

Abstract
Oral squamous cell carcinomas (OSCCs) are considered to arise from human oral keratinocytes. DNAs of human papillomaviruses (HPVs), predominantly types 16 and 18, etiological agents of cervical cancer, have been detected in approximately 25% of OSCCs. In accordance with the established role of E6 and E7 in inactivating p53 and pRB, respectively, mutations of p53 and inactivation of p16INK4a are frequently observed in HPV-negative OSCCs. In addition, other alterations such as overexpression of epidermal growth factor receptor (EGFR) are often observed in both HPV-positive and -negative OSCCs. However, causal-relationships between accumulation of these abnormalities and multi-step carcinogenesis are not fully understood. To elucidate underlying processes, we transduced either HPV16 E6/E7 or mutant CDK4 (CDK4R24C), cyclin D1 and human telomerase reverse transcriptase (TERT) into primary human tongue keratinocytes (HTK), and obtained immortal cell populations, HTK-16E6E7 and HTK-K4DT. Additional transduction of oncogenic HRAS or EGFR together with MYC into the HTK-16E6E7 and dominant-negative p53 expressing HTK-K4DT resulted in anchorage-independent growth and subcutaneous tumor formation in nude mice. These results indicate that either HRAS mutation or activation of EGFR in cooperation with MYC overexpression play critical roles in transformation of HTKs on a background of inactivation of the pRB and p53 pathways and telomerase activation. This in vitro model system recapitulating the development of OSCCs should facilitate further studies of mechanisms of carcinogenesis in the oral cavity.
Keywords: Oral squamous cell carcinoma, HPV, carcinogenesis, human tongue keratinocytes, EGFR, HRAS, MYC
Introduction
Oral cancers are the 6th most common human neoplasms accounting for 3% of all newly diagnosed cancers[1], with about 300,000 new cases being diagnosed every year worldwide [2, 3]. Despite efforts to improve the survival rates, these have basically remained unchanged for the last 20 years. Since 50 to 70% of patients die within 5 years due to local recurrence, invasion or metastasis to the cervical lymph nodes and/or lung, or second primary cancers, generally elsewhere in the oral cavity (in line with the ‘field cancerization’ theory), the prognosis is poor. Moreover, oral cancers have a severe impact on the quality of life of patients and survivors. In spite of the clinical importance, we are far away from having a complete understanding of the molecular mechanisms of initiation and progression of oral cancers.
The main accepted risk factors are tobacco usage and alcohol consumption but recently, human papillomaviruses (HPV) have also been postulated to play roles [4-6]. While more than 95% of cervical squamous cell carcinomas are linked to persistent HPV infection, the presence of the HPV genome in oral cancers is reported to range from 10 to 70%, depending on the area, the ethnicity of the patients, the type of specimen and the detection method [4]. Several studies have provided evidence that chronic infection in basal cells of the oral mucosa with high-risk HPVs, especially type 16 and 18, can promote oral carcinogenesis [7]. Two viral onco-proteins, E6 and E7, are thought to contribute to tumor progression by inactivating p53 and retinoblastoma tumor suppressor (pRB), respectively [8, 9]. E6 facilitates the degradation of p53 through its association with an accessory protein, E6-AP, a component of the ubiquitin proteolytic pathway [9]. E7 proteins of the high-risk types bind to pRB [10], leading to altered activity of this cell-cycle regulator. However, epidemiological studies and experimental data indicate that the viral presence is not enough to induce cancers even in the cervix and the requirement of additional cellular factors are especially suggested in the case of oral carcinogenesis, the roles of HPV are still under estimation.
More than 90% of oral cancers are histopathologically squamous cell carcinomas (SCCs). The development of oral squamous cell carcinomas (OSCCs) is a multistep process, starting from hyperplasia and dysplasia, and finally progressing to neoplasms (benign and malignant) [11, 12]. During these steps, multiple geneti-calterations may occur, including chromosomal aberrations, DNA mutations, amplification or deletions and/or epigenetic alterations. Numerous studies have revealed that oncogenes such as EGFR, ERBB2, HRAS, KRAS, and c-MYC (MYC) are often activated by overexpression, amplification, and/or mutation [1, 13-20]. As with other carcinomas, telomerase activation is also common in oral cancers [21, 22]. In addition, mutations of p53 and disruption of the pRB pathway (p16INK4a-CDK4/cyclin D1-pRB) are frequently observed [23-27]. Although such genetic changes have been identified, how they individually contribute to oral carcinogenesis has yet to be clarified in detail.
Recently, we have established in vitro multi-step carcinogenesis models for cervical cancer and epithelial ovarian cancer, respectively with and without HPV16 E6/E7 as transgenes [28, 29]. In the present study, taking advantage of this background, we could successfully induce tumorigenic transformation of normal human tongue keratinocytes with defined genetic elements so as to establish in vitro multistep carcinogenesis models for both HPV-positive and -negative OSCCs.
Materials and methods
Isolation of human tongue keratinocytes (HTKs)
Tongues were obtained from two tongue mucocele patients undergoing cystectomy at Hyogo College of Medicine Hospital, Japan. The Ethics Committee of Hyogo College of Medicine and National Cancer Center approved this study and the subjects gave informed consent for participation. The tongues were grossly normal and no pathological lesions were observed on subsequent histological examination. After colla-genase digestion under aseptic conditions, HTK cells were obtained by scraping with a surgical blade and maintained in Epilife (Invitrogen, Carlsbad, CA).
Viral vector construction and viral transduction
Construction of the retroviral expression vectors, pCLXSN-16E6E7, pCLXSH-TERT, pCMSCVpuro-MYC, -MYCT58A, pCMSCVbsd-MYC, -MYCT58A, pCMSCVbsd-HRASG12V, pCMSCVbsd was described previously [28-30] Wild type EGFR (EGFRWT) and a constitutive active form of EGFR (EGFRd746-750; deletion from E746 to A750) generated by site-directed mutagenesis were similarly recombined with the retroviral vector pDEST-PQCXIP by the LR reaction (Invitrogen) to generate pQCXIP-EGFRWT and pQCXIP-EGFRd746-750. The production of recombinant retroviruses was as described previously [31]. Construction of lentiviral vectors, CSII-CMV-TERT, CSII-CMV-cyclin D1, CSII-CMV-CDK4R24C and CSII-CMV-DNp53 and the production of recombinant lenti-viruses with the vesicular stomatitis virus G gly-coprotein (VSV-G) were as detailed earlier [29, 32]. Following the addition of recombinant viral fluid to cells in the presence of 4 mg/ml poly-brene, infected cells were selected in the presence of 50 mg/ml of G418, 1 mg/ml of puromycin, 1 mg/ml of blasticidine-S or 50 mg/ml of hygromycin-B.
Telomerase activity
Telomerase activity was detected using a non-radioisotopic method with a TRAPeze telomerase detection kit (Intergen, Burlington, MA), as previously described [33].
Western blot analysis
Western blotting was conducted as described prviously [33]. Antibodies against cyclin D1 (clone G124-326), CDK4(clone 97), p16INK4a (clone G175-405; BD Biosciences, Franklin Lakes, NJ), p53 (clone DO-1; Merck, Darmstadt, Germany), p21WAF1 (12D1; Oncogene Research Products, Cambridge, MA), MYC (sc-42), β-actin (sc-1616; Santa Cruz, CA), HPV16 E6 (clone 47A4)[34], HPV16 E7 (clone 8C9; Invitrogen) and keratin 14 (AF14; Covance, Princeton, NJ) were used as probes, and horseradish peroxidase-conjugated anti-mouse, anti-rabbit (Jackson Immunoresearch Laboratories, West Grove, PA) or anti-goat (sc-2033; Santa Cruz) immunoglobulins were employed as secondary antibodies.
Colony formation in soft agar (anchorage-independent growth)
Cells were seeded at 5×104 cells per 35-mm plate (BD Biosciences) in Epilife with 0.4 % aga-rose. Colonies over 50μm in diameter were counted after a lapse of 3 weeks. Five photographs of randomly selected areas in each dish were taken at the magnification of ×40. The numbers of colonies were measured with the COLONY program (Fujifilm, JAPAN). The experiments were performed in triplicate.
Tumorigenesis in nude mice
All surgical procedures and care administered to the animals were in accordance with institutional guidelines. Cells were resuspended in 50% Matrigel (BD Biosciences) and injected subcutaneously into a flank or orthotopically into female 6 to 7-week old BALB/c nude mice (Clea Japan Inc., Tokyo, Japan).
Immunohistochemical examination
Formalin-fixed and paraffin-embedded tissue sections (4 micrometer-thick) were deparaf-finized in xylene and rehydrated through a series of graded ethanols (100–70%). For antigen retrieval, slides were immersed in a citrate buffer (pH6.4) and heated for 15 minutes in a microwave. The slides were then incubated in methanol containing 0.3% H2O2 to inhibit endogenous peroxidase activity. After washing, primary antibody against keratin 14 (1:500, SP53, Spring Bioscience, Pleasanton, CA) was applied for 1 h and binding was detected using an Envision Kit (Dako Cytomation; K4006). Color development was achieved with 3, 3-diaminobenzine (DAB) as chromogen and hematoxylin counterstaining. As a negative control, we used normal non-immune serum from the same source as the primary antibody.
Results
Immortalization of HTK cells with or without viral oncogenes
To establish an in vitro model system for HPV-positive OSCCs, two independent batches of primary HTK cells (HTK1 and HTK3T) were transduced with retroviral vectors expressing HPV16 E6 and E7 (HTK1-E6E7). HTK3 cells were transduced with TERT first since HPV16 E6 and E7 are not sufficient to avoid telomere erosion. Pooled populations of these HTK cells were named HTK1-E6E7 and HTK3-TE6E7, respectively. Expression of the transgenes was confirmed by immunoblotting (Figure 1A). As expected, decreased levels of p53 were observed in these cells (Figure 1A). We have shown that both telomerase activation and inactivation of the p16INK4a/pRB pathway are required for immortalization of human primary epithelial cells. Disruption of the pRB pathway, such as inactivation of p16INK4a and overexpression of cyclin D1, are also frequently observed in OSCCs. In order to establish an in vitro model system for HPV16-negative OSCCs, a mutant form of CDK4 (CDK4R24C), which cannot be inactivated by p16, and cyclin D1 as well as TERT were transduced into HTK cells (HTK1 and HTK3) with lentiviral vectors (Figure 1B). Pooled populations of these HTK cells were named HTK1-K4DT and HTK3-K4DT, respectively. Expression of the transgenes was confirmed by immunoblotting (Figure 1B) and the TRAP assay (Figure 1C). As expected, the combination of HPV16 E6E7 or CDK4R24C, cyclin D1 and TERT resulted in extended life span and virtual immortalization of HTK cells (Figure 1D). Both the primary and immortalized cell lines expressed keratin 14, a marker of keratinocytes (data not shown). HTK1 cells showed normal diploidy and HTK1-K4DT cells was almost diploid though HTK3-K4DT cells tended to be tetraploid with some chromosomal abnormalities.
Figure 1.

Immortalization of primary human tongue keratinocytes. (A) Two batches of primary human tongue keratino-cyte (HTK), termed HTK1 and HTK3, were transduced with retroviruses expressing HPV 16 E6 and E7 for the HPV-positive OSCC model. After selection, cells were harvested and subjected to SDS-PAGE. Western blotting confirmed expression of the two transgenes in the resultant cell populations and showed endogenous expression of p53. (B) HTK1 and HTK3 were infected with lentiviruses expressing mutant CDK4, cyclin D1 and TERT for the HPV-negative OSCC model. Western blotting confirmed expression of the two transgenes in the resultant cell populations and showed endogenous expression of p16INK4a. (C) Telomerase activity of primary and immortalized HTK cells was measured by the TRAP assay. Hela cells, eight tandem repeats of the teiomeric sequence (TSR8) and CHAPS buffer alone (NC) were used as controls. (D) Growth curves of HTK1 and HTK3 cell lines. Day 0 is the time when the immortalizing genes (mutant CDK4, cyclin D1 and TERT) or HPV 16 E6E7 were transduced.
Combined transduction of HRAS and MYC into HTK1-K4DT-DNp53 and HTK1-E6E7 cells induces anchorage-independent growth and tumor-forming ability in nude mice
In HPV-negative OSCCs, overexpression of MYC and mutation of HRAS and p53 are frequently observed especially in tobacco chewing individuals for HRAS [35]. Thus, a dominant negative form of p53 (DNp53), HRASG12V and MYC were serially transduced into HTK1-K4DT cells. Expression of these transgenes together with accumulation of p53 and downregulation of p21WAF1 was confirmed by immunoblotting (Figure 2A). Then we assessed the effects of oncogenic HRASG12V and MYC on cell growth. HTK1-K4DT-DNp53 cells with HRASG12V and MYC grew faster than those with an empty vector (Figure 2B), and formed numerous and much larger colonies in soft agar medium than those with HRASG12V alone, whereas cells with empty vector formed no colonies (Figure 2C). HTK1-K4DT-DNp53 cells with HRASG12V and MYC or a mutant form of MYC (MYCT58A), which is resistant to proteosomal degradation, formed tumors in nude mice, whereas those without MYC failed to form tumors (Table 1). HTK1-K4DT-HRASG12V-MYC cells, which did not express a dominant negative form of p53 developed tumors less efficiently and with a long latent period, while HTK1-K4DT-DNp53 cells with MYC alone did not form tumors (Table 1).
Figure 2.

Anchorage-dependent and -independent growth of HTK1-E6E7-HRASG12V-MYC and HTK1-K4DT-DNp53-HRASG12V-MYC cells. (A) HTK1-K4DT cells were serially infected with lentiviruses encoding DNp53 and retroviruses encoding HRASG12V, MYC or empty vectors (-). After selection, cells were harvested and subjected to SDS-PAGE. Western blots show expression of the three transgenes and suppression of p21WAF1. (B) Growth curves for DNp53-vector, DNp53-HRASG12V or DNp53-HRASG12V-MYC expressing HTK1-K4DT cells. HTK1-K4DT-DNp53-HRASG12V cells showed the fastest growth rate. Cells (2 × 104) were cultured in triplicate 12-well plates and counted every 3 days. The graphs illustrate means + s.d. (C) Anchorage independent growth of HTK1-K4DT cells expressing different transgenes. Cells (5 × 104) were seeded in 35-mm plates. After 3 weeks, colonies was counted when sized > 50 μm in diameter. The experiments were performed in triplicate and the total number of colonies in a 15 mm2 area was counted. The graphs illustrate means + s.d. Scale bars, 250 μm. (D) HTK1-E6E7 cells were serially infected with retroviruses encoding HRASG12V, MYC or empty vectors (-). After selection, cells were harvested and subjected to SDS-PAGE. Western blots show expression of the two transgenes. (E) Growth curves for vector, HRASG12V or HRASG12V-MYC expressing HTK1-E6E7 cells. HTK1-E6E7-HRASG12V cells showed the fastest growth rate. Cells were grown as described in (B). (F) Anchorage independent growth of HTK1-E6E7 cells expressing different transgenes performed as for (C). Scale bars, 250 μm.
Table 1.
Summary of data for tumorigenic potential of HTK1 and HTK3 cells with various transgenes (1×106 cells/site).
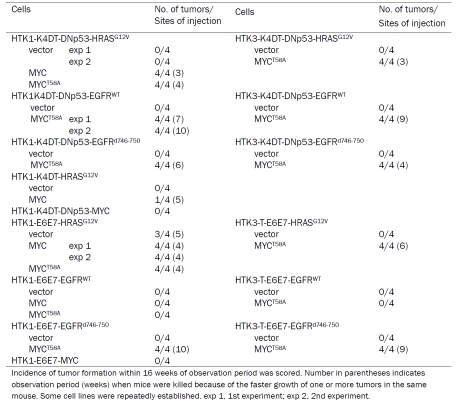 |
For the HPV-positive OSCC model, we transduced HRASG12V and MYC serially into HTK1-E6E7 cells and confirmed expression of transgenes by immunoblotting (Figure 2D). HTK1-E6E7 cells expressing HRASG12V and MYC or HRASG12V alone grew faster than those with empty vectors (Figure 2E). HTK1-E6E7 cells expressing HRASG12V and MYC formed numerous large colonies and those expressing HRASG12V alone formed some small colonies, whereas those with empty vectors formed no colonies (Figure 2F). HTK1-E6E7- HRASG12V cells (3/4) as well as HTK1-E6E7- HRASG12V-MYC cells formed tumors (8/8) in nude mice, whereas those expressing MYC alone failed to do so (Table 1). This is consistent with our previous results that a combination of E6E7 and oncogenic HRAS without MYC can confer tumorigenicity on human cervical keratinocytes and MYC substantially enhances the tumorigenicity. These results indicate that a combination of oncogenic HRAS and MYC can cooperately confer anchorage-independent growth and tumorigenicity on HTK cells expressing either E6E7 or CDK4/cyclin D1/TERT and DNp53.
Combined transduction of a constitutively active form of EGFR and a degradation-resistant form of MYC into HTK1-K4DT-DNp53 and HTK1-E6E7 cells induces anchorage-independent growth and tumor-forming ability in nude mice
Excluding cases in tobacco chewers, overexpression of EGFR or activating mutations of EGFR are observed more frequently than activating mutations in the RAS oncogenes [17, 35]. To determine a role of enhanced EGFR signaling in the development of OSCCs, wild type EGFR (EGFRWT) or a constitutively active form of EGFR (EGFRd746-750) instead of HRAS was transduced into HTK1-K4DT and HTK1-E6E7 cells as expected if HRAS and EGFR are acting in the same pathway. Expression of the transgenes was confirmed by immunoblotting (Figure 3A in HTK1-K4DT and Figure 4A in HTK1-E6E7). Total and the phosphorylated form of EGFR in HTK1-K4DT-DNp53-EGFRWT cells and HSC2 (human OSCC cell line with EGFR amplification) cells were higher than those in vector transduced cells and the phophorylation levels were further increased by the addition of EGF. As expected, the phosphorylation levels of EGFRWT in EGFRd746-750 expressing cells were much higher without addition of EGF, indicating ligand-independent activation of the EGFRd746-750 (Figure 3A, P-EGFR). Exogenous EGFR expression levels in HTK1-K4DT or HTK1-E6E7 cells were comparable to that of HSC2.
Figure 3.

Anchorage-dependent and -independent growth of HTK1-K4DT-DNp53-EGFRWT (or EGFRd746-750)-MYCT58A cells. (A) HTK1-K4DT-DNp53 cells were serially infected with retroviruses expressing EGFRWT, EGFRd746-750, MYCT58A or empty vectors (-). After selection, cells were harvested and subjected to SDS-PAGE. Western blots show expression of transgenes. EGFRd746-750; constitutively active mutant of EGFR. Cells were first starved in medium without bovine pituitary extract and EGF for 72 hours and some of them were stimulated with EGF for 30 min before harvesting as indicated. HSC2; human OSCC cell line with EGFR amplification. (B) Growth curves of HTK1-K4DT-DNp53 cells expressing different transgenes as described in (A). Cells were grown as described in Fig 2(B). (C) Anchorage-independent growth of HTK1-K4DT-DNp53 cells expressing different transgenes performed as for Fig. 2(C). Scale bars, 250 μn.
Figure 4.

Anchorage-dependent and -independent growth of HTK1-E6E7-EGFRWT (or EGFRd746-750)-MYCT58A cells. (A) HTK1-E6E7 cells were serially infected with retroviruses expressing EGFRWT, EGFRd746-750, MYCT58A or empty vectors (-). After selection, cells were harvested and subjected to SDS-PAGE. Western blots show expression of transgenes. HSC2; human OSCC cell line with EGFR amplification. (B) Growth curves of HTK1-E6E7 cells expressing different transgenes as described in (A). Cells were grown as described in Fig 2(B). (C) Anchorage-independent growth of HTK1-K4DT-E6E7 cells expressing different transgenes performed as for Fig. 2(C). Scale bars, 250 μm.
HTK1-K4DT cells expressing EGFRWT or EGFRd746-750 grew faster than those with an empty vector (Figure 3B), and additional transduction of MYCT58A only slightly enhanced proliferation in culture (Figure 3B). HTK1-K4DT-DNp53 cells expressing EGFRWT or EGFRd746-750 exhibited anchorage-independent growth, enhanced by additional MYCT58A transduction, whereas those with empty vector formed no colonies (Figure 3C). HTK1-K4DT-DNp53 cells expressing EGFRWT or EGFRd746-750 were able to form tumors only when MYCT58A was co-expressed (Table 1) and those with EGFRd746-750 formed tumors faster than those with EGFRWT (Table 1).
HTK1-E6E7 cells expressing EGFRd746-750 grew faster than those expressing EGFRWT (Figure 4B), and showed increased anchorage-independent growth regardless of the exogenous expression of MYCT58A, though those expressing EGFRWT showed poor anchorage-independent growth without MYCT58A (Figure 4C). In tumorigenic assay, HTK1-E6E7 cells expressing EGFRd746-750 cells formed tumors only when MYC or MYCT58A was coexpressed, whereas those expressing EGFRWT did not form tumors even with addition of MYC or MYCT58A (Table 1).
Histopathological findings for tumors derived from HTK1 cells
Isolated xenograft tumors were examined by histopathologically (Figure 5). HTK1-K4DT-DNp53 -HRASG12V-MYCT58A and HTK1-K4DT-DNp53 - EGFRd746-750-MYCT58A tumors mainly comprised poorly differentiated SCCs. On the other hand, tumors of HTK1-E6E7-HRASG12V-MYCT58A and HTK1-E6E7-EGFRd746-750-MYCT58A cells were well differentiated SCCs with keratin pearls. Both HTK1-K4DT-DNp53 - HRASG12V-MYCT58A and HTK1-E6E7-HRASG12V-MYCT58A cells were positive for keratin 14 proved to be carcinomatous in structure (Figure 5, insets).
Figure 5.

Histopathological findings for tumors derived from HTK1 cells. Histopathology of subcutaneous tumors of HTK1-E6E7- HRASG12V-MYCT58A, HTK1-E6E7- EGFRd746-750-MYCT58A HTK1-K4DT-DNp53-HRASG12V-MYCT58A and HTK1-K4DT-DNp53-EGFRd746-750-MYCT58A cells. The insets show immunohistochemical staining of keratin 14. Scale bars, 200 μm.
Confirmation of the multistage nature of carcinogenesis with the HTK3T cell line
It is possible that additional alterations (genetic and/or epigenetic) occurring during the process of introducing oncogenic genes could contribute to their tumorigenic phenotype. To address this possibility, we repeatedly transduced EGFRWT, EGFRd746-750 or HRASG12V plus MYCT58A into HTK3-TE6E7 and HTK3-K4DT-DNp53 cells, another independent batch of HTKs derived from a different patient. All these cells reproducibly developed subcutaneous tumors in nude mice, whereas those transduced with HRASG12V alone without MYCT58A and carrying empty vectors failed to form tumors (Table 1). The histological appearance of HTK3-K4DT-DNp53-HRASG12V-MYCT58A and HTK1-TE6E7-HRASG12V-MYCT58A tumors was similar to that of corresponding tumors with HTK1-K4DT and HTK1-E6E7 (data not shown). These results indicate that the combination of multiple genetic elements we applied can reproducibly fully transform HTK cells.
Discussion
Our goal is to develop an appropriate in vitro model for recapitulating development and progression of both HPV-positive and -negative human OSCCs. OSCCs are thought to arise from basal layer epithelial cells of oral mucosa, which regenerates stratified epithelium through terminal differentiation of keratinocytes. The fact that human tongue keratinocytes (HTKs) were here obtained from two patients without smoking histories or cancer, allowed us to explore inter-individual variation in the tumor formation process. In this report, we document establishment of an in vitro model system for HPV16-positive and -negative multistep carcinogenesis using normal HTKs.
As also shown earlier, overexpression of HPV16 E6 and E7 themselves could immortalize HTKs but did not support anchorage-independent growth. We next tried to immortalize HTKs without viral oncogenes to establish an in vitro model system for HPV-negative multistep carcinogenesis. By CDK4 and cyclin D1 transduction in combination with TERT, we here established novel HTK cell lines, termed HTK1-K4DT and HTK3-K4DT. The pRB pathway is frequently disrupted in OSCCs by p16INK4a inactivation and/or abnormal expression of CDK4/cyclin D1 [18, 20, 36, 37]. OSCCs, like many other carcinomas, maintain telomere length with telomerase activation [21, 22]. Immortality is one of the important characteristics of malignancy and ectopic expression of these genes thus could mimic the events that occur during development of OSCCs. Alterations of p53 have been detected in approximately 50% of OSCCs [23, 36-39] and some authors suggest that p53 alterations might represent an early step in the oral carcinogenesis, especially for HPV-negative OSCCs.
OSCCs often overexpress the epidermal growth factor receptor (EGFR) and some of its active variants or harbor activating mutations in the RAS oncogene with a rate ranging from 3% to 5% in Western countries and up to 50% in India and Southeast Asia. Importantly, EGFR mutations and KRAS mutations are mutually exclusively observed in non-small cell lung carcinomas[40]. A similar tendency has also been observed in a smaller number of OSCCs [19]. Recently, complications of chewing betel quid in oral cancer development have been found clinically meaningful and important in India and Southeast Asia. For example, EGFR amplification or a high frequency of mutations in codons 12 and 61 of the HRAS were reported to be associated with heavy betel quid users [16, 35, 37, 41]. EGFR, a key cancer-driving gene during OSCC development, belongs to the type I receptor tyrosine kinase (ERBB) subfamily, and appears more important than other ERBB members for oral cancer development [19]. In addition to gene amplification, activating mutation of genes in kinase signaling pathways is another common genetic event during cancer development. EGFR and its downstream effectors have diverse cellular functions, impacting on cell proliferation, differentiation, motility, survival, and tissue development [42]. The RAS-RAF-MAPK cascade is particularly active when cancer cells overexpress EGFR [43]. Schulze and colleagues have further shown that the majority of RAS-RAF -MAPK-induced changes in gene expression are dependent on the status of EGFR [44], highlighting the critical roles of the signal networking among different oncogenes in cancers [45]. In addition, Raimondi et al have shown that, in spite of ras being likely activated in many K14-expressing squamous epithelia in their animal system, those animals develop benign tumors only in the oral mucosa. This suggests that oral epithelial cells might be particularly sensitive to RAS-induced aberrant cell proliferation. Here, transduction of oncogenic HRAS or wild type EGFR or mutant EGFR (EGFRd746-750) into HTK-E6E7 and HTK-K4DT-DNp53 cells resulted in enhanced anchorage-independent growth but no tumor forming ability, except when oncogenic HRAS was transduced into HTK1-E6E7. Therefore we tried to define essential genetic alterations that cooperate with HRASG12V or wild type EGFR or EGFRd746-750 to induce a fully transformed phenotype. Amplification and/or overexpression of the MYC gene, an oncogene but also a strong inducer of apoptosis, are found in 26-40% of all oral cancers [15, 20]. Furthermore, amplification of MYC is a common finding in advanced stages, which may suggest a critical role in progression. In this study, transduction of wild-type or a mutant MYC strongly enhanced anchorage-independent growth of HTK-E6E7 and HTK-K4DT-DNp53 cells expressing HRASG12v, EGFRWT, or EGFRd746-750, and resulted in tumor formation, except when MYC or MYCT58A was transduced into HTK-E6E7-EGFRWT cells (Table 1). In addition, we have focused on development of an orthotopic model of OSCC through injecting HTK cell lines into the tongues of nude mice. HTK1-K4DT-DNp53-HRASG12V-MYCT58A and HTK1-K4DT-DNp53-EGFRWT-MYCT58A cells formed orthotopic tumors (Figure 6A) and showed SCC-like features histopathologically (Figure 6B). Furthermore, one out of the four tumors yielded regional metastases in 2-3 weeks (data not shown). Further investigation with the orthotopic model should be useful to identify genes and other factors involving in regional metastases.
Figure 6.
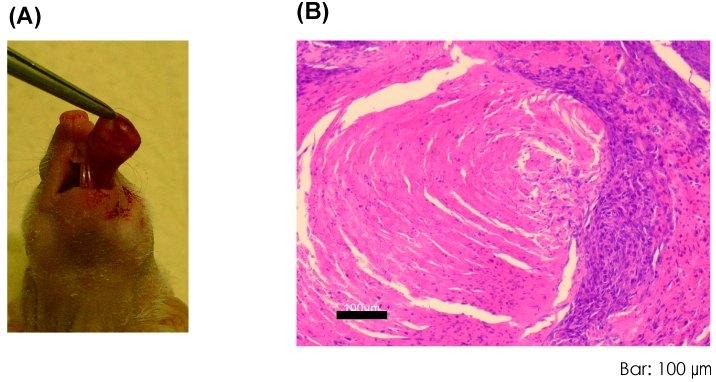
Orthotopic xenografts of HTK1-K4DT-DNp53-HRASG12V-MYCT58A cells (A) A representative tumor at day 63 after 1 × 105 cells were transplanted into the tongue of mice. (B) HE staining of the tumor showed SCC-like features histopathologically. Scale bar, 100 μm.
Human cancer cell lines, even though they are derived from well-differentiated carcinomas, rarely mimic the original histology when inoculated into mice. In our study, HTK1-K4DT-DNp53 cells expressing HRASG12V or EGFRd746-750 and MYC (HPV-negative model) formed tumors faster than HTK1-E6E7 cells expressing the same additional oncogenes (HPV-positive model), and HTK1-E6E7 cells expressing EGFRWT and MYC or MYCT58A failed to form tumors unlike the corresponding HTK1-K4DT-DNp53 cells (Table 1). In addition, isolated sub-cutaneously xenograft tumors of HTK1-E6E7 cells expressing either HRASG12V plus MYC T58A or EGFRd746-750 -MYCT58A (HPV-positive model) showed histological features of well-differentiated SCCs, but the corresponding HTK1-K4DT-DNp53 cells (HPV-negative model) did not (Figure 5). These differences of tumorigenicity and histopathology between our HPV-positive and HPV-negative models might reflect the favorable outcome associated with HPV in oropharyngeal cancer [17, 46]. Further efforts to clarify critical pathways in carcinogenesis with each histological subtype should help provide best targets for early detection and effective molecular therapies.
In summary, we newly immortalized primary HTK cells with cellular genes (CDK4, cyclin D1 and TERT) or viral oncogenes (HPV16 E6E7). With these non-tumorigenic cell lines, we have developed for the first time an in vitro culture model faithfully recapitulating the development of HPV-positive and -negative OSCCs with genetically defined elements. Our results provide evidence that either HRAS mutation or activation of EGFR in cooperation with MYC overexpression is a strong set of drivers sufficient for transformation of HTKs which have acquired inactivation of the pRB and p53 pathways and telomerase activation by either HPV16E6E7 or equivalent genetic alterations. Our experimental model should facilitate further studies to understand genesis of OSCCs and hopefully will assist in the evaluation of new therapies.
Acknowledgments
We would like to express our appreciation to Yuki Inagawa, Shin-ichi Ohno and Takako Ishi-yama for their expert technical assistance. This work was supported in part by Grants-in-Aid for Cancer Research from the Ministry of Health Labor and Welfare, for Cancer Research from the Yasuda Medical Foundation to T.K., and for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan to Y.Z., K.N., M.U., and T.K.
Conflict of interest
Authors have no conflict of interest.
References
- 1.Choi S, Myers JN. Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dent Res. 2008;87:14–32. doi: 10.1177/154405910808700104. [DOI] [PubMed] [Google Scholar]
- 2.Pereira MC, Oliveira DT, Landman G, Kowalski LP. Histologic subtypes of oral squamous cell carcinoma: prognostic relevance. J Can Dent Assoc. 2007;73:339–344. [PubMed] [Google Scholar]
- 3.Sudbo J, Bryne M, Mao L, Lotan R, Reith A, Kildal W, Davidson B, Soland TM, Lippman SM. Molecular based treatment of oral cancer. Oral Oncol. 2003;39:749–758. doi: 10.1016/s1368-8375(03)00098-8. [DOI] [PubMed] [Google Scholar]
- 4.Campisi G, Panzarella V, Giuliani M, Lajolo C, Di Fede O, Falaschini S, Di Liberto C, Scully C, Lo Muzio L. Human papillomavirus: its identity and controversial role in oral oncogenesis, premalignant and malignant lesions (review) Int J Oncol. 2007;30:813–823. [PubMed] [Google Scholar]
- 5.Psyrri A, DiMaio D. Human papillomavirus in cervical and head-and-neck cancer. Nat Clin Pract Oncol. 2008;5:24–31. doi: 10.1038/ncponc0984. [DOI] [PubMed] [Google Scholar]
- 6.Ragin CC, Modugno F, Gollin SM. The epidemiology and risk factors of head and neck cancer: a focus on human papillomavirus. J Dent Res. 2007;86:104–114. doi: 10.1177/154405910708600202. [DOI] [PubMed] [Google Scholar]
- 7.Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, Shah KV, Sidransky D. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92:709–720. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 8.Galloway DA, McDougall JK. The disruption of cell cycle checkpoints by papillomavirus oncoproteins contributes to anogenital neoplasia. Semin Cancer Biol. 1996;7:309–315. doi: 10.1006/scbi.1996.0040. [DOI] [PubMed] [Google Scholar]
- 9.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 10.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 11.Hunter KD, Parkinson EK, Harrison PR. Profiling early head and neck cancer. Nat Rev Cancer. 2005;5:127–135. doi: 10.1038/nrc1549. [DOI] [PubMed] [Google Scholar]
- 12.Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W, Sidransky D. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. 1996;56:2488–2492. [PubMed] [Google Scholar]
- 13.Ryott M, Wangsa D, Heselmeyer-Haddad K, Lindholm J, Elmberger G, Auer G, Lundqvist EV, Ried T, Munck-Wikland E. EGFR protein overexpression and gene copy number increases in oral tongue squamous cell carcinoma. Eur J Cancer. 2009;45:1700–1708. doi: 10.1016/j.ejca.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiang WF, Liu SY, Yen CY, Lin CN, Chen YC, Lin SC, Chang KW. Association of epidermal growth factor receptor (EGFR) gene copy number amplification with neck lymph node metastasis in areca-associated oral carcinomas. Oral Oncol. 2008;44:270–276. doi: 10.1016/j.oraloncology.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Vora HH, Shah NG, Trivedi TI, Goswami JV, Shukla SN, Shah PM. Expression of C-Myc mRNA in squamous cell carcinoma of the tongue. J Surg Oncol. 2007;95:70–78. doi: 10.1002/jso.20675. [DOI] [PubMed] [Google Scholar]
- 16.Sathyan KM, Nalinakumari KR, Kannan S. H-Ras mutation modulates the expression of major cell cycle regulatory proteins and disease prognosis in oral carcinoma. Mod Pathol. 2007;20:1141–1148. doi: 10.1038/modpathol.3800948. [DOI] [PubMed] [Google Scholar]
- 17.Na II, Kang HJ, Cho SY, Koh JS, Lee JK, Lee BC, Lee GH, Lee YS, Yoo HJ, Ryoo BY, Yang SH, Shim YS. EGFR mutations and human papillomavirus in squamous cell carcinoma of tongue and tonsil. Eur J Cancer. 2007;43:520–526. doi: 10.1016/j.ejca.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 18.Freier K, Joos S, Flechtenmacher C, Devens F, Benner A, Bosch FX, Lichter P, Hofele C. Tissue microarray analysis reveals site-specific prevalence of oncogene amplifications in head and neck squamous cell carcinoma. Cancer Res. 2003;63:1179–1182. [PubMed] [Google Scholar]
- 19.Sheu JJ, Hua CH, Wan L, Lin YJ, Lai MT, Tseng HC, Jinawath N, Tsai MH, Chang NW, Lin CF, Lin CC, Hsieh LJ, Wang TL, Shih Ie M, Tsai FJ. Functional genomic analysis identified epidermal growth factor receptor activation as the most common genetic event in oral squamous cell carcinoma. Cancer Res. 2009;69:2568–2576. doi: 10.1158/0008-5472.CAN-08-3199. [DOI] [PubMed] [Google Scholar]
- 20.Akervall J, Bockmuhl U, Petersen I, Yang K, Carey TE, Kurnit DM. The gene ratios c-MYC:cyclin-dependent kinase (CDK)N2A and CCND1:CDKN2A correlate with poor prognosis in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2003;9:1750–1755. [PubMed] [Google Scholar]
- 21.Freier K, Pungs S, Flechtenmacher C, Bosch FX, Lichter P, Joos S, Hofele C. Frequent high telomerase reverse transcriptase expression in primary oral squamous cell carcinoma. J Oral Pathol Med. 2007;36:267–272. doi: 10.1111/j.1600-0714.2007.00531.x. [DOI] [PubMed] [Google Scholar]
- 22.Pannone G, De Maria S, Zamparese R, Metafora S, Serpico R, Morelli F, Rubini C, Farina E, Carteni M, Staibano S, De Rosa G, Lo Muzio L, Bufo P. Prognostic value of human telomerase reverse transcriptase gene expression in oral carcinogenesis. Int J Oncol. 2007;30:1349–1357. doi: 10.3892/ijo.30.6.1349. [DOI] [PubMed] [Google Scholar]
- 23.Karsai S, Abel U, Roesch-Ely M, Affolter A, Hofele C, Joos S, Plinkert PK, Bosch FX. Comparison of p16(INK4a) expression with p53 alterations in head and neck cancer by tissue microarray analysis. J Pathol. 2007;211:314–322. doi: 10.1002/path.2100. [DOI] [PubMed] [Google Scholar]
- 24.Shaw R. The epigenetics of oral cancer. Int J Oral Maxillofac Surg. 2006;35:101–108. doi: 10.1016/j.ijom.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 25.Ogi K, Toyota M, Ohe-Toyota M, Tanaka N, Noguchi M, Sonoda T, Kohama G, Tokino T. Aberrant methylation of multiple genes and clinicopathological features in oral squamous cell carcinoma. Clin Cancer Res. 2002;8:3164–3171. [PubMed] [Google Scholar]
- 26.Hasegawa M, Nelson HH, Peters E, Ringstrom E, Posner M, Kelsey KT. Patterns of gene promoter methylation in squamous cell cancer of the head and neck. Oncogene. 2002;21:4231–4236. doi: 10.1038/sj.onc.1205528. [DOI] [PubMed] [Google Scholar]
- 27.Papadimitrakopoulou V, Izzo J, Lippman SM, Lee JS, Fan YH, Clayman G, Ro JY, Hittelman WN, Lotan R, Hong WK, Mao L. Frequent inactivation of p16INK4a in oral premalignant lesions. Oncogene. 1997;14:1799–1803. doi: 10.1038/sj.onc.1201010. [DOI] [PubMed] [Google Scholar]
- 28.Narisawa-Saito M, Yoshimatsu Y, Ohno S, Yugawa T, Egawa N, Fujita M, Hirohashi S, Kiyono T. An in vitro multistep carcinogenesis model for human cervical cancer. Cancer Res. 2008;68:5699–5705. doi: 10.1158/0008-5472.CAN-07-6862. [DOI] [PubMed] [Google Scholar]
- 29.Sasaki R, Narisawa-Saito M, Yugawa T, Fujita M, Tashiro H, Katabuchi H, Kiyono T. Oncogenic transformation of human ovarian surface epithelial cells with defined cellular oncogenes. Carcinogenesis. 2009;30:423–431. doi: 10.1093/carcin/bgp007. [DOI] [PubMed] [Google Scholar]
- 30.Takeda Y, Mori T, Imabayashi H, Kiyono T, Gojo S, Miyoshi S, Hida N, Ita M, Segawa K, Ogawa S, Sakamoto M, Nakamura S, Umezawa A. Can the life span of human marrow stromal cells be prolonged by bmi-1, E6, E7, and/or telomerase without affecting cardiomyogenic differentiation? J Gene Med. 2004;6:833–845. doi: 10.1002/jgm.583. [DOI] [PubMed] [Google Scholar]
- 31.Naviaux RK, Costanzi E, Haas M, Verma IM. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J Virol. 1996;70:5701–5705. doi: 10.1128/jvi.70.8.5701-5705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haga K, Ohno S, Yugawa T, Narisawa-Saito M, Fujita M, Sakamoto M, Galloway DA, Kiyono T. Efficient immortalization of primary human cells by p16INK4a-specific short hairpin RNA or Bmi-1, combined with introduction of hTERT. Cancer Sci. 2007;98:147–154. doi: 10.1111/j.1349-7006.2006.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yugawa T, Handa K, Narisawa-Saito M, Ohno S, Fujita M, Kiyono T. Regulation of Notch1 gene expression by p53 in epithelial cells. Mol Cell Biol. 2007;27:3732–3742. doi: 10.1128/MCB.02119-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang SE, Bhatia P, Johnson NW, Morgan PR, McCormick F, Young B, Hiorns L. Ras mutations in United Kingdom examples of oral malignancies are infrequent. Int J Cancer. 1991;48:409–412. doi: 10.1002/ijc.2910480318. [DOI] [PubMed] [Google Scholar]
- 36.Koontongkaew S, Chareonkitkajorn L, Chanvitan A, Leelakriangsak M, Amornphimoltham P. Alterations of p53, pRb, cyclin D(1) and cdk4 in human oral and pharyngeal squamous cell carcinomas. Oral Oncol. 2000;36:334–339. doi: 10.1016/s1368-8375(99)00093-7. [DOI] [PubMed] [Google Scholar]
- 37.Xu J, Gimenez-Conti IB, Cunningham JE, Collet AM, Luna MA, Lanfranchi HE, Spitz MR, Conti CJ. Alterations of p53, cyclin D1, Rb, and H-ras in human oral carcinomas related to tobacco use. Cancer. 1998;83:204–212. doi: 10.1002/(sici)1097-0142(19980715)83:2<204::aid-cncr2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 38.Wiest T, Schwarz E, Enders C, Flechtenmacher C, Bosch FX. Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene. 2002;21:1510–1517. doi: 10.1038/sj.onc.1205214. [DOI] [PubMed] [Google Scholar]
- 39.Chiba I, Shindoh M, Yasuda M, Yamazaki Y, Amemiya A, Sato Y, Fujinaga K, Notani K, Fukuda H. Mutations in the p53 gene and human papillomavirus infection as significant prognostic factors in squamous cell carcinomas of the oral cavity. Oncogene. 1996;12:1663–1668. [PubMed] [Google Scholar]
- 40.Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64:8919–8923. doi: 10.1158/0008-5472.CAN-04-2818. [DOI] [PubMed] [Google Scholar]
- 41.Murugan AK, Hong NT, Cue TT, Hung NC, Munirajan AK, Ikeda MA, Tsuchida N. Detection of two novel mutations and relatively high incidence of H-RAS mutations in Vietnamese oral cancer. Oral Oncol. 2009;45:e161–166. doi: 10.1016/j.oraloncology.2009.05.638. [DOI] [PubMed] [Google Scholar]
- 42.Wang K, Yamamoto H, Chin JR, Werb Z, Vu TH. Epidermal growth factor receptor-deficient mice have delayed primary endochondral ossification because of defective osteoclast recruitment. J Biol Chem. 2004;279:53848–53856. doi: 10.1074/jbc.M403114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 44.Schulze A, Nicke B, Warne PH, Tomlinson S, Downward J. The transcriptional response to Raf activation is almost completely dependent on Mitogen-activated Protein Kinase Kinase activity and shows a major autocrine component. Mol Biol Cell. 2004;15:3450–3463. doi: 10.1091/mbc.E03-11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 46.Hong AM, Dobbins TA, Lee CS, Jones D, Harnett GB, Armstrong BK, Clark JR, Milross CG, Kim J, O'Brien CJ, Rose BR. Human papillomavirus predicts outcome in oropharyngeal cancer in patients treated primarily with surgery or radiation therapy. Br J Cancer. 2010;103:1510–1517. doi: 10.1038/sj.bjc.6605944. [DOI] [PMC free article] [PubMed] [Google Scholar]