

Abstract
Rotenone, an organic pesticide and potent mitochondrial complex I inhibitor, causes Parkinson-like neurodegeneration in rodents and is implicated in human Parkinson’s disease. In this rapid report, rotenone induced a dose-dependent decrease in succinyl-coenzyme A (CoA) and increase in β-hydroxybutyryl-CoA in multiple human cell lines (IC50 < 100 nM). Rotenone also inhibited [U-13C6]-glucose-derived [13C]-acetyl-CoA and [13C]-succinyl-CoA biosynthesis in SH-SY5Y neuroblastoma cells. These changes are compatible with a compensatory metabolic rearrangement. Stable isotope dilution liquid chromatography–mass spectrometry and CoA thioester isotopomer analysis provided insight into mechanisms of rotenone toxicity, which will facilitate the development of new biomarkers of mitochondrial dysfunction.
Numerous studies have suggested a link between Parkinson’s disease (PD) and pesticide exposure,(1) particularly to rotenone and paraquat, two naturally occurring organic pesticides. A recently reported nested case-control study demonstrated a 2.5-fold increase in the relative risk of developing PD among farmers exposed to either rotenone or paraquat.(2) Although both pesticides are mitochondrial toxins, they appear to have distinct toxicological mechanisms.3,4 The focus of this rapid report is on how rotenone affects central metabolic pathways in the cell. Rotenone is known to bind to complex I of the electron transport chain, preventing the transfer of electrons from iron sulfur clusters to ubiquinone,(5) reducing oxidative phosphorylation, and generating reactive oxygen species (ROS).(6) Chronic low dose administration of rotenone to rats causes selective nigrostriatal degeneration and formation of α-synuclein-positive cytoplasmic inclusions, similar to Lewy bodies, in nigral dopaminergic neurons. This results in neurodegenerative features that are comparable to those seen in PD,7,8 although the exact mechanism involved in rotenone mediated-neurotoxicity remains unclear.
The majority of recent research on rotenone toxicity has focused on complex I inhibition and ROS formation.5,9 However, rotenone has effects that are independent of complex I inhibition,(10) which significantly alters cellular metabolism, including changes in a number of intermediates and end-products of major bioenergetic pathways.11,12 Technological developments in both mass spectrometry (MS) and NMR have produced a growing number of metabolomic assays to assess changes in intracellular and extracellular metabolites.13−15 In this study, we further explored metabolic changes induced by rotenone by analyzing its effects on intracellular levels of various short chain acyl-CoA thioesters, which are known to be involved in a number of metabolic pathways.(16) This was accomplished using stable isotope dilution liquid chromatography (LC)-MS, which provides the most specific methodology for rigorous quantification of biomarkers and endogenous metabolites.(17) Stable isotope labeling by essential nutrients in cell culture (SILEC), a technique in which cells are grown in the presence of [13C315N1]-pantothenate (a CoA precursor), was used to generate isotopically labeled short chain acyl-CoA thioesters.18,19 The extracted labeled CoA species were used as stable isotope internal standards in LC-selected reaction monitoring (SRM)/MS analyses to quantify changes in intracellular levels of short chain acyl-CoA species caused by subcytotoxic doses of rotenone in various human cell lines, including neuroblastoma (SH-SY5Y), hepatocellular carcinoma (HepG2), and bronchoalveolar carcinoma (H358) cells. To further characterize the temporal nature of these changes in a neuronal cell type, a time course study was performed in the SH-SY5Y cell line. Finally, isotopomer analysis using [U-13C6]-glucose was performed in SH-SY5Y cells in the presence or absence of rotenone.
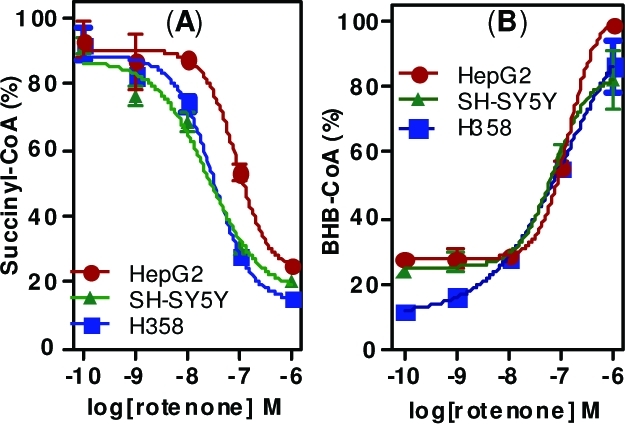
Rotenone was found to induce dose-dependent changes in the short chain acyl-CoA profiles of SH-SY5Y, HepG2, and H358 cells. Most notably, there was a significant decrease in levels of succinyl-CoA (Figure 1A) and a concomitant increase in β-hydroxybutyryl (BHB)-CoA (Figure 1B) after 1 h of treatment with an IC50 of <100 nM in all cell types. There were also significant changes in a number of other acyl-CoA thioesters, which varied among different cell types (Supporting Information, Table 1). To characterize the temporal nature of these changes, SH-SY5Y cells were treated with 100 nM rotenone and harvested at time points up to 6 h and analyzed for CoA thioesters. The observed changes seen in succinyl-CoA and BHB-CoA persisted for at least 6 h (Figure 1C). Overall, these results show that rotenone induces a rapid and persistent metabolic rearrangement in a variety of cell types, similar to changes observed during fasting.(20)
Figure 1.
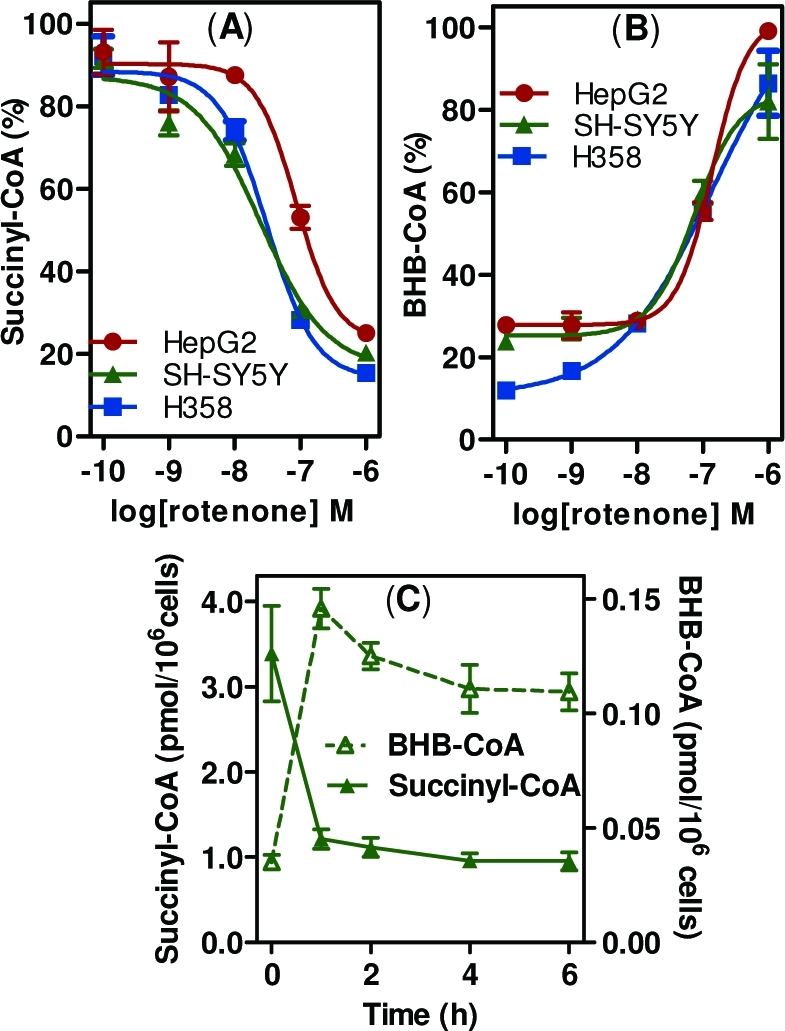
Rotenone-mediated changes in intracellular CoA thioester levels. (A) Succinyl-CoA. (B) BHB-CoA extracted from SH-SY5Y cells, HepG2 cells, and H358 cells treated with rotenone for 1 h, in triplicate. Values are shown as % of the maximal level for each cell line. (C) Time course of rotenone-mediated changes in intracellular succinyl-CoA and BHB-CoA. SH-SY5Y cells were treated with 100 nM rotenone, harvested at time points up to 6 h, and processed for CoAs. Error bars show SEMs for triplicate determinations.
Although the observed changes indicate that rotenone induces a significant metabolic disturbance, absolute CoA measurements represent only a snapshot of this metabolic state. To gain additional information on the cumulative flux through different pathways, cells were incubated with [U-13C6]-glucose, and isotopomer distribution of the resulting CoA derivatives was analyzed. [U-13C6]-glucose (M6) taken up by cells is converted during glycolysis into two [13C3]-pyruvates (M3), which can be converted by pyruvate dehydrogenase (PDH) into [13C2]-acetyl-CoA (M2). Both labeled carbons from acetyl-CoA can then be incorporated into citrate and eventually form [13C2]-succinyl-CoA (M2) in the first turn of the Krebs cycle. Additional cycles can result in the M3 and M4 succinyl-CoA isotopomers. It should be noted that isotopic tracer analysis is complex, with numerous intersecting metabolic pathways. For our purposes, however, this simplified interpretation made it possible to assess major changes that were occurring in the cells. SH-SY5Y cells were washed and treated with glucose-free media supplemented with either unlabeled glucose or [U-13C6]-glucose with or without 100 nM rotenone. The unlabeled cells were used to determine isotopic contributions of unlabeled CoA species to each isotopomer, which were subtracted from the labeled CoA species using a matrix analysis.(21)
Rotenone significantly decreased the incorporation of glucose-derived carbon atoms into acetyl-CoA, from 23% to 5%, demonstrating that rotenone inhibited the glucose-derived biosynthesis of acetyl-CoA (Figure 2). Interestingly, while there was a decrease in the % labeled acetyl-CoA (Figure 2), the overall concentration of acetyl-CoA did not change significantly (Supporting Information, Table 1). Rotenone also significantly decreased the M2, M3, and M4 isotopomers of succinyl-CoA, indicating decreased conversion of glucose to succinyl-CoA. Therefore, the decrease in absolute succinyl-CoA concentrations was due, in part, to decreased glucose-derived flux through the Krebs cycle (Figure 2). Lastly, there was no isotopic labeling of CoASH, confirming that the labeling occurred only in the acyl moiety.
Figure 2.

Effect of rotenone on the biosynthesis of glucose-derived acetyl-CoA and succinyl-CoA. SH-SY5Y cells were incubated in media containing [U-13C6]-glucose and 100 nM rotenone or DMSO (vehicle) for 6 h. Isotopomer distributions are shown in relation to the Krebs cycle. Error bars are SEMs for n = 5, *p < 0.05 compared with the DMSO vehicle.
In this study, we have shown for the first time that rotenone not only inhibits glucose-derived formation of both acetyl-CoA and succinyl-CoA but also induces an increase in the formation of BHB-CoA in a number of human cell lines. Increased ROS and NADH/NAD+ ratio resulting from rotenone-mediated complex I inhibition would decrease PDH activity,(22) explaining the decrease in acetyl-CoA labeling. This is consistent with a recent finding that rotenone causes increased shunting of glycolytic intermediates into lactate.(11) The absence of a decrease in the absolute concentration of acetyl-CoA indicates either an alternate source of acetyl-CoA or an inhibition of acetyl-CoA utilization.
The decrease in succinyl-CoA levels is consistent with a recent study in Arabidopsis cells, in which a rapid 50% decrease in α-ketoglutarate, the precursor of succinyl-CoA (Figure 2), was observed after 1 h of rotenone treatment.(12) This may be, in part, due to an inhibition of aconitase, which is known to be inhibited by rotenone and ROS formation.23,24 The dramatic decrease in intracellular succinyl-CoA (Figure 1A) would cause decreased protein succinylation, a post-translational modification with important regulatory implications. For example, 3-hydroxy-3-methyl-glutaryl (HMG)-CoA synthase, the rate-determining enzyme in ketogenesis is inactivated by succinylation, which is, in turn, regulated by succinyl-CoA concentrations.(25) In addition, a recent study has shown that succinylation of lysine residues occurs as a post-translational regulatory modification to certain enzymes such as isocitrate dehydrogenase.(26)
The rotenone-mediated increase in BHB-CoA (Figure 1B), together with the minimal incorporation of [U-13C6]-glucose into BHB-CoA (<5%) is indicative of a compensatory metabolic rearrangement. Specifically, BHB-CoA formed by increased fatty acid β-oxidation,(27) could be metabolized by 3-hydroxyacyl-CoA dehydrogenase to form acetoacetyl-CoA,(28) followed by thiolase (ACAT1)-mediated formation of acetyl-CoA.(29) This would allow the cell to maintain cellular bioenergetics while bypassing the inhibition of glucose-derived acetyl-CoA. Whether these changes are a pathological result of rotenone toxicity, or whether they represent a physiological compensation by known or unknown metabolic pathways, remains to be determined. However, the prolonged reliance on compensatory pathways could play an important role in rotenone toxicity in vivo due to differences in the abilities of different tissues or cell types to up-regulate or maintain these metabolic rearrangements.
In summary, we have shown that SILEC-based stable isotope dilution LC-MS methodology coupled with isotopomer analysis of CoA thioesters can be used in vitro to gain insight into the mechanisms involved in the toxicity of environmental agents such as rotenone. This methodology will also make it possible to further explore the role of mitochondrial dysfunction in metabolic and neurodegenerative diseases.
Glossary
Abbreviations
- BHB
β-hydroxybutyryl
- CoA
coenzyme A
- CoASH
reduced CoA
- HMG
3-hydroxy-3-methyl-glutaryl
- LC-MS
liquid chromatography–mass spectrometry
- PD
Parkinson’s disease
- ROS
reactive oxygen species
- SILEC
stable isotope labeling by essential nutrients in cell culture
- SRM
selected reaction monitoring
Supporting Information Available
Experimental procedures and supplementary table. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by NIH Grants U01ES016004, P30ES013508, and T32HL007439.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brown T. P.; Rumsby P. C.; Capleton A. C.; Rushton L.; Levy L. S. (2006) Environ. Health Perspect. 114, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner C. M.; Kamel F.; Ross G. W.; Hoppin J. A.; Goldman S. M.; Korell M.; Marras C.; Bhudhikanok G. S.; Kasten M.; Chade A. R.; Comyns K.; Richards M. B.; Meng C.; Priestley B.; Fernandez H. H.; Cambi F.; Umbach D. M.; Blair A.; Sandler D. P.; Langston J. W. (2011) Environ. Health Perspect. 119, 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez C.; Bandez M. J.; Navarro A. (2007) Front. Biosci. 12, 1079–1093. [DOI] [PubMed] [Google Scholar]
- Cicchetti F.; Drouin-Ouellet J.; Gross R. E. (2010) Trends Pharmacol. Sci. 31, 142. [DOI] [PubMed] [Google Scholar]
- Koopman W. J.; Nijtmans L. G.; Dieteren C. E.; Roestenberg P.; Valsecchi F.; Smeitink J. A.; Willems P. H. (2010) Antioxid. Redox. Signal 12, 1431–1470. [DOI] [PubMed] [Google Scholar]
- Tahara E. B.; Navarete F. D.; Kowaltowski A. J. (2009) Free Radical Biol. Med. 46, 1283–1297. [DOI] [PubMed] [Google Scholar]
- Betarbet R.; Sherer T. B.; MacKenzie G.; Garcia-Osuna M.; Panov A. V.; Greenamyre J. T. (2000) Nat. Neurosci. 3, 1301–1306. [DOI] [PubMed] [Google Scholar]
- Cannon J. R.; Tapias V.; Na H. M.; Honick A. S.; Drolet R. E.; Greenamyre J. T. (2009) Neurobiol. Dis. 34, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman D. L.; Brookes P. S. (2009) J. Biol. Chem. 284, 16236–16245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W. S.; Kruse S. E.; Palmiter R. D.; Xia Z. (2008) Proc. Natl. Acad. Sci. U.S.A 105, 15136–15141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q.; Vu H.; Liu L.; Wang T. C.; Schaefer W. H. (2011) J. Biomol. NMR 49, 207–219. [DOI] [PubMed] [Google Scholar]
- Garmier M.; Carroll A. J.; Delannoy E.; Vallet C.; Day D. A.; Small I. D.; Millar A. H. (2008) Plant Physiol. 148, 1324–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham O.; Slate N. G.; Goldberger O.; Xu Q.; Ramanathan A.; Souza A. L.; Clish C. B.; Sims K. B.; Mootha V. K. (2010) Proc. Natl. Acad. Sci. U.S.A 107, 1571–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor S. C.; Hansen M. K.; Corner A.; Smith R. F.; Ryan T. E. (2010) Mol. Biosyst. 6, 909–921. [DOI] [PubMed] [Google Scholar]
- Balcke G. U.; Kolle S. N.; Kamp H.; Bethan B.; Looser R.; Wagner S.; Landsiedel R.; van Ravenzwaay B. (2011) Toxicol. Lett. 203, 200–209. [DOI] [PubMed] [Google Scholar]
- Li L. O.; Klett E. L.; Coleman R. A. (2010) Biochim. Biophys. Acta 1801, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccimaro E.; Blair I. A. (2010) Bioanalysis 2, 311–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S. S.; Mesaros C.; Gelhaus S. L.; Blair I. A. (2011) Anal. Chem. 83, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S. S.; Blair I. A.. Nat. Protoc. 2011, not supplied. [DOI] [PMC free article] [PubMed]
- Gao L.; Chiou W.; Tang H.; Cheng X.; Camp H. S.; Burns D. J. (2007) J. Chromatogr. B 853, 303–313. [DOI] [PubMed] [Google Scholar]
- Fernandez C. A.; Des R. C.; Previs S. F.; David F.; Brunengraber H. (1996) J. Mass Spectrom. 31, 255–262. [DOI] [PubMed] [Google Scholar]
- Bunik V. I. (2003) Eur. J. Biochem. 270, 1036–1042. [DOI] [PubMed] [Google Scholar]
- Tretter L.; Adam-Vizi V. (2000) J. Neurosci. 20, 8972–8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulteau A. L.; Ikeda-Saito M.; Szweda L. I. (2003) Biochemistry 42, 14846–14855. [DOI] [PubMed] [Google Scholar]
- Lowe D. M.; Tubbs P. K. (1985) Biochem. J. 232, 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Tan M.; Xie Z.; Dai L.; Chen Y.; Zhao Y. (2011) Nat. Chem. Biol. 7, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten S. M.; Wanders R. J. (2010) J. Inherited Metab. Dis. 33, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. Y.; He X. Y.; Schulz H. (2005) FEBS J. 272, 4874–4883. [DOI] [PubMed] [Google Scholar]
- Korman S. H. (2006) Mol. Genet. Metab. 89, 289–299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.