

Background: Sld2 is required for the initiation of DNA replication.
Results: Sld2 inhibits assembly of replication fork helicase, but this inhibition is alleviated by origin single-stranded DNA and phosphorylation of Sld2.
Conclusion: Phosphorylation of Sld2 and origin single-stranded DNA may be important for assembly of the replication fork helicase.
Significance: We propose a new model for the assembly of the replication fork helicase.
Keywords: Cyclins, DNA Helicase, DNA Replication, DNA-Protein Interaction, Protein Kinases, Yeast, Origin
Abstract
The Cdc45-Mcm2–7-GINS (CMG) complex is the replication fork helicase in eukaryotes. Synthetic lethal with Dpb11-1 (Sld2) is required for the initiation of DNA replication, and the S phase cyclin-dependent kinase (S-CDK) phosphorylates Sld2 in vivo. We purified components of the replication initiation machinery and studied their interactions in vitro. We found that unphosphorylated or CDK-phosphorylated Sld2 binds to the mini chromosome maintenance (Mcm)2–7 complex with similar efficiency. Sld2 interaction with Mcm2–7 blocks the interaction between GINS and Mcm2–7. The interaction between CDK-phosphorylated Sld2 and Mcm2–7 is substantially inhibited by origin single-stranded DNA (ssDNA). Furthermore, origin ssDNA allows GINS to bind to Mcm2–7 in the presence of CDK-phosphorylated Sld2. However, unphosphorylated Sld2 blocks the interaction between GINS and Mcm2–7 even in the presence of origin ssDNA. We identified a mutant of Sld2 that does not bind to DNA. When this mutant is expressed in yeast cells, cell growth is severely inhibited with very slow progression into S phase. We propose a model wherein Sld2 blocks the interaction between GINS and Mcm2–7 in vivo. Once origin ssDNA is extruded from the Mcm2–7 ring and CDK phosphorylates Sld2, the origin ssDNA binds to CDK-phosphorylated Sld2. This event may allow the interaction between GINS and Mcm2–7 in vivo. Thus, CDK phosphorylation of Sld2 may be important to release Sld2 from Mcm2–7, thereby allowing GINS to bind Mcm2–7. Furthermore, origin ssDNA may stimulate the formation of the CMG complex by alleviating inhibitory interactions between Sld2 with Mcm2–7.
Introduction
The initiation of DNA replication is a highly regulated event in eukaryotes, and central to the start of DNA synthesis is activation of the replication fork helicase (1–5). The replication fork helicase in eukaryotes consists of the Cdc45 protein, the mini chromosome maintenance (Mcm)2 2–7 heterohexameric ring assembly, and the GINS tetramer (composed of Psf1, Psf2, Psf3, and Sld5), which form the CMG (Cdc45-Mcm2–7-GINS) complex (6–10). The CMG complex forms in S phase, and unwinding of DNA by the CMG complex is coordinated with DNA synthesis of the leading and lagging strands (9, 10).
Three proteins required for the initiation of DNA replication in budding yeast, Sld2, Sld3, and Dpb11, do not travel with the replication fork (11–13). Sld2 and Sld3 are phosphorylated by the cyclin-dependent kinase (CDK) during S phase, and phosphorylation of Sld2 and Sld3 by CDK promotes the association of Sld2 and Sld3 with Dpb11 (14–16). The essential requirement for CDK in the cell can be bypassed by an Sld3-Dpb11 fusion construct and a phosphor-mimicking mutant in Sld2 (Sld2T84D) (15). RecQL4 is a putative homolog of Sld2 (17–20), and Treslin/TICRR is a putative homolog of Sld3 (21–25).
The Mcm2–7 complex is loaded as a double hexamer to encircle double-stranded DNA (dsDNA) in late M phase and during G1 (26–28). Sld3 and Cdc45 bind to one another in vivo, and the Sld3-Cdc45 complex binds to early replication origins in G1 (29, 30). Interaction of Sld3-Cdc45 with replication origins depends on the Dbf4-dependent kinase (DDK) in an in vitro reconstitution assay (31). DDK phosphorylates multiple subunits of the Mcm2–7 complex, and DDK phosphorylation of Mcm4 alleviates an inhibitory activity of Mcm4 (32–35).
In vitro, Sld3-Cdc45 can form a stable ternary complex with Mcm2–7 (Fig. 1A) (36). Furthermore, binding of Sld3 to Mcm2–7 blocks the association of GINS with Mcm2–7, suggesting a mechanism to regulate replication fork helicase assembly (36). Although the Mcm2–7 complex surrounds dsDNA in G1, the CMG complex binds to single-stranded DNA (ssDNA), not dsDNA (7, 8). These data suggest that a single strand of DNA is extruded from the central channel of the Mcm2–7 ring prior to CMG complex formation. The Mcm2–7 complex may crack open to allow one strand of DNA to be extruded from the central channel of the Mcm2–7 complex (Fig. 1, B and C) prior to CMG complex assembly (8, 37). In vitro studies have shown that Sld3 binds tightly to discrete single-stranded regions of replication origins (38). Binding of Sld3 to origin ssDNA releases Sld3 from the Mcm2–7 complex, allowing GINS to bind to Mcm2–7 (Fig. 1D) (38). Formation of the CMG complex leads to DNA unwinding coupled to polymerase replication of the leading and lagging strands (Fig. 1E).
FIGURE 1.

Assembly of the replication fork helicase. A, Sld3-Cdc45 binds to Mcm2–7 at early origins in G1 (29). Sld3 blocks the interaction between GINS and Mcm2–7, thereby inhibiting premature activation of the helicase (36). B, the Mcm2–7 ring opens (8, 37). C, exposure of thymines in origin ssDNA promotes the association of Sld3 with origin ssDNA (38). D, interaction between Sld3 and ssDNA dissociates Sld3 from Mcm2–7, allowing GINS to bind to Mcm2–7 (38). E, CMG complex unwinds DNA ahead of the replicative polymerases (7, 9, 10).
Sld2 forms a transient preloading complex with Dpb11, GINS, and polymerase ϵ that is dependent upon CDK activity (30). This preloading complex may be important for the recruitment of GINS or polymerase ϵ to replication origins. In vitro studies have shown that CDK-phosphorylated Sld2 binds tightly to certain single-stranded regions of replication origins (39). The phosphor-mimetic mutant Sld2T84D also binds tightly to origin ssDNA, although Sld2 binds weakly to origin ssDNA (39). At the origin ARS1, CDK-phosphorylated Sld2 and Sld3 bind to complementary single-stranded regions of DNA in vitro (38, 39). The physiologic role of CDK-phosphorylated Sld2 binding to ssDNA is presently not known.
We purified components of the budding yeast replication initiation machinery and studied their interactions in vitro. We report here that unphosphorylated or CDK-phosphorylated Sld2 binds to the Mcm2–7 complex. Binding of CDK-phosphorylated Sld2 or Sld3 to Mcm2–7 is competitive. Furthermore, binding of CDK-phosphorylated Sld2 to Mcm2–7 inhibits the interaction between GINS and Mcm2–7. CDK-phosphorylated Sld2 in combination with Sld3 inhibits the interaction between GINS and Mcm2–7 in a concentration-dependent manner. In the presence of origin ssDNA, CDK-phosphorylated Sld2 or Sld3 is released from the Mcm2–7 complex. Origin ssDNA also allows GINS to bind to Mcm2–7 in the presence of CDK-phosphorylated Sld2 and Sld3. We identified a mutant of Sld2 that does not bind DNA. When this mutant is expressed in budding yeast, cell growth is severely inhibited, and cells progress very slowly into S phase. We propose that when budding yeast origin DNA is melted prior to the onset of DNA replication, the exposed ssDNA releases CDK-phosphorylated Sld2 and Sld3 from Mcm2–7, allowing GINS to bind to Mcm2–7. Thus, CDK phosphorylation of Sld2 may be important to release Sld2 from Mcm2–7, and origin ssDNA may help trigger the assembly of the CMG complex.
EXPERIMENTAL PROCEDURES
Proteins
GST-Sld2, PKA-Sld2, Sld2, GST-Sld2T84D, and Sld2T84D were purified as described (39). GINS, PKA-GINS, GST-Sld3, and Sld3 were purified as described (38). CDK was purified as described (14). PKA-Mcm2–7 (PKA tag at the N terminus of Mcm3), GST-Mcm2–7 (GST tag at the N terminus of Mcm2), and Mcm2–7 were purified as described (40). Protein kinase A was a generous gift from Susan Taylor.
Kinase Labeling
PKA labeling was performed as described (40). CDK labeling was performed in a 50-μl reaction containing 20 nm Sld2, 20 ng of CDK, 5 mm unlabeled ATP, 5 μCi of [γ-32P]ATP (6000 Ci/nmol; PerkinElmer Life Sciences) in 5 mm Tris-HCl, pH 8.5, 10 mm MgCl2, and 1 mm DTT. Reactions were incubated at 30 °C for 1 h.
Analytical Size Exclusion Chromatography
Unlabeled protein was mixed with radiolabeled protein as described in the figure legends in a final volume of 200 μl and incubated at 30 °C for 1 h in column buffer (50 mm Tris, pH 7.5, 100 mm NaCl, 1 mm EDTA, 5% glycerol). The protein mixture was then subjected to 24 ml of Superose 6 (GE Healthcare) size exclusion chromatography in the same column buffer. Each 250-μl fraction was then subjected to SDS-PAGE analysis and quantified using PhosphorImager analysis.
GST Pulldown Assays
Each GST pulldown reaction was in a volume of 100 μl and contained GST-tagged protein in GST-binding buffer (40 mm Tris-HCl, pH 7.5, 100 mm NaCl, 0.1 mm EDTA, 10% glycerol, 0.1% Triton X-100, 1 mm DTT, 0.7 μg/ml pepstatin, 0.1 mm PMSF, 0.1 mg/ml BSA) and varying amounts of radiolabeled protein as described in each figure. Reactions were incubated for 1 h at room temperature. Following incubation, reactions were added to 40 μl of prepared glutathione-Sepharose and gently mixed. Binding of GST-tagged protein to the beads was performed for 20 min with gentle mixing every few minutes. Once the binding was complete, the reaction mixture was aspirated, and the beads were washed two times with 0.5 ml of GST-binding buffer. After the last wash, 30 μl of 5× SDS sample buffer was added to each reaction, and the samples were boiled for 10 min. Samples (20 μl) were then analyzed by SDS-PAGE.
Construction of Sld2-td Degron Yeast Strain
The haploid strain and plasmid used to prepare the Sld2-degron were obtained from EUROpean Saccharomyces Cerevisiae ARchive for Functional Analysis (EUROSCARF). Primers were designed according to the strategy described previously, and degron cassette was amplified from pKLV plasmid (41). The amplified degron cassette was transformed into the haploid strain, and transformants were selected on YPD plates containing G418. Correct positioning of the degron cassette at the endogenous Sld2 native promoter was confirmed by PCR analysis. A positive clone was transformed with a PRS415 vector containing either an empty vector, Sld2, or the DNA binding mutant described in the figures under the control of the native Sld2 promoter. Positive transformants were selected on CSM-leu plates.
Serial Dilution Analysis
One colony from a freshly streaked CSM-leu plate was inoculated into 5 ml of CSM-leu liquid medium containing raffinose and grown at 24 °C. After 9 h, the cells were harvested and then resuspended in 5 ml of YP plus galactose liquid medium and grown at 24 °C for an additional 16 h. After 16 h, the OD of each culture was measured, and each culture was diluted to an approximate density of 106 cells with fresh YP plus galactose. Six additional serial dilutions were made, and 2 μl from each of the seven dilutions was spotted onto both a plate containing YPDCu and a plate containing YPGal. The YPDCu plate was kept at 24 °C (permissive conditions) for 2 days, and the YPGal plate was kept at 37 °C (restrictive conditions) for 2 days.
RESULTS
Sld2 or CDK-phosphorylated Sld2 Binds to Mcm2–7
The interaction of Sld2 with components of the prereplication complex has not yet been explored using purified proteins, and we therefore examined whether Sld2 binds to the Mcm2–7 complex in vitro. The purification of Sld2, S-CDK, and Mcm2–7 has been described in other publications from our laboratory (36, 38, 39). The purified Sld2 protein has a PKA tag at its N terminus; this tag is not physiologic, it is used to radiolabel the protein with PKA and [γ-32P]ATP (39). We also radiolabeled Sld2 with the physiologically relevant kinase S-CDK and [γ-32P]ATP (39). We purified and assembled a GST-Mcm2–7 complex, which is Mcm2–7 with a GST tag at the N terminus of Mcm2 (36).
We incubated GST-Mcm2–7 or GST with radiolabeled Sld2 and performed a GST pulldown assay. Bound protein was analyzed by SDS-PAGE followed by phosphorimaging (Fig. 2A). The results were quantified, and repeat experiments were averaged and plotted (Fig. 2B). We found that GST-Mcm2–7 pulled down more than half of the input radiolabeled Sld2 protein, whereas GST alone pulled down very little Sld2. Sld2 and CDK-phosphorylated Sld2 exhibited similar levels of binding, suggesting that CDK phosphorylation of Sld2 does not influence its association with Mcm2–7. We also performed the experiment with 5 mm ATP, ATPγS, ADP, or no nucleotide to determine the influence of nucleotides on the interaction between CDK-phosphorylated Sld2 and Mcm2–7 (Fig. 2C). We could not detect any difference between the various experiments, suggesting that nucleotide binding to Mcm2–7 does not affect the interaction between Mcm2–7 and CDK-phosphorylated Sld2.
FIGURE 2.

Sld2 and CDK-phosphorylated Sld2 bind to Mcm2–7 with similar affinity. A, 2 pmol of GST-Mcm2–7 (GST-Mcm2–7 has a GST tag at the N terminus of Mcm2), or GST was used to pull down various concentrations of radiolabeled Sld2 or CDK-phosphorylated Sld2. Bound protein was analyzed by SDS-PAGE and quantified with phosphorimaging. For detailed methods, see “Experimental Procedures.” B, results similar to the gels shown in A were averaged and plotted. C, experiments similar to A were conducted with 5 mm ATP, ATPγS, ADP, or no nucleotide. D, 2 pmol of GST-Sld2 or CDK-phosphorylated GST-Sld2 was used to pull down various concentrations of radiolabeled Mcm2–7 (radiolabeled Mcm2–7 has a radiolabeled PKA tag at the N terminus of Mcm3). E, the results were quantified, averaged, and plotted. F, experiments similar to D were conducted with 5 mm ATP, ATPγS, ADP, or no nucleotide.
We next performed a GST pulldown with GST-Sld2, CDK-phosphorylated GST-Sld2, or GST and radiolabeled Mcm2–7 (Fig. 2D). Radiolabeled Mcm2–7 has a PKA tag at the N terminus of Mcm3 for phosphorylation with PKA and [γ-32P]ATP. We found that GST-Sld2 or CDK-phosphorylated GST-Sld2 binds to Mcm2–7 with similar efficiency (Fig. 2, D and E), confirming that Sld2 and CDK-phosphorylated Sld2 bind to Mcm2–7 with similar efficiency. Very little binding is observed for GST alone, suggesting that the interaction is specific for Sld2. We next incubated CDK-phosphorylated GST-Sld2 with radiolabeled Mcm2–7 in the presence of 5 mm ATP, ATPγS, ADP, or no nucleotide (Fig. 2F). Similar levels of binding were observed for each of these conditions, suggesting that the interaction is not modified by nucleotide binding.
Sld2 or CDK-phosphorylated Sld2 Competes with GINS for Mcm2–7 Binding
We previously found that GINS binds to the Mcm2–7 complex (36), and we next investigated whether Sld2 and GINS compete with one another for Mcm2–7 binding. GST-Mcm2–7 was used to pull down radiolabeled GINS in the presence of difference amounts of unlabeled Sld2 or CDK-phosphorylated Sld2 (Fig. 3A). Radiolabeled GINS contains a PKA tag at the N terminus of Psf1 to allow for radiolabeling with PKA and [γ-32P]ATP. GST-Mcm2–7 pulled down a substantial fraction of GINS in the absence of Sld2, as expected (Fig. 3A). As the concentration of unlabeled Sld2 or CDK-phosphorylated Sld2 was increased, the level of GINS binding to Mcm2–7 decreased. Sld2 or CDK-phosphorylated Sld2 does not bind to GINS under the conditions of this assay (data not shown). These data suggest that Sld2 or CDK-phosphorylated Sld2 inhibits the interaction between GINS and Mcm2–7.
FIGURE 3.

Sld2 or CDK-phosphorylated Sld2 competes with GINS for Mcm2–7 binding. A, 2 pmol of GST-Mcm2–7 was incubated with 2 pmol of radiolabeled GINS (radiolabeled GINS has a radiolabeled PKA tag at the N terminus of Psf1) in the presence of various concentrations of unlabeled Sld2 or CDK-phosphorylated Sld2. The results were quantified, averaged, and plotted. For detailed methods, see “Experimental Procedures.” B, 2 pmol of GST-Sld2 or CDK-phosphorylated GST-Sld2 was incubated with 2 pmol of radiolabeled Mcm2–7 in the presence of various concentrations of unlabeled GINS. The results were quantified, averaged, and plotted.
We next used GST-Sld2 or GST-CDK-Sld2 to pull down radiolabeled Mcm2–7 in the presence of increasing concentrations of unlabeled GINS (Fig. 3B). Increasing concentrations of unlabeled GINS inhibited the interaction between Mcm2–7 and Sld2 or CDK-phosphorylated Sld2 (Fig. 3B). These data suggest that the binding of Sld2 or GINS to Mcm2–7 is competitive.
Sld2 or CDK-phosphorylated Sld2 Competes with Sld3 for Mcm2–7 Binding
We previously found that Sld3 and GINS compete with one another for Mcm2–7 binding (36). Because Sld2 and GINS also compete with one another for Mcm2–7 binding (Fig. 3), we next investigated whether Sld3 and Sld2 compete with one another for Mcm2–7 binding. GST-Sld3 was used to pull down radiolabeled Mcm2–7 in the presence of increasing amounts of unlabeled Sld2 or CDK-phosphorylated Sld2 (Fig. 4A). Unlabeled Sld2 or CDK-phosphorylated Sld2 inhibited the interaction between Sld3 and Mcm2–7 (Fig. 4, A and B). Moreover, increasing the concentration of Sld3 inhibited the interaction between Sld2 and Mcm2–7 in a concentration-dependent manner (Fig. 4, C and D). These data suggest that Sld2 or CDK-phosphorylated Sld2 competes with Sld3 for Mcm2–7 binding.
FIGURE 4.
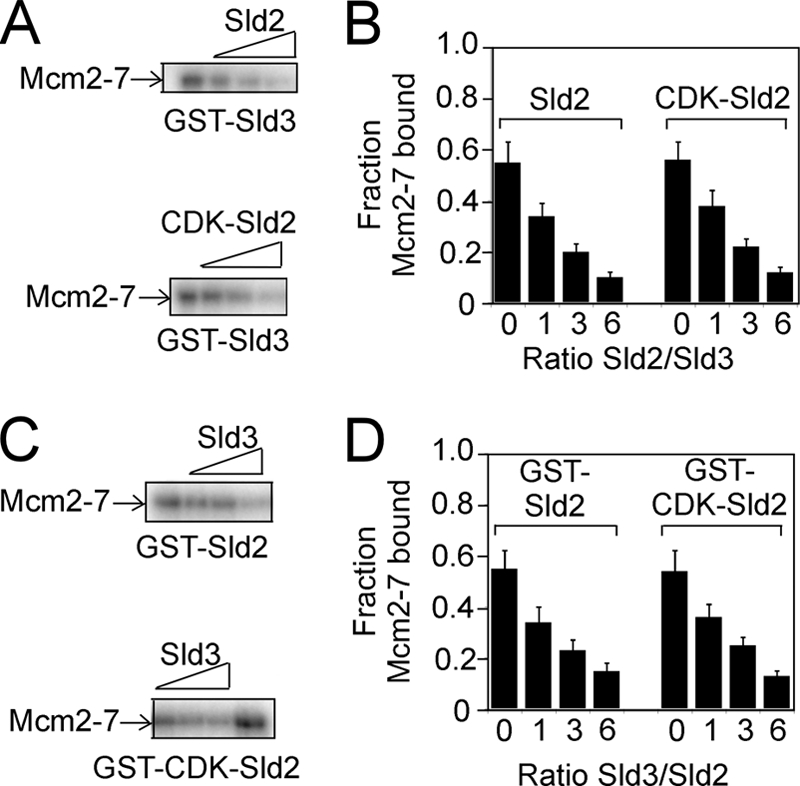
Sld2 or CDK-phosphorylated Sld2 competes with Sld3 for Mcm2–7 binding. A, 2 pmol GST-Sld3 was incubated with 2 pmol of radiolabeled Mcm2–7 in the presence of various concentrations of unlabeled Sld2 or CDK-phosphorylated Sld2. B, the results were quantified, averaged, and plotted. C, 2 pmol of GST-Sld2 or CDK-phosphorylated GST-Sld2 was incubated with 2 pmol of radiolabeled Mcm2–7 in the presence of various concentrations of unlabeled Sld3. D, the results were quantified, averaged, and plotted.
CDK-phosphorylated Sld2 and Sld3 Inhibit the Interaction between GINS and Mcm2–7 in a Concentration-dependent Manner
Sld2, CDK-phosphorylated Sld2, or Sld3 each independently inhibits the interaction between GINS and Mcm2–7. We next examined the effect of CDK-phosphorylated Sld2 and Sld3 in combination on the interaction between GINS and Mcm2–7. GST-Mcm2–7 was used to pull down radiolabeled GINS in the presence of increasing concentrations of CDK-phosphorylated Sld2, Sld3, or CDK-phosphorylated Sld2 and Sld3 (Fig. 5). The inhibition caused by 2 pmol of CDK-phosphorylated Sld2 or 2 pmol of Sld3 was similar to that caused by 1 pmol of CDK-phosphorylated Sld2 and 1 pmol of Sld3 (Fig. 5, A and B). Furthermore, the inhibition caused by 4 pmol of CDK-phosphorylated Sld2 or 4 pmol of Sld3 was similar to that caused by 2 pmol of CDK-phosphorylated Sld2 and 2 pmol of Sld3. Thus, CDK-phosphorylated Sld2 and Sld3 inhibit the interaction between GINS and Mcm2–7 in a concentration-dependent manner.
FIGURE 5.

CDK-phosphorylated Sld2 and Sld3 additively inhibit GINS binding to Mcm2–7. A, 1 pmol of GST-Mcm2–7 pulldown of 1 pmol of radiolabeled GINS in the presence of various amounts of unlabeled CDK-phosphorylated Sld2, unlabeled Sld3, or both CDK-phosphorylated Sld2 and unlabeled Sld3. The pulldown was analyzed by SDS-PAGE followed by phosphorimaging. B, results from gels like A were averaged and plotted.
CDK-phosphorylated Sld2 Forms an Equimolar Complex with Mcm2–7
We next used size exclusion chromatography to analyze the interactions among CDK-phosphorylated Sld3, GINS, and Mcm2–7. For these experiments, no GST tags were used, eliminating the effect of GST tags on the protein interactions detected. Furthermore, the experiments were performed with radiolabeled protein to quantify the amount of protein eluting in each fraction. CDK-phosphorylated Sld2 eluted with a peak corresponding to a Stokes radius of a ∼100-kDa protein (Fig. 6A). These data suggest that Sld2 exists as a low molecular mass species in solution. In the presence of equimolar Mcm2–7, the CDK-phosphorylated peak shifted to that of a ∼750-kDa protein, suggesting that CDK-phosphorylated Sld2 is stably bound to Mcm2–7 (Fig. 6A). When radiolabeled CDK-phosphorylated Sld2 is incubated with equimolar unlabeled GINS there is no effect on the elution profile, suggesting that Sld2 does not stably interact with GINS (Fig. 6B). When radiolabeled CDK-phosphorylated Sld2 is incubated with equimolar GINS and Mcm2–7, a mixture of small (∼100 kDa) and large (∼750 kDa) molecular mass species is observed (Fig. 6C). These data suggest that GINS is competing with Sld2 for interaction with Mcm2–7, as observed in Fig. 3 using the GST pulldown assay.
FIGURE 6.

CDK-phosphorylated Sld2 forms an equimolar complex with Mcm2–7. A–C, 100 pmol of [32P]CDK-phosphorylated Sld2 was incubated with equimolar Mcm2–7 (A), GINS (B), or GINS and Mcm2–7 (C) and subjected to size exclusion chromatography as described under “Experimental Procedures.” The radioactive counts in each fraction were used to calculate the pmol of CDK-phosphorylated Sld2 in each fraction. CDK-phosphorylated Sld2 (pmol) was then plotted versus the elution of molecular mass standards. D–F, 100 pmol of 32P-labeled GINS was incubated with equimolar Mcm2–7 (D), CDK-phosphorylated Sld2 (E), or Mcm2–7 and CDK-phosphorylated Sld2 (F) and subjected to size exclusion chromatography as described under “Experimental Procedures.” The radioactive counts in each fraction were used to calculate the pmol of GINS in each fraction. GINS (pmol) was then plotted versus the elution of molecular mass standards. G–I, 100 pmol of 32P-labeled Mcm2–7 was incubated with equimolar CDK-phosphorylated Sld2 (G), GINS (H), or CDK-phosphorylated Sld2 and GINS (I) and subjected to size exclusion chromatography as described under “Experimental Procedures.” The radioactive counts in each fraction were used to calculate the pmol of Mcm2–7 in each fraction. Mcm2–7 (pmol) was then plotted versus the elution of molecular mass standards.
Radiolabeled GINS alone elutes as a low molecular mass species (∼158 kDa), but in the presence of equimolar unlabeled Mcm2–7 it elutes as a high molecular mass species (∼750 kDa), suggesting a stable interaction between GINS and Mcm2–7 (Fig. 6D). Radiolabeled GINS does not shift when unlabeled CDK-phosphorylated Sld2 is added, suggesting that GINS does not interact stably with Sld2 (Fig. 6E). When radiolabeled GINS is incubated with equimolar unlabeled CDK-phosphorylated Sld2 and Mcm2–7, there is a mixture of low (∼158 kDa) and high (∼750 kDa) molecular mass species observed, suggesting that GINS and CDK-phosphorylated Sld2 compete with one another for Mcm2–7 binding (Fig. 6F). Radiolabeled Mcm2–7 is slightly shifted by equimolar unlabeled CDK-phosphorylated Sld2 or GINS (Fig. 6, G–I). Furthermore, the data establish a stoichiometry of binding of one Mcm2–7 hexamer to one Sld2 monomer or one GINS tetramer.
ssARS1-1 Inhibits the Interaction between CDK-phosphorylated Sld2 and Mcm2–7
It was previously observed that CDK-phosphorylated Sld2 or Sld2T84D binds tightly to a single-stranded T-rich region of ARS1 containing the A and B1 elements and encompassing nucleotides 810–889 (ssARS1-1) (39) (Fig. 7A). We next investigated whether ssARS1-1 affects the interaction between CDK-phosphorylated Sld2 and Mcm2–7. CDK-phosphorylated Sld2 was used to pull down radiolabeled Mcm2–7 in the presence of increasing concentrations of unlabeled ssARS1-1 (Fig. 7B). ssARS1-1 inhibited the interaction between CDK-phosphorylated Sld2 and Mcm2–7 in a concentration-dependent manner (Fig. 7, B and C). When the experiment was performed with Sld2 instead of CDK-phosphorylated Sld2, the level of inhibition was modest, consistent with the weaker affinity of Sld2 for ssARS1-1 compared with CDK-phosphorylated Sld2 (Fig. 7, B and C) (39). In contrast, ssARS1-2 did not inhibit the interaction between CDK-phosphorylated Sld2 and Mcm2–7, consistent with the negligible affinity of CDK-phosphorylated Sld2 and ssARS1-2 (Fig. 7, B and C) (39). ssARS1-1 also inhibited the interaction between Sld2T84D and Mcm2–7, consistent with the high affinity of Sld2T84D for ssARS1-1 (Fig. 7, B and C) (39).
FIGURE 7.

ssARS1-1 induces the dissociation of CDK-phosphorylated Sld2 from Mcm2–7. A, 2 pmol of CDK-phosphorylated GST-Sld2, GST-Sld2, or GST-Sld2T84D was incubated with 2 pmol of radiolabeled Mcm2–7 in the presence of varying concentrations of ssARS1-1 or ssARS1-2. The results were analyzed by SDS-PAGE. B, results from experiments like A were averaged and plotted.
ssARS1-1 Allows GINS to Bind to Mcm2–7 in the Presence of CDK-phosphorylated Sld2
ssARS1-1 inhibits the interaction between CDK-phosphorylated Sld2 and Mcm2–7 (Fig. 7), and we next investigated whether ssARS1-1 allows GINS to bind to Mcm2–7 in the presence of CDK-phosphorylated Sld2. GST-Mcm2–7 was used to pull down radiolabeled GINS in the presence of excess unlabeled CDK-phosphorylated Sld2 (Fig. 8A). The addition of unlabeled ssARS1-1 allowed for the interaction between GINS and Mcm2–7 in a concentration-dependent manner (Fig. 8A). As a control, ssARS1-2, a sequence that does not bind to CDK-phosphorylated Sld2, did not allow the interaction between GINS and Mcm2–7 (Fig. 8A) (39). These data suggest that the observed effect is specific for the DNA sequence used. When CDK-phosphorylated Sld2 is replaced with unphosphorylated Sld2 and ssARS1-1 is added to the GST pulldown reaction, the interaction between GINS and Mcm2–7 remains weak (Fig. 8B). These data are consistent with the weak interaction between unphosphorylated Sld2 and ssARS1-1 (39). In contrast, when Sld2T84D is used to block the interaction between GINS and Mcm2–7, the inhibition is alleviated by increasing concentrations of ssARS1-1, consistent with the high affinity of Sld2T84D for ssARS1-1 (Fig. 8B) (39).
FIGURE 8.

ssARS1-1 allows the interaction between GINS and Mcm2–7 in the presence of CDK-phosphorylated Sld2. A, 2 pmol of GST-Mcm2–7 was used to pull down 2 pmol of radiolabeled GINS in the presence or absence of 10 pmol of CDK-phosphorylated Sld2 and varying concentrations of origin ssDNA (ssARS1-1 or ssARS1-2). B, 2 pmol of GST-Mcm2–7 was used to pull down 2 pmol of radiolabeled GINS in the presence or absence of 10 pmol of Sld2 or Sld2T84D and varying concentrations of origin ssDNA (ssARS1-1).
Using size exclusion chromatography, we next examined whether ssARS1-1 allows GINS to bind to Mcm2–7 in the presence of CDK-phosphorylated Sld2 (Fig. 9). ssARS1-1 has no effect on the migration of CDK-phosphorylated Sld2 (Fig. 9A). However, ssARS1-1 shifts the elution profile of CDK-phosphorylated Sld2 and Mcm2–7 to a lower molecular mass peak (∼100 kDa), suggesting that ssARS1-1 dislodges CDK-phosphorylated Sld2 from Mcm2–7 (Fig. 9, B and C), consistent with the GST pulldown data of Fig. 7. ssARS1-1 also has no effect on the elution of GINS, either alone or in combination with Mcm2–7 (Fig. 9, D and E). However, when radiolabeled GINS is incubated with CDK-phosphorylated Sld2 and Mcm2–7 and ssARS1-1, the radiolabeled GINS elutes as a high molecular mass peak (∼750 kDa, Fig. 9F). These data are consistent with the GST pulldown data showing that ssARS1-1 allows GINS to bind Mcm2–7 in the presence of CDK-phosphorylated Sld2 (Fig. 8). As an additional control, ssARS1-1 does not affect the elution profile of Mcm2–7 (Fig. 9, G and H).
FIGURE 9.

ssARS1-1 releases CDK-phosphorylated Sld2 from Mcm2–7, allowing GINS to bind Mcm2–7. A–C, 100 pmol of [32P]CDK-phosphorylated Sld2 was incubated with 200 pmol of ssARS1-1 (A), 100 pmol of Mcm2–7 + 200 pmol of ssARS1-1 (B), or 100 pmol of Mcm2–7 + 100 pmol of GINS + 200 pmol of ssARS1-1 (C) and subjected to size exclusion chromatography as described under “Experimental Procedures.” The radioactive counts in each fraction were used to calculate the pmol of CDK-phosphorylated Sld2 in each fraction. CDK-phosphorylated Sld2 (pmol) was then plotted versus the elution of molecular mass standards. D–F, 100 pmol of 32P-labeled GINS was incubated with 200 pmol of ssARS1-1 (D), 100 pmol of Mcm2–7 + 200 pmol of ssARS1-1 (E), or 100 pmol of Mcm2–7 + 100 pmol of CDK-phosphorylated Sld2 + 200 pmol of ssARS1-1 (F) and subjected to size exclusion chromatography as described under “Experimental Procedures.” The radioactive counts in each fraction were used to calculate the pmol of GINS in each fraction. GINS (pmol) was then plotted versus the elution of molecular mass standards. G and H, 100 pmol of 32P-labeled Mcm2–7 was incubated with 200 pmol of ssARS1-1 (G) or 100 pmol of GINS + 100 pmol of CDK-phosphorylated Sld2 + 200 pmol of ssARS1-1 (H) and subjected to size exclusion chromatography as described under “Experimental Procedures.” The radioactive counts in each fraction were used to calculate the pmol of Mcm2–7 in each fraction. Mcm2–7 (pmol) was then plotted versus the elution of molecular mass standards.
ssARS1-1 and ssARS1-2 Allow GINS to Bind to Mcm2–7 in the Presence of CDK-phosphorylated Sld2 and Sld3
CDK-phosphorylated Sld2 and Sld3 compete with GINS for Mcm2–7 binding in a concentration-dependent manner. It was previously shown that ssARS1-2 allows GINS to bind Mcm2–7 in the presence of Sld3 (38). Furthermore, Fig. 8 shows that ssARS1-1 allows GINS to bind Mcm2–7 in the presence of CDK-phosphorylated Sld2. We next examined whether ssARS1-1 and ssARS1-2 allow GINS to bind to Mcm2–7 in the presence of CDK-phosphorylated Sld2 and Sld3. ssARS1-1 and ssARS1-2 will rapidly anneal with one another in solution in the presence of CDK-phosphorylated Sld2, and CDK-phosphorylated Sld2 and Sld3 bind very weakly to dsDNA (38, 39). Thus, we modified ssARS1-2 to eliminate the ability of this strand to anneal to ssARS1-1 while still retaining its high affinity for Sld3. If the adenines in ssARS1-2 are replaced with cytosines, the modified ssARS1-2-A-to-C sequence binds to Sld3 with wild-type affinity (Fig. 10A) (38). Thus, to avoid DNA annealing during the course of the reaction, we added ssARS1-1 and the modified ssARS1-2-A-to-C to a GST pulldown reaction containing GST-Mcm2–7, radiolabeled GINS, and unlabeled CDK-phosphorylated Sld2 and Sld3 (Fig. 10B). The addition of both ssARS1-1 and ssARS1-2-A-to-C allows GST-Mcm2–7 to pull down radiolabeled GINS in the presence of CDK-phosphorylated Sld2 and Sld3 (Fig. 10, B and C). In contrast, the addition of either ssARS1-2 or ssARS1-2-A-to-C is insufficient to allow GINS to bind to Mcm2–7 (Fig. 10, B and C). These data suggest that both DNA sequences are necessary to allow the association of GINS with Mcm2–7 in the presence of CDK-phosphorylated Sld2 and Sld3.
FIGURE 10.

ssARS1-1 and ssARS1-2 allow the interaction between GINS and Mcm2–7 in the presence of CDK-phosphorylated Sld2 and Sld3. A, DNA sequences used in this figure. CDK-phosphorylated Sld2 binds tightly to ssARS1-1, but not ssARS1-2 or ssARS1-2-A-to-C (39). Sld3 binds tightly to ssARS1-2 or ssARS1-2-A-to-C, but not ssARS1-1 (38). B, 2 pmol of GST-Mcm2–7 was used to pull down 2 pmol of radiolabeled GINS in the presence or absence of 10 pmol of CDK-phosphorylated Sld2 and Sld3 and varying concentrations of origin ssDNA (ssARS1-1 and/or ssARS1-2-A-to-C). C, results from experiments like B were averaged and plotted.
Identification of an Sld2 Triple Mutant That Does Not Bind Origin ssDNA
We next identified mutations of CDK-phosphorylated Sld2 that inhibit the interaction with origin ssDNA (ssARS1-1) by studying the interaction of CDK-phosphorylated Sld2 fragments and mutants with ssARS1-1. We first found that there are two binding sites in CDK-phosphorylated Sld2 for ssARS1-1: one at the N terminus (1–79), and a second within amino acids 79–453 (Fig. 11A). Within the N-terminal region, we further dissected the amino acid region to residues 41–79 (Fig. 11B). Mutations of positively charged and polar residues within this region revealed that two mutations, Sld2K44E and Sld2K50E, each reduce the binding of CDK-phosphorylated Sld2-(1–106) for ssARS1-1. However, these mutations do not affect the interaction between CDK-phosphorylated Sld2-(1–106) and Dpb11 (Fig. 11D), suggesting that the mutant fragments are properly expressed and folded. We next found that region -(390–453) of CDK-phosphorylated Sld2 binds to ssARS1-1 (Fig. 12A). We mutated the positively charged residues in this C-terminal region and found that one mutant, CDK-phosphorylated Sld2-(79–453)-K438E, does not bind to ssARS1-1 (Fig. 12B). CDK-phosphorylated Sld2-(79–453)-K438E binds to Dpb11 like wild-type CDK-phosphorylated Sld2-(79–453) (Fig. 12C).
FIGURE 11.

N-terminal mutations of CDK-phosphorylated Sld2 that inhibit binding to ssARS1-1. A and B, 3 pmol of CDK-phosphorylated GST-Sld2, full-length or fragments, was used to pull down varying amounts of ssARS1-1. The results were analyzed by SDS-PAGE followed by phosphorimaging. C, 3 pmol of CDK-phosphorylated GST-Sld2-1-106, wild-type or mutants, was used to pull down varying amounts of ssARS1-1. D, 2 pmol of CDK-phosphorylated GST-Sld2-1-106, wild-type or mutants, was used to pull down varying amounts of radiolabeled Dpb11.
FIGURE 12.

C-terminal mutation of CDK-phosphorylated Sld2 that inhibits binding to ssARS1-1. A, 3 pmol of CDK-phosphorylated GST-Sld2 fragments were used to pull down varying amounts of ssARS1-1. The results were analyzed by SDS-PAGE followed by phosphorimaging. B, 3 pmol of CDK-phosphorylated GST-Sld2–79-453, wild-type or mutants, was used to pull down varying amounts of ssARS1-1. Mutants 1-7 are: 1, K416E; 2, K420E,R421E,K422E; 3, R424E,K425E,K426E; 4, R435E,R436E; 5, K441E,K442E; 6, R444E; 7, R449E,R452E,R453E. C, 2 pmol of CDK-phosphorylated GST-Sld2–79-453, wild-type or mutants, was used to pull down varying amounts of radiolabeled Dpb11.
We isolated three mutations that inhibit the binding of CDK-phosphorylated Sld2 for ssARS1-1, and we next studied the ability of the full-length triple mutant, CDK-phosphorylated Sld2-K44E,K50E,K438E, to bind ssARS1-1, Dpb11, or Mcm2–7 (Fig. 13). CDK-phosphorylated Sld2-K44E,K50E,K438E does not bind to ssARS1-1 (Fig. 13A); however, CDK-phosphorylated Sld2-K44E,K50E,K438E binds to Dpb11 (Fig. 13B) and Mcm2–7 (Fig. 13C) like wild-type full-length CDK-phosphorylated Sld2. ssARS1-1 does not inhibit the interaction between CDK-phosphorylated GST-Sld2-K44E,K50E,K438E and Mcm2–7 (Fig. 13D), and ssARS1-1 does not allow the interaction between GINS and GST-Mcm2–7 in the presence of excess CDK-phosphorylated Sld2-K44E,K50E,K438E (Fig. 13E). These data confirm that ssARS1-1 inhibition of CDK-phosphorylated Sld2-Mcm2–7 interaction is mediated by contact between CDK-phosphorylated Sld2 and ssARS1-1.
FIGURE 13.

Triple mutant of CDK-phosphorylated Sld2 that does not bind to ssARS1-1. A, 3 pmol of CDK-phosphorylated GST-Sld2-full-length, wild-type, or triple mutant (K44E,K50E,K438E), was used to pull down varying amounts of ssARS1-1. The results were analyzed by SDS-PAGE followed by phosphorimaging. B, 2 pmol of CDK-phosphorylated GST-Sld2-full-length, wild-type or triple mutant (K44E,K50E,K438E), was used to pull down varying amounts of Dpb11. C, 2 pmol of CDK-phosphorylated GST-Sld2-full-length, wild-type or triple mutant (K44E,K50E,K438E), was used to pull down varying amounts of Mcm2–7. D, 2 pmol of CDK-phosphorylated GST-Sld2-full-length, wild-type or triple mutant, was incubated with 2 pmol of radiolabeled Mcm2–7 in the presence of varying concentrations of ssARS1-1. E, 2 pmol of GST-Mcm2–7 was used to pull down 2 pmol of radiolabeled GINS in the presence or absence of 10 pmol of CDK-phosphorylated Sld2, wild-type or triple mutant, and varying concentrations of ssARS1-1.
We next constructed a conditional degron yeast strain of SLD2 (sld2-td) using a standard technique (41). When sld2-td cells are shifted to 37 °C in the presence of galactose, wild-type Sld2 is degraded. We transformed sld2-td cells with a plasmid expressing SLD2 or sld2-K44E,K50E,K438E under control of its native promoter. When these cells are studied for cell growth by 10-fold serial dilutions and plating, the results show that cells expressing sld2-K44E,K50E,K438E exhibit a severe growth defect under restrictive conditions (Fig. 14A).
FIGURE 14.

Cells expressing sld2-K44E,K50E,K438E exhibit a severe growth defect with very slow progression into S phase. A, 10-fold serial dilutions of sld2-td cells expressing SLD2 or sld2-K44E,K50E,K438E at the restrictive (37 °C) or permissive temperature (24 °C). B, Western blot analysis of SLD2 and sld2-K44E,K50E,K438E whole cell extracts showing equivalent levels of Sld2 expression under restrictive conditions. C, sld2-td cells expressing SLD2 or sld2-K44E,K50E,K438E cultured in YP medium with 2% raffinose (YPRaff) to logarithmic phase at 24 °C and synchronized at G1 phase by incubation with α-factor. Cells were cultured first in YPRaff for 1 h at 24 °C then in YP medium with 2% galactose (YPGal) for 1 h at 24 °C, and finally in the same medium for 2 h at 37 °C in the presence of α-factor. Cells were washed and released into the fresh YPGal and cultured at 37 °C. Upper, experimental outline and timing of sampling. Lower, FACS analysis.
The Sld2 protein is expressed at equivalent levels in either cell strain, suggesting that the mutant protein is properly transcribed and expressed (Fig. 14B). We also studied the progression of cells into S phase by arresting the cells in G1 with α-factor and then releasing the cells into medium that lacked α-factor for 30–150 min. Cells expressing wild-type SLD2 accumulated 2C DNA content within 60 min after α-factor release; however, cells expressing sld2-K44E,K50E,K438E exhibited a severe defect in progression into S phase, with little 2C DNA content within 150 min after α-factor release (Fig. 14C). These data suggest that Sld2 binding to origin ssDNA is important for DNA replication.
DISCUSSION
We report that Sld2 binds directly to the Mcm2–7 complex, and CDK phosphorylation of Sld2 or ATP does not affect this interaction. Sld2 binding to Mcm2–7 blocks the interaction between GINS and Mcm2–7, and Sld2 is also competitive with Sld3 for Mcm2–7 binding. CDK-phosphorylated Sld2 and Sld3 inhibit the interaction between GINS and Mcm2–7. CDK-phosphorylated Sld2 binding to Mcm2–7 is stable to size exclusion chromatography, and the stoichiometry of binding is one Sld2 monomer bound to one Mcm2–7 hexamer. We previously found that CDK-phosphorylated Sld2 binds tightly to ssARS1-1, a single-stranded region of the origin ARS1, whereas unphosphorylated Sld2 binds less tightly to ssARS1-1 (39). Here, we show that ssARS1-1 substantially inhibits the interaction between CDK-phosphorylated Sld2 and Mcm2–7, whereas ssARS1-1 inhibition of the interaction between unphosphorylated Sld2 and Mcm2–7 is more modest. The interaction between GINS and Mcm2–7 is inhibited by the presence of Sld2, but this inhibition is alleviated by the presence of ssARS1-1 and CDK phosphorylation of Sld2. We previously found that ssARS1-2-A-to-C, a modified version of the origin sequence ssARS1-2, binds to Sld3. Here, we show that ssARS1-1 and ssARS1-2-A-to-C together allow the interaction between GST-Mcm2–7 and GINS in the presence of both CDK-phosphorylated Sld2 and Sld3. CDK-phosphorylated Sld2-K44E,K50E,K438E does not bind to ssARS1-1, but CDK-phosphorylated Sld2-K44E,K50E,K438E binds to Dpb11 and Mcm2–7 like wild-type CDK-phosphorylated Sld2. Expression of sld2-K44E,K50E,K438E in a sld2-td conditional degron strain results in a severe defect in growth and progression into S phase under restrictive conditions, suggesting that Sld2 interaction with origin-ssDNA is important for DNA replication.
Model for GINS Binding to Mcm2–7
Based on the data presented here and published elsewhere, we propose a new model for GINS binding to Mcm2–7 (Fig. 15). In G1, Sld3-Cdc45 binds to early replication origins (29, 30). This interaction may be mediated by direct contact between Sld3 and Mcm2–7 (Fig. 15A) (36). Sld3 binding to Mcm2–7 will block the interaction between GINS and Mcm2–7, preventing the premature activation of the replication fork helicase (36). During S phase, the DNA will be remodeled, with Mcm2–7 encircling one strand of origin DNA, whereas the second strand of DNA is positioned outside the Mcm2–7 ring (7, 26, 27). Sld3 may bind to the single-stranded origin DNA (38), allowing for the interaction between Sld2 and Mcm2–7 (Fig. 15B). Sld2 now blocks the interaction between GINS and Mcm2–7, preventing the interaction between GINS and Mcm2–7. Sld2 is then phosphorylated by the S-CDK in S phase, promoting the interaction between CDK-phosphorylated Sld2 and origin ssDNA (Fig. 15C). This interaction allows for the interaction between GINS and Mcm2–7, and the replication fork helicase is now assembled. There may be an additional mechanism that we have not yet discovered that helps ensure that Sld2 binds to Mcm2–7 before GINS binds to Mcm2–7.
FIGURE 15.

Model for activation of the replication fork helicase. A, Sld3-Cdc45 binds to Mcm2–7 in G1, and Sld3 blocks the interaction between GINS and Mcm2–7 (36). B, Mcm2–7 ring opens, and ssDNA is extruded from the interior of the Mcm2–7 ring (7, 8, 42). The ssDNA exposed binds to Sld3, releasing Sld3 from Mcm2–7 (38). This event allows Sld2 to bind to the Mcm2–7 complex, and Sld2 blocks GINS from binding to Mcm2–7. C, CDK phosphorylates Sld2, and CDK-phosphorylated Sld2 binds to origin ssDNA, releasing Sld2 from Mcm2–7. This event allows GINS to bind to Mcm2–7, and the replication fork helicase (CMG complex) is assembled.
Two Mechanisms to Block GINS Interaction with Mcm2–7
We found that Sld2 and Sld3 both block the interaction between GINS and Mcm2–7 (36) (Fig. 3). Furthermore, the presence of Sld2 and Sld3 blocks the interaction between GINS and Mcm2–7 more efficiently than Sld2 or Sld3 alone (Fig. 5). Why would the cell employ two different proteins to execute the same function, the blocking of GINS interaction with Mcm2–7? It may be that GINS interaction with Mcm2–7 is a key step that leads to activation of the replication fork helicase (7, 10). It is critical for replication fork unwinding to be precisely timed with DNA replication, and the premature activation of the replication fork helicase may result in uncoupled DNA unwinding. Thus, with Sld2 and Sld3 both blocking the interaction between GINS and Mcm2–7, there are two mechanisms to ensure that the replication fork helicase is not prematurely activated.
CDK Phosphorylation of Sld2 Is Important to Allow GINS Interaction with Mcm2–7
We find that unphosphorylated and CDK-phosphorylated Sld2 bind to Mcm2–7 with similar efficiency (Fig. 2). However, origin ssDNA inhibits the interaction between CDK-phosphorylated Sld2 and Mcm2–7 more efficiently than between unphosphorylated Sld2 and Mcm2–7 (Fig. 7). These data suggest that in the cell, unphosphorylated Sld2 will block the interaction between GINS and Mcm2–7. Once S-CDK has phosphorylated Sld2, the Sld2 protein will bind to origin ssDNA, allowing the interaction between GINS and Mcm2–7. This mechanism will couple the action of S-CDK at a replication origin with assembly of the replication fork helicase. This mechanism will ensure that the replication fork helicase will not be assembled until after S-CDK acts upon Sld2 at a replication origin.
Single-stranded DNA Is a Trigger to Allow GINS Interaction with Mcm2–7
Sld3 and Sld2 both block the interaction between GINS and Mcm2–7 (36) (Fig. 3). However, origin ssDNA inhibits the interaction between Sld3 and Mcm2–7 (38), and origin ssDNA also inhibits the interaction between CDK-phosphorylated Sld2 and Mcm2–7 (Fig. 7). Furthermore, origin ssDNA allows the interaction between GINS and Mcm2–7 in the presence of Sld3 and CDK-phosphorylated Sld2 (Fig. 10). These data suggest that origin ssDNA may be required to allow the direct interaction between GINS and Mcm2–7. Origin ssDNA may be extruded from the interior of the Mcm2–7 ring during S phase because Mcm2–7 encircles dsDNA during G1 phase (26, 27), and the CMG complex binds to ssDNA (7). This ssDNA exposure may bind to Sld3 and CDK-phosphorylated Sld2, allowing GINS to bind directly to Mcm2–7. If this model is correct, then the extrusion of ssDNA from the center of the Mcm2–7 ring may allow for the direct interaction between GINS and Mcm2–7. Thus, in this sense, the extrusion of ssDNA from the center of the Mcm2–7 ring may act as a trigger to allow the formation of the CMG complex, a key step in the initiation of DNA replication.
Acknowledgments
We thank Mike O'Donnell for the expression construct for GINS, Hiroyuki Araki for the expression construct for S-CDK, and Susan Taylor for purified PKA.
This work was supported by American Cancer Society Research Scholar Grant RSG-08-124-01-CCG (to D. L. K. and I. B.), National Science Foundation Grant 1121534 (to D. L. K. and I. B.), a Vanderbilt University Discovery grant (to D. L. K.), and a Vanderbilt Ingram Cancer Center grant (to D. L. K.).
- Mcm
- mini chromosome maintenance
- ATPγS
- adenosine 5′-3-O-(thio)triphosphate
- CDK
- cyclin-dependent kinase
- CMG
- Cdc45-Mcm2–7-GINS
- dsDNA
- double-stranded DNA
- S-CDK
- S phase CDK
- ssDNA
- single-stranded DNA.
REFERENCES
- 1. Diffley J. F. (2010) Cold Spring Harbor Symp. Quant. Biol. 75, 135–142 [DOI] [PubMed] [Google Scholar]
- 2. Labib K. (2010) Genes Dev. 24, 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Araki H. (2010) Curr. Opin. Cell Biol. 22, 766–771 [DOI] [PubMed] [Google Scholar]
- 4. MacNeill S. A. (2010) Biochem. J. 425, 489–500 [DOI] [PubMed] [Google Scholar]
- 5. Pospiech H., Grosse F., Pisani F. M. (2010) Subcell. Biochem. 50, 79–104 [DOI] [PubMed] [Google Scholar]
- 6. Moyer S. E., Lewis P. W., Botchan M. R. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 10236–10241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ilves I., Petojevic T., Pesavento J. J., Botchan M. R. (2010) Mol. Cell 37, 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Costa A., llves I., Tamberg N., Petojevic T., Nogales E., Botchan M. R., Berger J. M. (2011) Nat. Struct. Mol. Biol. 18, 471–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pacek M., Tutter A. V., Kubota Y., Takisawa H., Walter J. C. (2006) Mol. Cell 21, 581–587 [DOI] [PubMed] [Google Scholar]
- 10. Gambus A., Jones R. C., Sanchez-Diaz A., Kanemaki M., van Deursen F., Edmondson R. D., Labib K. (2006) Nat. Cell Biol. 8, 358–366 [DOI] [PubMed] [Google Scholar]
- 11. Masumoto H., Sugino A., Araki H. (2000) Mol. Cell. Biol. 20, 2809–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanemaki M., Labib K. (2006) EMBO J. 25, 1753–1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kamimura Y., Masumoto H., Sugino A., Araki H. (1998) Mol. Cell. Biol. 18, 6102–6109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tak Y. S., Tanaka Y., Endo S., Kamimura Y., Araki H. (2006) EMBO J. 25, 1987–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zegerman P., Diffley J. F. (2007) Nature 445, 281–285 [DOI] [PubMed] [Google Scholar]
- 16. Tanaka S., Umemori T., Hirai K., Muramatsu S., Kamimura Y., Araki H. (2007) Nature 445, 328–332 [DOI] [PubMed] [Google Scholar]
- 17. Masai H. (2011) J. Biochem. 149, 629–631 [DOI] [PubMed] [Google Scholar]
- 18. Sangrithi M. N., Bernal J. A., Madine M., Philpott A., Lee J., Dunphy W. G., Venkitaraman A. R. (2005) Cell 121, 887–898 [DOI] [PubMed] [Google Scholar]
- 19. Matsuno K., Kumano M., Kubota Y., Hashimoto Y., Takisawa H. (2006) Mol. Cell. Biol. 26, 4843–4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu J., Capp C., Feng L., Hsieh T. S. (2008) Dev. Biol. 323, 130–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanchez-Pulido L., Diffley J. F., Ponting C. P. (2010) Curr. Biol. 20, R509–510 [DOI] [PubMed] [Google Scholar]
- 22. Kumagai A., Shevchenko A., Shevchenko A., Dunphy W. G. (2010) Cell 140, 349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sansam C. L., Cruz N. M., Danielian P. S., Amsterdam A., Lau M. L., Hopkins N., Lees J. A. (2010) Genes Dev. 24, 183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boos D., Sanchez-Pulido L., Rappas M., Pearl L. H., Oliver A. W., Ponting C. P., Diffley J. F. (2011) Curr. Biol. 21, 1152–1157 [DOI] [PubMed] [Google Scholar]
- 25. Kumagai A., Shevchenko A., Shevchenko A., Dunphy W. G. (2011) J. Cell Biol. 193, 995–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Remus D., Beuron F., Tolun G., Griffith J. D., Morris E. P., Diffley J. F. (2009) Cell 139, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Evrin C., Clarke P., Zech J., Lurz R., Sun J., Uhle S., Li H., Stillman B., Speck C. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 20240–20245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gambus A., Khoudoli G. A., Jones R. C., Blow J. J. (2011) J. Biol. Chem. 286, 11855–11864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kamimura Y., Tak Y. S., Sugino A., Araki H. (2001) EMBO J. 20, 2097–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Muramatsu S., Hirai K., Tak Y. S., Kamimura Y., Araki H. (2010) Genes Dev. 24, 602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heller R. C., Kang S., Lam W. M., Chen S., Chan C. S., Bell S. P. (2011) Cell 146, 80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Randell J. C., Fan A., Chan C., Francis L. I., Heller R. C., Galani K., Bell S. P. (2010) Mol. Cell 40, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sheu Y. J., Stillman B. (2010) Nature 463, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sheu Y. J., Stillman B. (2006) Mol. Cell 24, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Masai H., Taniyama C., Ogino K., Matsui E., Kakusho N., Matsumoto S., Kim J. M., Ishii A., Tanaka T., Kobayashi T., Tamai K., Ohtani K., Arai K. (2006) J. Biol. Chem. 281, 39249–39261 [DOI] [PubMed] [Google Scholar]
- 36. Bruck I., Kaplan D. L. (2011) J. Biol. Chem. 286, 14157–14167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bochman M. L., Schwacha A. (2008) Mol. Cell 31, 287–293 [DOI] [PubMed] [Google Scholar]
- 38. Bruck I., Kaplan D. L. (2011) J. Biol. Chem. 286, 18602–18613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanter D. M., Kaplan D. L. (2011) Nucleic Acids Res. 39, 2580–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bruck I., Kaplan D. (2009) J. Biol. Chem. 284, 28823–28831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanchez-Diaz A., Kanemaki M., Marchesi V., Labib K. (2004) Sci. STKE 2004, PL8. [DOI] [PubMed] [Google Scholar]
- 42. Bochman M. L., Bell S. P., Schwacha A. (2008) Mol. Cell. Biol. 28, 5865–5873 [DOI] [PMC free article] [PubMed] [Google Scholar]