

Background: β-Propiolactone is applied for inactivation of viruses.
Results: A systematic overview of β-propiolactone modifications on the building blocks of nucleic acids and proteins.
Conclusion: Proteins are more extensively modified by than nucleic acids during inactivation of viruses with β-propiolactone.
Significance: The study provides detailed knowledge that can be utilized to elucidate the chemical modifications occurring in viruses after inactivation with β-propiolactone.
Keywords: Chromatography, Mass Spectrometry (MS), NMR, Vaccine Development, Virus, β-Propiolactone, Inactivation
Abstract
β-Propiolactone is often applied for inactivation of viruses and preparation of viral vaccines. However, the exact nature of the reactions of β-propiolactone with viral components is largely unknown. The purpose of the current study was to elucidate the chemical modifications occurring on nucleotides and amino acid residues caused by β-propiolactone. Therefore, a set of nucleobase analogues was treated with β-propiolactone, and reaction products were identified and quantified. NMR revealed at least one modification in either deoxyguanosine, deoxyadenosine, or cytidine after treatment with β-propiolactone. However, no reaction products were found from thymidine and uracil. The most reactive sides of the nucleobase analogues and nucleosides were identified by NMR. Furthermore, a series of synthetic peptides was used to determine the conversion of reactive amino acid residues by liquid chromatography-mass spectrometry. β-Propiolactone was shown to react with nine different amino acid residues. The most reactive residues are cysteine, methionine, and histidine and, to a lesser degree, aspartic acid, glutamic acid, tyrosine, lysine, serine, and threonine. Remarkably, cystine residues (disulfide groups) do not react with β-propiolactone. In addition, no reaction was observed for β-propiolactone with asparagine, glutamine, and tryptophan residues. β-Propiolactone modifies proteins to a larger extent than expected from current literature. In conclusion, the study determined the reactivity of β-propiolactone with nucleobase analogues, nucleosides, and amino acid residues and elucidated the chemical structures of the reaction products. The study provides detailed knowledge on the chemistry of β-propiolactone inactivation of viruses.
Introduction
Modern vaccines are based on sound scientific knowledge and rational design of all the development stages. Nevertheless, chemical inactivation of viral vaccines is still a matter of trial and error without much detailed knowledge of the chemistry involved. β-Propiolactone and formaldehyde are compounds that are frequently used for the inactivation of viruses and the production of vaccines, such as influenza, polio, and rabies vaccines (1–12). A considerable problem in the production of vaccines is the loss of immunogenicity during the inactivation process (13–15). To prevent destruction of important epitopes and receptor sites in viruses, chemicals with a different specific reactivity have been utilized for inactivation (8, 10, 11, 13, 16–19). According to the literature, formaldehyde reacts primarily with proteins (19, 20), whereas β-propiolactone modifies mainly DNA or RNA (21–23). Therefore, it is expected that β-propiolactone will maintain a high immunogenicity during the inactivation of viruses.
β-Propiolactone is a hazardous compound that may cause cancer (24–30). It consists of an almost planar highly strained four-membered ring (Fig. 1). Due to this strain, β-propiolactone reacts readily with nucleophiles. Extensive studies have been performed in the past to determine the reactivity with many nucleophiles, e.g. water, inorganic salts, amines, alcohols, thiols, and carboxylic acids (31–38). The reaction of β-propiolactone is a typical example of an Sn2 reaction in which the rate of disappearance depends on the concentrations of a particular nucleophile and β-propiolactone according to Equation 1.
In aqueous media, water is an important nucleophile for β-propiolactone, which has at room temperature a half-life of about 3–4 h (39). The half-life can be decreased in the presence of other nucleophiles. Both the concentration and nature of the nucleophile are important for the conversion rate (40). When a mixture of nucleophiles is used, the rate of disappearance of β-propiolactone equals the summation (41, 42),
 |
The situation becomes more complicated in the case of multiple nucleophilic sites present in one molecule, e.g. in a DNA or protein molecule (43, 44). The reactivity of a particular nucleophilic group in such molecules is influenced by global parameters, e.g. pH and temperature, and by local circumstances, e.g. the accessibility and non-covalent interactions (45, 46).
FIGURE 1.

Two possible reaction paths of a nucleophile (Nu) with β-propiolactone leading to (I) acylated and (II) alkylated products.
β-Propiolactone can react with nucleophiles in two ways, resulting in alkylated and acylated products (Fig. 1). According to the Hard Soft Acids Bases theory, hard nucleophiles, such as primary amino groups (RNH2), will react with the hard acyl carbon, whereas soft nucleophiles, such as thiol groups (RSH), will give alkylated products (47). Hardness or softness of a nucleophile is also influenced by local parameters. For example, hydrogen bonding can increase softness of primary amino or thiol groups, which may result in increased alkylation. Also, pH plays a role; when a thiol group becomes deprotonated, its softness decreases. The same holds for water, which becomes a harder nucleophile upon deprotonation. Generally, the microenvironment of the nucleophile influences the hardness and, by that, the reactivity of the nucleophilic group (48).
Not all reactions with β-propiolactone will lead to inactivation of a virus. The main reaction during inactivation will be the reaction with water to give a non-toxic compound: 3-hydroxypropionic acid. Also, salts and buffer components have a large effect on the conversion of β-propiolactone (34). Virus inactivation will only occur after reaction of β-propiolactone with viral constituents e.g. DNA/RNA and protein (23, 38, 49–53). Although a number of chemical modifications induced by β-propiolactone have been elucidated, the nature of all possible reactions is still largely unknown. Current knowledge is not sufficient to develop an accurate inactivation procedure with β-propiolactone to produce a viral vaccine that preserves the highest immunogenicity.
The purpose of the present work was to elucidate by using model compounds the reactions that occur during inactivation of viruses with β-propiolactone. The reactivity of β-propiolactone with (i) plain buffers, (ii) with nucleobase analogues and nucleosides, and (iii) with amino acid residues were investigated in detail. The reaction conditions used in this study were based on inactivation processes applied for viral vaccines, such as influenza and rabies vaccines (10, 54, 55). Frequently used buffers during inactivation of viruses are phosphate, citrate, and HEPES. The kinetics of hydrolysis of β-propiolactone was examined in these buffers. Also products formed during the reaction with buffer components were identified by nuclear magnetic resonance spectroscopy (NMR). Second, a systematic study was performed to determine the reactivity of particular reactive sites in nucleotides. A set of nucleobase analogues and nucleosides was used to identify and quantify by NMR the different chemical modifications that occur during β-propiolactone treatment. Furthermore, the reaction of β-propiolactone was investigated with various amino acid derivatives and synthetic peptides. The peptides synthesized had an amino acid sequence Ac-VTLXVTR-NH2 in which one amino acid residue (X) varies. The reaction of amino acid derivatives was determined by NMR, whereas the conversion of peptides was monitored by tandem reversed-phase liquid chromatography mass spectrometry (LC/MS). In this paper we give an overview of the major conversion products that can be formed during inactivation of viruses by β-propiolactone.
EXPERIMENTAL PROCEDURES
Chemicals
Nucleic acid analogues 2-aminopyridine, 2-aminopyrimidine, aniline, 7-azaindole, benzimidazole, cytidine, deoxyguanosine, deoxyadenosine, hypoxanthine, imidazole, indole, thymidine, pyridine, pyrimidin-4(3H)-one, 2-pyridone, and pyrrole were obtained from Sigma. Amino acid analogues N-acetylarginine methyl ester (Ac-Arg-OMe), N-acetylaspartic acid methyl ester (Ac-Asp-OMe), N-acetylcystine methyl ester ((Ac-Cys-OMe)2), N-acetylglutamic acid methyl ester (Ac-Glu-OMe), N-acetylhistidine methylamide (Ac-His-NHMe), N-acetyllysine methyl ester (Ac-Lys-OMe), N-acetylmethionine methyl ester (Ac-Met-OMe), N-acetyltyrosine amide (Ac-Tyr-NH2), and N-acetyltryptophan (Ac-Trp-OH) were purchased from Bachem Ag (Bubendorf, Switzerland). N-Acetylcysteine methyl ester (Ac-Cys-OMe) was from Sigma NMR. HEPES, sodium dihydrogen phosphate (NaH2PO4·2H2O), disodium hydrogen phosphate (Na2HPO4), trisodium citrate dihydrate, citric acid, and 3-iodopropionic acid were from Sigma. β-Propiolactone was obtained from Acros (Geel, Belgium). β-Propiolactone is reasonably anticipated to be a human carcinogen (56). Handling of this compound should be performed using an efficient hood while wearing adequate protection.
Reactions with Buffers
Three citrate buffers with a pH of 6.6, 7.2, or 7.8 were prepared by mixing 0.125 m sodium citrate with 0.125 m citric acid, three HEPES buffers with a pH of 6.6, 7.2, or 7.8 by adjusting 0.1 m HEPES solution with 1 m sodium hydroxide, and three phosphate buffers with a pH of 6.6, 7.2, or 7.8 combining 0.1 m Na2HPO4 with 0.1 m NaH2PO4. A phosphate-buffered saline (PBS) solution (155 mm NaCl, 2.71 mm Na2HPO4, and 1.54 mm KH2PO4, pH 7.2) was obtained from Invitrogen. The reaction was started by adding 1 μl of β-propiolactone to 990 μl of a buffer. The resulting mixture was briefly vortexed, and 500 μl was diluted with 200 μl of the internal standard trimethylsilyl d4 propionic acid sodium salt (0.075% in D2O,). 1H NMR spectra were recorded at 735-s intervals during 6–15 h. Relevant signals were integrated. From the integrals, the half-life of β-propiolactone was calculated using an exponential decay model (based upon Equation 3).
Reactions of β-Propiolactone with Nucleobase Analogues and Nucleosides
The nucleic acid analogues were dissolved in D2O to final concentrations of 20 mm. A reaction mixture was prepared by successively adding 1500 μl of a nucleobase analog solution, 460 μl of D2O, and 40 μl of 1.0 m potassium phosphate, pH 9.3. The pH of the samples was adjusted to 9.3 using sodium hydroxide of 1.0 m (the high pH was chosen to increase the amount of reaction products). The reaction was started by adding 2 μl of β-propiolactone. After mixing, the solution was incubated for 16 h at 22 °C. Samples were stored at 4 °C before analysis by NMR.
Reactions of β-Propiolactone with Amino Acid Derivatives
The amino acid analogues were dissolved in D2O to final concentrations of 20 mm. A reaction mixture was prepared by successively adding 1500 μl of an amino acid solution, 460 μl of D2O, 40 μl of 1.0 m potassium phosphate, pH 9.0, and 2 μl of β-propiolactone. After the addition of potassium phosphate and β-propiolactone, the solution was homogenized by gentle mixing. A sample of 500 μl was immediately taken and mixed 200 μl of trimethylsilyl d4 propionic acid. 1H NMR spectra were recorded at 735-s intervals during 6–15 h.
Peptides
Peptides were synthesized as described previously (19). In brief, the peptides were produced on a 30-mmol scale by using an automated multiple peptide synthesizer equipped with a 96-column reaction block (SYRO II, Fa. MultiSynTech, Gmbh, Witten, Germany). Couplings were performed with Fmoc2-amino acid (90 mmol), benzotriazolyloxy-tris-[N-pyrrolidino]phosphonium hexafluorophosphate (90 mmol), and N-methylmorpholine (180 mmol). The Fmoc group was cleaved with piperidine/N,N-dimethylacetamide (2/8, v/v). Side-chain deprotection and cleavage from the solid support was affected with trifluoroacetic acid (TFA)/water (19/1, v/v), except for cysteine-, methionine- and tryptophan-containing peptides, which were treated with TFA/ethane thiol (19/1, v/v). The peptides were purified by reversed-phase (C8 column) high performance liquid chromatography, and their identity was confirmed by LC/MS. Before use, peptides were dissolved in water at a final concentration of 1 mm.
Reactions of β-Propiolactone with Peptides
For the reaction of peptides with β-propiolactone, 10 μl of a 1 mm peptide solution and 20 μl of 0.1 m potassium phosphate of pH 3.0, 5.0, 7.0, or 9.0 were added to 70 μl of water. The reaction was started by adding 5 μl of 320 mm β-propiolactone in water (freshly prepared). After mixing, the solution was incubated for 16 h at 4 °C and then 2 h at 37 °C. Samples were stored at −20 °C before analysis by LC/MS.
Reactions of Iodopropionic Acid with Peptides
For the reaction of peptides with 3-iodopropionic acid, 10 μl of a 1 mm peptide solution and 20 μl of 0.1 m potassium phosphate of pH of 9.0 were added to 60 μl of water. The reaction was started by adding 10 μl of 400 mm 3-iodopropionic acid in water. After mixing, the solution was incubated for 14 days at 35 °C. Samples were stored at −20 °C before analysis by LC/MS.
Hydrolysis of β-Propiolactone-modified Residues
After the reaction of peptides with β-propiolactone, 90 μl of 10 mm sodium hydroxide solution was added to 10 μl of the reaction mixture. After mixing, the solution was incubated for 1 h at 37 °C to hydrolyze the base-sensitive modifications. Samples were diluted in 5% (v/v) DMSO and 5% (v/v) formic acid to stop the hydrolysis. Before analysis by LC/MS, samples were stored at −20 °C.
NMR Measurement
The samples were analyzed using an NMR spectrometer (JEOL JNM ECP400 spectrometer) operating at 25 °C and at 400.15 MHz for the 1H frequency. 1H spectra were collected using presaturation to saturate the residual proton signal of water. For each spectrum 16 scans were collected over a spectral width of 15 ppm. Relevant signals were integrated. From the integrals, half-life of β-propiolactone was calculated using an exponential decay model (Equation 4)
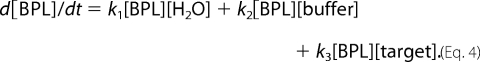 |
Concentration data obtained from the NMR experiments were fitted to the integrated differential equations using a numerical solution (calculated using Runge-Kutta method (42) to give reaction rate constants k1, k2, and k3. From these constants, the half-life of β-propiolactone was calculated (Equation 5).
with t½ = ln 2/Ktotal.
LC/MS
Peptide samples were analyzed by nano-scale reversed-phase liquid chromatography (1200 Series LC system; Agilent Technologies Deutschland GmbH, Waldbronn, Germany) coupled to electrospray mass spectrometry (LTQtm Orbitrap XL), essentially as previously described by Meiring et al. (57). Briefly, each peptide sample was diluted to a concentration of 0.1 μm in water containing 5% (v/v) DMSO and 5% (v/v) formic acid. An injection volume of 10 μl was used for analysis. To desalt the samples for MS analysis, analytes were trapped on a 23-mm length × 50-μm inner diameter trapping column with Reprosil pur C18-AQ (5 μm; Dr. Maisch HPLC GmbH, Ammerbruch-Entringen, Germany) at a flow rate of 3 μl/min and by using 100% solvent A (0.1 m acetic acid in water) as eluent for 10 min. Then analytes were separated by reversed-phase chromatography by using a 25-cm length × 25-μm inner diameter analytical column with Reprosil pur C18-AQ (3 μm) at a flow rate between 20–30 nl/min. A linear gradient was started from 10% solvent B (0.1 m acetic acid in acetonitrile) to 60% solvent B in 25 min. Next, the columns were equilibrated in 100% solvent A for 10 min. The peptides were measured in the MS1 mode (m/z 300–1500) to determine the mass increase and conversion after incubation with β-propiolactone. The heated capillary was set to 150 °C and electrospray voltage to 1.7–1.9 kV. The conversion of a peptide was calculated as a ratio of the chromatographic peak area of the modified peptide and the summed areas of all modified and unmodified peptide peaks. The conversion of peptides by the reaction of β-propiolactone was calculated after LC-MS analysis. Therefore, the multiply charged ESI mass spectra were deconvoluted to MH+ traces. Relevant chromatograms were smoothed according to the boxcar algorithm using a smoothing factor 5. The conversion of a peptide was determined as peak area of unmodified peptide divided by the sum of peak areas of unmodified and modified peptides. A second measurement was performed to obtain detailed sequence information. Peptides were analyzed comprising an MS1 scan (m/z 300–1500) followed by collision-induced dissociation or multistage activation of the selected ions in the MS1 spectra. The collision energy was set on 35%.
RESULTS AND DISCUSSION
Half-life of β-Propiolactone in Buffer Solutions
The inactivation of viruses is normally performed with β-propiolactone concentrations between 0.025 and 1% (4–160 mm). In this experiment we determined the half-life of 16 mm β-propiolactone in water or buffers containing citrate, phosphate, PBS, and HEPES (Table 1). The half-life of β-propiolactone in water was 225 min at 25 °C, which is in good agreement with the value of 3.5 h reported previously (39). An example of NMR spectra is given in Fig. 2, showing the conversion of 16 mm β-propiolactone in a citrate buffer (pH 7.8, 125 mm). β-Propiolactone (NMR peaks at 4.3 and 3.6 ppm) forms 3-hydroxypropionic acid (2.45 and 3.8 ppm) and at least two citrate adducts (AB systems at 2.9–2.75 and 2.75–2.6 and signals at 4.35–4.25 ppm). Fig. 3 shows the kinetics of the degradation of β-propiolactone in the citrate buffer and the formation of β-propiolactone products. The half-life of β-propiolactone in citrate buffer was short if compared with other buffers but independent of the tested pH values (Table 1). In contrast, β-propiolactone does not react with HEPES; its half-life is equal to β-propiolactone in water (Table 1), and no reaction products were detected by NMR. Phosphate buffers showed intermediate behavior with an effect of pH. β-Propiolactone had a half-life of almost 3 h at a pH value of 6.6, whereas at a pH of 7.8 this was reduced to little over 2 h. When the reaction was performed in phosphate buffer, monoalkylated phosphate molecules were also observed. NMR spectra showed a characteristic pseudo quartet at δ3.94. In PBS buffers, the presence of chloride anions (at 150 mm) causes the formation of 3-chloropropionic acid (characteristic NMR triplets at 2.78 and 3.78 ppm). In conclusion, the choice of the buffer and pH is important for availability of β-propiolactone to inactivate viruses.
TABLE 1.
Reaction rate constants and half-life of β-propiolactone in different buffer systems
| Buffer | pH | k1 (H2O) | k2 (buffer) | [Buffer] | Ktotala | t½ |
|---|---|---|---|---|---|---|
| m−1min | m−1min | m | s−1 | min | ||
| Citrate | 6.6 | 5.60E − 05 | 4.89E − 02 | 0.125 | 9.19E − 03 | 75 |
| Citrate | 7.2 | 5.60E − 05 | 5.11E − 02 | 0.125 | 9.46E − 03 | 73 |
| Citrate | 7.8 | 5.60E − 05 | 5.20E − 02 | 0.125 | 9.58E − 03 | 72 |
| HEPES | 6.6 | 5.60E − 05 | 2.05E − 04 | 0.1 | 3.10E − 03 | 224 |
| HEPES | 7.2 | 5.60E − 05 | 4.54E − 03 | 0.1 | 3.53E − 03 | 196 |
| HEPES | 7.8 | 5.60E − 05 | 1.96E − 03 | 0.1 | 3.28E − 03 | 212 |
| Phosphate | 6.6 | 5.60E − 05 | 9.87E − 03 | 0.1 | 4.07E − 03 | 170 |
| Phosphate | 7.2 | 5.60E − 05 | 1.84E − 02 | 0.1 | 4.92E − 03 | 141 |
| Phosphate | 7.8 | 5.60E − 05 | 2.38E − 02 | 0.1 | 5.46E − 03 | 127 |
| PBS | 7.2 | 5.60E − 05 | 4.7E − 03 | 0.15 | 3.80E − 03 | 182 |
| Water | 5.60E − 05 | 3.08E − 03 | 225 |
a Ktotal = k1*[H2O] + k2*[buffer] + k3*[target] and [H2O] = 55 M.
FIGURE 2.

NMR spectra after the reaction of β-propiolactone (16 mm) in citrate buffer (125 mm, pH 7. 8). Spectra were recorded (1–7) after 3, 15, 40, 89, 187, 387, and 775 min. Citrate adducts (C) and 3-hydroxypropionic acid (D) are indicated.
FIGURE 3.

Conversion of β-propiolactone in citrate buffer pH 7. 8, 125 mm at 25 °C; (⧫) β-propiolactone, (□) 3-hydroxy propionate and (△) citrate adducts.
Conversion and Structural Analyses of Nucleobase Analogues Treated with β-Propiolactone
Many nucleophilic sites with an unknown reactivity are present in DNA and RNA, which might react with β-propiolactone. Randerath et al. (51) demonstrated by using 32P-postlabeling 15 distinct adducts in DNA after treatment with high concentration of β-propiolactone. However, their chemical structures are not identified. As a model for nucleic acids, a set of nucleobase analogues and nucleosides was used in this study to determine the reactivity with β-propiolactone The conversion of the analogues revealed that principles, e.g. electron delocalization and tautomerization, affect the reactivity of β-propiolactone with particular nucleophilic sites in nucleobases.
Pyrrole and Pyridine
In heterocyclic compounds such as pyrrole and pyridine, reactivity with β-propiolactone is influenced by aromaticity. In case of pyrrole, the electron pair on the nitrogen atom is needed to sustain aromaticity. Therefore, this electron pair is less available for the reaction with β-propiolactone. Indeed, no reaction products were observed by NMR when treating an aqueous solution of pyrrole with β-propiolactone. According to theory, pyrrole will react at strong conditions with β-propiolactone at the C-3 carbon atom but not at the nitrogen atom (58). On the other hand, the lone pair on the pyridine nitrogen reacted fairly readily with β-propiolactone, because this electron pair is not involved in the aromaticity of pyridine. Sixty-five percent of the pyridine molecules was converted into N-(2-carboxyethyl) pyridine (NMR: δ 8.88, d, 2H, 8.56, dt, 1H, 8.08, t, 2H, 4.80, t, 2H, 2.92, t, 2H). The proposed structure is given in Table 2. In the case of indole, like pyrrole, no reaction products were observed after treatment of indole with β-propiolactone because the single electron pair of indole is needed for aromaticity (Table 2). As expected, the pyridine-like nitrogen atom N-7 in 7-azaindole did react with β-propiolactone, whereas the pyrrole like nitrogen atom N-1 was not modified (Table 2).
TABLE 2.
Products of nuclcobase analogues and nuclcosides obtained by the reaction with β-propiolactone


Conversion is the sum of all products compared to the residual amount of the nucleobase analogue or nucleoside.
Imidazole
In contrast to pyrrole, imidazole was alkylated by β-propiolactone, giving mono- and bis-alkylated products (Table 2). This result is reported previously in the literature (59). Both nitrogen atoms of imidazole, N-1 and N-3, were involved in the alkylation reaction. As a result of tautomerization, the nitrogen N-1 is deprotonated after alkylation at the nitrogen N-3. The electron pair at N-1, which becomes available in the mono-alkylated imidazole, reacted with β-propiolactone to form the bis-alkylated product. Moreover, acylation of imidazole occurred, which resulted in 3-hydroxypropionyl imidazole. However, the acylated compound became hydrolyzed in water providing 3-hydroxypropionic acid and imidazole. This phenomenon is the basis of the catalytic effect of imidazole, increasing the rate of hydrolysis of β-propiolactone (Fig. 4). When the reaction of imidazole with β-propiolactone (66 mm imidazole and 36 mm β-propiolactone in 20 mm phosphate buffer, pH 7.8) was recorded by NMR every 5 min, the transient appearance of N-(3- hydroxypropionyl) imidazole could be observed (NMR: δ 3.28, t, 2H, 4.05, t, 2H, 7.12, bs, 1H, 7.7, bs, 1H, 8.42, bs, 1H) together with the mono-alkylated (NMR: δ 2.7, t, 2H, 4.33, t, 2H, 7.32, s, 1H, 7.42, s, 1H, 8.42, s, 1H) and bis-alkylated (NMR: δ 2.72, t, 2H, 4.42, t, 2H, 7.48, d, 2H, 8.76, m, 1H) products. From the NMR spectra, the concentrations of β-propiolactone and its reaction products with imidazole and water were obtained (Fig. 4). Using a model depicted in Fig. 4, reaction rate constants (k1–5) were calculated with k1 = 5.6 − 10−5, k2 = 6.73 × 10−2, k3 = 9.89 × 10−3, k4 = 6.8 × 10−5, and k5 = 6.41 × 10−2.
FIGURE 4.

Reaction of imidazole with β-propiolactone. I, measured and calculated concentrations of β-propiolactone and products (+), hydroxypropionyl imidazole (⧫), 2-carboxyethyl imidazole (−), 1,3-bis(2-carboxyethyl) imidazole (○), and 3-hydroxypropionic acid (□), based on (II), the model with k1 = 0.000056, k2 = 0.067, k3 = 0.0098, k4 = 0.0000679, and k5 = 0.064.
The reaction of β-propiolactone with benzimidazole also afforded mono-alkylated (NMR: δ 8.5, s, 1H, 7.78, m, 2H, 7.5, m, 2H, 4.6, t, 2H, 2.81, t, 2H) and bis-alkylated (δ 9.28, s, 1H, 7.91, m, 2H, 7.7, m, 2H, 2.86, t, 2H) products as was observed by NMR. The yields of mono-alkylated and bis-alkylated benzimidazole were high, comparable with that of the alkylated imidazole products (Table 2).
Aniline, 2-Aminopyridine, and 2-Aminopyrimidine
Reaction of β-propiolactone with aniline resulted in mono-alkylation (NMR: δ 2.48, t, 2H, 3.38, t, 2H, 6.9, m, 3H, 7.3, m, 2H) and bis-alkylation (NMR: δ 2.55, t, 4H, 3.46, t, 4H, 7.05, m, 3H, 7.4–7.6 m, 2H). The primary amino group of aniline was converted substantially after the reaction, but no acylated product was observed (Table 2). The same products were found after treatment of aniline with 3-iodopropionic acid. 2-Aminopyridine reacted with β-propiolactone to gave two distinct products in a 2:1 ratio (Table 2): an alkylated pyridine nitrogen (NMR: δ 4.39, 2H, t, 2.74, 2H, t) and an alkylated amino group (NMR: δ 3.56, 2H, t, 2.56, 2H, t). 2-Aminopyrimidine reacted with β-propiolactone to give only a pyrimidine-N-alkylated product with a yield of about 12%. The NMR spectrum clearly showed the formation of an unsymmetrical product (NMR: aromatic proton signals at δ 7.1, dd, 1H, 8.38, dd, 1H, and 8.78, dd, 1H, and two aliphatic of CH2 groups at 2.79 and 4.39). No conversion was observed of the primary amino group in 2-aminopyrimidine.
2-Pyridone, Uracil, Pyrimidin-3(4H)-one, and Hypoxanthine
Amide groups that are not nucleophilic do not react with β-propiolactone under conditions tested here. However, it remained uncertain if β-propiolactone can react with the nitrogen atoms of a pyridine ring, such as N-1 of 2-pyridone, N-1 and N-3 of uracil, N-3 of pyrimidin-3(4H)-one, and N-1 of hypoxanthine (Table 2). Therefore, 2-pyridone was treated with β-propiolactone. No reaction products of 2-pyridone were detected by NMR. Also, uracil was not modified by β-propiolactone. In contrast, two alkylated products were formed by the reaction of β-propiolactone with pyrimidin-4(3H)-one (Table 2). The NMR spectrum showed two overlapping triplet signals around 4.2 ppm and two triplets at δ 2.71 and 2.66 ppm, indicative for two distinct NCH2 and CH2CO groups. The total conversion into alkylated nitrogen atoms N-1 and N-3 of pyrimidin-3(4H)-one was 25%. β-Propiolactone reacted with hypoxanthine to give a mixture of alkylated products (8% conversion). The NMR spectrum suggested at least three products; three triplets were observed around δ 4.4–4.6 (NCH2) together with three triplets at δ 2.68 and around δ 2.7–2.9 (CH2CO). In the aromatic part of the spectrum, six additional singlets appeared at δ 8.42, 8.35, 8.28, 8.2, 8.15, and 8.11, respectively. The tentative structures are given in Table 2. In conclusion, nitrogen atoms in a pyridone or pyrimidinone ring can react with β-propiolactone, but to a lesser degree than pyridine and pyrimidine.
Reaction of β-Propiolactone with Nucleosides
Based on the reactions with nucleobase analogues, we predicted that position N-7 and N-3 react with β-propiolactone, whereas the nitrogen atom N-1 and the exocyclic nitrogen C2N will probably hardly react.
The nitrogen atoms N-1 and C2N have homology with a pyridone ring or with the primary amino group of 2-aminopyrimidine, respectively. In the case of deoxyguanosine, additional signals were observed for the 1′H proton at 6.4 (t, 1H) and the H-8 proton at 8.88 ppm, indicating alkylation at position N-7 (Fig. 5). This product was also identified by Roberts and Warwick (23). At prolonged reaction times, additional H-8 signals at 7.92, 7.96, and 8.00 ppm were observed, indicating an unexpected product derived from the N-7 adduct. The proposed structure of the product is given in Fig. 6 in which the imidazole ring is hydrolyzed. As was shown previously, alkylation at N-7 of guanosine stimulates opening of the imidazole ring during the reaction of chloroethyl ethyl sulfide with 2′-deoxyguanosine (60). No indications were found for modifications at positions N-1, N-3, and C2N. For deoxyadenosine, modifications were expected at four positions i.e. N-1, N-3, C6N, and N-7. Only one modification was observed by NMR, consistent with alkylation at N-1, as reported by Chen et al. (61). It showed down-field shifts for the H-2 (8.57, s,1H) and H-8 (8.50, s, 1H) protons, indicating a charged ring system. In the NMR spectrum (Fig. 5) of the reaction mixture with β-propiolactone and cytidine, new signals were observed at δ 8.1 (d, 1H), 6.29 (d, 1H), and 5.89 (d, 1H). These signals indicate the formation of N-(2-carboxyethyl) cytidine (Table 2). As expected from reactions with nucleobase analogues, deoxythymidine did not react with β-propiolactone. If compared with other studies (23, 51), the reactions were preformed with relatively low concentrations of β-propiolactone (16 mm), which might also explain the little number of modifications observed. The concentration of β-propiolactone was comparable with the concentrations normally used for viral inactivation.
FIGURE 5.

NMR spectra of nucleosides treated with β-propiolactone. I, deoxyguanosine. II, deoxyadenosine. III, cytidine. Unmodified nucleosides cause off-scale peaks, whereas adducts show downfield-shifted peaks.
FIGURE 6.

Ring opening reaction of desoxyguanosine-N-7-carboxyethylated adduct.
Reaction of β-Propiolactone with Peptides
A set of 12 synthetic peptides (Ac-VTLXVTR-NH2, in which X = Ala, Cys, Asp, Glu, His, Lys, Met, Asn, Gln, Ser, Trp, or Tyr) was used to investigate the reactivity of β-propiolactone with specific amino acid residues (Table 3). The reactions were performed in phosphate buffers with a pH of 3, 5, 7, or 9. Peptide 1 was designed to be inert for β-propiolactone. Indeed, LC/MS analyses revealed that peptide 1 was not modified after incubation with β-propiolactone in buffers adjusted to a pH of 3, 5, or 7. However, a minor conversion was observed at pH 9 (Table 3). LC/MS2 analyses showed that peptide 1 gave two products with a mass increment of 72 Da located on either the residue Thr2 or the Thr6 (Fig. 7). Other peptides containing a cysteine (peptide 2), aspartic acid (peptide 3), glutamic acid (peptide 4), lysine (peptide 6), methionine (peptide 7), serine (peptide 10), or tyrosine residue (peptide 12) were found to be modified by β-propiolactone. The conversion of these peptides depended on the pH (Table 3); the higher the pH, the higher the conversion of the peptides, except for the methionine residue. Substantial conversion of the methionine residue was measured at pH 3. This latter result is in agreement with data published previously (52). Peptides containing asparagine, glutamine, or tryptophan residues (peptide 8, 9, 11) did not react with β-propiolactone. Based on the LC/MS analyses, the conversion rate of other amino acid residues was at pH 7 in decreasing order: cysteine > methionine > histidine > aspartic or glutamic acid > tyrosine > lysine > serine > threonine.
TABLE 3.
Peptides treated with β-propiolactone at different pH values
| Peptide | Sequence | Mass | Adductsa | ΔMb | Conversionc |
|||
|---|---|---|---|---|---|---|---|---|
| pH 3.0 | pH 5.0 | pH 7.0 | pH 9.0 | |||||
| Da | Da | % | ||||||
| 1 | Ac-VTLAVTR-NH2 | 799.492 | 0d | –e | –e | – | – | 0.3 ± 0.1 |
| 2 | Ac-VTLCVTR-NH2 | 831.464 | 4 | 72/144 | – | 8.4 ± 2.2 | 98.5 ± 0.4 | 98.6 ± 00 |
| 3 | Ac-VTLDVTR-NH2 | 843.481 | 2 | 72/144 | 1.8 ± 0.3 | 4.2 ± 0.2 | 9.2 ± 1.2 | 8.0 ± 0.7 |
| 4 | Ac-VTLEVTR-NH2 | 857.497 | 2 | 72/144 | 0.8 ± 0.1 | 2.8 ± 0.1 | 7.7 ± 1.1 | 10.8 ± 2.3 |
| 5 | Ac-VTLHVTR-NH2 | 865.513 | 2 | 72/144 | – | 0.3 ± 0.0 | 15.5 ± 0.4 | 25.0 ± 0.7 |
| 6 | Ac-VTLKVTR-NH2 | 856.549 | 2 | 72 | – | – | 0.7 ± 0.2 | 5.8 ± 0.4 |
| 7 | Ac-VTLMVTR-NH2 | 859.495 | 2 | 72/144 | 58.7 ± 2.8 | 50.7 ± 3.2 | 37.6 ± 1.2 | 36.4 ± 4.0 |
| 8 | Ac-VTLNVTR-NH2 | 842.497 | 0 | – | – | – | – | 0.5 ± 0.1 |
| 9 | Ac-VTLQVTR-NH2 | 856.513 | 0 | – | – | – | – | 1.0 ± 0.1 |
| 10 | Ac-VTLSVTR-NH2 | 815.487 | 1 | 72 | – | – | 0.2 ± 0.0 | 2.2 ± 0.1 |
| 11 | Ac-VTLWVTR-NH2 | 914.534 | 0 | – | – | – | – | 0.2 ± 0.0 |
| 12 | Ac-VTLYVTR-NH2 | 891.518 | 1 | 72 | – | – | 2.7 ± 0.1 | 14.7 ± 0.3 |
a The number of β-propiolactone adducts attached to the residue in bold (X) are given in bold. Reaction products were identified based on the number of peaks in HPLC chromatograms and MS2 data.
b Mass increments of peptides in phosphate buffers of pH 7.0 are observed after treatment with β-propiolactone.
c Data are the mean ± S.D.; n = 3.
d The alanine residue is not modified. However, two reaction products were found in which one of the threonine residues was slightly modified by β-propiolactone at a pH of 9.0.
e No reaction products of β-propiolactone were detected.
FIGURE 7.

LC/MS chromatograms and MS2 spectra were obtained from peptide 1 after treatment with β-propiolactone at pH of 9. 0. Peptide 1 was slightly converted in two reaction products with a β-propiolactone attached to Thr6 (I) and Thr2 (II) residues, resulting in a mass increment of 72 Da. MS2 spectra revealed also a characteristic fragment with a neutral loss of 90 Da (C3H6O3).
Structural Analysis of Converted Amino Acid Residues
Cysteine
The treatment of cysteine residues (peptide 2) with β-propiolactone resulted in four reaction products that could be separated chromatographically (Fig. 8). LC/MS revealed two adducts with a mass increment of 72 Da (product I and II) and two adducts with an increase of 144 Da (product III and IV). LC/MS2 analysis was used to verify that adducts of β-propiolactone were attached to cysteine residue, although the interpretation of MS2 spectra is rather complex (Fig. 8). Fragmentation of products I and II started with a characteristic neutral loss of 106 Da (C3H6O2S) followed by the fragmentation of the peptide backbone (Table 4). The neutral loss resulted in a truncated cysteine side chain (ΔM of −34 Da = 72–106 Da). MS2 spectra obtained from products I and II are comparable, except for the amount the neutral loss fragment formed (neutral loss of product I > product II). MS2 analysis of product III and IV containing β-propiolactone adducts resulted also in fragments with neutral losses. The spectrum of product III showed a fragment with neutral loss of 178 Da. Neutral loss of 178 Da can be explained by truncation of the cysteine residue (C6H10O4S). However, inadequate backbone fragmentation occurred. The spectrum of product IV demonstrated neutral losses of 72 Da (C3H4O2) and 178 Da (C6H10O4S), and also, backbone fragmentation occurred.
FIGURE 8.

LC/MS analyses performed on peptide 2 treated with β-propiolactone. Four products (I–IV) were identified by LC/MS in which the cysteine residue was modified by β-propiolactone. MS2 spectra I-IV revealed the peptide sequence with a truncated cysteine side chain (−34 Da). Fragmentation during MS2 analysis resulted in a neutral loss fragment of 106 Da (C3H6O2S) or 178 Da (C6H10O4S) if one or two β-propiolactone molecules were attached to the cysteine residue respectively.
TABLE 4.
Typical neutral loss fragments of β-propiolactone-modified amino acid residues
| Residue | Product | ΔM | Neutral loss fragments | |
|---|---|---|---|---|
| Da | ||||
| Cysteine | I | 106 | HS-CO-CH2-CH2OH | C3H6O2S |
| II | 106 | HS-CH2-CH2-COOH | C3H6O2S | |
| III | 178 | HOOC-CH2-CH2-S-CH2-CH2-COOH | C6H10O4S | |
| IV | 72 | H2C CH-COOH CH-COOH |
C3H4O2 | |
| IV | 178 | HS-CH2-CH2-(CO)-O-CH2-CH2-COOH | C6H10O4S | |
| Aspartate | I | 72 | H2CCH-COOH |
C3H4O2 |
| II | 72 | H2CCH-COOH |
C3H4O2 | |
| II | 144 | H2CCH-(CO)-O-CH2-CH2-COOH |
C6H8O4 | |
| Glutamate | I | 72 | H2CCH-COOH |
C3H4O2 |
| II | 72 | H2CCH-COOH |
C3H4O2 | |
| II | 144 | H2CCH-(CO)-O-CH2-CH2-COOH |
C6H8O4 | |
| Histidine | –a | |||
| Lysine | – | |||
| Methionine | I | 120 | H3C-S-CH2-CH2-COOH | C4H8O2S |
| II | 192 | H3C-S-CH2-CH2-(CO)-O-CH2-CH2-COOH | C7H12O4S | |
| Serine | I | 90 | HO-CH2-CH2-COOH | C3H6O3 |
| Threonine | I | 90 | HO-CH2-CH2-COOH | C3H6O3 |
| Tyrosine | – |
a No neutral loss fragments were detected.
The two products with a mass increment of 72 Da can be explained as acylation (product I) and alkylation (product II) of the cysteine residue. The assignment of both peaks was based on the results obtained from the reaction of peptide 2 with 3-iodopropionic acid. This compound can only alkylate reactive amino acid residues. Alkylated products of β-propiolactone and 3-iodopropionic acid have the same structure. As expected treatment with 3-iodopropionic acid resulted in only one product with a mass increment of 72 Da. The retention time of this product was comparable with product II of the peptide 2 treated with β-propiolactone. MS2 analysis confirmed that the cysteine residue of peptide 2 was modified by 3-iodopropionic acid, and the MS2 spectrum was exactly the same as the MS2 spectrum of the product in product II (Fig. 8). In addition, the treatment of peptide 2 with 3-iodopropionic acid gave two products with an increase of 144 Da. The retention times on the column, mass increments, and MS2 spectra of these two products were the same as for products III and IV from β-propiolactone-treated peptide 2. A mixture of β-propiolactone and 3-iodopropionic acid-treated peptide 2 was prepared (in 1:1 molar ratio) and analyzed by LC/MS. The measurement confirmed that the products II-IV were identical because of the elution times and masses (supplemental Fig. S1). In a different experiment, the reaction of β-propiolactone with a cysteine analog (Ac-Cys-OMe) was monitored by NMR. The analysis revealed that cysteine reacts very fast with β-propiolactone at a pH of 9.4 (bicarbonate buffer 100 mm) to give both alkylated (NMR: δ 2.07, 3H, s, 2.47, 2H, t, 2.79, 2H, t, 2.95–3.12, m, 2H, 3.79, 3H, s) and acylated (NMR: δ 2.03, 3H, s, 2.9, 2H, t, 3.26–3.53, 2H, m, 3.78, 3H, s, 3.89, 2H, t) products in about a 2:1 ratio, respectively (Table 5). The formation of acylated product is somewhat unexpected, because Hard Soft Acids Bases theory predicts alkylated product (the thiol group is considered to be a prototypically soft nucleophile (47)). When a 10× excess of β-propiolactone was used, bis-alkylated product was also formed. The proposed structures of modified cysteine residues are given in Table 5.
TABLE 5.
Proposed structures of β-propiolactone-modified amino acid residues

Cystine
According to the literature, β-propiolactone reacts also with disulfide groups (62–64). However, this finding could not be supported in the present study. Peptide 2 containing a cysteine residue was treated with iron(III) chloride to oxidize peptide 2. LC/MS analysis revealed that the oxidation resulted in the formation of a disulfide bridge between two peptide molecules (product MH+ = 1661.92 ± 0.02 Da). Based on the peak areas, 97% of peptide 2 was converted into a dipeptide containing a disulfide bridge. Subsequently, the oxidized peptide was treated with β-propiolactone. The expected products (MH+ = 1733.9 and 1806.0 Da) were not detected by LC/MS, whereas the residual amount of peptide 2 (thiol) was substantially converted by β-propiolactone (MH+ = 904.5 Da). A second experiment confirmed that disulfide groups do not react with β-propiolactone. A cystine derivative (Ac-Cys-OMe)2 was treated with β-propiolactone and monitored by NMR at regular intervals. No reaction products were generated. We conclude that cystine residues do not react with β-propiolactone.
Aspartic and Glutamic Acid
The reaction of β-propiolactone with peptides containing an aspartic or glutamic acid residue (peptides 3 and 4, respectively) resulted in a major product with a mass increment of 72 Da and a minor component with an increase of 144 Da (conversions of about 8 and 0.1%, respectively). LC/MS2 analyses confirmed that one or two β-propiolactone molecules was attached to the aspartic or glutamic acid residue (supplemental Fig. S2). The same products were obtained, albeit in low yields by treating peptides 3 and 4 with 3-iodopropionic acid. Retention times, mass increments, and MS2 spectra were identical for either the β-propiolactone- or 3-iodopropionic acid-modified peptides (peptides 3 or 4). Based on these results, we conclude that aspartic or glutamic acid residues are probably alkylated by the reaction with β-propiolactone. The proposed structures of β-propiolactone-modified aspartic or glutamic acid residues are given in Table 5.
Histidine
Two reaction products were found after incubation of a peptide 5 (containing a histidine residue) with β-propiolactone. Two products of peptide 5 were chromatographically separated and detected in significant amounts by LC/MS with a mass increment of 72 and 144 Da (Fig. 9). Two distinct products with a mass increase of 72 Da were expected, because it has been postulated that histidine can be alkylated at position N-1 and N-3 of the imidazole group (65). However LC/MS revealed only one chromatographically separated product with a mass increment of 72 Da. If histidines were alkylated at position N-1 or at position N-3, the peptides eluted simultaneously. LC/MS2 analysis showed that β-propiolactone (ΔM of 72 Da) was present at the histidine residue (Fig. 9). Unfortunately, LC/MS2 analysis cannot reveal which nitrogen of the histidine residue is modified: position N-1 and/or N-3. The product with mass increase of 144 Da showed poor fragmentation during MS2 analysis. Observed neutral loss fragments of 17, 44, and 61 Da were ascribed as truncation of the arginine side chain. Multistage activation performed on fragment ions revealed the peptide sequence and the modified histidine residue (Fig. 9). Treatment of peptide 5 with 3-iodopropionic acid did not result in significant conversion of the histidine residue. The experiment with 3-iodopropionic acid did not support that alkylation of histidine residues occurs during the reaction with β-propiolactone. In addition, the reaction of a histidine derivative (Ac-His-NHMe) with β-propiolactone was recorded by NMR. The reaction resulted in mono-alkylation and bis-alkylation (NMR: δ 6.8, s, 1H, 7.6, s, 1H and 7.3, s, 1H, 7.7, s, 1H, respectively). Also, NMR demonstrated the presence of only one monoalkylated product, which is in accordance with the results obtained from LC/MS analysis. Because the nitrogen atom N-1 of histidine is less accessible than the nitrogen atom N-3, we assume that the histidine residues will be monoalkylated at position N-3. The proposed structures of β-propiolactone-modified histidine residues are given in Table 5.
FIGURE 9.

LC/MS analyses performed on peptide 5 treated with β-propiolactone. Two products (I and II) were observed by LC/MS containing one or two β-propiolactone molecules attached to the histidine residue, respectively. MS2 analyses revealed the peptide sequence and the attachment of β-propiolactone (+72 Da) (I). IIa, two dominant fragment ions (483.8 and 497.3) were observed during fragmentation of product II, which contains double attachment (+144 Da). IIb, multistage activation performed on fragment 497.3 provided full peptide sequence and the attribution of the modified histidine residue.
Lysine
Two reaction products with only one attachment of β-propiolactone (ΔM of 72 Da) were detected on peptide 6. Both products were widely separated by chromatography (Fig. 10). MS2 measurement confirmed the modification of the lysine residue. The two products can be explained by alkylation and acylation reactions of β-propiolactone (Table 5). Both structures were described previously as N-(2-carboxyethyl)lysine and N-(3-hydroxypropionyl)lysine (66). Alkylation of the lysine side chain forms a secondary amine group that is protonated at a low pH. The polarity of the modified peptide and retention times on the column will not change drastically if compared with the non-modified peptide. Therefore, product I is probably peptide 6 with an alkylated lysine residue (Fig. 10), because the peptide was detected as a double-charged ion with a slightly longer retention time (Δt = ∼0.5 min). In the case of acylation of lysine residues, an amide (peptide) bond is formed that resulted in neutral moiety at a low pH. Therefore, acylation of peptides reduces the polarity and increases the retention times. Indeed, product II mainly consist of single-charged ions with a prolonged retention time (Δt = ∼2.5 min). The reaction of β-propiolactone with a lysine derivative (Ac-Lys-OMe) resulted in both acylated and alkylated products (Table 5). The alkylated product showed characteristic triplets for the secondary amine group (-CH2NHCH2-) at δ 3.18 and 3.20 ppm, whereas the acylated product gave triplets for -N(CO)CH2- at δ 2.57 and 3.84 ppm.
FIGURE 10.

LC/MS analyses performed on peptide 6 treated with β-propiolactone. Two products (I and II) were observed by LC/MS containing one attachment of β-propiolactone. MS2 analyses revealed the peptide sequence of both products and the β-propiolactone modification of lysine residue (I). Product I is probably peptide 6 with an alkylated lysine residue and product II with an acylated lysine residue.
Methionine
LC/MS analysis revealed two adducts with one or two β-propiolactone molecules attached to peptide 7 (Fig. 11). The product with a single attachment of β-propiolactone (ΔM of 72 Da) eluted 5 min earlier than the native peptide, indicating strongly increased polarity. The adduct containing two β-propiolactone molecules (ΔM of 144 Da) eluted 0.5 min later than the product with a single attachment. The structure of a single attachment to methionine has been described in the literature (52) and explains the increased polarity due to the positively charged sulfur atom. MS2 measurement did not provide any sequence information. Modified methionines present in peptides resulted in particular neutral losses of 120 Da for the single attachment (C4H8O2S) and 192 Da for the double attachment of β-propiolactone (C7H12O4S). However, multistage activation on the peptide fragment with a neutral loss (MH22+ of 406.8) provided the full peptide sequence (Fig. 11) with a truncated methionine side chain (ΔM of −48 Da = 72–120 Da). The reaction of 3-iodopropionic acid with peptide 7 resulted in two reaction products with the same mass increments, retention times, and MS2 spectra as the products of β-propiolactone-treated peptide 7, indicating that β-propiolactone only alkylates methionine residues. A mixture of β-propiolactone and 3-iodopropionic acid-treated peptide 7 (in 1:1 ratio) demonstrated by LC/MS the same elution times and masses for the products I and II (supplemental Fig. S3). NMR analysis of Ac-Met-OMe treated with β-propiolactone revealed that an alkylated product was formed (Table 5). In the NMR spectrum, two characteristic singlets were observed corresponding to the diastereomers (around 2.95 ppm) of the S-methyl group together with additional signals of the alkylated product (NMR: δ 4.65, m, 1H, 3.58–3.32, m, 4H, 2.75, dt, 2H, 2.08, s, 3H). When the reaction was carried out with a 10-fold excess of β-propiolactone, another sulfonium adduct was observed (NMR: δ 2.97, ds, 3H, 3.04, dt, 2H, 4.41, t, 2H). The NMR data obtained are in accordance with a biscarboxyethoxylated methionine. Both MS and NMR analyses confirmed that methionine residues become alkylated by the reaction of β-propiolactone. The proposed structures of both products are given in Table 5.
FIGURE 11.

LC/MS analyses performed on β-propiolactone-treated peptide 7. LC/MS chromatograms demonstrated two reaction products (I and II) with a single and double β-propiolactone attachment. Ia, the MS2 spectrum obtained from peptide 7 with a single attachment of β-propiolactone provided only a fragment ion (406.8) with a neutral loss of 120 Da. Ib, multistage activation performed on the fragment ion (406.8) revealed the peptide sequence with a truncated methionine residue (−48 Da). The second product (II) with two attachments of β-propiolactone resulted in a comparable (Ia) MS2 spectrum and (Ib) multistage fragmentation spectrum.
Serine and Tyrosine
Peptides containing a serine or tyrosine residue (peptide 10 and 12, respectively) were slightly converted at pH 7 to a product with a mass increment of 72 Da, corresponding to a single attachment of β-propiolactone (Table 5). MS2 analysis confirmed that β-propiolactone was attached to the serine or tyrosine residue (supplemental Figs. 4 and 5). β-Propiolactone probably reacts with the hydroxyl group of serine or tyrosine. The MS2 spectrum of the peptide 10 with a modified serine residue showed a typical fragment ion with a neutral loss of 90 Da (C3H6O3). A fragment ion with a similar neutral loss of 90 Da was not observed for peptide 12 with a modified tyrosine residue (Table 4).
From the literature it remains unclear if serine, threonine, and tyrosine residues are acylated or alkylated by β-propiolactone. Determann and Joachim (67) found only an acid labile product from tyrosine and concluded that acylation occurred. This was in contrast with results obtained by Gresham et al. (68). They observed alkylation of the phenol during the reaction with β-propiolactone, although acylation of the phenol occurred under acid catalysis. Also, theoretical predictions made by Zhang and Yang (69) indicate that alkylation of tyrosine residue will occur by treatment of β-propiolactone. For unequivocal identification of the reaction products, three different experiments were performed. Peptides 1, 10, and 12 treated with β-propiolactone were also incubated at an increased pH (∼12). The modified serine, threonine, and tyrosine residues were completely hydrolyzed, indicating that they were acylated by β-propiolactone instead of alkylated. The hydrolysis of these modified peptides resulted in the reversion to the original peptides. Also, treatment of peptide 10 and 12 with 3-iodopropionic acid did not result in significant alkylation of the serine and tyrosine residues. In addition, a tyrosine derivative (Ac-Tyr-NH2) was treated with β-propiolactone, and the reaction was monitored by NMR, confirming that tyrosine is acylated by β-propiolactone (NMR: δ 7.32 and 7.1 (AB, 4H), 3.86, t, 2H, 2.63, t, 2H). Based on the results, we conclude that serine, threonine, and tyrosine residues are acylated during the reaction with β-propiolactone (proposed structures are given in Table 5).
Conclusions
β-Propiolactone is extensively used for the production of inactivated viral vaccines against influenza and rabies. Despite decades of large scale use of these vaccines, the chemical modifications that occur during viral inactivation were only partially known. Here we present a detailed and systematic overview of β-propiolactone modifications on the building blocks of nucleic acids and proteins: nucleobase analogues, nucleosides, and amino acid residues. The nucleosides deoxyadenine, cytosine, and deoxyguanine were modified by β-propiolactone, alkylating on positions N-1 of deoxyadenine, on exocyclic amino group of cytosine, and on position N-7 of deoxyguanine. Furthermore, nine amino acid residues were shown to react with β-propiolactone (Table 3). In many cases the amino acid residues were alkylated, but serine, threonine, and tyrosine residues were exclusively acylated. Cysteine and lysine residues were partially acylated and alkylated. Unexpectedly, it was shown that disulfide groups in cystine residues do not react with β-propiolactone. On the other hand, highest conversions were observed for cysteine and methionine residues. Less, but significant, conversions were determined for the nucleosides deoxyadenosine, cytidine, and deoxyguanosine. The results indicate that proteins are more extensively modified by than nucleic acids during inactivation of viruses with β-propiolactone. The amount and nature of modifications in viral components, i.e. proteins, DNA, or RNA, will be dependent on concentration of β-propiolactone, type of buffer, pH of the mixture, and intrinsic reactivity of individual nucleophilic groups. Conditions for inactivation should be optimized to result in a complete inactivation and a sustained high immunogenicity of the virus.
In this study the relative reactivity of nucleosides and amino acid residues for β-propiolactone was elucidated. The data can be utilized to predict or to elucidate through LC/MS or NMR studies the chemical modifications occurring in viral components during the inactivation of viruses with β-propiolactone.
Acknowledgment
We thank Dr. Gideon Kersten for critically reading the manuscript.
This work was supported by the World Health Organization using a grant from the Bill and Melinda Gates Foundation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
- Fmoc
- fluorenyl methoxycarbonyl
- BPL
- β-propiolactone
- ΔM
- mass increments.
REFERENCES
- 1. Campbell T. M., Studdert M. J., Blackney M. H. (1982) Vet. Microbiol. 7, 535–544 [DOI] [PubMed] [Google Scholar]
- 2. Frösner G. G., Stephan W., Dichtelmüller H. (1983) Eur. J. Clin. Microbiol. 2, 355–357 [DOI] [PubMed] [Google Scholar]
- 3. Horowitz B. (1991) Biotechnology 19, 417–430 [DOI] [PubMed] [Google Scholar]
- 4. Logrippo G. A. (1960) Ann. N.Y. Acad. Sci. 83, 578–594 [DOI] [PubMed] [Google Scholar]
- 5. Race E., Stein C. A., Wigg M. D., Baksh A., Addawe M., Frezza P., Oxford J. S. (1995) Vaccine 13, 1567–1575 [DOI] [PubMed] [Google Scholar]
- 6. Scheidler A., Rokos K., Reuter T., Ebermann R., Pauli G. (1998) Biologicals 26, 135–144 [DOI] [PubMed] [Google Scholar]
- 7. Tidke R., Préhaud C., Coulon P., Blancou J., Flamand A. (1987) Vaccine 5, 229–233 [DOI] [PubMed] [Google Scholar]
- 8. Budowsky E. I., Friedman E. A., Zheleznova N. V., Noskov F. S. (1991) Vaccine 9, 398–402 [DOI] [PubMed] [Google Scholar]
- 9. Polley J. R., Guerin M. M. (1957) Can. J. Microbiol. 3, 871–877 [DOI] [PubMed] [Google Scholar]
- 10. Kalbfuss B., Genzel Y., Wolff M., Zimmermann A., Morenweiser R., Reichl U. (2007) Biotechnol. Bioeng. 97, 73–85 [DOI] [PubMed] [Google Scholar]
- 11. Jones R. L., Froeschle J. E., Atmar R. L., Matthews J. S., Sanders R., Pardalos J., Moeller L., Chin J. E., Famula M., Briggs D. J., Lang J. (2001) Vaccine 19, 4635–4643 [DOI] [PubMed] [Google Scholar]
- 12. Salk J. E., Krech U., Youngner J. S., Bennett B. L., Lewis L. J., Bazeley P. L. (1954) Am. J. Public Health Nations Health 44, 563–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rappuoli R. (1997) in New Generation Vaccines (Levine M. M., Woodrow G. C., Kaper J. B., Cobon G. S. eds) 2nd Ed., pp. 417–435, Marcel Dekker, Inc., New York [Google Scholar]
- 14. Metz B., Jiskoot W., Hennink W. E., Crommelin D. J., Kersten G. F. (2003) Vaccine 22, 156–167 [DOI] [PubMed] [Google Scholar]
- 15. Delrue I., Delputte P. L., Nauwynck H. J. (2009) Vet. Res. 40, 62. [DOI] [PubMed] [Google Scholar]
- 16. Ada G. L. (1997) in New Generation Vaccines (Levine M. M., Woodrow G. C., Kaper J. B., Cobon G. S. eds) 2nd Ed., pp. 13–23, Marcel Dekker, Inc., New York [Google Scholar]
- 17. Fu Z. F., Dietzschold B., Plotkin S. A., Rupprecht C. E. (1997) in New Generation Vaccines (Levine M. M., Woodrow G. C., Kaper J. B., Cobon G. S. eds) 2nd Ed., pp. 607–617, Marcel Dekker, Inc., New York [Google Scholar]
- 18. André F. E. (1995) J. Infect. Dis. 171, S33–S39 [DOI] [PubMed] [Google Scholar]
- 19. Metz B., Kersten G. F., Hoogerhout P., Brugghe H. F., Timmermans H. A., de Jong A., Meiring H., ten Hove J., Hennink W. E., Crommelin D. J., Jiskoot W. (2004) J. Biol. Chem. 279, 6235–6243 [DOI] [PubMed] [Google Scholar]
- 20. Metz B., Kersten G. F., Baart G. J., de Jong A., Meiring H., ten Hove J., van Steenbergen M. J., Hennink W. E., Crommelin D. J., Jiskoot W. (2006) Bioconjug. Chem. 17, 815–822 [DOI] [PubMed] [Google Scholar]
- 21. Colburn N. H., Richardson R. G., Boutwell R. K. (1965) Biochem. Pharmacol. 14, 1113–1118 [DOI] [PubMed] [Google Scholar]
- 22. Maté U., Solomon J. J., Segal A. (1977) Chem.-Biol. Interact. 18, 327–336 [DOI] [PubMed] [Google Scholar]
- 23. Roberts J. J., Warwick G. P. (1963) Biochem. Pharmacol. 12, 1441–1442 [DOI] [PubMed] [Google Scholar]
- 24. Colburn N. H. (1969) Vox Sang. 17, 64. [PubMed] [Google Scholar]
- 25. Garte S. J., Hochwalt A. E. (1989) Environ. Health Perspect. 81, 29–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hochwalt A. E., Solomon J. J., Garte S. J. (1988) Cancer Res. 48, 556–558 [PubMed] [Google Scholar]
- 27. Hochwalt A. E., Wirgin I., Felber M., Currie D. D., Garte S. J. (1988) Mol. Carcinog. 1, 4–6 [DOI] [PubMed] [Google Scholar]
- 28. Searle C. E. (1961) Br. J. Cancer 15, 804–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wattenberg L. W., Hochalter J. B., Galbraith A. R. (1987) Cancer Res. 47, 4351–4354 [PubMed] [Google Scholar]
- 30. Woodworth B. A., Scribner J. D., Scribner N. K. (1986) Cancer Lett. 31, 293–297 [DOI] [PubMed] [Google Scholar]
- 31. Long F. A., Purchase M. (1950) J. Am. Chem. Soc. 72, 3267–3273 [Google Scholar]
- 32. Gresham T. L., Jansen J. E., Shaver F. W. (1948) J. Am. Chem. Soc. 70, 1003–1004 [Google Scholar]
- 33. Gresham T. L., Jansen J. E., Shaver F. W., Bankert R. A., Fiedorek F. T. (1951) J. Am. Chem. Soc. 73, 3168–3171 [Google Scholar]
- 34. Gresham T. L., Jansen J. E., Shaver F. W., Gregory J. T. (1948) J. Am. Chem. Soc. 70, 999–1001 [Google Scholar]
- 35. Gresham T. L., Jansen J. E., Shaver F. W., Gregory J. T., Beears W. L. (1948) J. Am. Chem. Soc. 70, 1004–1006 [Google Scholar]
- 36. Bartlett P. D., Rylander P. N. (1951) J. Am. Chem. Soc. 73, 4273–4274 [Google Scholar]
- 37. Bartlett P. D., Small G., Jr. (1950) J. Am. Chem. Soc. 72, 4867–4869 [Google Scholar]
- 38. Dijkstra J. (1975) Chem.-Biol. Interact. 10, 115–121 [DOI] [PubMed] [Google Scholar]
- 39. Hoffman R. K., Buchanan L. M., Spiner D. R. (1966) Appl. Microbiol. 14, 989–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Swain C. G., Scott C. B. (1953) J. Am. Chem. Soc. 75, 141–147 [Google Scholar]
- 41. Davis R. E., Suba L., Klimishin P., Carter J. (1969) J. Am. Chem. Soc. 91, 104–107 [Google Scholar]
- 42. Manso J. A., Pérez-Prior M. T., García-Santos, Mdel P., Calle E., Casado J. (2005) Chem. Res. Toxicol. 18, 1161–1166 [DOI] [PubMed] [Google Scholar]
- 43. Carlson R. M. (1990) Environ. Health Perspect. 87, 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lopachin R. M., Decaprio A. P. (2005) Toxicol. Sci. 86, 214–225 [DOI] [PubMed] [Google Scholar]
- 45. Baugh C. M., Shaw E. N. (1966) Biochim. Biophys. Acta 114, 213–221 [PubMed] [Google Scholar]
- 46. Lyle T. A., Royer R. E., Daub G. H., Vander Jagt D. L. (1980) Chem.-Biol. Interact. 29, 197–207 [DOI] [PubMed] [Google Scholar]
- 47. Ho T. L. (1975) Chem. Rev. 75, 1–20 [Google Scholar]
- 48. Roos G., Loverix S., Brosens E., Van Belle K., Wyns L., Geerlings P., Messens J. (2006) Chembiochem 7, 981–989 [DOI] [PubMed] [Google Scholar]
- 49. Brown A., Consigli R. A., Zabielski J., Weil R. (1974) J. Virol. 14, 840–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hemminki K. (1981) Chem.-Biol. Interact. 34, 323–331 [DOI] [PubMed] [Google Scholar]
- 51. Randerath K., Reddy M. V., Gupta R. C. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 6126–6129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Taubman M. A., Atassi M. Z. (1968) Biochem. J. 106, 829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Atassi M. Z. (1969) Immunochemistry 6, 801–810 [DOI] [PubMed] [Google Scholar]
- 54. Polley J. R., Guerin M. M. (1957) Can. J. Microbiol. 3, 863–870 [DOI] [PubMed] [Google Scholar]
- 55. Budowsky E. I., Zalesskaya M. A. (1991) Vaccine 9, 319–325 [DOI] [PubMed] [Google Scholar]
- 56. IARC (1999) Monographs on the Evaluation of Carcinogenic Risks to Humans, International Agency for Research on Cancer, Vol. 71, p. 1103, Lyon, France [Google Scholar]
- 57. Meiring H. D., Heeft E. v. d., Hove G. J. t., Jong A. d. (2002) J. Sep. Sci. 25, 557–568 [Google Scholar]
- 58. Katritzky A. R., Pozharskii A. F. (2000) Handbook of Heterocyclic Chemistry, Pergamon, Amsterdam [Google Scholar]
- 59. Blackburn G. M., Duce D. (1977) J. Chem. Soc. Perkin Trans. 2, 1492–1498 [Google Scholar]
- 60. Sack G. H., Fenselau C., Kan M.-N. N., Kan L. S., Wood G. W., Lau P.-Y. (1978) J. Org. Chem. 43, 3932–3936 [Google Scholar]
- 61. Chen R., Mieyal J. J., Goldthwait D. A. (1981) Carcinogenesis 2, 73–80 [DOI] [PubMed] [Google Scholar]
- 62. Searle C. E. (1961) Biochim. Biophys. Acta 52, 579–582 [DOI] [PubMed] [Google Scholar]
- 63. Brusick D. J. (1977) Mutat. Res. 39, 241–255 [DOI] [PubMed] [Google Scholar]
- 64. Perrin P., Morgeaux S. (1995) Biologicals 23, 207–211 [DOI] [PubMed] [Google Scholar]
- 65. Jones J. B., Hysert D. W. (1971) Can. J. Chem. 49, 3012–3019 [Google Scholar]
- 66. Segal A., Garte S. J. (1976) Chem.-Biol. Interact. 15, 319–326 [DOI] [PubMed] [Google Scholar]
- 67. Determann H., Joachim H. U. (1971) Z. Klin. Chem. Klin. Biochem. 9, 398–401 [PubMed] [Google Scholar]
- 68. Gresham T. L., Jansen J. E., Shaver F. W., Bankert R. A., Beears W. L., Prendergast M. G. (1949) J. Am. Chem. Soc. 71, 661–663 [Google Scholar]
- 69. Zhang Y. L., Yang Z. Z. (2000) J. Mol. Struct. Theochem. 496, 139–144 [Google Scholar]