

Abstract
Efficient striated muscle contraction requires precise assembly and regulation of diverse actin filament systems, most notably the sarcomeric thin filaments of the contractile apparatus. By capping the pointed ends of actin filaments, tropomodulins (Tmods) regulate actin filament assembly, lengths, and stability. Here, we explore the current understanding of the expression patterns, localizations, and functions of Tmods in both cardiac and skeletal muscle. We first describe the mechanisms by which Tmods regulate myofibril assembly and thin filament lengths, as well as the roles of closely related Tmod family variants, the leiomodins (Lmods), in these processes. We also discuss emerging functions for Tmods in the sarcoplasmic reticulum. This paper provides abundant evidence that Tmods are key structural regulators of striated muscle cytoarchitecture and physiology.
1. Introduction
Striated muscle cells are composed of densely packed myofibrils, which are, in turn, composed of large numbers of sarcomeres that repeat in series. In sarcomeres, actin (thin) filaments slide past myosin (thick) filaments to produce sarcomere shortening and, thus, contractile force. The precise temporal and spatial orchestration of actin filament assembly and architecture is critical for muscle contractile function [1–3]. Notably, precise regulation of actin filament lengths is a key feature of striated muscle sarcomeres and confers muscle-specific biomechanical and contractile properties (sarcomere length-tension relationships) [4–6]. Such finely tuned actin assembly is made possible by the regulatory actions of actin-binding proteins. While a host of proteins are known to nucleate filament assembly, cap the fast-growing (barbed) ends of actin filaments, or bind along the sides of filaments, only the tropomodulin (Tmod) family of proteins caps the slow-growing (pointed) filament ends [7–12]. Tmods (~40 kDa) are present in all metazoans, including flies and worms [13–15], with four Tmod isoforms expressed in mammalian cells; Tmod1 is predominantly expressed in terminally differentiated, postmitotic cells (such as erythrocytes, lens fiber cells, neurons, and striated muscle), Tmod2 is in neuronal tissues, Tmod3 is nearly ubiquitous, and Tmod4 is restricted to skeletal muscle fibers [16–26]. Thus, the Tmod isoforms relevant to actin filament regulation in mammalian striated muscles are Tmod1, Tmod3, and Tmod4. Tmods are dynamic caps that inhibit actin monomer association and dissociation from actin filament pointed ends [12, 27–30]; for a review, see [8]. Tmods also bind the terminal tropomyosins (TMs) of TM-coated actin filaments, and, through TM-isoform-specific binding [16–18, 31–39], they regulate the tightness of actin filament pointed-end capping and, thus, actin filament stability and lengths [12, 15, 19, 30, 36, 37, 40–42]; for reviews, see [1, 8, 43]. Aside from their pointed-end capping activities, some Tmods also possess actin monomer-binding and nucleation activities [40, 44, 45]. In addition to Tmods, striated muscles also contain leiomodin2 (Lmod2), a larger Tmod family variant (~65 kDa) with a potent actin-nucleating activity that can be regulated by TM [21, 46]. It is also noteworthy that the closely related Lmod1, which is predominantly expressed in smooth muscles, is additionally present in a subset of extraocular striated muscle fibers [47, 48].
The amino acid sequences of Tmods share ~80% similarity, and Tmods share a common domain structure with two major domains: an unstructured and extended N-terminal half, the TM/pointed-end actin capping (TM-Cap) domain, and a compactly folded C-terminal half, the leucine-rich repeat/pointed-end actin capping (LRR-Cap) domain (Figure 1). The N-terminal TM-Cap domain is disordered in solution [32, 49–51], except for one α-helix (residues 24–35), which provides the first TM-binding site, and two downstream predicted α-helices (residues 65–75 and 126–135), which bind actin or provide the second TM-binding site, respectively [22, 34, 36–39, 42, 45, 51–53] (Figure 1). The C-terminal LRR-Cap domain of Tmods consists of a series of five leucine-rich repeats (LRRs) that are composed of tandem repeats of a parallel α-helix/β-sheet pair, followed by a nonhomologous C-terminal α-helix (α6) and C-terminal tail, which, together, provide an actin pointed-end capping site [40, 54–56] (Figure 1). Tmod's TM-Cap is the unique TM-regulated actin pointed-end capping domain of the Tmod family, with TM enhancing the capping activity of the TM-Cap domain several thousand-fold [12, 30, 36–38, 40, 42]. On the other hand, Tmod's LRR-Cap domain provides a unique actin pointed-end capping domain for TM-free actin filaments [40, 54, 56]. The TM-Cap domain can also bind actin monomers, and the TM-Cap and the LRR-Cap domains are both required for actin nucleation [44, 45]. These distinct actin-regulatory activities of the TM-Cap and LRR-Cap domains and their constituent α-helices, with respect to monomer-binding, filament capping, and filament nucleation [40, 42, 44, 45] (Figure 1), impart Tmods with isoform-specific actin-regulatory functions. Lmod1 and Lmod2 contain the α1- and α2-helices, but not the α3-helix, of the TM-Cap domain, as well as the complete LRR-Cap domain of Tmods, followed by a nonhomologous, ~150-residue-long C-terminal extension [21, 46]. This extension contains a polyproline region, two predicted α-helices, a basic region, and a WASP homology 2 (WH2) domain, which, in Lmod2, imparts strong actin-nucleating activity [46] (Figure 1).
Figure 1.
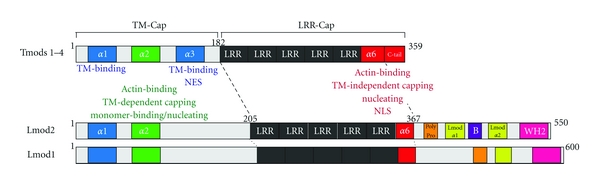
Schematic of the molecular sequence and domain organization of Tmods and Lmods found in vertebrate striated muscle. Tmods contain an N-terminal TM-Cap domain and a C-terminal LRR-Cap domain, whose α-helices possess distinct combinations of TM-binding and actin-regulatory activities, as shown. The α-helices are numbered sequentially within each domain, and the NES and NLS of Tmods are highlighted. Lmods also contain a C-terminal extension with a polyproline region, two predicted α-helices, a basic region, and a WH2 domain.
Tmod family members play important roles in myofibril assembly and thin filament length regulation and are essential for striated muscle development and contractile function in both vertebrates and invertebrates. Tmod1 (possibly in combination with Lmod2 [46, 57, 58]) caps the pointed ends of the sarcomeric thin filaments in cardiac muscle, whereas a combination of Tmod1 and/or Tmod4 caps thin filament pointed ends in skeletal muscle [11, 19, 31, 59–62] (Table 1, Figures 2 and 3). The relative proportions of Tmod1 and Tmod4 in skeletal muscle have not been measured directly but may depend on the muscle twitch speed, with Tmod4 predominating in fast skeletal muscle in chickens [19]. By contrast, Tmod3 is normally not associated with the sarcomeric thin filaments but, instead, appears to cap and stabilize cytoplasmic (nonmuscle) γ-actin filaments in the sarcoplasmic reticulum (SR) of skeletal muscle [63] (Table 1, Figure 3). In mice, an SR-associated (or T-tubule) compartment flanking the Z-line also contains Tmod1 and Tmod4 [63]. Tmod1 is also associated with costameric α2-spectrin at the sarcolemma of fast chicken skeletal muscle fibers [19] (Table 1). Two recent reviews have discussed the roles of Tmods in regulating actin dynamics in myofibril assembly and thin filament length regulation in some detail [1, 64]. Therefore, in this paper, we will cover areas that have not been previously discussed and emphasize new and recent discoveries.
Table 1.
Expression and localization of Tmods and Lmods in striated muscle.
| Protein | Striated muscle expression | Localization in striated muscle | References |
|---|---|---|---|
| Tmod1 | Cardiac and skeletal muscle | Thin filament pointed ends | [11, 28, 31, 59, 60, 62, 65–67] |
| SR- or T-tubule-associated compartment* | [63] | ||
| Sarcolemma | [19] | ||
| Tmod3 | Cardiac and skeletal muscle | SR** | [63] |
| Tmod4 | Skeletal muscle | Thin filament pointed ends | [19, 31] |
| SR- or T-tubule-associated compartment | [63] | ||
| Lmod1 | Slow extraocular muscle fibers | A-band | [47] |
| Lmod2 | Cardiac muscle | Thin filament pointed ends | [46, 57, 58] |
| A-band | [57] | ||
*To date, the putative SR-associated or T-tubule localization of Tmod1 has only been observed in skeletal muscle.
Figure 2.
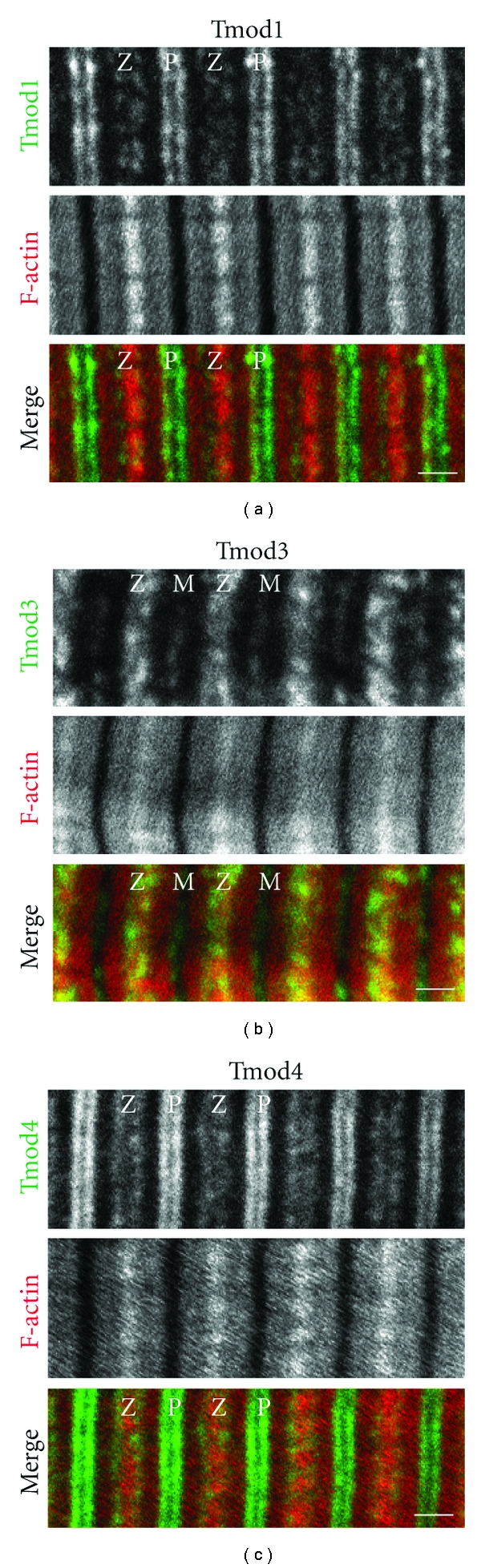
Immunofluorescence localization of Tmods in skeletal muscle. Panels depict longitudinal cryosections of mouse tibialis anterior muscle immunostained for (a) Tmod1, (b) Tmod3, or (c) Tmod4, phalloidin-stained for F-actin, and imaged by confocal microscopy, prepared as in [31, 63]. Note the predominant localization of Tmod1 and Tmod4 at the pointed ends of the phalloidin-stained thin filaments. Tmod1 and Tmod4 also exhibit Z-line-flanking localizations, corresponding to an SR- or T-tubule-associated compartment. By contrast, the M-line and Z-line-flanking localization of Tmod3 is a signature of the SR [63]. Z, Z-line; M, M-line; P, thin filament pointed ends. Bars, 1 μm.
Figure 3.
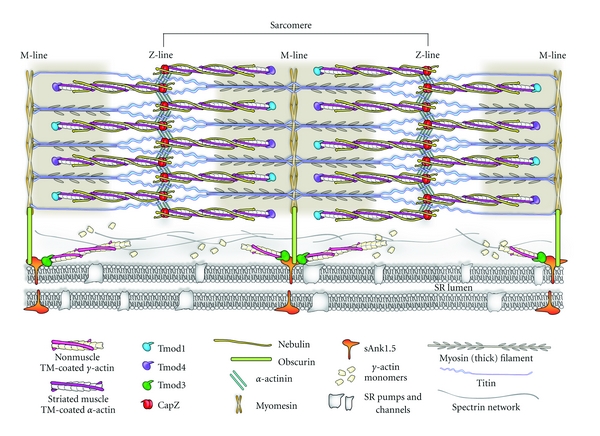
Model diagram of Tmods in the sarcomeres and SR of skeletal muscle. Thin filaments, consisting of α-actin subunits, are capped at their pointed ends by a combination of Tmod1 and Tmod4 in skeletal muscle, whereas Tmod3 is associated with cytoplasmic (nonmuscle) γ-actin filaments in the SR. The exact stoichiometries and distributions of Tmod1 versus Tmod4 on the thin filament pointed ends are unknown. Tmod-capped thin filaments extend past the distal N-terminus of nebulin into the middle of the sarcomere. By contrast, in cardiac muscle (not shown here), thin filament pointed ends are capped solely by Tmod1 and do not contain nebulin. Filaments composed of γ-actin, nonmuscle TMs, and Tmod3 are anchored to the SR membrane via sAnk1.5 and laterally connected by a spectrin network. Tmod3/nonmuscle TM/γ-actin filaments are connected to myofibrils via Tmod3 binding to sAnk1.5, which links to obscurin, which, in turn, is linked to myomesin and titin at the M-line. To date, Tmod3's localization with respect to the cardiac SR remains undetermined. Note that for clarity, the thick filaments are not drawn to scale with respect to the thin filaments.
2. Tmods in Muscle Development and Myofibril Assembly
Tmod1 is one of the earliest sarcomeric proteins expressed during the development of cardiac and skeletal muscle, matching the expression patterns of the basic structural components of sarcomeres. During mouse embryonic development, Tmod1 mRNA and protein are first detected at E8.0 in the developing heart tube as well as in the blood islands of the yolk sac [23, 59, 68], coinciding with sarcomeric α-actinin and α-myosin heavy chain (α-MHC) expression [59, 65, 73]. Tmod1 persists throughout mouse heart development, with expression especially high in the atrium and lower in the ventricle at E15.5 in mid-gestation [65], similar to α-MHC expression [73]. In chicken heart development, Tmod1 transcripts are expressed at Hamburger-Hamilton stage 11 in the looping heart tube, earlier than those of Lmod2 [58], and Tmod1 protein is detected coordinately with cardiac actin and myosin in cardiomyocytes differentiating from explants of precardiac mesoderm [74]. In mouse skeletal muscle development, Tmod1 mRNA is first detected in the developing somites at E9.5, progressing in a caudal-to-rostral fashion during embryonic development, resembling the expression patterns of α-MHC and other sarcomeric proteins [23, 59]. By E14.5-15.5, Tmod1 mRNA and protein are abundant in the developing skeletal muscles of the trunk and limbs, as well as the diaphragm [23, 31]. In chickens, Tmod1 mRNA and protein are expressed in embryonic skeletal muscle at 12 and 18 days of development, while Tmod4 mRNA and protein are not expressed in skeletal muscle until after hatching [19]. The timing and patterns of Tmod3 and Tmod4 mRNA expression during mammalian muscle development have not been investigated although Tmod3 and Tmod4 proteins are detected at E15.5 in developing mouse hindlimb and back muscles [31]. Thus, in summary, Tmod1 expression patterns are consistent with the notion that Tmod1 is an integral structural component of the sarcomeric contractile machinery in both cardiac and skeletal muscle.
To understand how sarcomeric actin filament (thin filament) assembly and lengths are coordinated with the precisely orchestrated spatiotemporal assembly of myofibril components during development, Tmod1 assembly into myofibrils has been studied in the developing embryonic mouse and chick heart and in cultured cardiomyocytes. In the developing mouse heart, Tmod1 associates with nascent myofibrils containing nonstriated actin (i.e., with unregulated lengths) during initial stages of myofibrillogenesis and gradually becomes striated coordinately with actin, as myofibrils mature and precise thin filament lengths are specified [59] (R. B. Nowak and V. M. Fowler, unpublished data). This mirrors the results of experiments on cultured chick skeletal myotubes, in which Tmod1 assembles into puncta in nascent myofibrils in lamellipodial extensions containing nonstriated actin early in myofibrillogenesis and gradually becomes striated along with actin as myofibrils mature [61]. By contrast, in whole-mount preparations of embryonic chicken hearts, in chick cardiac mesoderm explants, and in developing axolotl hearts, Tmod1 localizes to puncta at the cell membrane, where myofibril assembly is initiated early in development, but as myofibrillogenesis nears completion later in development, thin filaments appear to become striated (i.e., length-regulated) prior to capping by Tmod1 [66, 67, 74]. Similarly, in embryonic chick cardiomyocyte cultures, Tmod1 assembles onto thin filaments after TM and all other major sarcomeric proteins have assembled, and after actin becomes striated, suggesting that Tmod1 capping is not required for thin filament length regulation during myofibril assembly [60]. These differences in the timing of Tmod1 assembly are not explained by variations in Tmod isoforms, because Tmod1 caps the thin filament pointed ends in all of these systems [19, 59, 61, 66, 67, 74]. Instead, alterations in Tmod1 conformation and epitope accessibility at various stages of myofibril assembly may play a role, because a Tmod1 polyclonal antibody or a Tmod1 monoclonal antibody (mAb95) detects Tmod1 in nonstriated actin structures at the membrane and in mature striated myofibrils in embryonic chicken hearts, skeletal myotubes, and axolotl hearts [61, 66, 67, 74], whereas another antibody, mAb9, did not detect any Tmod1 associated with nonstriated actin structures near cell membranes in either chick or axolotl hearts [66, 67]. Since mAb9 binds to the actin-capping site in the LRR-Cap domain of Tmod1 [28, 40], Tmod1's actin-capping activity at thin filament pointed ends may vary during myofibril assembly, thus masking or revealing this epitope. Based on these studies, Tmod1 may have an early function in regulating actin pointed-end dynamics in the initial stages of myofibril assembly at the membrane and a later function in maintaining thin filament lengths in mature myofibrils [60, 65, 66].
An essential requirement for Tmod1 in cardiac development and myofibril assembly is demonstrated by the phenotype of Tmod1-null mice [59, 68] (Table 2). Deletion of Tmod1 is embryonic-lethal, with Tmod1-null embryos appearing morphologically normal through E7.5 but exhibiting defects in cardiac looping morphogenesis, chamber formation, and myofibrillogenesis by E8.5, which result in aborted establishment of the circulation and death by E9.5-10.5 [59, 65, 68]. The primary defect of Tmod1-null mice is in the myocardium, because cardiomyocyte-specific expression of Tmod1, via a Tmod1 transgene expressed using an α-MHC promoter, can completely reverse essentially all of the pathological phenotypes observed in the Tmod1-null mouse, including the cardiac looping defect, aberrant myofibril assembly, defects in yolk sac vasculogenesis, and embryonic lethality [65]. In the absence of Tmod1, myofibrils in the developing mouse heart are unable to mature from primitive I-Z-I bodies with nonstriated actin, resulting in incompletely assembled thin filaments and impaired contractility [59, 65, 68]. However, these results are distinct from an in vitro model of myofibril assembly in cardiomyocytes differentiating from mouse embryonic stem cells in Tmod1-null embryoid bodies, where occasional thin and wispy myofibrils do assemble with regulated thin filament lengths [69]. Similar to the Tmod1-null embryonic heart, contractility is also impaired in this in vitro model, due to reduced numbers and defective maturation of myofibrils and cardiomyocytes [69]. The unexpected presence of some myofibrils with regulated thin filament lengths in absence of Tmod1 is unlikely to be due to compensation by Tmod4, since Tmod4 mRNA is not expressed in the embryonic stem cells or embryoid bodies [69]. However, Tmod3 mRNA is present, and while Tmod3 mRNA levels are unchanged upon deletion of Tmod1 [69], Tmod3 protein may have assembled onto the thin filament pointed ends, structurally compensating for absence of Tmod1, similar to compensation by Tmod3 protein in Tmod1-null skeletal muscles [31].
Table 2.
Summary of myofibril assembly and thin filament length phenotypes in Tmod1 perturbation experiments.
| Perturbation | Model system | Experimental strategy | Resulting phenotype | References |
|---|---|---|---|---|
| Reduced Tmod1 levels | Mouse embryonic heart | Gene knockout | Myofibril disorganization, nonstriated actin | [59, 65, 68] |
| Mouse embryonic stem cells | Gene knockout | Myofibril disorganization, nonstriated actin | [69] | |
| Mouse skeletal muscle | Gene knockout | No effect* | [31] | |
| Rat cardiomyocytes | Antisense cDNA expression | Thin filament elongation, nonstriated actin | [70] | |
| Drosophila primary muscle cells** | RNAi knockdown | Sarcomere length irregularity, thin filament elongation | [71] | |
| C. elegans body wall muscle*** | RNAi knockdown, unc94/tmd-1 mutant alleles | Myofibril disorganization, nonstriated actin, myofibril attachment defects | [14, 15] | |
| Inhibition of Tmod1 function | Chick cardiomyocytes | Antibody inhibition of actin capping | Thin filament elongation, nonstriated actin | [28] |
| Chick cardiomyocytes | Antibody inhibition of TM binding | Thin filament disassembly | [41] | |
| Tmod1 overexpression | Chick cardiomyocytes | cDNA expression | Thin filament shortening | [29] |
| Rat cardiomyocytes | cDNA expression | Myofibril disorganization, thin filament shortening | [70] | |
| Adult mouse heart | α-Myosin heavy chain promoter-driven Tmod1 transgene | Myofibril disorganization, thin filament shortening, intercalated disc defects | [72] | |
| Drosophila indirect flight muscle** | HSP-90-driven Tmod transgene | Thin filament shortening | [13] | |
| Drosophila primary muscle cells** | Dmef-GAL4, UAS-Tmod transgene | Thin filament shortening | [71] | |
*No myofibril assembly or thin filament length alterations occur in Tmod1-null mouse skeletal muscle, but extrasarcomeric SR defects do occur due to Tmod3 translocation from the SR to the thin filament pointed ends.
**Drosophila Tmod was knocked down or overexpressed.
***C. elegans UNC94/TMD-1 was knocked down or overexpressed.
In contrast to cardiac muscle, deletion of Tmod1 from mouse skeletal muscle does not appear to affect skeletal muscle development or myofibril assembly in vivo [31] (Table 2). This is most likely due to the additional presence of Tmod4 at the thin filament pointed ends, which may protect embryonic skeletal muscles from the myofibril assembly defects observed in Tmod1-deficient cardiac myofibrils (Figures 2 and 3). In addition, in adult Tmod1-null muscles, Tmod3 translocates from extrasarcomeric sites to cap thin filament pointed ends together with Tmod4, thus maintaining correct thin filament lengths and sarcomere structure, structurally compensating for the absence of Tmod1 as skeletal muscle matures [31, 63]. Taken together, these studies indicate that, in mice, complete removal of pointed-end capping by Tmods severely disrupts both muscle development and myofibril assembly (in cardiac muscle), but partial removal has no obvious effects (in skeletal muscle). The minimum stoichiometry of Tmods that are required to sustain the normal developmental program and myofibrillogenesis in skeletal muscle remains unclear.
An intriguing question raised by these studies is the relationship between Tmod1's role in myofibril assembly and Tmod1's role in muscle development [31, 59, 65, 68, 69]. Namely, is Tmod1's primary role to regulate myofibrillogenesis, with defects in cardiac looping and chamber formation in Tmod1-null hearts as secondary consequences of impaired contractility? Alternatively, could Tmod1 also function in an independent pathway to regulate cardiac morphogenesis and development? Interestingly, Tmod1 is trafficked through the nucleus in cultured skeletal myocytes and cardiomyocytes, as well as fibroblasts, and Tmod1, Tmod2, and Tmod3 all contain a pattern-4 nuclear localization signal (NLS; residues 340–343 of Tmod1; Figure 1), whereas Tmod4 and the Drosophila Tmod homologue contain arginine- and lysine-containing motifs that are similar to pattern-4 NLSs [75]. Similarly, Tmod1 contains a nuclear export signal (NES; residues 134–136; Figure 1) that, when mutated, results in Tmod1 accumulation in the nucleus [75]. Tmod1 accumulation in the nuclei of differentiating C2C12 mouse myoblasts results in depressed expression of muscle-specific genes and delayed myogenic differentiation in vitro [75]. Tmod1's NES is contained within the TM-binding α3-helix (Figure 1), and mutation of these residues to eliminate TM binding leads to nuclear accumulation of Tmod1, indicating that the TM-binding and nuclear export activities of Tmod1 may function coordinately [52, 75]. Tmod1 may also influence muscle development via Tmod1 capping and stabilization of TM-actin filaments, thereby depleting the actin monomer pool and regulating muscle gene expression via the actin/myocardin-related transcription factor/serum response factor circuitry [76].
Recent studies of Tmod function in invertebrates provide clues to a network of actin regulatory proteins that orchestrate actin filament organization in myofibril assembly and control thin filament lengths in sarcomeres [64] (Table 2). In the obliquely striated body-wall muscle of C. elegans, mutation or RNAi depletion of the Tmod homolog UNC94/TMD-1 leads to severe actin filament disorganization of actin filament bundles with accumulation near cell-cell boundaries and impaired motility [14, 15]. In addition, defects are also observed in the organization of muscle dense bodies, the myofibril attachment sites at the membrane [14]. The actin organization and myofibril assembly defects in unc94/tmd-1 mutants are strikingly similar to those observed in C. elegans mutants for actin-depolymerizing factor (ADF)/cofilin, actin-interacting protein 1, and profilin, which also regulate sarcomeric actin filament assembly and stability [15, 77–79]. ADF/cofilin enhances dissociation of actin subunits from the pointed end [80], while TMD-1 inhibits ADF/cofilin-induced actin depolymerization by a TM-dependent pointed-end capping mechanism in vitro [15]. These biochemical interactions imply that TMD-1 would be antagonistic to ADF/cofilin function in vivo. However, unexpectedly, TMD-1 cooperates synergistically with ADF/cofilin to control sarcomeric actin organization and motility, with double mutants more severely affected than single mutants [15]. These in vivo genetic interactions could be explained if, in muscle cells, TMD-1 and ADF/cofilin both prevent excessive actin elongation at the thin filament pointed end by TMD-1 capping the pointed end and ADF/cofilin inducing pointed-end disassembly. The identification of cofilin2 gene mutations in patients with nemaline myopathy, a skeletal muscle disease characterized by accumulations of aberrant actin filament bundles and muscle weakness [81], suggests conserved mechanisms to regulate sarcomeric actin assembly and organization that may also involve Tmods [15]; for reviews, see [1, 64].
3. Tmods and Thin Filament Length Regulation in Striated Muscle Sarcomeres
Once thin filaments are assembled into sarcomeres and myofibrils during muscle development, Tmods function to regulate thin filament lengths by controlling actin filament elongation and shortening at pointed ends [1] (Table 2). In cardiac myofibrils, where thin filament pointed ends are capped by Tmod1, actin dynamics and thin filament lengths are inversely proportional to Tmod1 levels [29, 70]. This was observed directly in experiments in which rhodamine-labeled skeletal muscle α-actin (rho-α-actin) was microinjected into living chick cardiomyocytes, allowing direct visualization of actin monomer incorporation at both the barbed and pointed ends [29]. Inhibition of actin dynamics at the barbed and pointed ends has distinct effects: overexpression of GFP-Tmod1 reduces actin incorporation at pointed ends and leads to thin filament shortening, but cytochalasin D inhibition of barbed-end dynamics has no effect on lengths [29]. Furthermore, microinjection of an antibody that inhibits Tmod1's actin pointed-end capping activity by binding to the actin capping site in the C-terminal α6-helix of the LRR-Cap [28, 40] results in elongation of thin filaments from their pointed ends and inhibition of cardiomyocyte beating [28]. Thus, while actin monomer exchange occurs at both barbed and pointed ends in striated muscle sarcomeres, only subunit exchange at pointed ends directly determines thin filament length. Moreover, both rho-α-actin microinjection into cardiomyocytes [29] and GFP-α-actin expression in living mouse skeletal muscle [82] revealed preferential incorporation at thin filament pointed ends, indicating that the pointed ends are more dynamic than the barbed ends. The interaction between Tmod1 and cardiac TM is essential for thin filament length regulation by Tmod1; monoclonal antibody inhibition of the TM-binding site in the TM-Cap α1-helix causes Tmod1 dissociation and thin filament depolymerization from the pointed end [41]. Indeed, Tmod1 targeting to pointed ends in cardiomyocytes depends on prior assembly of TM [60] and on both the α1- and α3-helices in the TM-Cap domain [56]. Thus, actin, Tmod1, and TM synergize to maintain thin filament lengths in cardiomyocytes.
While cultured cardiomyocytes provide a useful system to study Tmod targeting to thin filament pointed ends and length regulation, they provide less insight into the physiological ramifications of altered thin filament regulation when Tmod1 levels are perturbed. α-MHC-driven transgenic overexpression of Tmod1 in the in vivo mouse heart leads to shorter thin filaments, consistent with increased pointed-end capping in these hearts [72] (Table 2). Mice with Tmod1-overexpressing hearts develop a dilated cardiomyopathy between P14 and P28, which occurs concomitantly with a loss of myofibrillar organization, featuring defects that reflect the in vitro scenario with striking similarity [70, 72]. The onset of dilation does not appear to alter the extent of Tmod1 overexpression, indicating that these processes are not directly coupled to one another, but rather, overexpression of Tmod1 appears to trigger a cascade of compensatory responses, including myocardial remodeling, changes in intercalated discs, and myocardial Ca2+ handling that lead to dilated cardiomyopathy [83–88].
The notion that the extent of pointed-end capping by Tmods is inversely proportional to thin filament length is not unique to cardiomyocytes and is supported by studies in Drosophila melanogaster indirect flight muscle (Table 2). Transient overexpression of Drosophila Tmod during indirect flight muscle development via the HSP-90 promoter inhibits actin elongation from thin filament pointed ends and reduces thin filament lengths [13]. Note that the Drosophila Tmod gene was originally misidentified as responsible for the Sanpodo (spdo) asymmetric cell division phenotype [89, 90], but spdo was later determined to be a tetraspanin membrane protein in the Notch signaling pathway [90–92]. Thin filament lengths in Drosophila are also regulated by sarcomere length short (SALS), a Drosophila WH2-domain-containing protein that is required for GFP-actin incorporation at pointed ends and thin filament elongation during myofibril assembly in developing larval muscles [71]. Genetic manipulation of SALS and Tmod levels in primary embryonic muscle cells revealed that SALS promotes the lengthening of thin filaments by antagonizing the pointed-end capping activity of Tmod [71], analogous to a proposed function of Lmod2 in cardiomyocytes (see Section 5) [58], which do not contain the insect-specific SALS protein [71]. However, similar to Tmod, SALS appears to cap actin filament pointed ends in vitro, which is puzzling based on its in vivo function in promoting pointed-end elongation [71]. Further biochemical studies are required to clarify cooperative and/or competitive actin regulation by SALS and Tmod. Presumably, Drosophila indirect flight muscle cells are programmed to maintain a balance between SALS and Tmod to optimize thin filament lengths in a manner that promotes muscle contraction and flight, as flies with excess Tmod and shortened thin filaments are unable to fly [13].
In the case of myofibrils in mammalian skeletal muscle, thin filament length regulation is further complicated by the presence of a combination of Tmod isoforms (Tmod1 and Tmod4) at the pointed ends [11, 19, 31] (Figures 2 and 3). Partially depleting the Tmod isoform inventory of skeletal muscle by deleting Tmod1 does not alter thin filament lengths in vivo, presumably because Tmod3 can structurally substitute for the absence of Tmod1 by leaving its SR compartment (see Section 6) to cap thin filament pointed ends in conjunction with Tmod4 [31, 63]. The switch from Tmod1/Tmod4-based capping to Tmod3/Tmod4-based capping of thin filament pointed ends produces physiological changes despite unchanged thin filament lengths; these changes include depressed isometric stress production, impaired locomotor activity, and fiber type reprogramming toward a faster phenotype [31]. Given that this Tmod1-null skeletal muscle phenotype is distinct from the more striking thin filament length misregulation phenotypes observed as a consequence of Tmod1 perturbations in cardiac muscle (Table 2), the minimum Tmod isoform types and levels that are required to maintain correct thin filament lengths in skeletal muscle sarcomeres remain unclear. Indeed, the stoichiometry of Tmods at thin filament pointed ends in rat psoas skeletal muscle, which likely contains Tmod1 and Tmod4 [11, 20, 21, 31] (V. M. Fowler, unpublished data), has been determined to be between 1.2–1.6 Tmods/pointed end, based on quantitative immunoprecipitation using Tmod1 antibodies [11], but this may have been an underestimate due to antibody cross-reactivity with Tmod4 in the absence of prior cross-adsorption [31]. To fully address this quandary in vivo, Tmod1 and Tmod4 isoform stoichiometries at thin filament pointed ends need to be measured for various skeletal muscles, and Tmod3-null, Tmod4-null, and Tmod1/Tmod3/Tmod4-double- and triple-null mice need to be developed for future studies.
4. Tmod/Nebulin Interactions and Thin Filament Length Regulation in Skeletal Muscle
In addition to being capped by both Tmod1 and Tmod4, skeletal muscle thin filaments are also distinct from cardiac thin filaments because they contain giant (~600–900 kDa) nebulin molecules that coextend with actin along their lengths (Figure 3). Several recent reviews have discussed the roles of nebulin in thin filament length regulation and actomyosin crossbridge activity [93–96], but, here, we will focus on aspects of nebulin relevant to Tmods. Nebulin's N-terminal domain is located near thin filament pointed ends and contains an interaction site for Tmods [97], while nebulin's C-terminal domain is located at the barbed filament end in the Z-line, containing an interaction site for CapZ [98, 99]. Tmod1, Tmod4, and the C-terminal LRR-Cap domain of Tmod1 bind to an N-terminal module (M1M2M3) of nebulin in vitro, based on solid-phase ELISA-binding assays with nebulin fragments and blot overlay assays with full-length nebulin [54, 97]. Binding of native nebulin and Tmods purified from mammalian skeletal muscle has also been demonstrated in blot overlay assays, but this interaction is weak [97, 100]. Tmod4 binds to nebulin more strongly than Tmod1, and TM does not appear to affect the interaction between Tmods and nebulin [97]. However, the effect of the nebulin M1M2M3 module on Tmod's ability to cap the pointed ends of TM-coated or TM-free actin filaments has not been investigated in vitro.
Previous models of nebulin's layout within the thin filament have asserted that nebulin is a “molecular ruler” that orchestrates thin filament assembly and lengths. The ruler hypothesis argues that nebulin specifies the number of actin monomers that can polymerize and fill the space between its C-terminal CapZ-binding site and its N-terminal Tmod-binding M1M2M3 module, which would presumably localize to their respective thin filament ends [1, 3, 93, 101, 102]. Such an arrangement predicts that shorter or longer nebulin splice variants [103–105] produce shorter or longer thin filaments, respectively, and account for the correlation between nebulin isoform size and thin filament length [106, 107]. However, surprisingly, recent data have demonstrated instead that the nebulin M1M2M3 module is located only ~0.9-1.0 μm away from the Z-line in all mature mammalian muscles studied, whereas the thin filament pointed ends extend from ~0.1 μm up to ~0.4 μm further from the Z-line, well past the nebulin M2M3M3 module [1, 31, 108] (Figure 3). Moreover, despite the interactions between Tmods and the nebulin M1M2M3 module in vitro, Tmod1 and Tmod4 are located at the extreme pointed ends of the thin filaments and do not colocalize with the nebulin M1M2M3 module in mature mouse, rabbit, and chicken muscles [31, 108], as well as in human muscles (D. S. Gokhin and V. M. Fowler, unpublished data). Thus, Tmods likely do not interact with nebulin in mature mammalian muscles in vivo [31, 108]. Furthermore, in neonatal nebulin-null mice, Tmod1 is correctly localized to the pointed ends of the thin filaments, whose lengths are now uniformly ~1.0 μm, up to ~30% shorter than in wild-type muscles (as determined by Tmod1 localization and phalloidin staining) [109]. Thin filament lengths in the absence of nebulin are identical across all muscles and, curiously, are similar to the distance at which the nebulin M1M2M3 module normally resides with respect to the Z-line [31, 108]. In addition, thin filaments in nebulin-null mouse skeletal muscles appear to partially degenerate during muscle use as the mice develop, until lethality occurs between P7 and P21 [99, 109], explaining the broad Tmod localization observed across the I-bands at P10 [6]. Irregular thin filament disassembly during muscle development or use may also explain the broad Tmod localization pattern associated with the nonuniform and shorter filament lengths in the muscles of human nemaline myopathy patients that contain reduced nebulin levels [6]. Likewise, thin filaments in nebulin siRNA-treated chick skeletal myotubes are unusually prone to latrunculin A-induced actin depolymerization [110]. Based on these observations, nebulin appears to both mechanically and molecularly stabilize a large, ~1.0-μm-long core region of the thin filament, whose length defines the minimum thin filament length that a skeletal muscle sarcomere can stably maintain. In conjunction, actin dynamics at the thin filament pointed ends (regulated by Tmod1 and Tmod4) are proposed to specify the lengths of the nebulin-free, ~0.1 to ~0.4-μm-long thin filament extensions beyond the M1M2M3 module of nebulin [1, 29, 31, 108, 110]. However, the actin dynamics properties and actomyosin crossbridge-regulatory characteristics of these nebulin-free extensions (compared to those of the “canonical” nebulin-coated thin filament cores) have not been studied directly.
It remains uncertain how the nebulin M1M2M3 module, or other nebulin modules, could regulate Tmod-mediated capping “at a distance” in order to specify precise muscle-specific thin filament lengths determined by the variable lengths of the nebulin-free thin filament extensions [1, 31, 108]. This conundrum is highlighted by an elegant recent experiment in chick skeletal myotubes, in which endogenous nebulin was replaced by a shorter mininebulin containing the N- and C-termini of human nebulin but lacking 18 (out of 22) central superrepeats [110]. Even in the presence of mininebulin, the thin filaments still extended well past the N-terminal end of mininebulin and were capped by Tmod1 at their normal location in the center of the sarcomere [110]. Nevertheless, the dynamics of GFP-α-actin, GFP-Tmod1, and GFP-TM were enhanced in the absence of nebulin, and dynamics were reduced (rescued) by introducing mininebulin, as measured by fluorescence recovery after photobleaching (FRAP). This suggests that nebulin may alter the long-range conformation of the TM polymer and/or actin molecules in the thin filament, thereby indirectly stabilizing Tmod capping of actin and TM at the pointed end some distance away from the nebulin molecule itself [110]. The maximum distance from which nebulin can affect Tmod dynamics remains elusive, but it is most likely on the order of ~0.3-0.4 μm, which is the longest thin filament pointed-end extension measured to date [108] (D. S. Gokhin and V. M. Fowler, unpublished data). It is also possible that the nebulin M1M2M3 module may interact directly with Tmod at as-yet-unidentified developmental junctures or during the early stages of myofibril assembly when thin filaments may be shorter [61, 111, 112]. Thus, in skeletal muscle, thin filament lengths are likely regulated in a complex fashion by the dynamic properties of Tmods, nebulin, actin, and TM in combination. Heterogeneous expression of thin filament-associated protein isoforms may determine muscle-specific thin filament lengths that are essential for specifying muscle-specific sarcomere length-tension relationships for joint motion and mammalian motility [4–6].
5. Lmods in Actin Nucleation, Thin Filament Assembly, and Length Regulation
A plethora of actin nucleators have been identified in nonmuscle cells that promote assembly of actin filaments into lamellipodia, filopodia, or stress fibers via actin assembly at barbed filament ends [113]. In striated muscle, the formin-homology-domain protein FHOD3 regulates actin assembly and myofibril integrity in rat cardiomyocytes [114, 115], while the C-terminal SH3 domain of nebulin activated by N-WASP promotes de novo assembly of thin filaments at Z-lines during sarcomerogenesis [116]. The larger Tmod family member, Lmod2, which is localized to stripes flanking the M-line in cultured cardiomyocytes [46, 57, 58], also exhibits a potent actin nucleating activity in vitro, which is enhanced by TM [46]. In rat cardiomyocytes, siRNA knockdown of Lmod2 results in defective myofibril assembly with disorganized, thin, and wispy myofibrils, implicating Lmod2 in the assembly of thin filaments and myofibrils [46]. Based on the in vitro properties of Lmod2, de novo Lmod2-nucleated filaments assembling into sarcomeres could grow from their barbed ends with Lmod2 remaining at their pointed ends [46, 57, 58]. However, this mechanism would be somewhat surprising, since previous studies have shown that thin filaments in sarcomeres elongate from their pointed ends (see Section 3) [13, 29, 71]. In agreement with the latter, overexpression of Lmod2 in embryonic chick cardiomyocytes displaced Tmod1 from pointed ends and led to thin filament lengthening [58]. By analogy to SALS function in Drosophila myocytes (see Section 3) [71], Lmod2 has been proposed to “fine-tune” thin filament lengths in mature cardiomyocyte sarcomeres by competing with and antagonizing the activity of Tmod1 at the thin filament pointed ends [58]. Alternatively, since the Lmod2 WH2 nucleation domain is required for thin filament elongation in this system [58], it is possible that this filament lengthening could be due to de novo nucleation and elongation of new filaments that are then incorporated into existing sarcomeres. Testing these models and integrating Lmod2 functions with those of the FHOD3 and nebulin-SH3 actin nucleators will require the determination of the sites of actin incorporation and filament elongation during thin filament assembly and turnover during myofibrillogenesis, as compared to myofibril maturation and maintenance during muscle contraction and adaptation in vivo.
Lmod2 has also been suggested to play a role in regulating actomyosin contractility in mature sarcomeres. In neonatal rat cardiomyocytes, Lmod2 colocalizes with myosin in the A-band (i.e., in broad stripes flanking Tmod1 at the pointed ends) [57]. This A-band localization of Lmod2 was only observed in mature myofibrils and depended on myosin activity as well as on the availability of free actin monomers [57]. Indeed, the myofibril disruption observed upon siRNA knockdown of Lmod2 [46] could potentially be due to effects on myosin activity, which influences myofibril assembly and organization under some conditions [57, 117–120]. The relationship of the A-band localization of Lmod2 in rat cardiomyocytes [57] to the pointed-end localization of Lmod2 in embryonic chick cardiomyocytes [58] is unclear, since full-length Lmod2 is a strong actin nucleator that produces barbed-end elongation in vitro and does not cap actin filament pointed ends [46, 57, 58]. However, truncated Lmod2 missing the WH2 domain caps pointed ends in a TM-dependent manner [58], suggesting that additional regulatory factors that inhibit nucleation activity may convert Lmod2 to pointed-end capping in cells. Further studies will be required to determine whether the disparate localizations reported in different studies are due to Lmod isoform or species differences or differences in myofibril maturation in rat as opposed to chicken cardiomyocytes. Additional work will also be required to resolve the reported discrepancies in Lmod2 functions, including knockout models for Lmods to determine in vivo functions.
Considerably less is known about Lmod1, which is predominantly expressed in smooth muscle cells, in which thin filament lengths are not precisely regulated or aligned. In hypercontracted smooth muscle, which reveals a subdomain organization of α-actinin-containing dense bodies alternating with actin filament-rich regions, both Lmod1 and Tmod1 are concentrated in the actin filament-rich regions between the dense bodies [48]. This suggests that Lmod1 and Tmod1 may be associated with the pointed ends of variable-length smooth muscle thin filaments extending from the dense bodies. However, in extraocular striated muscle, Lmod1 colocalizes with the typical myosin-staining doublet in the A-bands of slow muscle fibers [47], similar to Lmod2 in rat cardiomyocytes [57]. This contrasts with Tmod1, which localizes to a single stripe in the middle of the sarcomere in these unstretched muscles, as expected from the close proximity of the locations of the thin filament pointed ends from each half-sarcomere [47, 48]. This suggests that, unlike Tmod1, which caps the thin filament pointed ends, Lmod1 may interact with the regions of thin filaments that interact with myosin in the A-band, or Lmod1 may possibly bind directly to myosin. Lmod1 binds striated muscle TM in vitro [48], but its function has otherwise not been studied. (A third Lmod gene, Lmod3, has been identified (human gene ID, 56203; mRNA, NM_198271), but its protein localization and function remain unknown).
6. Tmods and the SR
The SR is a membrane system that wraps around myofibrils and serves as the Ca2+ reservoir for muscle contraction. The skeletal muscle SR contains an actin filament network composed of cytoplasmic γ-actin (in contrast to myofibrils, which contain skeletal muscle α-actin) [63]. γ-Actin is thought to be involved in the compensatory remodeling response of skeletal muscle to muscular dystrophy, based on its dramatic upregulation in diverse animal models of the disease [121, 122]. While the filament-level architecture of the γ-actin network is unknown, it likely shares features with Tmod-capped actin filament architectures in nonmuscle cells, such as epithelial cells, lens fiber cells, and red blood cells [123–125]. In mouse skeletal muscle, the predominant Tmod isoform associated with γ-actin in the SR is Tmod3, which shares a localization pattern with γ-actin and nonmuscle TMs in an SR microdomain at the M-line and flanking the Z-line [63] (Figures 2 and 3). Tmod3 appears to be tethered to the SR at the M-line via an interaction with small ankyrin 1.5 (sAnk1.5) [63], a 17-kDa splice variant of ankyrin-R (ankyrin 1) with a hydrophobic transmembrane segment [126–128]. The Tmod3/sAnk1.5 complex in the SR contains γ-actin and nonmuscle TMs (TM5NM1 and TM4) and is not associated with sarco/endoplasmic reticulum Ca2+-ATPase or the ryanodine receptor. Instead, Tmod3-capped γ-actin filaments may be components of an SR-to-myofibril mechanical linking system, the best-characterized component of which is the giant (~720 kDa) protein obscurin, which links sAnk1.5 in the SR to myomesin and titin in the M-line [129, 130]. The mouse skeletal muscle SR also contains a minor Z-line-flanking microdomain containing Tmod1 and Tmod4, which, based on its localization pattern, may be associated with the SR or T-tubule system [63].
Counterintuitively, deletion of Tmod1 in mice enables the study of the functional significance of Tmod3 in the SR, because Tmod3 vacates the SR and moves to the thin filament pointed ends in adult Tmod1-null muscle [31, 63]. Thus, Tmod1 deletion appears to be a proxy for “conditional” Tmod3 deletion from the SR. In this scenario, loss of Tmod3 from the SR selectively destabilizes its associated complex with sAnk1.5 and perturbs the localizations of γ-actin, nonmuscle TMs, and sAnk1.5. This leads to aberrant SR swelling, depressed Ca2+ release, and myofibril misalignment [63]. Notably, previously identified SR-associated membrane skeleton proteins, β2-spectrin and ankyrin-B, which are involved in targeting SR-associated ion channels in the heart [131–134], are not associated with the Tmod3/sAnk1.5 complex, and are not perturbed by deletion of Tmod1 and redistribution of Tmod3 from the SR to the thin filament pointed ends [63]. This suggests that Tmod3, γ-actin, and nonmuscle TMs define a novel membrane skeleton-like network associated with sAnk1.5 in the SR, which mechanically stabilizes the SR, facilitates SR Ca2+ release, and provides connectivity among adjacent myofibrils via linkages between sAnk1.5 and obscurin [63, 130, 135]. Certainly, creation of a Tmod3-null mouse is required to demonstrate rigorously that the SR phenotype is indeed due to the redistribution of Tmod3 from the SR to the thin filament pointed ends, rather than due to deletion of Tmod1 from a different SR compartment or from the T-tubules. In addition, by analogy to nebulin-null mice [136], it is also possible that effects of Tmod1 deletion on actin pointed-end dynamics and thin filament stability may lead to secondary effects on SR Ca2+ handling (see Section 7). Future work needs to focus on the molecular architecture of the SR-associated γ-actin cytoskeletal network at the Z- and M-lines and the mechanisms that drive isoform-specific sorting of actins and Tmods in striated muscle. Furthermore, whether Tmod3 plays a role in the cardiac SR analogous to its role in the skeletal muscle SR remains to be determined. It is tempting to speculate that the upregulation of γ-actin that occurs in muscular dystrophy might influence Ca2+ handling in the SR in addition to its better-characterized role in binding to dystrophin and mechanically fortifying the sarcolemma; such a model would be consistent with evidence implicating aberrant SR Ca2+ handling in the pathogenesis of muscular dystrophy [137–139].
7. Potential Relevance of Tmods to Hereditary Muscle Diseases
A persistent question is whether or not Tmods are directly involved in the pathogenesis of human nemaline myopathy, a skeletal muscle wasting disease that afflicts ~1 : 50,000 individuals, arises from mutations in thin filament-associated proteins, and is marked by the presence of aberrant actin bundles and so-called “nemaline bodies” consisting of thin filament material [140–142]. In human skeletal muscles, a combination of Tmod1 and Tmod4 caps the pointed ends of the thin filaments (D. S. Gokhin and V. M. Fowler, unpublished data), identical to mouse muscles [31, 63], and mutations in almost all known thin filament-associated proteins, including α-actin (ACTA1), β- and γ-TM isoforms (TPM2 and TPM3), troponin I and T isoforms (TNNI2, TNNT1, and TNNT3), and nebulin (NEB) [140–142], have been found to produce various degrees of hereditary nemaline myopathy. A nemaline myopathy-linked ADF/cofilin (CFL2) mutation has also been identified, implying that aberrant regulation of actin dynamics and turnover in thin filaments can lead to muscle pathologies (see Section 2) [81]. While Tmod1 deletion in mice produces mild muscle pathology with depressed isometric stress production, nemaline bodies are not observed, most likely due to structural compensation by Tmod3, which caps the thin filament pointed ends with Tmod4 in the absence of Tmod1, thus preserving thin filament stability and length regulation [31, 63]. Nevertheless, it is possible that weakened and more dynamic Tmod3/Tmod4 binding to the thin filament pointed ends may alter TM-troponin regulation of actomyosin crossbridge activity and thereby contribute to muscle weakness [31]; similar changes in productive actomyosin crossbridge formation have been observed in nebulin-null mice as well as in human nemaline myopathy [143–147]. However, the phenotype of Tmod1-null skeletal muscle is distinct from classical nemaline myopathy, because Tmod1-null muscle is not overtly myopathic [31], and Tmod3 translocation from the SR to thin filament pointed ends is accompanied by SR defects and myofibril misalignment that can account for the muscle weakness [63]. Furthermore, no Tmod mutations have yet been identified as a basis for nemaline myopathy.
Clearly, Tmod perturbations can cause muscle pathology in mice, and mutations in the proteins that comprise the Tmod-associated thin filament structures (i.e., disease “hotspots”) in skeletal muscle cause hereditary diseases in humans. However, despite Tmods being integral structural components of thin filaments, they are, thus far, conspicuously absent from the list of thin filament proteins that cause nemaline myopathy. Indeed, Tmods appear to be the only thin filament proteins with no as-yet-identified mutations that cause nemaline myopathy. Why might this be the case? The most likely answer is that Tmods are essential genes in humans. Deletion of Tmod1 is embryonic lethal in mice due to defects in cardiac looping morphogenesis and myofibril assembly, resulting in aborted development early in embryogenesis [59, 68]; indeed, studies of Tmod1's functions in noncardiac tissues in vivo require cardiac-specific rescue of Tmod1 to permit animal development and progression into adulthood [31, 63, 65, 124, 125]. Thus, it would be reasonable to predict that wholesale Tmod1 deletion would be lethal in humans as well. However, it is conceivable that Tmod1 mutations resulting in milder Tmod1 functional deficits or partially reduced Tmod1 levels could be associated with hereditary myopathies in humans. Tmod4 mutations are another possibility; in humans, Tmod4 is only present in skeletal muscle [20, 21] and is a good candidate gene for skeletal muscle disease, because its perturbation would not be expected to cause cardiac defects. Nonetheless, the most probable reason that no myopathy-linked Tmod1 or Tmod4 mutations have yet been identified is compensation by other Tmod isoforms (i.e., Tmod3), as shown in mice [31, 63]. Additional studies on the individuals who are diagnosed with a nemaline myopathy of unknown genetic etiology [140] are required for the identification of putative Tmod mutations associated with nemaline myopathy or other skeletal muscle myopathies.
In addition to the potential involvement of Tmod1 and Tmod4 capping of α-actin thin filaments in nemaline myopathy, putative Tmod3 capping of γ-actin filaments in the SR [63] may play a role in muscular dystrophy. Tmod3 capping of γ-actin filaments occurs in conjunction with nonmuscle TM5NM1 and TM4 binding to the sides of γ-actin filaments, which is essential for normal excitation-contraction coupling, SR function, and maintaining a nondystrophic condition [148–150]. γ-Actin is dramatically upregulated in dystrophic muscle [121, 122], but whether this affects SR structure or function or specific SR-associated γ-actin-regulatory proteins (e.g., Tmod3 and nonmuscle TM5NM1 and TM4) remains unknown. Indeed, γ-actin upregulation without compensatory alterations in the pool of γ-actin-regulatory proteins may result in uncontrolled γ-actin polymerization, which may adversely affect the structure, stability, and function of the SR and/or sarcolemma. Another possibility is that muscular dystrophy-induced misregulation of γ-actin-regulatory proteins may alter γ-actin turnover in a manner that is toxic to the muscle cell via mechanisms involving aberrant Ca2+ handling [137, 138]. It is important to recognize that such proposals are speculative at this point. However, given this clinical context, they highlight the fact that the future of the “Tmods in muscle” field lies in dissecting the molecular mechanisms by which Tmods cap and stabilize actin filaments during development and disease, exploring the abilities of Tmods to regulate muscle physiology and contractility, and uncovering potential links between Tmod perturbations and muscle pathology.
Acknowledgments
This work was supported by NIH Grant no. R01-HL083464 (to V. M. Fowler) and NHLBI vascular biology training grant T32-HL007195-34 (to D. S. Gokhin). The authors gratefully acknowledge Sawako Yamashiro for assistance with preparing Figure 1 and for contributions to the initial version of this paper text. They also acknowledge Roberta B. Nowak for assistance with preparing Figure 3.
References
- 1.Littlefield RS, Fowler VM. Thin filament length regulation in striated muscle sarcomeres: pointed-end dynamics go beyond a nebulin ruler. Seminars in Cell and Developmental Biology. 2008;19(6):511–519. doi: 10.1016/j.semcdb.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark KA, McElhinny AS, Beckerle MC, Gregorio CC. Striated muscle cytoarchitecture: an intricate web of form and function. Annual Review of Cell and Developmental Biology. 2002;18:637–706. doi: 10.1146/annurev.cellbio.18.012502.105840. [DOI] [PubMed] [Google Scholar]
- 3.Littlefield R, Fowler VM. Defining actin filament length in striated muscle: rulers and caps or dynamic stability? Annual Review of Cell and Developmental Biology. 1998;14:487–525. doi: 10.1146/annurev.cellbio.14.1.487. [DOI] [PubMed] [Google Scholar]
- 4.Gokhin DS, Bang ML, Zhang J, Chen J, Lieber RL. Reduced thin filament length in nebulin-knockout skeletal muscle alters isometric contractile properties. American Journal of Physiology—Cell Physiology. 2009;296(5):C1123–C1132. doi: 10.1152/ajpcell.00503.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Granzier HLM, Akster HA, Ter Keurs HEDJ. Effect of thin filament length on the force-sarcomere length relation of skeletal muscle. American Journal of Physiology—Cell Physiology. 1991;260(5):C1060–C1070. doi: 10.1152/ajpcell.1991.260.5.C1060. [DOI] [PubMed] [Google Scholar]
- 6.Ottenheijm CAC, Witt CC, Stienen GJ, Labeit S, Beggs AH, Granzier H. Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Human Molecular Genetics. 2009;18(13):2359–2369. doi: 10.1093/hmg/ddp168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coluccio LM. An end in sight: tropomodulin. Journal of Cell Biology. 1994;127(6):1497–1499. doi: 10.1083/jcb.127.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer RS, Fowler VM. Tropomodulins: life at the slow end. Trends in Cell Biology. 2003;13(11):593–601. doi: 10.1016/j.tcb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Fowler VM. Regulation of actin filament length in erythrocytes and striated muscle. Current Opinion in Cell Biology. 1996;8(1):86–96. doi: 10.1016/s0955-0674(96)80052-4. [DOI] [PubMed] [Google Scholar]
- 10.Fowler VM. Capping actin filament growth: tropomodulin in muscle and nonmuscle cells. Society of General Physiologists Series. 1997;52:79–89. [PubMed] [Google Scholar]
- 11.Fowler VM, Sussmann MA, Miller PG, Flucher BE, Daniels MP. Tropomodulin is associated with the free (pointed) ends of the thin filaments in rat skeletal muscle. Journal of Cell Biology. 1993;120(2):411–420. doi: 10.1083/jcb.120.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber A, Pennise CR, Babcock GG, Fowler VM. Tropomodulin caps the pointed ends of actin filaments. Journal of Cell Biology. 1994;127(6):1627–1635. doi: 10.1083/jcb.127.6.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mardahl-Dumesnil M, Fowler VM. Thin filaments elongate from their pointed ends during myofibril assembly in Drosophila indirect flight muscle. Journal of Cell Biology. 2001;155(6):1043–1053. doi: 10.1083/jcb.200108026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevenson TO, Mercer KB, Cox EA, et al. unc-94 encodes a tropomodulin in Caenorhabditis elegans. Journal of Molecular Biology. 2007;374(4):936–950. doi: 10.1016/j.jmb.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamashiro S, Cox EA, Baillie DL, Hardin JD, Ono S. Sarcomeric actin organization is synergistically promoted by tropomodulin, ADF/cofilin, AIP1 and profilin in C. elegans. Journal of Cell Science. 2008;121(23):3867–3877. doi: 10.1242/jcs.040477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fowler VM. Identification and purification of a novel M(r) 43,000 tropomyosin-binding protein from human erythrocyte membranes. Journal of Biological Chemistry. 1987;262(26):12792–12800. [PubMed] [Google Scholar]
- 17.Fowler VM. Tropomodulin: a cytoskeletal protein that binds to the end of erythrocyte tropomyosin and inhibits tropomyosin binding to actin. Journal of Cell Biology. 1990;111(2):471–482. doi: 10.1083/jcb.111.2.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watakabe A, Kobayashi R, Helfman DM. N-tropomodulin: a novel isoform of tropomodulin identified as the major binding protein to brain tropomyosin. Journal of Cell Science. 1996;109, part 9:2299–2310. doi: 10.1242/jcs.109.9.2299. [DOI] [PubMed] [Google Scholar]
- 19.Almenar-Queralt A, Lee A, Conley CA, De Pouplana LR, Fowler VM. Identification of a novel tropomodulin isoform, skeletal tropomodulin, that caps actin filament pointed ends in fast skeletal muscle. Journal of Biological Chemistry. 1999;274(40):28466–28475. doi: 10.1074/jbc.274.40.28466. [DOI] [PubMed] [Google Scholar]
- 20.Cox PR, Zoghbi HY. Sequencing, expression analysis, and mapping of three unique human tropomodulin genes and their mouse orthologs. Genomics. 2000;63(1):97–107. doi: 10.1006/geno.1999.6061. [DOI] [PubMed] [Google Scholar]
- 21.Conley CA, Fritz-Six KL, Almenar-Queralt A, Fowler VM. Leiomodins: larger members of the tropomodulin (Tmod) gene family. Genomics. 2001;73(2):127–139. doi: 10.1006/geno.2000.6501. [DOI] [PubMed] [Google Scholar]
- 22.Sung LA, Fowler VM, Lambert K, Sussman MA, Karr D, Chien S. Molecular cloning and characterization of human fetal liver tropomodulin. A tropomyosin-binding protein. Journal of Biological Chemistry. 1992;267(4):2616–2621. [PubMed] [Google Scholar]
- 23.Ito M, Swanson B, Sussman MA, Kedes L, Lyons G. Cloning of tropomodulin cDNA and localization of gene transcripts during mouse embryogenesis. Developmental Biology. 1995;167(1):317–328. doi: 10.1006/dbio.1995.1026. [DOI] [PubMed] [Google Scholar]
- 24.Fischer RS, Lee A, Fowler VM. Tropomodulin and tropomyosin mediate lens cell actin cytoskeleton reorganization in vitro. Investigative Ophthalmology and Visual Science. 2000;41(1):166–174. [PubMed] [Google Scholar]
- 25.Sussman MA, McAvoy JW, Rudisill M, et al. Lens tropomodulin: developmental expression during differentiation. Experimental Eye Research. 1996;63(2):223–232. doi: 10.1006/exer.1996.0111. [DOI] [PubMed] [Google Scholar]
- 26.Woo MK, Fowler VM. Identification and characterization of tropomodulin and tropomyosin in the adult rat lens. Journal of Cell Science. 1994;107, part 5:1359–1367. doi: 10.1242/jcs.107.5.1359. [DOI] [PubMed] [Google Scholar]
- 27.Fischer RS, Fritz-Six KL, Fowler VM. Pointed-end capping by tropomodulin3 negatively regulates endothelial cell motility. Journal of Cell Biology. 2003;161(2):371–380. doi: 10.1083/jcb.200209057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gregorio CC, Weber A, Bondad M, Pennise CR, Fowler VM. Requirement of pointed-end capping by tropomodulin to maintain actin filament length in embryonic chick cardiac myocytes. Nature. 1995;377(6544):83–86. doi: 10.1038/377083a0. [DOI] [PubMed] [Google Scholar]
- 29.Littlefield R, Almenar-Queralt A, Fowler VM. Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nature Cell Biology. 2001;3(6):544–551. doi: 10.1038/35078517. [DOI] [PubMed] [Google Scholar]
- 30.Weber A, Pennise CR, Fowler VM. Tropomodulin increases the critical concentration of barbed end-capped actin filaments by converting ADP.P(i)-actin to ADP-actin at all pointed filament ends. Journal of Biological Chemistry. 1999;274(49):34637–34645. doi: 10.1074/jbc.274.49.34637. [DOI] [PubMed] [Google Scholar]
- 31.Gokhin DS, Lewis RA, McKeown CR, et al. Tropomodulin isoforms regulate thin filament pointed-end capping and skeletal muscle physiology. Journal of Cell Biology. 2010;189(1):95–109. doi: 10.1083/jcb.201001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenfield NJ, Fowler VM. Tropomyosin requires an intact N-terminal coiled coil to interact with tropomodulin. Biophysical Journal. 2002;82(5):2580–2591. doi: 10.1016/S0006-3495(02)75600-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vera C, Sood A, Gao KM, Yee LJ, Lin JJC, Sung LA. Tropomodulin-binding site mapped to residues 7-14 at the N-terminal heptad repeats of tropomyosin isoform 5. Archives of Biochemistry and Biophysics. 2000;378(1):16–24. doi: 10.1006/abbi.2000.1802. [DOI] [PubMed] [Google Scholar]
- 34.Babcock GG, Fowler VM. Isoform-specific interaction of tropomodulin with skeletal muscle and erythrocyte tropomyosins. Journal of Biological Chemistry. 1994;269(44):27510–27518. [PubMed] [Google Scholar]
- 35.Sussman MA, Fowler VM. Tropomodulin binding to tropomyosins. Isoform-specific differences in affinity and stoichiometry. European Journal of Biochemistry. 1992;205(1):355–362. doi: 10.1111/j.1432-1033.1992.tb16787.x. [DOI] [PubMed] [Google Scholar]
- 36.Kostyukova AS, Choy A, Rapp BA. Tropomodulin binds two tropomyosins: a novel model for actin filament capping. Biochemistry. 2006;45(39):12068–12075. doi: 10.1021/bi060899i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kostyukova AS, Hitchcock-DeGregori SE. Effect of the structure of the N terminus of tropomyosin on tropomodulin function. Journal of Biological Chemistry. 2004;279(7):5066–5071. doi: 10.1074/jbc.M311186200. [DOI] [PubMed] [Google Scholar]
- 38.Kostyukova AS, Hitchcock-DeGregori SE, Greenfield NJ. Molecular Basis of Tropomyosin Binding to Tropomodulin, an Actin-capping Protein. Journal of Molecular Biology. 2007;372(3):608–618. doi: 10.1016/j.jmb.2007.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uversky VN, Shah SP, Gritsyna Y, Hitchcock-DeGregori SE, Kostyukova AS. Systematic analysis of tropomodulin/tropomyosin interactions uncovers fine-tuned binding specificity of intrinsically disordered proteins. Journal of Molecular Recognition. 2011;24:647–655. doi: 10.1002/jmr.1093. [DOI] [PubMed] [Google Scholar]
- 40.Fowler VM, Greenfield NJ, Moyer J. Tropomodulin contains two actin filament pointed end-capping domains. Journal of Biological Chemistry. 2003;278(41):40000–40009. doi: 10.1074/jbc.M306895200. [DOI] [PubMed] [Google Scholar]
- 41.Mudry RE, Perry CN, Richards M, Fowler VM, Gregorio CC. The interaction of tropomodulin with tropomyosin stabilizes thin filaments in cardiac myocytes. Journal of Cell Biology. 2003;162(6):1057–1068. doi: 10.1083/jcb.200305031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kostyukova AS, Rapp BA, Choy A, Greenfield NJ, Hitchcock-DeGregori SE. Structural requirements of tropomodulin for tropomyosin binding and actin filament capping. Biochemistry. 2005;44(12):4905–4910. doi: 10.1021/bi047468p. [DOI] [PubMed] [Google Scholar]
- 43.Kostyukova AS. Tropomodulin/tropomyosin interactions regulate actin pointed end dynamics. Advances in Experimental Medicine and Biology. 2008;644:283–292. doi: 10.1007/978-0-387-85766-4_21. [DOI] [PubMed] [Google Scholar]
- 44.Fischer RS, Yarmola EG, Weber KL, et al. Tropomodulin 3 binds to actin monomers. Journal of Biological Chemistry. 2006;281(47):36454–36465. doi: 10.1074/jbc.M606315200. [DOI] [PubMed] [Google Scholar]
- 45.Yamashiro S, Speicher KD, Speicher DW, Fowler VM. Mammalian tropomodulins nucleate actin polymerization via their actin monomer binding and filament pointed end-capping activities. Journal of Biological Chemistry. 2010;285(43):33265–33280. doi: 10.1074/jbc.M110.144873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chereau D, Boczkowska M, Skwarek-Maruszewska A, et al. Leiomodin is an actin filament nucleator in muscle cells. Science. 2008;320(5873):239–243. doi: 10.1126/science.1155313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Conley CA, Fowler VM. Localization of the human 64 kD autoantigen D1 to myofibrils in a subset of extraocular muscle fibers. Current Eye Research. 1999;19(4):313–322. doi: 10.1076/ceyr.19.4.313.5304. [DOI] [PubMed] [Google Scholar]
- 48.Conley CA. Leiomodin and tropomodulin in smooth muscle. American Journal of Physiology—Cell Physiology. 2001;280(6):C1645–C1656. doi: 10.1152/ajpcell.2001.280.6.C1645. [DOI] [PubMed] [Google Scholar]
- 49.Kostyukova A, Maeda K, Yamauchi E, Krieger I, Maéda Y. Domain structure of tropomodulin: distinct properties of the N-terminal and C-terminal halves. European Journal of Biochemistry. 2000;267(21):6470–6475. doi: 10.1046/j.1432-1327.2000.01738.x. [DOI] [PubMed] [Google Scholar]
- 50.Kostyukova AS, Tiktopulo EI, Maéda Y. Folding properties of functional domains of tropomodulin. Biophysical Journal. 2001;81(1):345–351. doi: 10.1016/S0006-3495(01)75704-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greenfield NJ, Kostyukova AS, Hitchcock-DeGregori SE. Structure and tropomyosin binding properties of the N-terminal capping domain of tropomodulin 1. Biophysical Journal. 2005;88(1):372–383. doi: 10.1529/biophysj.104.051128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kong KY, Kedes L. Leucine 135 of tropomodulin-1 regulates its association with tropomyosin, its cellular localization the integrity of sarcomeres. Journal of Biological Chemistry. 2006;281(14):9589–9599. doi: 10.1074/jbc.M512064200. [DOI] [PubMed] [Google Scholar]
- 53.Vera C, Lao J, Hamelberg D, Amy Sung L. Mapping the tropomyosin isoform 5 binding site on human erythrocyte tropomodulin: further insights into E-Tmod/TM5 interaction. Archives of Biochemistry and Biophysics. 2005;444(2):130–138. doi: 10.1016/j.abb.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 54.Krieger I, Kostyukova A, Yamashita A, Nitanai Y, Maéda Y. Crystal structure of the C-terminal half of tropomodulin and structural basis of actin filament pointed-end capping. Biophysical Journal. 2002;83(5):2716–2725. doi: 10.1016/S0006-3495(02)75281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu S, Symersky J, Li S, et al. Structural genomics of Caenorhabditis elegans: crystal structure of the tropomodulin C-terminal domain. Proteins. 2004;56(2):384–386. doi: 10.1002/prot.10597. [DOI] [PubMed] [Google Scholar]
- 56.Tsukada T, Kotlyanskaya L, Huynh R, et al. Identification of residues within tropomodulin-1 responsible for its localization at the pointed ends of the actin filaments in cardiac myocytes. Journal of Biological Chemistry. 2011;286(3):2194–2204. doi: 10.1074/jbc.M110.186924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Skwarek-Maruszewska A, Boczkowska M, Zajac AL, et al. Different localizations and cellular behaviors of leiomodin and tropomodulin in mature cardiomyocyte sarcomeres. Molecular Biology of the Cell. 2010;21(19):3352–3361. doi: 10.1091/mbc.E10-02-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsukada T, Pappas CT, Moroz N, Antin PB, Kostyukova AS, Gregorio CC. Leiomodin-2 is an antagonist of tropomodulin-1 at the pointed end of the thin filaments in cardiac muscle. Journal of Cell Science. 2010;123(18):3136–3145. doi: 10.1242/jcs.071837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fritz-Six KL, Cox PR, Fischer RS, et al. Aberrant myofibril assembly in tropomodulin1 null mice leads to aborted heart development and embryonic lethality. Journal of Cell Biology. 2003;163(5):1033–1044. doi: 10.1083/jcb.200308164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gregorio CC, Fowler VM. Mechanisms of thin filament assembly in embryonic chick cardiac myocytes: tropomodulin requires tropomyosin for assembly. Journal of Cell Biology. 1995;129(3):683–695. doi: 10.1083/jcb.129.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Almenar-Queralt A, Gregorio CC, Fowler VM. Tropomodulin assembles early in myofibrillogenesis in chick skeletal muscle: evidence that thin filaments rearrange to form striated myofibrils. Journal of Cell Science. 1999;112, part 8:1111–1123. doi: 10.1242/jcs.112.8.1111. [DOI] [PubMed] [Google Scholar]
- 62.Sussman MA, Sakhi S, Barrientos P, Ito M, Kedes L. Tropomodulin in rat cardiac muscle: localization of protein is independent of messenger RNA distribution during myofibrillar development. Circulation Research. 1994;75(2):221–232. doi: 10.1161/01.res.75.2.221. [DOI] [PubMed] [Google Scholar]
- 63.Gokhin DS, Fowler VM. Cytoplasmic γ-actin and tropomodulin isoforms link to the sarcoplasmic reticulum in skeletal muscle fibers. Journal of Cell Biology. 2011;194(1):105–120. doi: 10.1083/jcb.201011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ono S. Dynamic regulation of sarcomeric actin filaments in striated muscle. Cytoskeleton. 2010;67(11):677–692. doi: 10.1002/cm.20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McKeown CR, Nowak RB, Moyer J, Sussman MA, Fowler VM. Tropomodulin1 is required in the heart but not the yolk sac for mouse embryonic development. Circulation Research. 2008;103(11):1241–1248. doi: 10.1161/CIRCRESAHA.108.178749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ehler E, Fowler VM, Perriard JC. Myofibrillogenesis in the developing chicken heart: role of actin isoforms and of the pointed end actin capping protein tropomodulin during thin filament assembly. Developmental Dynamics. 2004;229(4):745–755. doi: 10.1002/dvdy.10482. [DOI] [PubMed] [Google Scholar]
- 67.McLean MD, Zajdel RW, Dube S, Thurston H, Dube DK. Tropomodulin expression in developing hearts of normal and cardiac mutant Mexican axolotl. Cardiovascular Toxicology. 2006;6(2):85–98. doi: 10.1385/ct:6:2:85. [DOI] [PubMed] [Google Scholar]
- 68.Chu X, Chen J, Reedy MC, Vera C, Sung KLP, Sung LA. E-Tmod capping of actin filaments at the slow-growing end is required to establish mouse embryonic circulation. American Journal of Physiology—Heart and Circulatory Physiology. 2003;284(5):H1827–H1838. doi: 10.1152/ajpheart.00947.2002. [DOI] [PubMed] [Google Scholar]
- 69.Ono Y, Schwach C, Antin PB, Gregorio CC. Disruption in the tropomodulin1 (Tmod1) gene compromises cardiomyocyte development in murine embryonic stem cells by arresting myofibril maturation. Developmental Biology. 2005;282(2):336–348. doi: 10.1016/j.ydbio.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 70.Sussman MA, Baqué S, Uhm CS, et al. Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils. Circulation Research. 1998;82(1):94–105. doi: 10.1161/01.res.82.1.94. [DOI] [PubMed] [Google Scholar]
- 71.Bai J, Hartwig JH, Perrimon N. SALS, a WH2-domain-containing protein, promotes sarcomeric actin filament elongation from pointed ends during Drosophila muscle growth. Developmental Cell. 2007;13(6):828–842. doi: 10.1016/j.devcel.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 72.Sussman MA, Welch S, Cambon N, et al. Myofibril degeneration caused by tropomodulin overexpression leads to dilated cardiomyopathy in juvenile mice. Journal of Clinical Investigation. 1998;101(1):51–61. doi: 10.1172/JCI1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lyons GE, Schiaffino S, Sassoon D, Barton P, Buckingham M. Developmental regulation of myosin gene expression in mouse cardiac muscle. Journal of Cell Biology. 1990;111(6):2427–2436. doi: 10.1083/jcb.111.6.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rudy DE, Yatskievych TA, Antin PB, Gregorio CC. Assembly of thick, thin, and titin filaments in chick precardiac explants. Developmental Dynamics. 2001;221(1):61–71. doi: 10.1002/dvdy.1125. [DOI] [PubMed] [Google Scholar]
- 75.Kong KY, Kedes L. Cytoplasmic nuclear transfer of the actin-capping protein tropomodulin. Journal of Biological Chemistry. 2004;279(29):30856–30864. doi: 10.1074/jbc.M302845200. [DOI] [PubMed] [Google Scholar]
- 76.Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nature Reviews Molecular Cell Biology. 2010;11(5):353–365. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ono S. The Caenorhabditis elegans unc-78 gene encodes a homologue of actin-interacting protein 1 required for organized assembly of muscle actin filaments. Journal of Cell Biology. 2001;152(6):1313–1319. doi: 10.1083/jcb.152.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mohri K, Ono K, Yu R, Yamashiro S, Ono S. Enhancement of actin-depolymerizing factor/cofilin-dependent actin disassembly by actin-interacting protein 1 is required for organized actin filament assembly in the Caenorhabditis elegans body wall muscle. Molecular Biology of the Cell. 2006;17(5):2190–2199. doi: 10.1091/mbc.E05-11-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ono S, Mohri K, Ono K. Microscopic evidence that actin-interacting protein 1 actively disassembles actin-depolymerizing factor/cofilin-bound actin filaments. Journal of Biological Chemistry. 2004;279(14):14207–14212. doi: 10.1074/jbc.M313418200. [DOI] [PubMed] [Google Scholar]
- 80.Ono S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. International Review of Cytology. 2007;258:1–82. doi: 10.1016/S0074-7696(07)58001-0. [DOI] [PubMed] [Google Scholar]
- 81.Agrawal PB, Greenleaf RS, Tomczak KK, et al. Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. American Journal of Human Genetics. 2007;80(1):162–167. doi: 10.1086/510402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Papponen H, Kaisto T, Leinonen S, Kaakinen M, Metsikkö K. Evidence for γ-actin as a Z disc component in skeletal myofibers. Experimental Cell Research. 2009;315(2):218–225. doi: 10.1016/j.yexcr.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 83.Sussman MA, Welch S, Gude N, et al. Pathogenesis of dilated cardiomyopathy: molecular, structural, and population analyses in tropomodulin-overexpressing transgenic mice. American Journal of Pathology. 1999;155(6):2101–2113. doi: 10.1016/S0002-9440(10)65528-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ehler E, Horowits R, Zuppinger C, et al. Alterations at the intercalated disk associated with the absence of muscle LIM protein. Journal of Cell Biology. 2001;153(4):763–772. doi: 10.1083/jcb.153.4.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lim HW, De Windt LJ, Mante J, et al. Reversal of cardiac hypertrophy in transgenic disease models by calcineurin inhibition. Journal of Molecular and Cellular Cardiology. 2000;32(4):697–709. doi: 10.1006/jmcc.2000.1113. [DOI] [PubMed] [Google Scholar]
- 86.Welch S, Plank D, Witt S, et al. Cardiac-specific IGF-1 expression attenuates dilated cardiomyopathy in tropomodulin-overexpressing transgenic mice. Circulation Research. 2002;90(6):641–648. doi: 10.1161/01.res.0000013780.77774.75. [DOI] [PubMed] [Google Scholar]
- 87.Sussman MA, Welch S, Walker A, et al. Hypertrophic defect unmasked by calcineurin expression in asymptomatic tropomodulin overexpressing transgenic mice. Cardiovascular Research. 2000;46(1):90–101. doi: 10.1016/s0008-6363(99)00422-8. [DOI] [PubMed] [Google Scholar]
- 88.Sussman MA, Lim HW, Gude N, et al. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281(5383):1690–1693. doi: 10.1126/science.281.5383.1690. [DOI] [PubMed] [Google Scholar]
- 89.Dye CA, Lee JK, Atkinson RC, Brewster R, Han PL, Bellen HJ. The Drosophila sanpodo gene controls sibling cell fate and encodes a tropomodulin homolog, an actin/tropomyosin-associated protein. Development. 1998;125(10):1845–1856. doi: 10.1242/dev.125.10.1845. [DOI] [PubMed] [Google Scholar]
- 90.Skeath JB, Doe CQ. Sanpodo and Notch act in opposition to Numb to distinguish sibling neuron fates in the Drosophila CNS. Development. 1998;125(10):1857–1865. doi: 10.1242/dev.125.10.1857. [DOI] [PubMed] [Google Scholar]
- 91.Babaoglan AB, O’Connor-Giles KM, Mistry H, Schickedanz A, Wilson BA, Skeath JB. Sanpodo: a context-dependent activator and inhibitor of Notch signaling during asymmetric divisions. Development. 2009;136(24):4089–4098. doi: 10.1242/dev.040386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O’Connor-Giles KM, Skeath JB. Numb inhibits membrane localization of sanpodo, a four-pass transmembrane protein, to promote asymmetric divisions in Drosophila. Developmental Cell. 2003;5(2):231–243. doi: 10.1016/s1534-5807(03)00226-0. [DOI] [PubMed] [Google Scholar]
- 93.Pappas CT, Bliss KT, Zieseniss A, Gregorio CC. The Nebulin family: an actin support group. Trends in Cell Biology. 2011;21(1):29–37. doi: 10.1016/j.tcb.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Labeit S, Ottenheijm CAC, Granzier H. Nebulin, a major player in muscle health and disease. FASEB Journal. 2011;25(3):822–829. doi: 10.1096/fj.10-157412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ottenheijm CAC, Granzier H. New insights into the structural roles of nebulin in skeletal muscle. Journal of Biomedicine and Biotechnology. 2010;2010 doi: 10.1155/2010/968139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ottenheijm CAC, Granzier H. Lifting the nebula: novel insights into skeletal muscle contractility. Physiology. 2010;25(5):304–310. doi: 10.1152/physiol.00016.2010. [DOI] [PubMed] [Google Scholar]
- 97.McElhinny AS, Kolmerer B, Fowler VM, Labeit S, Gregorio CC. The N-terminal end of nebulin interacts with tropomodulin at the pointed ends of the thin filaments. Journal of Biological Chemistry. 2001;276(1):583–592. doi: 10.1074/jbc.M005693200. [DOI] [PubMed] [Google Scholar]
- 98.Pappas CT, Bhattacharya N, Cooper JA, Gregorio CC. Nebulin interacts with CapZ and regulates thin filament architecture within the Z-disc. Molecular Biology of the Cell. 2008;19(5):1837–1847. doi: 10.1091/mbc.E07-07-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Witt CC, Burkart C, Labeit D, et al. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO Journal. 2006;25(16):3843–3855. doi: 10.1038/sj.emboj.7601242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chitose R, Watanabe A, Asano M, et al. Isolation of nebulin from rabbit skeletal muscle and its interaction with actin. Journal of Biomedicine and Biotechnology. 2010;2010 doi: 10.1155/2010/108495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fowler VM, McKeown CR, Fischer RS. Nebulin: does it measure up as a ruler? Current Biology. 2006;16(1):R18–R20. doi: 10.1016/j.cub.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 102.McElhinny AS, Kazmierski ST, Labeit S, Gregorio CC. Nebulin: the nebulous, multifunctional giant of striated muscle. Trends in Cardiovascular Medicine. 2003;13(5):195–201. doi: 10.1016/s1050-1738(03)00076-8. [DOI] [PubMed] [Google Scholar]
- 103.Labeit S, Kolmerer B. The complete primary structure of human nebulin and its correlation to muscle structure. Journal of Molecular Biology. 1995;248(2):308–315. doi: 10.1016/s0022-2836(95)80052-2. [DOI] [PubMed] [Google Scholar]
- 104.Donner K, Sandbacka M, Lehtokari VL, Wallgren-Pettersson C, Pelin K. Complete genomic structure of the human nebulin gene and identification of alternatively spliced transcripts. European Journal of Human Genetics. 2004;12(9):744–751. doi: 10.1038/sj.ejhg.5201242. [DOI] [PubMed] [Google Scholar]
- 105.Buck D, Hudson BD, Ottenheijm CAC, Labeit S, Granzier H. Differential splicing of the large sarcomeric protein nebulin during skeletal muscle development. Journal of Structural Biology. 2010;170(2):325–333. doi: 10.1016/j.jsb.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kruger M, Wright J, Wang K. Nebulin as a length regulator of thin filaments of vertebrate skeletal muscles: correlation of thin filament length, nebulin size, and epitope profile. Journal of Cell Biology. 1991;115(1):97–107. doi: 10.1083/jcb.115.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Labeit S, Gibson T, Lakey A, et al. Evidence that nebulin is a protein-ruler in muscle thin filaments. FEBS Letters. 1991;282(2):313–316. doi: 10.1016/0014-5793(91)80503-u. [DOI] [PubMed] [Google Scholar]
- 108.Castillo A, Nowak R, Littlefield KP, Fowler VM, Littlefield RS. A nebulin ruler does not dictate thin filament lengths. Biophysical Journal. 2009;96(5):1856–1865. doi: 10.1016/j.bpj.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bang ML, Li X, Littlefield R, et al. Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. Journal of Cell Biology. 2006;173(6):905–916. doi: 10.1083/jcb.200603119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pappas CT, Krieg PA, Gregorio CC. Nebulin regulates actin filament lengths by a stabilization mechanism. Journal of Cell Biology. 2010;189(5):859–870. doi: 10.1083/jcb.201001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sanger JW, Chowrashi P, Shaner NC, et al. Myofibrillogenesis in skeletal muscle cells. Clinical Orthopaedics and Related Research. 2002;(403):S153–S162. doi: 10.1097/00003086-200210001-00018. [DOI] [PubMed] [Google Scholar]
- 112.Sanger JW, Wang J, Holloway B, Du A, Sanger JM. Myofibrillogenesis in skeletal muscle cells in zebrafish. Cell Motility and the Cytoskeleton. 2009;66(8):556–566. doi: 10.1002/cm.20365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Welch MD, Mullins RD. Cellular control of actin nucleation. Annual Review of Cell and Developmental Biology. 2002;18:247–288. doi: 10.1146/annurev.cellbio.18.040202.112133. [DOI] [PubMed] [Google Scholar]
- 114.Iskratsch T, Lange S, Dwyer J, Kho AL, Dos Remedios C, Ehler E. Formin follows function: a muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. Journal of Cell Biology. 2010;191(6):1159–1172. doi: 10.1083/jcb.201005060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Taniguchi K, Takeya R, Suetsugu S, et al. Mammalian formin Fhod3 regulates actin assembly and sarcomere organization in striated muscles. Journal of Biological Chemistry. 2009;284(43):29873–29881. doi: 10.1074/jbc.M109.059303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Takano K, Watanabe-Takano H, Suetsugu S, et al. Nebulin and N-WASP cooperate to cause IGF-1-induced sarcomeric actin filament formation. Science. 2010;330(6010):1536–1540. doi: 10.1126/science.1197767. [DOI] [PubMed] [Google Scholar]
- 117.Kagawa M, Sato N, Obinata T. Effects of BTS (N-benzyl-p-toluene sulphonamide), an inhibitor for myosin-actin interaction, on myofibrillogenesis in skeletal muscle cells in culture. Zoological Science. 2006;23(11):969–975. doi: 10.2108/zsj.23.969. [DOI] [PubMed] [Google Scholar]
- 118.Soeno Y, Shimada Y, Obinata T. BDM (2,3-butanedione monoxime), an inhibitor of myosin actin interaction, suppresses myofibrillogenesis in skeletal muscle cells in culture. Cell and Tissue Research. 1999;295(2):307–316. doi: 10.1007/s004410051237. [DOI] [PubMed] [Google Scholar]
- 119.Ramachandran I, Terry M, Ferrari MB. Skeletal muscle myosin cross-bridge cycling is necessary for myofibrillogenesis. Cell Motility and the Cytoskeleton. 2003;55(1):61–72. doi: 10.1002/cm.10113. [DOI] [PubMed] [Google Scholar]
- 120.Skwarek-Maruszewska A, Hotulainen P, Mattila PK, Lappalainen P. Contractility-dependent actin dynamics in cardiomyocyte sarcomeres. Journal of Cell Science. 2009;122(12):2119–2126. doi: 10.1242/jcs.046805. [DOI] [PubMed] [Google Scholar]
- 121.Hanft LM, Bogan DJ, Mayer U, Kaufman SJ, Kornegay JN, Ervasti JM. Cytoplasmic γ-actin expression in diverse animal models of muscular dystrophy. Neuromuscular Disorders. 2007;17(7):569–574. doi: 10.1016/j.nmd.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hanft LM, Rybakova IN, Patel JR, Rafael-Fortney JA, Ervasti JM. Cytoplasmic γ-actin contributes to a compensatory remodeling response in dystrophin-deficient muscle. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(14):5385–5390. doi: 10.1073/pnas.0600980103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weber KL, Fischer RS, Fowler VM. Tmod3 regulates polarized epithelial cell morphology. Journal of Cell Science. 2007;120(20):3625–3632. doi: 10.1242/jcs.011445. [DOI] [PubMed] [Google Scholar]
- 124.Nowak RB, Fischer RS, Zoltoski RK, Kuszak JR, Fowler VM. Tropomodulin1 is required for membrane skeleton organization and hexagonal geometry of fiber cells in the mouse lens. Journal of Cell Biology. 2009;186(6):915–928. doi: 10.1083/jcb.200905065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Moyer JD, Nowak RB, Kim NE, et al. Tropomodulin 1-null mice have a mild spherocytic elliptocytosis with appearance of tropomodulin 3 in red blood cells and disruption of the membrane skeleton. Blood. 2010;116(14):2590–2599. doi: 10.1182/blood-2010-02-268458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gallagher PG, Forget BG. An alternate promoter directs expression of a truncated, muscle-specific isoform of the human ankyrin 1 gene. Journal of Biological Chemistry. 1998;273(3):1339–1348. doi: 10.1074/jbc.273.3.1339. [DOI] [PubMed] [Google Scholar]
- 127.Gallagher PG, Tse WT, Scarpa AL, Lux SE, Forget BG. Structure and organization of the human Ankyrin-1 gene. Journal of Biological Chemistry. 1997;272(31):19220–19228. doi: 10.1074/jbc.272.31.19220. [DOI] [PubMed] [Google Scholar]
- 128.Porter NC, Resneck WG, O’Neill A, Van Rossum DB, Stone MR, Bloch RJ. Association of small ankyrin 1 with the sarcoplasmic reticulum. Molecular Membrane Biology. 2005;22(5):421–432. doi: 10.1080/09687860500244262. [DOI] [PubMed] [Google Scholar]
- 129.Fukuzawa A, Lange S, Holt M, et al. Interactions with titin and myomesin target obscurin and obscurin-like 1 to the M-band—implications for hereditary myopathies. Journal of Cell Science. 2008;121(11):1841–1851. doi: 10.1242/jcs.028019. [DOI] [PubMed] [Google Scholar]
- 130.Bagnato P, Barone V, Giacomello E, Rossi D, Sorrentino V. Binding of an ankyrin-1 isoform to obscurin suggests a molecular link between the sarcoplasmic reticulum and myofibrils in striated muscles. Journal of Cell Biology. 2003;160(2):245–253. doi: 10.1083/jcb.200208109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mohler PJ, Davis JQ, Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biology. 2005;3(12) doi: 10.1371/journal.pbio.0030423. Article ID e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mohler PJ, Yoon W, Bennett V. Ankyrin-B targets β2-spectrin to an intracellular compartment in neonatal cardiomyocytes. Journal of Biological Chemistry. 2004;279(38):40185–40193. doi: 10.1074/jbc.M406018200. [DOI] [PubMed] [Google Scholar]
- 133.Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421(6923):634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 134.Tuvia S, Buhusi M, Davis L, Reedy M, Bennett V. Ankyrin-B is required for intracellular sorting of structurally diverse Ca2+ homeostasis proteins. Journal of Cell Biology. 1999;147(5):995–1007. doi: 10.1083/jcb.147.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kontrogianni-Konstantopoulos A, Jones EM, Van Rossum DB, Bloch RJ. Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Molecular Biology of the Cell. 2003;14(3):1138–1148. doi: 10.1091/mbc.E02-07-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ottenheijm CAC, Fong C, Vangheluwe P, et al. Sarcoplasmic reticulum calcium uptake and speed of relaxation are depressed in nebulin-free skeletal muscle. FASEB Journal. 2008;22(8):2912–2919. doi: 10.1096/fj.07-104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tay JSH, Lai PS, Low PS, Lee WL, Gan GC. Pathogenesis of Duchenne muscular dystrophy: the calcium hypothesis revisited. Journal of Paediatrics and Child Health. 1992;28(4):291–293. doi: 10.1111/j.1440-1754.1992.tb02669.x. [DOI] [PubMed] [Google Scholar]
- 138.Hopf FW, Turner PR, Steinhardt RA. Calcium misregulation and the pathogenesis of muscular dystrophy. Sub-cellular Biochemistry. 2007;45:429–464. doi: 10.1007/978-1-4020-6191-2_16. [DOI] [PubMed] [Google Scholar]
- 139.Goonasekera SA, Lam CK, Millay DP, et al. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. Journal of Clinical Investigation. 2011;121(3):1044–1052. doi: 10.1172/JCI43844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sanoudou D, Beggs AH. Clinical and genetic heterogeneity in nemaline myopathy—a disease of skeletal muscle thin filaments. Trends in Molecular Medicine. 2001;7(8):362–368. doi: 10.1016/s1471-4914(01)02089-5. [DOI] [PubMed] [Google Scholar]
- 141.Ochala J. Thin filament proteins mutations associated with skeletal myopathies: defective regulation of muscle contraction. Journal of Molecular Medicine. 2008;86(11):1197–1204. doi: 10.1007/s00109-008-0380-9. [DOI] [PubMed] [Google Scholar]
- 142.Laing NG, Wallgren-Pettersson C. 161st ENMC International Workshop on nemaline myopathy and related disorders, Newcastle upon Tyne, 2008. Neuromuscular Disorders. 2009;19(4):300–305. doi: 10.1016/j.nmd.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 143.Chandra M, Manidi R, Ford S, et al. Nebulin alters cross-bridge cycling kinetics and increases thin filament activation. A novel mechanism for increasing tension and reducing tension cost. Journal of Biological Chemistry. 2009;284(45):30889–30896. doi: 10.1074/jbc.M109.049718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ottenheijm CAC, Hooijman P, DeChene ET, Stienen GJ, Beggs AH, Granzier H. Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2) Journal of Structural Biology. 2010;170(2):334–343. doi: 10.1016/j.jsb.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bang ML, Caremani M, Brunello E, et al. Nebulin plays a direct role in promoting strong actin-myosin interactions. FASEB Journal. 2009;23(12):4117–4125. doi: 10.1096/fj.09-137729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ochala J, Lehtokari VL, Iwamoto H, et al. Disrupted myosin cross-bridge cycling kinetics triggers muscle weakness in nebulin-related myopathy. FASEB Journal. 2011;25(6):1903–1913. doi: 10.1096/fj.10-176727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ochala J, Iwamoto H, Larsson L, Yagi N. A myopathy-linked tropomyosin mutation severely alters thin filament conformational changes during activation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(21):9807–9812. doi: 10.1073/pnas.1001733107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kee AJ, Schevzov G, Nair-Shalliker V, et al. Sorting of a nonmuscle tropomyosin to a novel cytoskeletal compartment in skeletal muscle results in muscular dystrophy. Journal of Cell Biology. 2004;166(5):685–696. doi: 10.1083/jcb.200406181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vlahovich N, Kee AJ, Van der Poel CD, et al. Cytoskeletal tropomyosin Tm5NM1 is required for normal Excitation- contraction coupling in skeletal muscle. Molecular Biology of the Cell. 2009;20(1):400–409. doi: 10.1091/mbc.E08-06-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vlahovich N, Schevzov G, Nair-Shaliker V, et al. Tropomyosin 4 defines novel filaments in skeletal muscle associated with muscle remodelling/regeneration in normal and diseased muscle. Cell Motility and the Cytoskeleton. 2008;65(1):73–85. doi: 10.1002/cm.20245. [DOI] [PubMed] [Google Scholar]