The resulting solution was lyophilized for 16 h at −50°C and 120 mT using an EZ-DRY lyophilizer (FTS Systems Inc., Stone Ridge, NY). Following lyophilization, the powder was ground manually and divided into two fractions by means of sieving. The first fraction (DPI-1) was sieved through 120- and 170-mesh sieves and the second fraction (DPI-2) was collected by passing the powder through 170- and 230-mesh sieves. These formulations were used for physical characterization and pulmonary absorption studies. The final formulation contained 0.5U of LMWH per 1 mg of powder. For the FTIR studies, the samples were prepared by mixing LMWH and lactose in a 1:1 (w/w) ratio; the amount of drug in these samples was 600 times than that used in preparing the other samples.
Characterization of dry powder formulations of LMWH
Different instrumental techniques were used to characterize the formulations for drug–excipient interaction, particle morphology, crystallinity, surface area. and particle size.
Fourier transform infrared (FTIR) spectroscopy. Interactions between the excipients and LWMH were studied by FTIR. The IR spectra of the drug solution, solid lactose, and lyophilized LMWH plus lactose powder were obtained in a Nexus 470 FTIR (Thermo Nicolet Corporation, Madison, WI). The spectra were recorded under an automatic atmosphere suppression mode, and the number of scans and resolution were 32 and 4, respectively. The scans were performed between wavelengths 4000 and 700 cm−1.
X-ray powder diffraction (XRD). The effect of increasing concentrations (5, 10, and 15%, w/v) of lactose on the crystal structure of dry powder of LMWH was investigated by XRD analysis. The data were generated in a Siemens D5000 Diffractometer (Bruker AXS Inc., Madison, WI) and collected using primary monochromated radiation (CuKα1, λ = 1.54056 Å), at 2θ of 0.05, and a dwell time of 1.0 sec per step.
Brunauer-Emmett-Teller (BET) specific surface area analysis. The specific surface area of the two optimized formulations with different particle size ranges was measured in a TriStar™ 3000 Surface Area Analyzer (Micromeritics Instrument Corporation, Norcross, GA). Weighed powder was placed onto a 12-mm bulb sample cell and degassed for a minimum of 3 h. The recorded data were analyzed using TriStars Confirm™ software (version 6.04), and the surface area was determined according to the BET principle of surface area.
Scanning electron microscopic (SEM) analysis. SEM images of the formulations were obtained in a Hitachi S-3400-II scanning electron microscope (Hitachi High Technologies America Inc, Schaumburg, IL) equipped with an image analyzer. The samples were prepared on a conductive, double-sided adhesive tape and then sputter-coated with gold under argon at an atmospheric pressure of 50 mPa.
Determination of particle size and aerodynamic diameters. The particle size distribution and mean volume-based diameter of the formulations were studied by using a Microtrac® S3500 (North Largo, FL) particle size analyzer. Samples for particle size analysis were prepared by dispersing the freeze-dried formulations in 500 μL water. One or two drops of Tween 80 was added to the mixture to produce an even dispersion of the particles in water. The aerodynamic diameter of the dry powder formulations was determined in an eight-stage Andersen cascade impactor (Westech Instruments Inc., Marietta, GA). The formulations were fired three times into the cascade impactor at a flow rate of 28.3 L/min. The deposited formulation at each stage was collected on filter papers and weighed to determine dry powder content. The aerodynamic diameter was calculated according to the instructions supplied by Westech Instruments Inc.
Pulmonary absorption studies
Male Sprague-Dawley rats (Charles River Laboratories, Charlotte, NC) weighing 250–350 g were used for the in vivo absorption experiments (n = 3–5). Prior to the experiment, the animals were anesthetized by an intramuscular injection of an anesthetic mixture containing xylazine (20 mg/mL) and ketamine (100 mg/mL). Anesthesia was maintained with additional doses of anesthetic solution as needed throughout the experiments. LMWH solution containing enoxaparin (50 U/kg) or dry powder formulations containing enoxaparin (75 U/kg) was administered intratracheally as described earlier.(9) Briefly, the trachea was localized by using a small animal laryngoscope, a MicroSprayer® tube (Penn Century Inc., Philadelphia, PA) attached to a syringe was inserted into the trachea and the formulation in the syringe was quickly sprayed. Similarly, pulmonary administration of lyophilized dry powder was performed by using a dry powder insufflator (Penn Century Inc., Philadelphia, PA). The amount of formulation administered was 3.5–5.5 mg depending on animal body weight. For bioavailability studies, formulations were administered subcutaneously (50 U/kg) as a single injection under the back skin. After pulmonary and subcutaneous administration, blood samples (∼300 μL) were collected from the tip of the rat tail at 0, 30, 60, 120, 240, and 360 min in citrated microcentrifuge tubes and placed on ice. Subsequently, plasma was separated by centrifugation (3000×g for 10 min) and the plasma samples thus obtained were stored at −20°C until further analysis. LMWH absorption was determined indirectly by measuring plasma antifactor Xa levels using a colorimetric assay kit (Chromogenix Coatest Heparin Kit®, Diapharma Group Inc., West Chester, OH). The assay can reproducibly measure 0.05–0.7 U/mL of antifactor Xa activity in plasma samples.(14)
All animal studies were approved by the Texas Tech University Health Sciences Center (TTUHSC) Animal Care and Use Committee and were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals (Protocol # 02004).
Data analysis
Standard noncompartmental analysis (Kinetica®, Version 4.0, Innaphase Corp. Philadelphia, PA) was performed for the enoxaparin absorption profiles. The area under the plasma concentration versus time curve (AUC0→360) was calculated by the trapezoidal method. The relative bioavailability was estimated by comparing the AUC0→360 for enoxaparin after pulmonary delivery with that of subcutaneously administered enoxaparin. One-way analysis of variance (ANOVA) was used to compare the data. When the differences in the means were significant, post hoc pair wise comparisons were performed using the Newman-Keuls multiple comparison test (GraphPad Prism, version 3.03, GraphPad Software, San Diego, CA). Differences in p-values < 0.05 were considered statistically significant.
Results and Discussion
FTIR spectroscopy
FTIR spectroscopy was used to characterize possible interactions between LMWH and lactose carrier occurring after lyophilization. Because the proportion of LMWH in the formulations used in the pulmonary absorption studies was negligible compared to the amount of lactose, for the FTIR analysis the lyophilized formulations of LMWH and lactose was prepared at a ratio of 1:1 (w/w). Upon lyophilization, it is possible that the carboxyl or sulfate groups of LMWH interact with the hydroxyl groups of lactose and any such interactions would be reflected in the IR spectra as a shift in the band vibrations. LMWH is composed of repeating disaccharide units of D-glucosamine and uronic acid linked by 1, 4-interglycosidic bonds. The main functional groups in the disaccharide unit of LMWH are COO− and 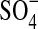 . LMWH showed a strong band for a carbonyl functional group at 1596 cm−1 that corresponds to the carboxylic acid group (COO−) present in LMWH. The peak at about 1222 cm−1 is probably from S = O stretching of the sulfate ion of LMWH (Fig. 1A). The spectrum of lyophilized LMWH:lactose (1:1) showed two distinct peaks at wavelengths 1596 and 1222 cm−1 that correspond to the carboxylic acid and sulfate groups of LMWH, respectively (Fig. 1A). Compared to plain LMWH, no shift in peak position was observed in the lyophilized LMWH-lactose formulations. The FTIR data suggest that a possible hydrogen bonding between the drug and lactose did not produce a significant change in dry powder formulation. However, the observed changes of peak intensity and stretching frequency may be a result of formation of H-bonds between the drug and excipient, although there was no shift in peak position (Fig. 1A). The FTIR spectra of LMWH matches those from our previous study wherein we reported an interaction between LMWH and dendrimer.(14) The FTIR data of lyophilized formulations also agree with previous IR spectra of freshly prepared solid dispersions of indomethacin, an acidic drug, with either lactose or Eudragit 100, in which no interactions between carboxyl groups of the drug and hydroxyl groups of the excipients were observed.(15)
. LMWH showed a strong band for a carbonyl functional group at 1596 cm−1 that corresponds to the carboxylic acid group (COO−) present in LMWH. The peak at about 1222 cm−1 is probably from S = O stretching of the sulfate ion of LMWH (Fig. 1A). The spectrum of lyophilized LMWH:lactose (1:1) showed two distinct peaks at wavelengths 1596 and 1222 cm−1 that correspond to the carboxylic acid and sulfate groups of LMWH, respectively (Fig. 1A). Compared to plain LMWH, no shift in peak position was observed in the lyophilized LMWH-lactose formulations. The FTIR data suggest that a possible hydrogen bonding between the drug and lactose did not produce a significant change in dry powder formulation. However, the observed changes of peak intensity and stretching frequency may be a result of formation of H-bonds between the drug and excipient, although there was no shift in peak position (Fig. 1A). The FTIR spectra of LMWH matches those from our previous study wherein we reported an interaction between LMWH and dendrimer.(14) The FTIR data of lyophilized formulations also agree with previous IR spectra of freshly prepared solid dispersions of indomethacin, an acidic drug, with either lactose or Eudragit 100, in which no interactions between carboxyl groups of the drug and hydroxyl groups of the excipients were observed.(15)
FIG. 1.

(A) FTIR analysis of a dry powder of enoxaparin solution, untreated lactose, and a lyophilized enoxaparin plus lactose (1:1 w/w) formulation; (B) X-ray diffractogram of a dry powder formulation of LMWH containing different concentrations of lactose: Formulation 1—LMWH in 5% lactose; Formulation 2—LMWH in 10% lactose; Formulation 3—LMWH in 15% lactose.
XRD
XRD analysis was performed to study the influence of lyophilization on the crystallinity of lactose and LMWH. Lactose shows crystalline structure when the concentration of lactose in the lyophilized formulation was 5% (Fig. 1B). With the increase in concentration of lactose from 5 to 10 or 15%, a reduction in peak intensity or no peak was observed suggesting that crystalline lactose has converted to an amorphous state. This change from crystalline to amorphous structure may stem from a number of factors including water content and concentration as discussed by Nail.(16) No peaks for LMWH were observed because the proportion of LMWH in the formulations was negligible compared to that of lactose. Moreover, the reduction in crystallinity was observed because of the masking of crystal surfaces by amorphous lactose. The degree of crystallinity can significantly influence the flow property of a pharmaceutical formulation. In fact, a favorable flow property is important for efficient delivery from the device and for desirable aerodynamic behavior and deposition patterns in the respiratory system. The flowability of moderately crystalline formulations is better compared to highly crystalline or amorphous materials. This assumption is based on the fact that in a moderately crystalline formulation, the proportion of amorphous material is relatively high. The amorphous fraction of the formulation coats the uneven surfaces of crystalline materials, filling the pores and crevices and thereby acts as glidants to increase the flowability by a phenomenon called the ball-bearing effect, which is observed during filling of hard gelatin capsules and compression of tablets.(17,18) However, purely amorphous substances tend to stick to plastic or metallic surfaces because of static electricity, and this phenomenon is probably responsible for poor flowability of amorphous substances. So it is reasonable to assume that because of the ball-bearing effect, moderately crystalline materials are likely to be emitted from the device with more ease compared to highly crystalline or amorphous formulations. As a result of enhanced flowability, a larger amount of drug will be deposited in the lungs and produce an improved absorption compared to highly crystalline or purely amorphous material. Based on the XRD data, we hypothesize that, in terms of flowability and based on the aerodynamic diameter and deposition patterns, Formulation 2 containing 10% lactose would be the better formulation for DPI.
SEM
The surface morphology of the particles was examined under a scanning electron microscope. SEMs of lactose revealed differences in particle size and morphology before and after lyophilization (Fig. 2A and B). Untreated lactose particles show rough surfaces and a well-known tomahawk shape, but upon lyophilization the particles assumed a more elongated and smooth surface structure (Fig. 2B). Because the proportion of LMWH in the actual formulation was negligible compared to the amount of lactose carrier, all of the observed particles are probably lactose particles. The particles of LMWH showed a clear crystalline structure after lyophilization (Fig. 2C). The lyophilized particles of the lactose–LMWH formulation showed small pores and crevices on their surface. Although a slight reduction in the crystallinity of the formulations was observed (Fig. 2D and E), any significant change in the crystal structure of the drug is unlikely to occur upon lyophilization. The porous nature of particles used in DPIs is important because increased porosity makes the particles lighter, which in turn, may result in a decrease in the aerodynamic diameter. Although the SEM analysis in the present study (Fig. 2) reveals the carrier's surface morphology, several other factors of a DPI formulation, including particle density and porosity, need to be studied further to obtain an optimized formulation for deeper lung deposition upon inhalation.
FIG. 2.

Scanning electron micrographs of (A) untreated lactose; (B) lyophilized lactose; (C) lyophilized LMWH; (D) DPI-1—sieved through 120- and 170-mesh sieves; (E) DPI-2—sieved through 170- and 230-mesh sieves.
Particle size, aerodynamic diameter and surface area analysis
The particle size data presented in Figure 3A show that the particle size of DPI-1 was 16.7 ± 5.5 μm, whereas that for DPI-2 was 11.6 ± 5.8 μm. The volume diameters of the particles agree with the SEM data, in that the particle size of DPI-2 is smaller than that of DPI-1. Although the mean volume diameter of the particles was > 1–5 μm, the aerodynamic diameter meets the criteria for respirability. In fact, the respirability of an inhaled formulation depends on the aerodynamic diameter, which dictates the deposition patterns of inhalable formulations. The differences between the mean volume and aerodynamic diameters stem from the fact that the aerodynamic diameter is influenced by particle density, shape factor, and geometric diameter, whereas volume diameter depends only on the physical size of the particle.(3) The particle morphology observed under SEM agrees with the differences observed between the actual particle size and the aerodynamic diameter. As the particles were elongated in shape with surface pores and crevices, the lyophilized particles showed favorable aerodynamic properties compared to the untreated particles. In fact, recent studies suggest that greater surface smoothness and elongation of lactose crystals aided in increasing the respirable fraction of sulbutamol.(19–21)
FIG. 3.

(A) Volume-based mean and aerodynamic diameter of dry powder formulations. Data represent mean ± SD, n = 3–5. (B) Surface area of LMWH dry powder formulations. Data represent mean ± SD, n = 3–5. *Results are significantly different, p < 0.05. DPI-1: sieved through 120- and 170-mesh sieves; DPI-2: sieved through 170- and 230-mesh sieves.
In addition to the particle size, the surface area of the formulations was determined. With a decrease in particle size there was an increase in surface area and subsequent increase in absorption. The surface area of the DPI-2 formulation (4.39 m2/g) was significantly larger than that of the DPI-1 formulation (1.55 m2/g) (Fig. 3B), suggesting that the particle size of DPI-2 is smaller than that of DPI-1, as was observed in the SEM and particle size analyses. In addition to the particle size, shape and surface morphology can have some influence on the surface area. For example, the surface area of smooth and nonporous particles is likely to be different from that of particles with uneven and porous particles. The greater surface area of the DPI-2 formulation suggests that this formulation will have a better dissolution profile in the respiratory fluid compared to the DPI-1 formulation.
Pulmonary absorption studies
The pharmacokinetics of LMWH solution and the dry powder formulations were studied by measuring plasma antifactor Xa levels after pulmonary administration of the formulations to anesthetized rats. Compared to subcutaneous injection of LMWH, LMWH solutions administered via the pulmonary route produced a relative bioavailability of ∼32.5% (Table 1), which is much higher than that administered via the nasal route.(8) Interestingly, administration of the two powder formulations led to different pulmonary absorption profiles (Fig. 4 and Table 1).The AUC0–360 value for DPI-2 with its smaller particle size was significantly higher than that of the liquid formulation of LMWH (p < 0.05). Furthermore, the AUC0–360 for the DPI-2 formulation was significantly higher than that for DPI-1 (p < 0.05). Although the relative bioavailability of DPI-1 was lower than that of the liquid formulation, DPI-2 produced an almost 1.5-fold increase in the relative bioavailability compared to the aerosolized solution of LMWH (F = 46.1% vs. 32.5%). Overall, when the absorption profiles of the dry powders were compared, the absorption of LMWH from dry powder of smaller particle size was significantly higher than from dry powder of larger size (Fig. 4 and Table 1).The differences between the pulmonary absorption profiles of the two formulations could be because of the differences in the respirable fraction of the particles. Although the aerodynamic diameter of both formulations was very similar, the volume diameter of DPI-2 was significantly smaller than that of DPI-1 (Fig. 3A). For this reason, the DPI-2 formulation is likely to contain a larger fraction of particles that are respirable compared to the DPI-1 formulation.
Table 1.
Pharmacokinetic Parameters of Different Enoxaparin Formulations after Subcutaneous or Pulmonary Administration
| Enoxaparin formulations | Cmax(U/mL) | T½(min) | AUC0→360(U · min/mL) | Frelative(%) |
|---|---|---|---|---|
| Solution | 0.166 ± 0.039 | 30 ± 0 | 31.9 ± 9.9 | 32.5 ± 10.0 |
| DPI-1 | 0.143 ± 0.030 | 30 ± 0 | 25.6 ± 12.4 | 22.1 ± 6.8 |
| DPI-2 | 0.279 ± 0.019 | 386 ± 81 | 67.9 ± 1.80 | 46.1 ± 1.2a |
| Subcutaneous | 0.349 ± 0.03 | 160 ± 61 | 98.24 ± 20.45 | — |
The dose administered was 50 U/kg and the data represent mean ± SD, n = 3–5.
Results are significantly different from those obtained with solution of LMWH, p < 0.05.
FIG. 4.

Changes in antifactor Xa activity after pulmonary administration of enoxaparin as dry powder or liquid formulations. Data represent mean ± SD, n = 3–5.
The results obtained in the present study are consistent with the findings reported by others,(9,22,23) that dry powders are more efficacious than a solution formulation for pulmonary delivery of insulin and calcitonin. However, reports on the relative rates of absorption of macromolecular drugs administered as dry powder inhalers and inhaler solutions are rather conflicting and the pharmacology is poorly understood. When human granulocyte colony-stimulating factor was administered as a dry powder via the pulmonary route, the bioavailability of a dry powder formulation was lower than that administered as an inhaled solution.(24) Similarly, we have shown that insulin administered as a dry powder is better absorbed compared to that administered as a solution.(9) In contrast to these reports, no differences in the bioavailability of several therapeutic proteins, including calcitonin, insulin, and thyrotropin stimulating hormone, were observed when administered as a dry powder versus an inhaled solution.(25)
We believe that dry powder is better absorbed compared to solution for a number of reasons. The pulmonary delivery of dry powder using insufflators (Penn Century Inc., Philadelphia, PA) requires a relatively larger amount of air (3 mL) to force the dry powder through the delivery device for inhalation into rats. This forceful delivery may cause deposition of powder formulations at deeper lung regions compared to drug solution administered by a Microsprayer® device. Alternatively, particles from dry powder formulations may have different deposition and distribution patterns compared to droplets from liquid formulations. Deposition upon pulmonary administration is largely controlled by the particle or droplet size of the dry powder and liquid formulations, respectively. Particle size can also play a role in absorption of poorly soluble drugs. For a given solid particle or liquid droplet of similar size, the number of drug molecules present in a solid particle is likely to be higher than that present in a liquid droplet because liquids in nebulizers or pMDIs are not usually a saturated solution of solute in solvent. Furthermore, as observed by others, elongated lactose crystals can provide for more efficient deposition of the drug compared to spherical liquid droplets. Presumably, unlike liquid droplets, solid particles must undergo dissolution before absorption can occur. For this reason, solid drug particles will have longer residence time in the lungs compared to liquid droplets, which may lead to an increase in absorption. Previously, it has been shown that increase in residence by means of mucoadhesion increases pulmonary absorption of calcitonin.(26) There are also reports that suggest that short residence time results in poor deposition of particles.(27) Contrasting these assumptions, it has also been hypothesized that short residence time increases systemic absorption.(28) On the other hand, it is possible that pulmonary absorption of LMWH involves certain transport pathways that are saturated at a faster rate when the drug is administered as droplets of an aqueous solution compared to when it is administered as particles of a dry powder formulation. This may eventually contribute to increased absorption by dry powder formulation. Because of apparently conflicting findings and hypotheses, more mechanistic studies are required to rule in or rule out any of the above hypotheses.
Taken together, the data presented above show that a lactose-based dry powder formulation of LMWH was effective in enhancing pulmonary absorption of drugs. The dry powder formulation of LMWH was at least 1.5-fold as effective as an inhaled solution in enhancing LMWH absorption through the pulmonary route. The study also shows that a dry powder inhaler dispensing a larger particle size may be less suitable than a dry powder inhaler dispensing a smaller particle size for pulmonary delivery. The increase in absorption by dry powder formulation may stem from the variability in deposition patterns of droplets or particles after administration of aerosolized solution or dry powder. Further, the delivery device used in the study was not perhaps optimal for generation of aerosolized particles with consistent size and characteristics. Therefore, extensive in vivo and in vitro comparative studies should be performed to determine why a dry powder formulation is better absorbed than a liquid formulation of the drug after pulmonary administration.
Acknowledgments
The work was supported in part by an NIH grant (R15HL7713302).
Author Disclosure Statement
All authors declare that no conflicting interests exist.
References
- 1.Crompton GK. Dry powder inhalers: advantages and limitations. J Aerosol Med. 1991;4:151–156. doi: 10.1089/jam.1991.4.151. [DOI] [PubMed] [Google Scholar]
- 2.Atkins PJ. Dry powder inhalers: an overview. Respir Care. 2005;50:1304–1312. discussion 1312. [PubMed] [Google Scholar]
- 3.Telko MJ. Hickey AJ. Dry powder inhaler formulation. Respir Care. 2005;50:1209–1227. [PubMed] [Google Scholar]
- 4.Agu RU. Ugwoke MI. Armand M. Kinget R. Verbeke N. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir Res. 2001;2:198–209. doi: 10.1186/rr58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang T. Arnold JJ. Ahsan F. Tetradecylmaltoside (TDM) enhances in vitro and in vivo intestinal absorption of enoxaparin, a low molecular weight heparin. J Drug Target. 2005;13:29–38. doi: 10.1080/10611860400020191. [DOI] [PubMed] [Google Scholar]
- 6.Mustafa F. Yang T. Khan MA. Ahsan F. Chain length-dependent effects of alkylmaltosides on nasal absorption of enoxaparin. J Pharm Sci. 2004;93:675–683. doi: 10.1002/jps.10579. [DOI] [PubMed] [Google Scholar]
- 7.Yang T. Mustafa F. Bai S. Ahsan F. Pulmonary delivery of low molecular weight heparins. Pharm Res. 2004;21:2009–2016. doi: 10.1023/b:pham.0000048191.69098.d6. [DOI] [PubMed] [Google Scholar]
- 8.Yang T. Hussain A. Paulson J. Abbruscato TJ. Ahsan F. Cyclodextrins in nasal delivery of low-molecular-weight heparins: in vivo and in vitro studies. Pharm Res. 2004;21:1127–1136. doi: 10.1023/b:pham.0000032998.84488.7a. [DOI] [PubMed] [Google Scholar]
- 9.Hussain A. Majumder QH. Ahsan F. Inhaled insulin is better absorbed when administered as a dry powder compared to solution in the presence or absence of alkylglycosides. Pharm Res. 2006;23:138–147. doi: 10.1007/s11095-005-8926-9. [DOI] [PubMed] [Google Scholar]
- 10.Steckel H. Bolzen N. Alternative sugars as potential carriers for dry powder inhalations. Int J Pharm. 2004;270:297–306. doi: 10.1016/j.ijpharm.2003.10.039. [DOI] [PubMed] [Google Scholar]
- 11.Baldrick P. Bamford DG. A toxicological review of lactose to support clinical administration by inhalation. Food Chem Toxicol. 1997;35:719–733. doi: 10.1016/s0278-6915(97)00041-0. [DOI] [PubMed] [Google Scholar]
- 12.Louey MD. Mulvaney P. Stewart PJ. Characterisation of adhesional properties of lactose carriers using atomic force microscopy. J Pharm Biomed Anal. 2001;25:559–567. doi: 10.1016/s0731-7085(00)00523-9. [DOI] [PubMed] [Google Scholar]
- 13.Zeng XM. Martin GP. Marriott C. Pritchard J. Lactose as a carrier in dry powder formulations: the influence of surface characteristics on drug delivery. J Pharm Sci. 2001;90:1424–1434. doi: 10.1002/jps.1094. [DOI] [PubMed] [Google Scholar]
- 14.Bai S. Thomas C. Ahsan F. Dendrimers as a carrier for pulmonary delivery of enoxaparin, a low-molecular weight heparin. J Pharm Sci. 2007;96:2090–2106. doi: 10.1002/jps.20849. [DOI] [PubMed] [Google Scholar]
- 15.Valizadeh H. Nokhodchi A. Qarakhani N. Zakeri-Milani P. Azarmi S. Hassanzadeh D. Löbenberg R. Physico-chemical characterization of solid dispersions of indomethacin with PEG 6000, Myrj 52, lactose, sorbitol, dextrin, and Eudragit E100. Drug Dev Ind Pharm. 2004;30:303–317. doi: 10.1081/ddc-120030426. [DOI] [PubMed] [Google Scholar]
- 16.Nail SL. Jiang S. Chongprasert S. Knopp SA. Fundamentals of freeze-drying. Pharm Biotechnol. 2002;14:281–360. doi: 10.1007/978-1-4615-0549-5_6. [DOI] [PubMed] [Google Scholar]
- 17.Teng Y. Qiu Z. Wen H. Systematical approach of formulation, process development using roller compaction. Eur J Pharm Biopharm. May 3; doi: 10.1016/j.ejpb.2009.04.008. [E-pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 18.Augsburger LL. Hard, soft shell capsules. In: Banker GS, editor; Rhodes CT, editor. Modern Pharmaceutics. 3rd. Marcel Dekker Inc.; New York: 1996. pp. 395–440. [Google Scholar]
- 19.Larhrib H. Martin GP. Marriott C. Prime D. The influence of carrier and drug morphology on drug delivery from dry powder formulations. Int J Pharm. 2003;257:283–296. doi: 10.1016/s0378-5173(03)00156-x. [DOI] [PubMed] [Google Scholar]
- 20.Larhrib H. Martin GP. Prime D. Marriott C. Characterisation and deposition studies of engineered lactose crystals with potential for use as a carrier for aerosolised salbutamol sulfate from dry powder inhalers. Eur J Pharm Sci. 2003;19:211–221. doi: 10.1016/s0928-0987(03)00105-2. [DOI] [PubMed] [Google Scholar]
- 21.Zeng XM. Martin AP. Marriott C. Pritchard J. The influence of carrier morphology on drug delivery by dry powder inhalers. Int J Pharm. 2000;200:93–106. doi: 10.1016/s0378-5173(00)00347-1. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi S. Kondo S. Juni K. Pulmonary delivery of salmon calcitonin dry powders containing absorption enhancers in rats. Pharm Res. 1996;13:80–83. doi: 10.1023/a:1016081301369. [DOI] [PubMed] [Google Scholar]
- 23.Okamoto H. Aoki M. Danjo K. A novel apparatus for rat in vivo evaluation of dry powder formulations for pulmonary administration. J Pharm Sci. 2000;89:1028–1035. doi: 10.1002/1520-6017(200008)89:8<1028::aid-jps7>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 24.Niven RW. Lott FD. Ip AY. Cribbs JM. Pulmonary delivery of powders and solutions containing recombinant human granulocyte colony-stimulating factor (rhG-CSF) to the rabbit. Pharm Res. 1994;11:1101–1109. doi: 10.1023/a:1018924512928. [DOI] [PubMed] [Google Scholar]
- 25.Komada F. Iwakawa S. Yamamoto N. Sakakibara H. Okumura K. Intratracheal delivery of peptide and protein agents: absorption from solution and dry powder by rat lung. J Pharm Sci. 1994;83:863–867. doi: 10.1002/jps.2600830621. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto H. Kuno Y. Sugimoto S. Takeuchi H. Kawashima Y. Surface-modified PLGA nanosphere with chitosan improved pulmonary delivery of calcitonin by mucoadhesion and opening of the intercellular tight junctions. J Control Release. 2005;102:373–381. doi: 10.1016/j.jconrel.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 27.Fink JB. Aerosol delivery to ventilated infant and pediatric patients. Respir Care. 2004;49:653–665. [PubMed] [Google Scholar]
- 28.Hochhaus G. New developments in corticosteroids. Proc Am Thorac Soc. 2004;1:269–274. doi: 10.1513/pats.200402-007MS. [DOI] [PubMed] [Google Scholar]