

Abstract
Drinking water supplies in many geographic areas contain chromium in the +3 and +6 oxidation states. Public health concerns are centered on the presence of hexavalent Cr that is classified as a known human carcinogen via inhalation. Cr(VI) has high environmental mobility and can originate from anthropogenic and natural sources. Acidic environments with high organic content promote the reduction of Cr(VI) to nontoxic Cr(III). The opposite process of Cr(VI) formation from Cr(III) also occurs, particularly in the presence of common minerals containing Mn(IV) oxides. Limited epidemiological evidence for Cr(VI) ingestion is suggestive of elevated risks for stomach cancers. Exposure of animals to Cr(VI) in drinking water induced tumors in the alimentary tract, with linear and supralinear responses in the mouse small intestine. Chromate, the predominant form of Cr(VI) at neutral pH, is taken up by all cells through sulfate channels and is activated nonenzymatically by ubiquitously present ascorbate and small thiols. The most abundant form of DNA damage induced by Cr(VI) is Cr-DNA adducts, which cause mutations and chromosomal breaks. Emerging evidence points to two-way interactions between DNA damage and epigenetic changes that collectively determine the spectrum of genomic rearrangements and profiles of gene expression in tumors. Extensive formation of DNA adducts, clear positivity in genotoxicity assays with high predictive values for carcinogenicity, the shape of tumor–dose responses in mice, and a biological signature of mutagenic carcinogens (multispecies, multisite, and trans-sex tumorigenic potency) strongly support the importance of the DNA-reactive mutagenic mechanisms in carcinogenic effects of Cr(VI). Bioavailability results and kinetic considerations suggest that 10–20% of ingested low-dose Cr(VI) escapes human gastric inactivation. The directly mutagenic mode of action and the incompleteness of gastric detoxification argue against a threshold in low-dose extrapolation of cancer risk for ingested Cr(VI).
1. Environmental Contamination with Chromium
Three thermodynamically stable Cr forms, Cr(0), Cr(III), and Cr(VI), are used commercially and are present in the environment. Cr(0) is found almost exclusively in its metallic form, most commonly as a component of iron-based alloys such as stainless steel. Only Cr(VI), but not Cr(0) or Cr(III), has been shown to cause cancers in laboratory animals and occupationally exposed workers.1−3 Environmental pollution with various forms of Cr results from its numerous uses in the chemical industry, production of dyes, wood preservation, leather tanning, chrome plating, manufacturing of various alloys, and many other applications and products.1,2 Stainless steel contains up to 20% Cr by weight and is the highest volume product containing this metal. The magnitude of Cr utilization is also evident from the estimated occupational exposure by more than 500,000 workers in the U.S. alone.(3) Incineration and emissions from cars create ambient pollution with small Cr(VI)- and Cr(III)-containing particles, which leads to low-level inhalation exposures by large segments of the general population and increases Cr levels in surface waters. The most serious cases of anthropogenic contamination of drinking water in the U.S. came from the discharges of toxic Cr(VI) by cooling towers.(4) Other large-scale environmental pollution with Cr(VI) involved improper disposal of millions of tons of incompletely processed chromite ore.(5) Hundreds of the largest toxic waste sites in the U.S. known as Superfund sites contain Cr as a major contaminant.(2) The presence of Cr(VI) in drinking water can also result from the oxidation of naturally occurring Cr(III) by Mn(III/IV) oxides in birnessite,(6) a common mineral that coats weathered grains and fractures in Cr-rich ultramafic rocks and serpentinites that are enriched with chromite [FeCr(III)2O4]. In addition to birnessite, the presence of two other Mn(IV) oxide-containing minerals, asbolane and lithiophorite, has also been associated with the formation of Cr(VI) from natural Cr(III).(7) Examination of four minerals made of Mn oxides (birnessite, cryptomelane, todorokite, and hausmannite) showed that birnessite had the highest ability to oxidize Cr(III) under laboratory conditions.(8)
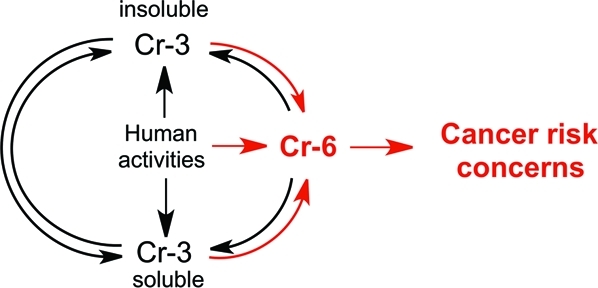
Cr(0)-containing products are generally highly resistant to corrosion; however, slow oxidation of Cr-containing alloys can result in the limited release of soluble Cr(III) into soil and water. Cr(VI) is the most mobile form of chromium in the environment. Mobility of Cr(III) strongly depends on the acidity and Cr(III)-binding properties of soluble and insoluble components at the contamination sites. Cr(VI) and Cr(III) have dramatically different toxicological properties,1,2 and all public health concerns are centered on the presence of toxic hexavalent Cr in drinking water. In 1991, the US-EPA set the current standard for Cr in drinking water (maximum contaminant level) at 100 μg/L,(9) which does not distinguish between the presence of toxic Cr(VI) and nontoxic Cr(III). Considering long-standing public concerns about Cr(VI), it is unclear why there are no requirements for specific monitoring of Cr(VI) levels. The analytical technologies for Cr speciation and detection were not very sensitive until the mid-1980s, and reliable measurements of Cr(VI) at environmental levels typically required preconcentration steps, which were more suitable for specialized academic laboratories than for routine monitoring. However, analytical methodologies for Cr(VI) underwent dramatic improvements a long time ago. The EPA’s own Method 218.6 for the detection of hexavalent Cr was developed approximately 20 years ago and affords a reliable monitoring of Cr(VI) at 1 μg/L. This methodology is not expensive and is based on a routine HPLC column separation of chromate anion followed by a colorimetric detection with diphenylcarbazide. Essentially all analytical laboratories are equipped with HPLC and should be able to perform this type of Cr(VI) measurements at relatively low cost. Recent improvements of chromatographic and postcolumn derivatization conditions in the EPA’s Method 218.6 allow the detection of Cr(VI) in drinking water at 3 ng/L (Application Update 179 from Dionex, Inc.). This sensitivity is more than sufficient to meet the most stringent regulations for Cr(VI), including those associated with the Public Health Goal of 20 ng/L in California. For many samples with very low Cr levels, Cr(VI) analyses by the modified Method 218.6 could be even more sensitive than measurements of total Cr. Although the presence of Cr(VI) near municipal water reservoirs is the obvious reason for its monitoring in drinking water supplies, environmental contamination with Cr(III) can also generate Cr(VI) through oxidation reactions with water chlorination products,(10) Ca and Mn oxides,11,12 and photoxidation.(13) Overall, industrial activities associated with the direct release of Cr(VI) into the soil and water is the most important source of Cr(VI) contamination in drinking water (Figure 1).
Figure 1.

Origin and interconversion of different forms of Cr in water. Route 1 is the most important overall source of Cr(VI), whereas route 2 describes the most important natural process of Cr(VI) formation.
2. Main Aqueous Forms of Cr(VI) AND Cr(III)
2.1. Chromium-3
Cr(III) forms hexacoordinate complexes with the octahedral arrangement of ligands. Hexaaquachromium(III) [Cr(H2O)63+] is the main Cr species in solutions of inorganic Cr(III) salts under strongly acidic pH. At pH 4 and higher, Cr(III)-bound H2O molecules undergo hydrolysis, which leads to the formation of soluble and insoluble oligomeric and polymeric products (Figure 2A). The reactions of hydrolysis and polymerization are accelerated by increasing pH >5, and adjustment of solutions to neutral pH results in a rapid precipitation of multinuclear Cr(III) hydroxides. In addition to pH, the generation of polymeric products is influenced by the Cr concentrations, composition, and “age” of the solutions. Surface waters commonly contain a mixture of soluble monomeric and oligomeric Cr(III) products.14,15 Mild acidity and the absence of significant amounts of organics in drinking water supplies limit the presence of dissolved Cr(III) due to its poor solubility. In contrast to its aqua complexes, Cr(III) compounds with small organic molecules such as organic acids, biological buffers, amino acids, and others are soluble and remain monomeric for a long time.(16) Thus, the description of Cr(III) insolubility at neutral pH is only correct for solutions lacking ligands that compete with H2O. Formation of stable Cr(III) complexes with small organic molecules at the primary source of emission or contamination sites can increase their environmental mobility and maintain the solubility of Cr(III) even at neutral pH. Binding of Cr(III) by Fe-oxyhydroxides and immobilized organic matter in the soil or sediments diminishes its soluble pool. Interactions between Cr(III) with components of environmental media involve continuous cycles of adsorption/desorption and precipitation/dissolution, which control the extent of its environmental mobility.
Figure 2.

Aqueous species of Cr(III) and Cr(VI). (A) Hydrolysis and polymerization of hexaaquachromium(III). Transition pH values are approximate. (B) Structures of sulfate, chromate, and dichromate.
Animal and epidemiological studies have consistently found inorganic Cr(III) to be nontoxic and noncarcinogenic,1,2 which has also been confirmed for the dietary supplement chromium picolinate.(17) The absence of toxic effects for Cr(III) complexes results from their poor ability to enter cells, lack of intracellular accumulation, and high stability of coordinated multidentate ligands, which prevents binding to cellular macromolecules.(16) Systemic absorption of Cr(III) taken by human volunteers in the form of dissolved inorganic salts is measurable but usually less than 0.5%.2,18 No elevation in urinary excretion of Cr(III) was detected from the ingestion of insoluble Cr2O3,(19) which raises the question as to why the use of inorganic salts resulted in Cr(III) absorption at all considering the formation of oligomeric products after the dissolution of these compounds in water. A plausible explanation for these results lies in highly acidic conditions of the human gastric environment (pH 1–3),(20) which is expected to promote dissociation of Cr(III) oligomers. Low pH also strongly enhances the exchange of H2O for organic ligands in the Cr(III) coordination sphere,(21) allowing the formation of new Cr(III) complexes that could remain soluble upon reaching neutral pH of the small intestine, where the absorption of metal ions takes place. A short stomach residency time for pure water and other simple liquids, however, is expected to limit the extent of ligand exchange reactions.
2.2. Chromium-6
Chromate anion (CrO42–) is the predominant form of Cr(VI) in dilute solutions at neutral pH. Chromate exists in equilibrium with its protonated form HCrO4– in approximately 3:1 ratio at these conditions.(22) HCrO4– becomes the most prevalent species of Cr(VI) at pH < 6. Concentrated solutions of Cr(VI) at highly acidic pH contain the dichromate anion Cr2O72– (Figure 2B). At levels below the current federal standard of 100 μg/L, the presence of dichromate in drinking water should be minimal. Both chromate and dichromate have tetrahedral arrangements of coordinated oxygen groups.
Cr(VI) is a potent oxidant at highly acidic pH when any organic molecule with oxidizable groups can promote its reduction to Cr(III). Increasing pH enhances the stability of Cr(VI), particularly at near neutral and alkaline pH. The strong pH effect on Cr(VI) reduction is demonstrated by the findings that human gastric juice was capable of reducing approximately 70% of Cr(VI) after a 30-min incubation at pH 1.4 but was completely ineffective at pH 7.0.(18) The presence of Fe2+ is important for abiotic reduction of Cr(VI) under anaerobic conditions in soil and underground water. Uptake and reduction by microorganisms is another detoxification process for Cr(VI), although it is limited by the ability of resistant bacteria striving in contaminated water to rapidly extrude Cr(VI).(23) Overall, Cr(VI) is a persistent contaminant in many water sources due to their low organic content and lack of sufficient acidity to catalyze reduction to Cr(III).
3. Cellular Metabolism and DNA Damage by Cr(VI)
3.1. Cellular Metabolism
Cr(VI) displays no ability to damage DNA directly and requires reductive activation for its genotoxic activity. Structural similarity of chromate ion to sulfate (Figure 2B) allows its easy entry through the general sulfate channels.(16) As with abiotic reactions, cellular reduction of Cr(VI) yields thermodynamically stable Cr(III).24,25 Efficient uptake of Cr(VI) followed by Cr(III) trapping via its binding to macromolecules leads to a massive accumulation of Cr relative to its extracellular concentrations, ranging from 10- to 20-fold after 3-h exposures to about 100-fold after 24-h exposures.26,27 With the apparent exception of bacteria producing hyperoxidized Mn(III/IV),(28) biological systems lack the ability to reoxidize Cr(III) to Cr(VI). Extracellular reduction of Cr(VI) is a detoxification process that produces poorly permeable nontoxic Cr(III) (Figure 3). Studies of reduction activities in tissue homogenates and biological fluids showed that ascorbate (Asc) was the principal biological reducer of Cr(VI), accounting for 80–95% of its metabolism.29−31 A combined activity of Asc and small thiols glutathione (GSH) and cysteine is responsible for >95% of Cr(VI) reduction in vivo. Tissue concentrations of GSH and Asc are not usually dramatically different, and the predominant role of Asc stems from its very high rate of Cr(VI) reduction. At physiological 1 mM concentration, t1/2 for Cr(VI) reduction by Asc was 1 min vs 60.7 min for GSH and 13.3 min for Cys.(32) Despite its slower rate of reduction, GSH is more important for Cr(VI) metabolism than Cys due to its higher cellular concentrations.
Figure 3.

Reduction of Cr(VI) and its genotoxic impact. Extracellular reduction is a detoxification process, generating poorly cell-permeable Cr(III). In contrast, cellular Cr(VI) reduction is the activation process, producing redox-active intermediates Cr(V/IV) and stable Cr(III) forming mutagenic Cr-DNA adducts. Asc is the dominant reducer of Cr(VI) in cells in vivo, whereas GSH is the most prominent reducer in cultured cells due to their Asc-deficiency.
Depending on the nature of the reducing agent, its concentration, and stoichiometry, Cr(VI) reduction reactions generate variable amounts of transient products such as Cr(V), Cr(IV), and sulfur- and carbon-based radicals.33−36 As expected for these important biological antioxidants, GSH-, cysteine-, and Asc-derived radicals formed in Cr(VI) reactions are unreactive toward DNA.32,37,38 In the presence of H2O2, intermediate Cr forms can catalyze Fenton-type reactions, generating highly reactive OH• radicals.39,40 Formation of reactive oxygen species (ROS) and direct oxidizing abilities of Cr(V)41,42 are the two main processes contributing to the induction of oxidative stress in Cr(VI)-treated cells. More detailed discussion of properties of intermediate Cr forms can be found in other reviews.43,44 Although all biological reducers convert Cr(VI) to Cr(III), their mechanisms of reduction are not the same. Kinetic analyses predict that Cys acts almost exclusively as a one-electron reducer,(45) which is consistent with the presence of strong Cr(V) signal in Cys-driven reactions.(46) Reduction by GSH can proceed through either one- or two-electron reactions.34,47,48 Asc is a highly efficient two-electron donor, yielding Cr(IV) as the first reduction intermediate and dehydroascorbic acid as the oxidized product. The presence of Cr(V) is only detectable under nonphysiological conditions of equimolar or higher ratio of Cr(VI) to Asc.35,49,50 Insufficient amounts of Asc for the completion of Cr(VI) reduction in these reactions were responsible for a transient appearance of Cr(V), likely resulting from comproportionation of Cr(IV) and Cr(VI). Severe Asc deficiency of human and nonhepatic rodent cells in standard cultures27,51−54 raises concerns that studies with cultured cells may not accurately recapitulate genotoxic properties of Cr(VI) in vivo. The Asc-depleted state of cultured cells results from the absence of this vitamin in the most common types of synthetic growth media, and typical additions of 10% serum to the media theoretically supplies only 10% of normal vitamin C levels. The actual levels of Asc in the growth media are lower due to its loss during the preparation and storage of serum. The half-life of Asc at 37 °C in cell culture media is 6–7 h.(55) Recently fed cells can contain up to 50–60 μM vitamin C, but in many cases, its levels are dramatically lower or undetectable. Even when cells start with 50–60 μM Asc, they become completely depleted of Asc after 24–48 h in culture.(54) Physiological concentrations of vitamin C in white blood cells and epithelial tissues are usually in the 1–2 mM range.55−57
3.2. Cr-DNA Damage
Cellular metabolism of Cr(VI) can cause both oxidative and nonoxidative forms of DNA damage.43,44,58 The most abundant and specific type of DNA damage is Cr-DNA binding (adducts), which has been detected in reduction reactions in vitro, in various cultured cells and in vivo.(16) Formation of Cr-DNA adducts in human cells was already clearly evident after brief exposures to 2 μM (current federal standard for Cr in water) and even lower concentrations of Cr(VI).27,59 The rapid loss of Cr-DNA adducts from normal but not nucleotide excision repair (NER)-deficient human cells(59) firmly demonstrated that Cr binding to chromosomal DNA occurred in intact cells and did not arise artificially during DNA purification. Cr-DNA adducts are a heterogeneous group that includes binary [Cr(III)-DNA] and several ternary [ligand-Cr(III)-DNA] adducts where the ligand can be Asc, GSH, cysteine, or histidine. All four ternary adducts have been detected in Cr(VI)-treated cells51,60 and are readily formed during in vitro Cr(VI) metabolism. DNA cross-linking of GSH,33,38,61 cysteine,(21) and Asc32,51 was observed in their corresponding reactions with Cr(VI), while DNA attachment of nonreducing amino acid histidine occurred efficiently during the reduction of Cr(VI) with Asc.(62) Cr(VI) also causes the formation of protein-Cr(III)-DNA cross-links,63,64 which are rare lesions and whose main toxicological significance could lie not necessarily in the contribution to genotoxic responses but rather in the inhibition of gene-specific expression.(65) Binary Cr-DNA adducts are the most frequent DNA modifications in the in vitro reductions of Cr(VI). In Cr(VI) reactions with its two main biological reducers, Asc(51) and GSH,(38) binary adducts accounted for 75–95% of the total DNA-bound Cr. When normalized for recovery, Asc-Cr-DNA cross-links have been calculated to comprise 6% of Cr-DNA adducts in human A549 cells with restored Asc levels.(51) Cys-Cr-DNA and GSH-Cr-DNA accounted for 24 and 17% of all DNA adducts in hamster CHO cells,(60) although these values have not been adjusted for recovery.
Protein-Cr-DNA cross-links constitute only about 0.1% of total adducts immediately after Cr(VI) exposures,16,66 but their relative amounts are likely higher at later postexposure times due to delayed formation(67) and slower repair(66) relative to those of small Cr-DNA adducts.(59) All small ternary adducts are formed via the conjugation of preformed ligand-Cr(III) complexes with DNA,51,62 whereas protein–DNA cross-linking proceeds through the initial formation of binary Cr(III)-DNA adducts followed by a slow reaction of protein capture.(67) Binary Cr(III)-DNA adducts can result from direct DNA binding by Cr(III), but the role of other Cr forms in their formation cannot be excluded.(16)
3.3. Mutagenicity of Cr-DNA Adducts
Complex metabolism of Cr(VI) and the formation of several types of DNA damage make it difficult to assess the importance of specific lesions in Cr(VI)-treated cells. These difficulties can be overcome using a shuttle-vector methodology, which allows for the examination of mutagenic and genotoxic properties of in vitro formed DNA damage during replication in intact human cells. Replication of adduct-carrying shuttle-vectors in human cells showed that the most abundant adduct, the binary Cr-DNA conjugate, was weakly mutagenic, whereas four ternary adducts containing DNA-cross-linked Asc,(32) GSH, cysteine, and histidine(68) were strongly mutagenic. Asc-Cr-DNA was the most potent mutagenic and replication-inhibiting adduct. In vitro reduction of Cr(VI) by purified Asc,(32) GSH,(38) or cysteine(37) also led to the production of mutagenic and replication-blocking DNA lesions, as revealed by analyses of replicated progeny of shuttle-vector plasmids propagated in human fibroblasts. Asc-driven metabolism of Cr(VI) in vitro resulted in the strongest mutagenic responses,(32) while reactions with GSH showed low yields of mutagenic damage and GSH-Cr-DNA adducts.(38) These findings were corroborated by a strong potentiation of Cr(VI) mutagenicity in cells with restored Asc levels.(27) Blocking of Cr-DNA binding during the reduction or dissociation of Cr-DNA adducts eliminated all mutagenic and replication-blocking responses in shuttle-vector plasmids incubated in Cr(VI) reactions containing Asc,32,69 GSH,(38) or Cys,(37) demonstrating a key role of Cr-DNA adducts in the mutagenicity and genotoxicity of Cr(VI) when metabolized by its three main biological reducers. In agreement with these results, in vitro reduction reactions employing iron-free reagents failed to generate detectable amounts of single-strand breaks and abasic sites in DNA.21,32,38,45 Inhibition of in vitro replication on DNA templates damaged in Cr(VI)-Asc reactions was also dependent on Cr-DNA binding.(70)
Involvement of Cr-DNA adducts in bacterial mutagenesis by Cr(VI) is indicated by higher yields of revertants in the Ames test using a NER-deficient Salmonella uvrA strain.(71) Cr(VI) mutagenicity in transgenic lacI mice was inhibited by GSH depletion,(72) which points to the importance of nonoxidative mechanisms and GSH-Cr-DNA adducts in mutagenic responses in vivo. Cr(VI)-induced fold changes in the number of HPRT mutants were reported to be lower in NER-deficient clones of CHO cells grown under the standard Asc-deficient conditions,(73) which contrasts the positive role of NER in the removal of adducts and survival of CHO(74) and human cells.(59) Thus, it is possible that NER operates differently on distinct Cr-DNA adducts. However, interpretation of the results with NER-null CHO clones was complicated by their greater background mutation frequencies, and these high denominator values offer an alternative explanation for an appearance of diminished mutagenic responses for Cr(VI) when data were expressed as a fold of change.(73)
3.4. Chromosomal Breakage by Cr-DNA Adducts
Ternary but not binary Cr-DNA adducts were strong inhibitors of replication in plasmids transfected into human cells.32,37 These results were unexpected given weak structural distortions in Cr-adducted DNA with 1–2° unwinding angle per Cr atom(75) and the inability of Cr-DNA adducts to block DNA replication in vitro.(61,70) The phosphate group is a primary site for Cr(III)-DNA binding,62,68 although additional coordination to N7-dG is also possible as evidenced by the ability of Cr(III) to bind to dG,(62) G/C-targeted mutagenesis32,37,68 and preferential incisions by a bacterial NER nuclease in Cr-adducted DNA.(76) Replication inhibition by ternary Cr-DNA adducts required the presence of mismatch repair (MMR) proteins,(77) which were found to recognize these modifications with an affinity that was comparable to that of their natural substrate, G/T mismatch.(78) Binary Cr-DNA adducts were poorly bound by MMR proteins, and their weak effects on vector replication were MMR-independent.(78) In a full agreement with plasmid replication experiments, MMR-null mouse and human cells were resistant to apoptosis and clonogenic toxicity by Cr(VI).52,77,78 Studies with human cells of different histological origin showed that the high genotoxicity and cytotoxicity of Cr-DNA adducts resulted from their misprocessing by MMR into DNA double-strand breaks (DSB).27,77,78 Importantly, DSB were detected by various techniques, including a direct methodology of pulse-field gel electrophoresis.(78) MMR was responsible for all DSB generated in Asc-restored normal human cells treated with both high and very low environmentally relevant (0.2–2 μM) concentrations of Cr(VI).(27) The Asc-Cr-DNA adduct was very efficiently recognized by MMR proteins in vitro,(78) and restoration of Asc levels in cells strongly increased cytotoxicity and DSB formation by Cr(VI) via MMR activity.27,52,78 Asc-deficiency of human lung BEAS-2B cells during their chronic exposures to Cr(VI) could explain the inability of this cell transformation model(79) to recapitulate a hallmark of Cr(VI) carcinogenesis in the lung, microsatellite instability resulting from inactive MMR.(58)
Formation of DSB by Cr-DNA adducts occurs in the G2 phase of the cell cycle,27,77,78 although their initial recognition by MMR proteins takes place in the preceding S phase and is dependent on ongoing replication.27,78 G2-specific DSB were observed for both soluble27,77 and particulate chromates.(80) A progression of G2 cells with incompletely repaired DNA breaks through the cell cycle could be responsible for the observed S-phase arrest and DSB during chronic exposures to Cr(VI).81,82 Cr-induced DSB were more persistent in human cells lacking WRN helicase,82,83 a protein mutated in the Werner syndrome of premature aging. WRN was specifically required for the repair of MMR-generated DSB through its involvement in the activation of the initial steps in homologous recombination, leading to the formation of RAD51 foci.(83) Cr(VI) treatments of WRN-deficient cells also led to higher levels of telomere damage,(84) although it remains unknown whether Cr-DNA adducts and aberrant MMR contribute to the observed telomeric abnormalities.
4. Carcinogenicity of Cr(VI) via Drinking Water
4.1. Epidemiological Studies
Occupational exposures to Cr(VI) via inhalation have consistently been found to increase the risk of cancers in the respiratory system. Both highly soluble and poorly soluble chromates were determined to be carcinogenic.1,2,85−87 Information about the carcinogenicity of ingested Cr(VI) is much more limited. A study by Zhang and Li(88) reported increased mortality from stomach cancers among rural residents in the Liaoning Province of China where drinking water was heavily contaminated with Cr(VI) released by the ore smelting facility. One recent reanalysis of this study confirmed the originally reported association between Cr(VI) contamination and cancer mortality,(89) while another study using a smaller control population did not.(90) A meta-analysis of studies among chromate workers did not find a link between inhalation exposures to Cr(VI) and cancers outside the respiratory system.(91) Neither the ecological report from China nor the meta-study of occupational exposures is strong in addressing the issue of Cr(VI) carcinogenicity in the digestive system. Inherent to all ecological studies is their inability to control for confounders and other risk factors, which is important for the analysis of stomach tumors in China with its well-known high incidence of this cancer. For human inhalation exposures, Cr(VI) ingestion was only assumed, and even if it occurred, ingested doses of Cr(VI) would be dramatically lower than doses in the respiratory tissues. Therefore, susceptible individuals would be expected to develop lethal lung cancers before the appearance of the late age-arising tumors in the GI system. Tobacco smoking is a risk factor for cancers in many tissues, and very high percentages of smokers in the industrial cohorts would further diminish the ability to detect any moderate carcinogenic effects of airborne Cr(VI) in the alimentary tract.
4.2. Animal Studies
The National Toxicology Program (NTP) has recently completed 2-year rodent studies of Cr(VI) carcinogenicity via drinking water exposure.(92) Both male and female F344/N rats showed significantly increased incidence of tumors in the oral cavity at the highest concentration of Cr(VI) (180 mg/L). B6C3F1 mice of both sexes developed tumors in the small intestine (duodenum and jejunum), with statistically significant increases at the two highest doses of Cr(VI). Plots of mouse tumor rates versus daily ingested doses of Cr(VI) showed a linear shape of dose dependence for males and supralinearity for females (Figure 4A). The duodenum of all exposed mice had elevated incidences of diffuse epithelial hyperplasia, which was graded from minimal to mild. No overt toxicity (necrosis) was found in the small intestine of any Cr(VI)-exposed groups of mice or rats.
Figure 4.

(A) Tumor rates in the small intestine of mice as a function of ingested daily dose of Cr(VI). Data are from the NTP bioassay.(92) (B) Expected shape of tumor dose-dependence for a carcinogenic mechanism based on cytotoxicity-induced regenerative proliferation.
Cr(VI) ingestion in the NTP study(92) has also led to some pro-inflammatory responses detected by the presence of the infiltrating histiocytes. Significant increases in histiocytic cell infiltrations were found in the upper portion of the small intestine (duodenum) and mesenteric lymph nodes in both sexes of mice and rats and in the liver of male and female rats and female mice. The pancreatic lymph nodes of female rats and male and female mice also had elevated amounts of histiocytic infiltrates. It has been recently hypothesized that inflammation and the resulting oxidative DNA damage could be important factors in the carcinogenic effects of ingested Cr(VI).93,94 However, the presence of histiocytic infiltrations at several sites in rats was not associated with Cr(VI) carcinogenesis in this species. In mice, the overlap between the appearance of histiocytic cells and tumorigenesis was found only in the small intestine but not in the liver or lymph nodes. The biological significance of histiocytic infiltrations and their ability to cause oxidative DNA damage in the host tissue cells are currently unknown.
A recent study utilizing 5 and 20 mg/L Cr(VI), concentrations that were below carcinogenic doses in the NTP rodent assay, did not find any increases in two forms of DNA damage in the duodenum of female SKH-1 mice after 9-month long exposure through drinking water.(95) These results were viewed as supportive of the threshold mechanism, questioning the human relevance of the observed carcinogenic effects in the mouse small intestine. However, the employed biomarkers were insensitive for the detection of DNA damage in the mouse duodenum. After a massive ex vivo exposure to 1.6 mM Cr(VI) (83.2 mg/L), only 2.5- and 3.8-fold increases were measured for protein–DNA cross-links and 8-oxo-dG, respectfully. Such a low responsiveness was clearly insufficient to detect DNA damage for exposures with 4.2- and 16.6-times lower Cr(VI) levels in the 20 mg/L and 5 mg/L test groups, even in the implausible situation of no reduction and no dilution of Cr(VI) in the stomach. The use of 8-oxo-dG as a biomarker of DNA damage has one severe limitation: its short lifetime. Repair of 50% 8-oxo-dG occurs within 30 min and is complete by 2 h.(96) This short lifetime would make it very difficult to detect 8-oxo-dG even after recently ingested water with a sufficiently high dose producing strong responses ex vivo.
4.3. Is Cytotoxicity-Driven Proliferation Involved in Mouse Intestinal Carcinogenesis?
The presence of diffuse epithelial hyperplasia in the duodenum of mice could raise the question of whether tumorigenesis in this tissue was a high-dose effect resulting from Cr(VI) cytotoxicity and subsequent compensatory proliferation. Although the NTP study has not found evidence of necrosis in the small intestine of Cr(VI)-exposed mice,(92) it is possible that the diffuse hyperplasia was a result of regenerative responses. Persistent hyperproliferation could lead to increased accumulation of spontaneous mutations and eventually cancer. This carcinogenic process should exhibit a steep sublinear, threshold-type dose dependence (Figure 4B), as it relies on the induction of cell death, and small nontoxic doses would be unable to initiate tumorigenesis.
The presence of Cr(VI)-induced hyperplasia could also be viewed as a manifestation of cancer-protective responses by the small intestine. Elimination of genetically damaged cells by apoptosis or another form of cell death is a firmly established protective mechanism against cancer. Thus, there are two opposing interpretations for the carcinogenic significance of the diffuse epithelial hyperplasia: one is pro-tumorigenic; the other is antitumorigenic. The supralinear shapes of dose–tumor responses in the NTP studies for female mice and combined male plus female results (Figure 4A) are more consistent with the activation of cancer-protective mechanisms. Tumor rates versus dose in male mice visually displayed a linear dose-dependence. Thus, experimental results do not support the engagement of cytotoxicity-based carcinogenic mechanisms with their expected dose–response sublinearity.
4.4. Cocarcinogenicity of Cr(VI)
The presence of Cr(VI) in drinking water has been found to increase the frequency of skin tumors in UV-irradiated hairless mice.97,98 Ingestion of Cr(VI) alone produced no skin tumors, indicating that synergistic effects resulted from the enhancement of UV-initiated tumorigenesis. Cocarcinogenic activity of Cr(VI) did not involve oxidative mechanisms, as supplementation with antioxidants vitamin E or selenomethionine showed no effect on its potentiating ability.(98) These two antioxidants effectively suppressed the ability of As(III) to enhance UV skin carcinogenesis in hairless mice.(99) A possible target for synergism between Cr(VI) and UV could be the repair of DNA photolesions by NER. Since abundant Cr-DNA adducts are good NER substrates,(59) they would compete with UV-DNA damage for NER proteins and consequently increase the persistence of premutagenic pyrimidine dimers in UV-irradiated keratinocytes. The biological plausibility of Cr–UV cocarcinogenesis through the competition for NER factors is supported by the comutagenicity of Cr(VI) and UV.(100)
4.5. Critical Factors for Low-Dose Extrapolation
A single ecological study linking the presence of high Cr(VI) concentrations in drinking water to elevated mortality from stomach cancers did not include a dose-dependence analysis,(88) making it unsuitable for risk assessment. Therefore, calculations of cancer risks from Cr(VI) ingestion must rely on the extrapolation of results from high-dose animal studies to low-dose human exposures. This situation is very common, as potential environmental exposures to carcinogens are always much lower than doses in positive epidemiological or animal studies. The use of high carcinogen doses in the NTP bioassay results from the need to obtain statistically robust tumor responses. As an illustration of this problem, the standard size of 50 animals/group was insufficient to reach statistical significance for a 4-fold increase in the tumor rate for female mice receiving 1.2 mg/kg daily Cr(VI) dose.(92)
A linear extrapolation of cancer risk to low environmental doses is a default regulatory approach for carcinogens with a mutagenic mode of action via DNA-reactive mechanisms.(101) A second critical consideration for linear extrapolations is whether environmental levels are sufficient to deliver a parental carcinogen or its reactive metabolite to a target organ. In the case of Cr(VI), the question of environmental relevance of the high-dose animal results is centered on the completeness of Cr(VI) reductive inactivation in the human stomach prior to reaching the small intestine. The next two sections will review evidence for Cr(VI) mutagenicity/genotoxicity and incompleteness of gastric reduction at low doses.
5. Mutagenic Mechanism in Cr(VI) Carcinogenesis
5.1. Mutagenicity of Cr(VI)
Cr(VI) compounds have been examined for mutagenicity in a large number of genotoxicity assays, from bacteria to laboratory rodents and human cells. Consistently positive results have been found in all standard mutagenicity tests,(102) including a reverse mutation assay in Salmonella,1,71,103HPRT mutagenesis in hamster V79 and CHO cells,(104)TK mutagenesis in human lymphoma cells,(105) and in vivo assays in transgenic animals.72,106 Cr(VI) is also known to induce clastogenic micronuclei in primary human cells,(27) chromosomal aberrations in various cell lines, and micronuclei in vivo.(102) Mutagenic responses in standard cultures of V79 and CHO cells were relatively moderate.(104) However, the mutagenicity of Cr(VI) in CHO and V79 cells was dramatically increased when their Asc levels were restored to physiological levels.(27) Slowly dividing kidney and lung cells of lacI transgenic mice(72) showed mutagenic responses to Cr(VI) that were comparable or even higher than those in rapidly dividing Asc-deficient rodent cells,(104) further pointing to the limitations of standard cell cultures to accurately recapitulate genotoxic effects. A high frequency of deletions in plasmids treated with Cr(VI) and Asc in vitro(32) and a poor recovery of deletion mutants at the HPRT gene in cultured cells and lacI gene in transgenic mice suggest that current in vivo models and Asc-restored cells could still underestimate the true levels of Cr(VI) mutagenesis. In vivo and in vitro mutagenicity tests are also incapable of detecting chromosomal translocations, which are expected to arise from a massive formation of Cr(VI)-induced DSB in Asc-normalized cells.27,78 Importantly, chromosomal translocations are a major mechanism for the activation of oncogenes in human cancers.(107)
Predictive values for rodent carcinogenicity are approximately 80% for the Ames test (reverse mutagenesis in Salmonella)(108) and even higher for mutation assays in transgenic rodents.(109) The micronucleus assay has lower predictive potential but it is complementary to Ames and in vivo mutagenesis due to its sensitivity in detecting deletion- and translocation-promoting DSB. A recent analysis of a large database of carcinogenicity and genotoxicity studies concluded that a combination of the Ames test and the in vitro micronucleus assay was able to predict all genotoxic rodent carcinogens.(110) Thus, the positive findings with Cr(VI) in Ames, rodent mutation, and clastogenic micronucleus assays discussed above strongly argue for the importance of a mutagenic (genotoxic) mechanism in Cr(VI) carcinogenicity. A general property of mutagenic chemicals is their multispecies, trans-sex, and multisite carcinogenesis.(101) Cr(VI) clearly fits this description of a mutagenic carcinogen as it is known to induce tumors in humans (unquestionably in the respiratory system), laboratory rodents, both sexes of mice and rats, and at different locations (the muscular system and different sites in the alimentary canal and respiratory tract).1,58,92
5.2. DNA-Reactive Mechanism in Cr(VI) Mutagenesis and Clastogenesis
Mutagenic responses can arise from direct chemical-DNA damage (DNA-reactive mechanism) or high dose-induced nucleotide misbalances, reactive oxygen species, and other metabolic alterations (indirect mutagenicity). Linear low-dose extrapolations in cancer risk assessment are only appropriate for DNA-reactive carcinogens.(101) Positive results in the Ames test are considered as evidence of direct mutagenicity, and they alone would represent a very difficult barrier to overcome during a regulatory process for drug approval.(111) The Ames test was consistently positive for Cr(VI)1,71,103 but negative for noncarcinogenic Cr(III) compounds.112,113 As discussed in section 3.2 and summarized in earlier reviews,16,114 Cr(VI) causes extensive formation of Cr-DNA adducts in both in vivo and in a variety of human and rodent cells in culture. Ternary Cr-DNA adducts were the principal mutagenic lesions generated during Cr(VI) metabolism by its three main reducers.32,37,38,69 Generation of DSB and chromosomal breaks by Cr(VI) was also dependent on ternary Cr-DNA adducts that are mistakenly recognized by MMR proteins as DNA mismatches and are then processed into DNA breaks.77,78 Importantly, Cr-DNA adducts and MMR-dependent DSB were detected at environmentally relevant doses both at and below the current federal standard for Cr in drinking water.(27) Collectively, these results demonstrate that DNA-reactive processes involving the formation of Cr-DNA adducts are major causes of Cr(VI) mutagenesis and clastogenesis. As argued earlier,(16) DNA binding by Cr(III) complexes predicts a linear dose-dependence between cellular Cr(VI) and mutagenic Cr-DNA adducts. Any oxidative or other forms of Cr-DNA damage arising from reactions of Cr(IV) should also display linear yields, as this intermediate is formed in all metabolic reactions of Cr(VI) (Figure 3).
5.3. Genotoxic vs Epigenetic Mechanisms
Cancer is a genetic disease caused by a combination of mutations activating oncogenes and inactivating tumor suppressor genes. Recent whole genome sequencing studies have found that human tumors contain between several dozens and several hundreds of cancer-promoting mutations.115,116 Cancer can also be described as an epigenetic disease since some tumor suppressor genes are silenced epigenetically, and cancer cells display globally different gene expression profiles that require corresponding chromatin remodeling at numerous promoters. Mutations in only two cancer genes, the p53 tumor suppressor and the KRAS oncogene, were sufficient to alter the expression of several hundred genes.(117) Mutations and other genomic rearrangements of histone-modifying proteins can convert them into oncogenes with ensuing abnormal chromatin remodeling.118−120 TET1 has recently been found to participate in CpG demethylation,(121) and this gene forms a leukemia-inducing fusion product with the MLL oncogene.(122) The opposite process of epigenetic changes causing mutations can also take place. For example, biallelic silencing of the MMR gene MLH1 via promoter hypermethylation dramatically increases mutation rates and eventually leads to colon cancer.(123)
Approximately 80% of lung cancers among chromate workers exhibited microsatellite instability (a hallmark of inactive MMR), which was associated with the loss of expression of key MMR proteins.124,125 This MMR-null phenotype was suggested to result from the selection of resistant cells during chronic exposures to cytotoxic doses of Cr(VI), as MMR-deficient cells are resistant to killing by Cr(VI).(77) Epigenetic repression of several genes (MLH1, p16, APC, and MGMT) in human chromate-associated cancers125−127 may reflect the presence of mutations in proteins regulating epigenetic stability or a direct ability of Cr(VI) to induce stable transcriptional silencing. Induction of epigenetic effects by Cr(VI) has been demonstrated but only in Asc-deficient cells.128,129 Sun et al.(129) have found that Asc supplementation blocked the induction of epigenetic changes by chromate in human A549 cells. The importance of Asc in epigenetic regulation stems from its role as a cofactor for dioxygenases, a group of enzymes that include histone demethylases and the 5mC demethylating protein TET1. Depletion of cellular Asc during Cr(VI) metabolism is expected to impede the removal of repressive DNA (methylated CpG) and histone H3 (Lys-9 di- and trimethylation) marks with the ensuing negative consequences for gene expression.
Although DNA damage and epigenetic responses are commonly viewed as separate events, DNA repair can induce large-scale chromatin remodeling, particularly in the case of DSB processed by homologous recombination.(130) DSB have also been shown to generate a rapid transcriptional repression in the adjacent regions,(131) and it is possible that this repressive chromatin state can sometimes be retained even after the completion of DSB repair. Cr(VI) is a potent inducer of DSB even at low doses,27,78 which could lead to the formation of transcriptionally repressed sites.
6. Is Human Gastric Detoxification of Low Cr(VI) Doses Complete?
The carcinogenic risk of ingested Cr(VI) at environmentally relevant doses has been questioned(94) on the basis of the reported high Cr(VI)-reducing capacity of gastric juice.132,133 High detoxification potential of human stomach and other tissues is also a key element of the threshold model of Cr(VI) carcinogenesis.(134) This model would argue that despite a mutagenic mode of action, the complete detoxification of moderate Cr(VI) doses in the stomach makes it inappropriate to perform linear extrapolations of cancer risks from animal tumor responses detected at high doses that could have exceeded the capacity for gastric reduction.
The ability of gastric juices to reduce/detoxify Cr(VI) is generally accepted in the field; however, studies with human volunteers and kinetic considerations of reduction and stomach emptying time do not support the completeness of the detoxification process. Three factors influence the extent of Cr(VI) reduction in the stomach: its reduction capacity, reduction rate, and stomach emptying time. On the basis of the reported high reduction capacity of the stomach (>80 mg/day),(133) the rate of reduction by gastric juice under fasting conditions could exhibit pseudo first-order kinetics in a broad range of low to moderate Cr(VI) concentrations. A fundamental property of first-order reactions is independence of the reaction half-time on concentration. This means that the extent of gastric reduction should be the same for both very small and very large amounts of Cr(VI). In agreement with first-order kinetics, the initial rates of reduction by human gastric juice were found to be independent of Cr(VI) concentrations.94,132 Reduction of 0.1 mg/L Cr(VI) (current EPA standard for total chromium) by artificial gastric juice was the first-order reaction.(135) A similar bioavailability of Cr(VI) for small and large doses further supports the first-order reaction kinetics of gastric reduction. For example, human subjects excreted 2.1% of the ingested 20 ng of Cr(VI) in the 24-h urine(18) and 1.43% of the ingested 5 mg of Cr(VI). The latter value is my very conservative estimate calculated as 1/4th of the average 4-day excretion of 5.7% reported in the original study.(136) Thus, the bioavailability of 20 ng and 5 mg of Cr(VI) (250,000-fold range) appears to be comparable. The approximately 10-fold higher bioavailability of ingested Cr(VI) compared to that of Cr(VI) reduced with orange juice prior to ingestion(137) suggests that the bulk of absorbed Cr from Cr(VI) was likely a cell-permeable chromate.
A classic study by Donaldson and Barreras(18) also performed a very important experiment on the bioavailability of 20 ng of Cr(VI) that was directly delivered into the duodenum of human subjects. In this case, 10.6% of Cr was excreted in the urine versus 2.1% for ingestion. Since the duodenal delivery used 100% Cr(VI), then the amount of Cr(VI) reaching the small intestine in their oral route experiment can be derived from the urinary excretion of 2.1% divided by 0.106 = 19.8%. For the study with the ingestion of 5 mg of Cr(VI) by human volunteers,(136) the same type of calculations using the estimated 1.43% urinary excretion predict 13.5% nonreduced Cr(VI) reaching the duodenum.
A second approach for the estimation of the percentage of Cr(VI) escaping stomach detoxification can be based on the considerations of the competing processes of stomach emptying and gastric reduction. Incubation with human gastric juice for 30 min at room temperature left 29.6% nonreduced Cr(VI) based on the diminished uptake by everted intestinal rings and 29.2% from a chromatographic profile (as determined by Image J).(18) The mean value of two measurements gives a rate with t1/2 =17 min, which should be twice as fast at physiological temperature (t1/2 = 8.5 min) due to a typical increase in the reactions rates by a factor of 2 for each 10 °C increase. A review by Paustenbach et al.(138) reported t1/2 = 7 min for the Cr(VI) reduction rate by human gastric juice at physiological temperature. A combination of t1/2 = 15.2 min for human stomach emptying (mean value from three studies with water)139−141 and a more protective rate of t1/2 = 7 min predicts that 22.2% Cr(VI) will reach the duodenum. Overall, both bioavailability and gastric reduction rate-based estimations suggest that 10–20% Cr(VI) ingested with water escapes gastric inactivation and reaches the small intestine, which is a site for its systemic absorption and a target of carcinogenic effects in mice. These estimates do not apply to the consumption of water with food, which is expected to promote Cr(VI) reduction through increased stomach residency time and delivery of additional reducers. Vitamin C-rich products are particularly beneficial for the enhancement of gastric detoxification of Cr(VI).
7. Conclusions
The presence of Cr(VI) in drinking water results from both anthropogenic and natural sources. Limited epidemiological studies are insufficient to establish carcinogenic risks of Cr(VI) ingestion in humans. Consumption of Cr(VI) through drinking water produced clear carcinogenic effects in both sexes of mice and rats, and Cr(VI) has a firmly established potential to cause human respiratory cancers. Since Cr(VI) is taken up via ubiquitously expressed transporters and metabolized by ubiquitously present cellular reducers, cells of the human digestive system are also expected to form cancer-promoting Cr-DNA damage. Multispecies and multisite carcinogenicity of Cr(VI) along with its broad genotoxicity provide a strong basis for a classification of Cr(VI) exposures through drinking water as likely to be carcinogenic to humans. Diverse lines of evidence demonstrate the importance of a DNA-reactive mutagenic mechanism in Cr(VI) carcinogenicity, lending mechanistic support for a linear low-dose extrapolation of cancer risks in humans. The bioavailability results and kinetic considerations indicate incomplete gastric detoxification of Cr(VI) at environmental levels of exposure, predicting its uptake and genotoxic metabolism in the small intestine (Figure 5).
Figure 5.

Genotoxic risks of ingested Cr(VI). Dose-independence of gastric detoxification is predicted to be applicable for a broad range of environmental Cr(VI) concentrations. A linear yield of Cr-DNA adducts and chromosomal breaks at environmental Cr(VI) doses has been demonstrated experimentally (section 5.2). Linearity of cancer risk at low levels of DNA damage is a default regulatory assumption for mutagenic carcinogens.(101) At low doses of Cr(VI), Cr-DNA adducts are a principal cause of chromosomal breaks in Asc-restored human cells and mutagenic DNA damage during Cr(VI) metabolism by its three main reducers. Sublinearity of direct and ROS-mediated DNA damage by Cr(V) stems from its detectable formation only under conditions of limited (depleted) Asc concentrations.
Glossary
Abbreviations
- Asc
ascorbate
- DSB
double-strand breaks
- GSH
glutathione
- MMR
mismatch repair
- NTP
National Toxicology Program
- NER
nucleotide excision repair
- ROS
reactive oxygen species.
This work was supported by NIEHS grants R01 ES008786 and P42 ES013660.
Funding Statement
National Institutes of Health, United States
References
- International Agency for Research on Cancer (1990) Chromium, Nickel and Welding, IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 49, pp 49–256, World Health Organization, Lyon, France. [PMC free article] [PubMed] [Google Scholar]
- Agency for Toxic Substances and Disease Registry (2000) Toxicological Profile for Chromium, U.S. Department of Health and Human Services, Washington, DC. [PubMed] [Google Scholar]
- Occupational Safety and Health Administration (OSHA), Department of Labor (2006) Occupational exposure to hexavalent chromium. Final rule. Fed. Regist. 71, 10099–10385. [PubMed] [Google Scholar]
- Pellerin C.; Booker S. M. (2000) Reflections on hexavalent chromium: health hazards of an industrial heavyweight. Environ. Health Perspect. 108, A402–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A. H.; Fagliano J. A.; Savrin J. E.; Freeman N. C.; Lioy P. J. (1998) The association of chromium in household dust with urinary chromium in residences adjacent to chromate production waste sites. Environ. Health Perspect. 106, 833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oze C.; Bird D. K.; Fendorf S. (2007) Genesis of hexavalent chromium from natural sources in soil and groundwater. Proc. Natl. Acad. Sci. U.S.A. 104, 6544–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fandeur D.; Juillot F.; Morin G.; Olivi L.; Cognigni A.; Webb S. M.; Ambrosi J. P.; Fritsch E.; Guyot F.; Brown G. E. Jr. (2009) XANES evidence for oxidation of Cr(III) to Cr(VI) by Mn-oxides in a lateritic regolith developed on serpentinized ultramafic rocks of New Caledonia. Environ. Sci. Technol. 43, 7384–7390. [DOI] [PubMed] [Google Scholar]
- Feng X. H.; Zhai L. M.; Tan W. F.; Liu F.; He J. Z. (2007) Adsorption and redox reactions of heavy metals on synthesized Mn oxide minerals. Environ. Pollut. 147, 366–373. [DOI] [PubMed] [Google Scholar]
- Environmental Protection Agency (EPA) (1991) National primary drinking water regulations; final rule. Fed. Regist. 56, 3536–3537. [Google Scholar]
- Clifford D., and Chau D. M. (1988) The fate of chromium(III) in chlorinated water. EPA document 600/S2-87/100 (Jan 1988).
- Apte A. D.; Tare V.; Bose P. (2006) Extent of oxidation of Cr(III) to Cr(VI) under various conditions pertaining to natural environment. J. Hazard. Mater. 128, 164–174. [DOI] [PubMed] [Google Scholar]
- Pillay K.; von Blottnitz H.; Petersen J. (2003) Ageing of chromium(III)-bearing slag and its relation to the atmospheric oxidation of solid chromium(III)-oxide in the presence of calcium oxide. Chemosphere 52, 1771–1779. [DOI] [PubMed] [Google Scholar]
- Dai R.; Yu C.; Liu J.; Lan Y.; Deng B. (2010) Photo-oxidation of Cr(III)-citrate complexes forms harmful Cr(VI). Environ. Sci. Technol. 44, 6959–6964. [DOI] [PubMed] [Google Scholar]
- Saleh F. Y.; Mbamalu G. E.; Jaradat Q. H.; Brungardt C. E. (1996) Ion chromatography-photodiode array UV-visible detection of Cr(III) hydrolytic polymerization products in pure and natural waters. Anal. Chem. 68, 740–745. [DOI] [PubMed] [Google Scholar]
- Stewart I. I.; Olesik J. W. (2000) Investigation of Cr(III) hydrolytic polymerisation products by capillary electrophoresis-inductively coupled plasma-mass spectrometry. J. Chromatogr., A 872, 227–246. [DOI] [PubMed] [Google Scholar]
- Zhitkovich A. (2005) Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI). Chem. Res. Toxicol. 18, 3–11. [DOI] [PubMed] [Google Scholar]
- Stout M. D.; Nyska A.; Collins B. J.; Witt K. L.; Kissling G. E.; Malarkey D. E.; Hooth M. J. (2009) Chronic toxicity and carcinogenicity studies of chromium picolinate monohydrate administered in feed to F344/N rats and B6C3F1 mice for 2 years. Food Chem. Toxicol. 47, 729–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson R. M. Jr.; Barreras R. F. (1966) Intestinal absorption of trace quantities of chromium. J. Lab. Clin. Med. 68, 484–493. [PubMed] [Google Scholar]
- Finley B. L.; Scott P. K.; Norton R. L.; Gargas M. L.; Paustenbach D. J. (1996) Urinary chromium concentrations in humans following ingestion of safe doses of hexavalent and trivalent chromium: implications for biomonitoring. J. Toxicol. Environ. Health 48, 479–499. [DOI] [PubMed] [Google Scholar]
- DeSesso J. M.; Jacobson C. F. (2001) Anatomical and physiological parameters affecting gastrointestinal absorption in humans and rats. Food Chem. Toxicol. 39, 209–228. [DOI] [PubMed] [Google Scholar]
- Zhitkovich A.; Messer J.; Shrager S. (2000) Reductive metabolism of Cr(VI) by cysteine leads to the formation of binary and ternary Cr-DNA adducts in the absence of oxidative DNA damage. Chem. Res. Toxicol. 13, 1114–1124. [DOI] [PubMed] [Google Scholar]
- Theopold K. H. (1997) Chromium: Inorganic and Coordination Chemistry, in Encyclopedia of Inorganic Chemistry (King B. R., Ed.) Vol. 2, pp 666–678, John Wiley & Sons, New York. [Google Scholar]
- Branco R.; Chung A. P.; Johnston T.; Gurel V.; Morais P.; Zhitkovich A. (2008) Chromate-inducible chrBACF operon from transposable element TnOtChr confers resistance to chromium(VI) and superoxide. J. Bacteriol. 190, 6996–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega R.; Fayard B.; Salome M.; Deves G.; Susini J. (2005) Chromium oxidation state imaging in mammalian cells exposed in vitro to soluble or particulate chromate compounds. Chem. Res. Toxicol. 18, 1512–1519. [DOI] [PubMed] [Google Scholar]
- Levina A.; Harris H. H.; Lay P. A. (2007) X-ray absorption and EPR spectroscopic studies of the biotransformations of chromium(VI) in mammalian cells. Is chromodulin an artifact of isolation methods?. J. Am. Chem. Soc. 129, 1065–1075. [DOI] [PubMed] [Google Scholar]
- Messer J.; Reynolds M.; Stoddard L.; Zhitkovich A. (2006) Causes of DNA single-strand breaks during reduction of chromate by glutathione in vitro and in cells. Free Radical Biol. Med. 40, 1981–1992. [DOI] [PubMed] [Google Scholar]
- Reynolds M.; Stoddard L.; Bespalov I.; Zhitkovich A. (2007) Ascorbate acts as a highly potent inducer of chromate mutagenesis and clastogenesis: linkage to DNA breaks in G2 phase by mismatch repair. Nucleic Acids Res. 35, 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray K. J.; Tebo B. M. (2007) Cr(III) is indirectly oxidized by the Mn(II)-oxidizing bacterium Bacillus sp. strain SG-1. Environ. Sci. Technol. 41, 528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y.; Fukuda K. (1990) Reduction of hexavalent chromium by ascorbic acid and glutathione with special reference to the rat lung. Arch. Toxicol. 64, 169–176. [DOI] [PubMed] [Google Scholar]
- Standeven A. M.; Wetterhahn K. E. (1991) Ascorbate is the principal reductant of chromium (VI) in rat liver and kidney ultrafiltrates. Carcinogenesis 12, 1733–1737. [DOI] [PubMed] [Google Scholar]
- Standeven A. M.; Wetterhahn K. E. (1992) Ascorbate is the principal reductant of chromium(VI) in rat lung ultrafiltrates and cytosols, and mediates chromium-DNA binding in vitro. Carcinogenesis 13, 1319–1324. [DOI] [PubMed] [Google Scholar]
- Quievryn G.; Peterson E.; Messer J.; Zhitkovich A. (2003) Genotoxicity and mutagenicity of chromium(VI)/ascorbate-generated DNA adducts in human and bacterial cells. Biochemistry 42, 1062–1070. [DOI] [PubMed] [Google Scholar]
- Borges K. M.; Boswell J. S.; Liebross R. H.; Wetterhahn K. E. (1991) Activation of chromium(VI) by thiols results in chromium(V) formation, chromium binding to DNA and altered DNA conformation. Carcinogenesis 12, 551–561. [DOI] [PubMed] [Google Scholar]
- Bose R. N.; Moghaddas S.; Gelerinter E. (1992) Long-lived chromium(IV), chromium(V) metabolites in the chromium(VI)-glutathione reaction: NMR, ESR, HPLC, and kinetic characterization. Inorg. Chem. 31, 1987–1994. [Google Scholar]
- Stearns D. M.; Wetterhahn K. E. (1994) Reaction of Cr(VI) with ascorbate produces chromium(V), chromium(IV), and carbon-based radicals. Chem. Res. Toxicol. 7, 219–230. [DOI] [PubMed] [Google Scholar]
- Lay P. A.; Levina A. (1998) Activation of molecular oxygen during the reactions of chromium(VI/V/IV) with biological reductants: implications for chromium-induced genotoxicities. J. Am. Chem. Soc. 120, 6704–6714. [Google Scholar]
- Zhitkovich A.; Song Y.; Quievryn G.; Voitkun V. (2001) Nonoxidative mechanisms are responsible for the induction of mutagenesis by reduction of Cr(VI) with cysteine: role of ternary DNA adducts in Cr(III)-dependent mutagenesis. Biochemistry 40, 549–560. [DOI] [PubMed] [Google Scholar]
- Guttmann D.; Poage G.; Johnston T.; Zhitkovich A. (2008) Reduction with glutathione is a weakly mutagenic pathway in chromium(VI) metabolism. Chem. Res. Toxicol. 21, 2188–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X. L.; Dalal N. S. (1990) ESR spin trapping detection of hydroxyl radicals in the reactions of Cr(V) complexes with hydrogen peroxide. Free Radical Res. Commun. 10, 17–26. [DOI] [PubMed] [Google Scholar]
- Luo H.; Lu Y.; Mao Y.; Shi X.; Dalal N. S. (1996) Role of chromium(IV) in the chromium(VI)-related free radical formation, dG hydroxylation, and DNA damage. J. Inorg. Biochem. 64, 25–35. [DOI] [PubMed] [Google Scholar]
- Sugden K. D.; Campo C. K.; Martin B. D. (2001) Direct oxidation of guanine and 7,8-dihydro-8-oxoguanine in DNA by a high-valent chromium complex: a possible mechanism for chromate genotoxicity. Chem. Res. Toxicol. 14, 1315–1322. [DOI] [PubMed] [Google Scholar]
- Slade P. G.; Priestley N. D.; Sugden K. D. (2007) Spiroiminodihydantoin as an oxo-atom transfer product of 8-oxo-2′-deoxyguanosine oxidation by chromium(V). Org. Lett. 9, 4411–4414. [DOI] [PubMed] [Google Scholar]
- Sugden K. D.; Stearns D. M. (2000) The role of chromium(V) in the mechanism of chromate-induced oxidative DNA damage and cancer. J. Environ. Pathol. Toxicol. Oncol. 19, 215–230. [PubMed] [Google Scholar]
- Levina A.; Lay P. A. (2005) Mechanistic studies of relevance to the biological activities of chromium. Coord. Chem. Rev. 42, 281–298. [Google Scholar]
- Quievryn G.; Goulart M.; Messer J.; Zhitkovich A. (2001) Reduction of Cr(VI) by cysteine: significance in human lymphocytes and formation of DNA damage in reactions with variable reduction rates. Mol. Cell. Biochem. 222, 107–118. [PubMed] [Google Scholar]
- Borges K. M.; Boswell J. S.; Liebross R. H.; Wetterhahn K. E. (1991) Activation of chromium(VI) by thiols results in chromium(V) formation, chromium binding to DNA and altered DNA conformation. Carcinogenesis 12, 551–561. [DOI] [PubMed] [Google Scholar]
- O’Brien P.; Wang G.; Wyatt P. B. (1992) Studies on the kinetics of the reduction of chromate by glutathione and related thiols. Polyhedron 11, 3211–3216. [Google Scholar]
- Levina A.; Zhang L.; Lay P. A. (2003) Structure and reactivity of a chromium(V) glutathione complex. Inorg. Chem. 42, 767–784. [DOI] [PubMed] [Google Scholar]
- Lay P. A.; Levina A. (1998) Activation of molecular oxygen during the reactions of chromium(VI/V/IV) with biological reductants: implications for chromium-induced genotoxicities. J. Am. Chem. Soc. 120, 6704–6714. [Google Scholar]
- Goodgame D. M. L.; Joy M. A. (1987) EPR study of the Cr(V) and radical species produced in the reduction of Cr(VI) by ascorbate. Inorg. Chem. Acta 135, 115–118. [Google Scholar]
- Quievryn G.; Messer J.; Zhitkovich A. (2002) Carcinogenic chromium(VI) induces cross-linking of vitamin C to DNA in vitro and in human lung A549 cells. Biochemistry 41, 3156–3167. [DOI] [PubMed] [Google Scholar]
- Reynolds M.; Zhitkovich A. (2007) Cellular vitamin C increases chromate toxicity via a death program requiring mismatch repair but not p53. Carcinogenesis 28, 1613–1620. [DOI] [PubMed] [Google Scholar]
- Salnikow K.; Donald S. P.; Bruick R. K.; Zhitkovich A.; Phang J. M.; Kasprzak K. S. (2004) Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J. Biol. Chem. 279, 40337–40344. [DOI] [PubMed] [Google Scholar]
- Karaczyn A.; Ivanov S.; Reynolds M.; Zhitkovich A.; Kasprzak K. S.; Salnikow K. (2006) Ascorbate depletion mediates up-regulation of hypoxia-associated proteins by cell density and nickel. J. Cell. Biochem. 97, 1025–1035. [DOI] [PubMed] [Google Scholar]
- Bergsten P.; Amitai G.; Kehrl J.; Dhariwal K. R.; Klein H. G.; Levine M. (1990) Millimolar concentrations of ascorbic acid in purified human mononuclear leukocytes. Depletion and reaccumulation. J. Biol. Chem. 265, 2584–2587. [PubMed] [Google Scholar]
- Slade R.; Stead A. G.; Graham J. A.; Hatch G. E. (1985) Comparison of lung antioxidant levels in humans and laboratory animals. Am. Rev. Respir. Dis. 131, 742–746. [DOI] [PubMed] [Google Scholar]
- Kojo S. (2004) Vitamin C: basic metabolism and its function as an index of oxidative stress. Curr. Med. Chem. 11, 1041–1064. [DOI] [PubMed] [Google Scholar]
- Salnikow K.; Zhitkovich A. (2008) Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic and chromium. Chem. Res. Toxicol. 21, 28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds M.; Peterson E.; Quievryn G.; Zhitkovich A. (2004) Human nucleotide excision repair efficiently removes chromium-DNA phosphate adducts and protects cells against chromate toxicity. J. Biol. Chem. 279, 30419–30424. [DOI] [PubMed] [Google Scholar]
- Zhitkovich A.; Voitkun V.; Costa M. (1995) Glutathione and free amino acids form stable adducts with DNA following exposure of intact mammalian cells to chromate. Carcinogenesis 16, 907–913. [DOI] [PubMed] [Google Scholar]
- O’Brien T.; Xu J.; Patierno S. R. (2001) Effect of glutathione on chromium-induced DNA crosslinking and DNA polymerase arrest. Mol. Cell. Biochem. 222, 173–182. [PubMed] [Google Scholar]
- Zhitkovich A.; Voitkun V.; Costa M. (1996) Formation of the amino acid-DNA complexes by hexavalent and trivalent chromium in vitro: importance of trivalent chromium and the phosphate group. Biochemistry 35, 7275–7282. [DOI] [PubMed] [Google Scholar]
- Costa M. (1991) DNA-protein complexes induced by chromate and other carcinogens. Environ. Health Perspect. 92, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitkovich A.; Voitkun V.; Kluz T.; Costa M. (1998) Utilization of DNA-protein crosslinks as a biomarker of chromium exposure. Environ. Health Perspect. 106, 969–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnekenburger M.; Talaska G.; Puga A. (2007) Chromium cross-links histone deacetylase 1-DNA methyltransferase 1 complexes to chromatin, inhibiting histone-remodeling marks critical for transcriptional activation. Mol. Cell. Biol. 27, 7089–7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecevic A.; Hagan E.; Reynolds M.; Poage G.; Johnston T.; Zhitkovich A. (2010) XPA impacts formation but not proteasome-sensitive repair of DNA-protein crosslinks induced by chromate. Mutagenesis 25, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfie A.; Hagan E.; Zhitkovich A. (2010) Mechanism of DNA-protein cross-linking by chromium. Chem. Res. Toxicol. 23, 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voitkun V.; Zhitkovich A.; Costa M. (1998) Cr(III)-mediated crosslinks of glutathione or amino acids to the DNA phosphate backbone are mutagenic in human cells. Nucleic Acids Res. 26, 2024–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quievryn G.; Messer J.; Zhitkovich A. (2006) Lower mutagenicity but higher stability of Cr-DNA adducts formed during gradual chromate activation with ascorbate. Carcinogenesis 27, 2316–2321. [DOI] [PubMed] [Google Scholar]
- O’Brien T.; Mandel H. G.; Pritchard D. E.; Patierno S. R. (2002) Critical role of chromium (Cr)-DNA interactions in the formation of Cr-induced polymerase arresting lesions. Biochemistry 41, 12529–12537. [DOI] [PubMed] [Google Scholar]
- Watanabe K.; Sakamoto K.; Sasaki T. (1998) Comparisons on chemically-induced mutation among four bacterial strains, Salmonella typhimurium TA102 and TA2638, and Escherichia coli WP2/pKM101 and WP2 uvrA/pKM101: collaborative study II. Mutat. Res. 412, 17–31. [DOI] [PubMed] [Google Scholar]
- Cheng L.; Sonntag D. M.; de Boer J.; Dixon K. (2000) Chromium(VI)-induced mutagenesis in the lungs of big blue transgenic mice. J. Environ. Pathol. Toxicol. Oncol. 19, 239–249. [PubMed] [Google Scholar]
- Brooks B.; O’Brien T. J.; Ceryak S.; Wise J. P. Sr.; Wise S. S.; Wise J. P. Jr.; Defabo E.; Patierno S. R. (2008) Excision repair is required for genotoxin-induced mutagenesis in mammalian cells. Carcinogenesis 29, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien T. J.; Brooks B. R.; Patierno S. R. (2005) Nucleotide excision repair functions in the removal of chromium-induced DNA damage in mammalian cells. Mol. Cell. Biochem. 279, 85–95. [DOI] [PubMed] [Google Scholar]
- Blankert S. A.; Coryell V. H.; Picard B. T.; Wolf K. K.; Lomas R. E.; Stearns D. M. (2003) Characterization of nonmutagenic Cr(III)-DNA interactions. Chem. Res. Toxicol. 16, 847–854. [DOI] [PubMed] [Google Scholar]
- Arakawa H.; Wu F.; Costa M.; Rom W.; Tang M. S. (2006) Sequence specificity of Cr(III)-DNA adduct formation in the p53 gene: NGG sequences are preferential adduct-forming sites. Carcinogenesis 27, 639–645. [DOI] [PubMed] [Google Scholar]
- Peterson-Roth E.; Reynolds M.; Quievryn G.; Zhitkovich A. (2005) Mismatch repair proteins are activators of toxic responses to chromium-DNA damage. Mol. Cell. Biol. 25, 3596–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds M. F.; Peterson-Roth E. C.; Johnston T.; Gurel V. M.; Menard H. L.; Zhitkovich A. (2009) Rapid DNA double-strand breaks resulting from processing of Cr-DNA crosslinks by both MutS dimers. Cancer Res. 69, 1071–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues C. F.; Urbano A. M.; Matoso E.; Carreira I.; Almeida A.; Santos P.; Botelho F.; Carvalho L.; Alves M.; Monteiro C.; Costa A. N.; Moreno V.; Alpoim M. C. (2009) Human bronchial epithelial cells malignantly transformed by hexavalent chromium exhibit an aneuploid phenotype but no microsatellite instability. Mutat. Res. 670, 42–52. [DOI] [PubMed] [Google Scholar]
- Xie H.; Holmes A. L.; Young J. L.; Qin Q.; Joyce K.; Pelsue S. C.; Peng C.; Wise S. S.; Jeevarajan A. S.; Wallace W. T.; Hammond D.; Wise J. P. Sr. (2009) Zinc chromate induces chromosome instability and DNA double strand breaks in human lung cells. Toxicol. Appl. Pharmacol. 234, 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha L.; Ceryak S.; Patierno S. R. (2004) Generation of S phase-dependent DNA double-strand breaks by Cr(VI) exposure: involvement of ATM in Cr(VI) induction of gamma-H2AX. Carcinogenesis 25, 2265–2274. [DOI] [PubMed] [Google Scholar]
- Liu F. J.; Barchowsky A.; Opresko P. L. (2009) The Werner syndrome protein functions in repair of Cr(VI)-induced replication-associated DNA damage. Toxicol. Sci. 110, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecevic A.; Menard H.; Gurel V.; Hagan E.; DeCaro R.; Zhitkovich A. (2009) WRN helicase promotes repair of DNA double-strand breaks caused by aberrant mismatch repair of chromium-DNA adducts. Cell Cycle 8, 2769–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F. J.; Barchowsky A.; Opresko P. L. (2010) The Werner syndrome protein suppresses telomeric instability caused by chromium (VI) induced DNA replication stress. PLoS One 5, e11152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso T. F. (1997) Chromium as an industrial carcinogen: Part I. Am. J. Ind. Med. 31, 1291–1239. [DOI] [PubMed] [Google Scholar]
- Sorahan T.; Burges D. C.; Hamilton L.; Harrington J. M. (1998) Lung cancer mortality in nickel/chromium platers, 1946–95. Occup. Environ. Med. 55, 236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb H. J.; Lees P. S.; Pinsky P. F.; Rooneym B. C. (2000) Lung cancer among workers in chromium chemical production. Am. J. Ind. Med. 38, 115–126. [DOI] [PubMed] [Google Scholar]
- Zhang J. D.; Li X. L. (1987) Chromium pollution of soil and water in Jinzhou. Zhonghua Yu Fang Yi Xue Za Zhi 21, 262–264. [PubMed] [Google Scholar]
- Beaumont J. J.; Sedman R. M.; Reynolds S. D.; Sherman C. D.; Li L. H.; Howd R. A.; Sandy M. S.; Zeise L.; Alexeeff G. V. (2008) Cancer mortality in a Chinese population exposed to hexavalent chromium in drinking water. Epidemiology 19, 12–23. [DOI] [PubMed] [Google Scholar]
- Kerger B. D.; Butler W. J.; Paustenbach D. J.; Zhang J.; Li S. (2009) Cancer mortality in Chinese populations surrounding an alloy plant with chromium smelting operations. J. Toxicol. Environ. Health A 72, 329–344. [DOI] [PubMed] [Google Scholar]
- Gatto N. M.; Kelsh M. A.; Mai D. H.; Suh M.; Proctor D. M. (2010) Occupational exposure to hexavalent chromium and cancers of the gastrointestinal tract: a meta-analysis. Cancer Epidemiol. 34, 388–399. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program (2008) NTP Toxicology and Carcinogenesis Studies of Sodium Dichromate Dihydrate (CAS No. 7789-12-0) in F344/N Rats and B6C3F1Mice (Drinking Water Studies). Natl. Toxicol. Program Tech. Rep. Ser. 546, 1–192. [PubMed] [Google Scholar]
- Nickens K. P.; Patierno S. R.; Ceryak S. (2010) Chromium genotoxicity: A double-edged sword. Chem.-Biol. Interact. 188, 276–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C. M.; Haws L. C.; Harris M. A.; Gatto N. M.; Proctor D. M. (2011) Application of the U.S. EPA mode of action Framework for purposes of guiding future research: a case study involving the oral carcinogenicity of hexavalent chromium. Toxicol. Sci. 119, 20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Flora S.; D’Agostini F.; Balansky R.; Micale R.; Baluce B.; Izzotti A. (2008) Lack of genotoxic effects in hematopoietic and gastrointestinal cells of mice receiving chromium(VI) with the drinking water. Mutat. Res. 659, 60–67. [DOI] [PubMed] [Google Scholar]
- Lan L.; Nakajima S.; Oohata Y.; Takao M.; Okano S.; Masutani M.; Wilson S. H.; Yasui A. (2004) In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 101, 13738–13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson T.; Kluz T.; Burns F.; Rossman T.; Zhang Q.; Uddin A.; Nadas A.; Costa M. (2004) Exposure to chromium (VI) in the drinking water increases susceptibility to UV-induced skin tumors in hairless mice. Toxicol. Appl. Pharmacol. 196, 431–437. [DOI] [PubMed] [Google Scholar]
- Uddin A. N.; Burns F. J.; Rossman T. G.; Chen H.; Kluz T.; Costa M. (2007) Dietary chromium and nickel enhance UV-carcinogenesis in skin of hairless mice. Toxicol. Appl. Pharmacol. 221, 329–338. [DOI] [PubMed] [Google Scholar]
- Uddin A. N.; Burns F. J.; Rossman T. G. (2005) Vitamin E and organoselenium prevent the cocarcinogenic activity of arsenite with solar UVR in mouse skin. Carcinogenesis 26, 2179–2186. [DOI] [PubMed] [Google Scholar]
- Hartwig A.; Beyersmann D. (1989) Comutagenicity and inhibition of DNA repair by metal ions in mammalian cells. Biol. Trace Elem. Res. 21, 359–365. [DOI] [PubMed] [Google Scholar]
- U.S. Environmental Protection Agency (2005) Guidelines for carcinogen risk assessment (http://www.epa.gov/cancerguidelines).
- McCarroll N.; Keshava N.; Chen J.; Akerman G.; Kligerman A.; Rinde E. (2010) An evaluation of the mode of action framework for mutagenic carcinogens case study II: chromium (VI). Environ. Mol. Mutagen. 51, 89–111. [DOI] [PubMed] [Google Scholar]
- Bucher J. (2007) NTP toxicity studies of sodium dichromate dihydrate (CAS No. 7789120) administered in drinking water to male and female F344/N rats and B6C3F1 mice and male BALB/c and am3-C57BL/6 mice. Toxicol. Rep. Ser. 72, 1–G4. [PubMed] [Google Scholar]
- Cohen M. D.; Kargacin B; Klein C. B.; Costa M. (1993) Mechanisms of chromium carcinogenicity and toxicity. Crit. Rev. Toxicol. 23, 255–281. [DOI] [PubMed] [Google Scholar]
- Chen J.; Thilly W. G. (1994) Mutational spectrum of chromium(VI) in human cells. Mutat. Res. 323, 21–27. [DOI] [PubMed] [Google Scholar]
- Itoh S.; Shimada H. (1998) Bone marrow and liver mutagenesis in lacZ transgenic mice treated with hexavalent chromium. Mutat. Res. 412, 63–67. [DOI] [PubMed] [Google Scholar]
- Rabbitts T. H. (2009) Commonality but diversity in cancer gene fusions. Cell 137, 391–395. [DOI] [PubMed] [Google Scholar]
- Zeiger E. (1998) Identification of rodent carcinogens and noncarcinogens using genetic toxicity tests: premises, promises, and performance. Regul. Toxicol. Pharmacol. 28, 85–95. [DOI] [PubMed] [Google Scholar]
- Lambert I. B.; Singer T. M.; Boucher S. E.; Douglas G. R. (2005) Detailed review of transgenic rodent mutation assays. Mutat. Res. 590, 1–280. [DOI] [PubMed] [Google Scholar]
- Kirkland D.; Reeve L.; Gatehouse D.; Vanparys P. (2011) A core in vitro genotoxicity battery comprising the Ames test plus the in vitro micronucleus test is sufficient to detect rodent carcinogens and in vivo genotoxins. Mutat. Res. 721, 27–73. [DOI] [PubMed] [Google Scholar]
- Dearfield K. L.; Thybaud V.; Cimino M. C.; Custer L.; Czich A.; Harvey J. S.; Hester S.; Kim J. H.; Kirkland D.; Levy D. D.; Lorge E.; Moore M. M.; Ouédraogo-Arras G.; Schuler M.; Suter W.; Sweder K.; Tarlo K.; van Benthem J.; van Goethem F.; Witt K. L. (2011) Follow-up actions from positive results of in vitro genetic toxicity testing. Environ. Mol. Mutagen. 52, 177–204. [DOI] [PubMed] [Google Scholar]
- Bianchi V.; Celotti L.; Lanfranchi G.; Majone F.; Marin G.; Montaldi A.; Sponza G.; Tamino G.; Venier P.; Zantedeschi A.; Levis A. G. (1983) Genetic effects of chromium compounds. Mutat. Res. 117, 279–300. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program (2010) NTP toxicology and carcinogenesis studies of chromium picolinate monohydrate (CAS No. 27882-76-4) in F344/N rats and B6C3F1 mice (feed studies). Natl. Toxicol. Program Tech. Rep. Ser. 556, 1–194. [PubMed] [Google Scholar]
- O’Brien T. J.; Ceryak S.; Patierno S. R. (2003) Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat. Res. 533, 3–36. [DOI] [PubMed] [Google Scholar]
- Jones S.; Zhang X.; Parsons D. W.; Lin J. C.; Leary R. J.; Angenendt P.; Mankoo P.; Carter H.; Kamiyama H; Jimeno A.; Hong S. M.; Fu B.; Lin M. T.; Calhoun E. S.; Kamiyama M.; Walter K.; Nikolskaya T.; Nikolsky Y.; Hartigan J.; Smith D. R.; Hidalgo M.; Leach S. D.; Klein A. P.; Jaffee E. M.; Goggins M.; Maitra A.; Iacobuzio-Donahue C.; Eshleman J. R.; Kern S. E.; Hruban R. H.; Karchin R.; Papadopoulos N.; Parmigiani G.; Vogelstein B.; Velculescu V. E.; Kinzler K. W. (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.; Jiang Z.; Liu J.; Haverty P. M.; Guan Y.; Stinson J.; Yue P.; Zhang Y.; Pant K. P.; Bhatt D.; Ha C.; Johnson S.; Kennemer M. I.; Mohan S.; Nazarenko I.; Watanabe C.; Sparks A. B.; Shames D. S.; Gentleman R.; de Sauvage F. J.; Stern H.; Pandita A.; Ballinger D. G.; Drmanac R.; Modrusan Z.; Seshagiri S.; Zhang Z. (2010) The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 465, 473–477. [DOI] [PubMed] [Google Scholar]
- McMurray H. R.; Sampson E. R.; Compitello G.; Kinsey C.; Newman L.; Smith B.; Chen S. R.; Klebanov L.; Salzman P.; Yakovlev A.; Land H. (2008) Synergistic response to oncogenic mutations defines gene class critical to cancer phenotype. Nature 453, 1112–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan C. G.; Zhang J.; Kasper L. H.; Lerach S.; Payne-Turner D.; Phillips L. A.; Heatley S. L.; Holmfeldt L.; Collins-Underwood J. R.; Ma J.; Buetow K. H.; Pui C. H.; Baker S. D.; Brindle P. K.; Downing J. R. (2011) CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L.; Dominguez-Sola D.; Chiarenza A.; Fabbri G.; Grunn A.; Trifonov V.; Kasper L. H.; Lerach S.; Tang H.; Ma J.; Rossi D.; Chadburn A.; Murty V. V.; Mullighan C. G.; Gaidano G.; Rabadan R.; Brindle P. K.; Dalla-Favera R. (2011) Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471, 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol C. J.; Houvras Y.; Jane-Valbuena J.; Bilodeau S.; Orlando D. A.; Battisti V.; Fritsch L.; Lin W. M.; Hollmann T. J.; Ferré F.; Bourque C.; Burke C. J.; Turner L.; Uong A.; Johnson L. A.; Beroukhim R.; Mermel C. H.; Loda M.; Ait-Si-Ali S.; Garraway L. A.; Young R. A.; Zon L. I. (2011) The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 471, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. U.; Su Y.; Zhong C.; Ming G. L.; Song H. (2011) Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M.; Koh K. P.; Shen Y.; Pastor W. A.; Bandukwala H.; Brudno Y.; Agarwal S.; Iyer L. M.; Liu D. R.; Aravind L.; Rao A. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchins M. P.; Ward R. L. (2009) Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J. Med. Genet. 46, 793–802. [DOI] [PubMed] [Google Scholar]
- Hirose T.; Kondo K.; Takahashi Y.; Ishikura H.; Fujino H.; Tsuyuguchi M.; Hashimoto M.; Yokose T.; Mukai K.; Kodama T.; Monden Y. (2002) Frequent microsatellite instability in lung cancer from chromate-exposed workers. Mol. Carcinog. 33, 172–180. [DOI] [PubMed] [Google Scholar]
- Takahashi Y.; Kondo K.; Hirose T.; Nakagawa H.; Tsuyuguchi M.; Hashimoto M.; Sano T.; Ochiai A.; Monden Y. (2005) Microsatellite instability and protein expression of the DNA mismatch repair gene, hMLH1, of lung cancer in chromate-exposed workers. Mol. Carcinog. 42, 150–158. [DOI] [PubMed] [Google Scholar]
- Kondo K.; Takahashi Y.; Hirose Y.; Nagao T.; Tsuyuguchi M.; Hashimoto M.; Ochiai A.; Monden Y.; Tangoku A. (2006) The reduced expression and aberrant methylation of p16(INK4a) in chromate workers with lung cancer. Lung Cancer 53, 295–302. [DOI] [PubMed] [Google Scholar]
- Ali A. H.; Kondo K.; Namura T.; Senba Y.; Takizawa H.; Nakagawa Y.; Toba H.; Kenzaki K.; Sakiyama S.; Tangoku A. (2011) Aberrant DNA methylation of some tumor suppressor genes in lung cancers from workers with chromate exposure. Mol. Carcinog. 50, 89–99. [DOI] [PubMed] [Google Scholar]
- Klein C. B.; Su L.; Bowser D.; Leszczynska J. (2002) Chromate-induced epimutations in mammalian cells. Environ. Health. Perspect. 110, 739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Zhou X.; Chen H.; Li Q.; Costa M. (2009) Modulation of histone methylation and MLH1 gene silencing by hexavalent chromium. Toxicol. Appl. Pharmacol. 237, 258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha M.; Peterson C. L. (2009) Chromatin dynamics during repair of chromosomal DNA double-strand breaks. Epigenomics 1, 371–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag N. M.; Rafalska-Metcalf I. U.; Balane-Bolivar C.; Janicki S. M.; Greenberg R. A. (2010) ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Flora S; Badolati G. S.; Serra D; Picciotto A; Magnolia M. R.; Savarino V. (1987) Circadian reduction of chromium in the gastric environment. Mutat. Res. 192, 169–174. [DOI] [PubMed] [Google Scholar]
- De Flora S; Camoirano A; Bagnasco M; Bennicelli C; Corbett G. E.; Kerger B. D. (1997) Estimates of the chromium(VI) reducing capacity in human body compartments as a mechanism for attenuating its potential toxicity and carcinogenicity. Carcinogenesis 18, 531–537. [DOI] [PubMed] [Google Scholar]
- De Flora S. (2000) Threshold mechanisms and site specificity in chromium(VI) carcinogenesis. Carcinogenesis 21, 533–541. [DOI] [PubMed] [Google Scholar]
- Gammelgaard B.; Jensen K.; Steffansen B. (1999) In vitro metabolism and permeation studies in rat jejunum: organic chromium compared to inorganic chromium. J. Trace Elem. Med. Biol. 13, 82–88. [DOI] [PubMed] [Google Scholar]
- Kerger B. D.; Finley B. L.; Corbett G. E.; Dodge D. G.; Paustenbach D. J. (1997) Ingestion of chromium(VI) in drinking water by human volunteers: absorption, distribution, and excretion of single and repeated doses. J. Toxicol. Environ. Health 50, 67–95. [DOI] [PubMed] [Google Scholar]
- Kuykendall J. R.; Kerger B. D.; Jarvi E. J.; Corbett G. E.; Paustenbach D. J. (1996) Measurement of DNA-protein cross-links in human leukocytes following acute ingestion of chromium in drinking water. Carcinogenesis 17, 1971–1977. [DOI] [PubMed] [Google Scholar]
- Paustenbach D. J.; Finley B. L.; Mowat F. S.; Kerger B. D. (2003) Human health risk and exposure assessment of chromium (VI) in tap water. J. Toxicol. Environ. Health A 66, 1295–1339. [DOI] [PubMed] [Google Scholar]
- Chang F. Y.; Lu C. I.; Chen C. Y.; Lee S. D.; Tsai D. S.; Fu S. E. (2001) The pharmacological effect of omeprazole on water gastric emptying: A study based on an impedance measure. Pharmacology 63, 50–57. [DOI] [PubMed] [Google Scholar]
- Chang F. Y.; Chen C. Y.; Lu C. L.; Luo J. C.; Jiun K. L.; Lee S. D.; Wu C. W. (2004) Undisturbed water gastric emptying in patients of stomach cancer. Hepatogastroenterology 51, 1219–1224. [PubMed] [Google Scholar]
- Umenai T; Arai N; Chihara E. (2009) Effect of the preliminary hydration on gastric emptying time for water in healthy volunteers. Acta Anaesthesiol. Scand. 53, 223–226. [DOI] [PubMed] [Google Scholar]