

Abstract
The effects of the nature and amount of base, substrate structure, amount of added water and solvent on the condensation of carbonyl compounds with cyclopentadiene in the presence of secondary amines were investigated. Based on these studies, a new efficient and green synthesis of fulvenes was developed.
Keywords: Fulvenes, LFER studies, Green synthesis, Catalysis, Cyclopentadiene
1. Introduction
Fulvenes constitute an important class of cross-conjugated olefins with unique electronic, spectroscopic and chemical properties.1 Though the first fulvene synthesis was reported by Thiele in 1900,2 considerable effort has been directed toward synthetic improvements,3–4 since Thiele’s original method based on the condensation of aldehydes and ketones with cyclopentadiene in the presence of alkoxides gives low yields with considerable amounts of resinous byproducts, presumably due to competing aldol condensations.
Neuenschwander developed an alternative synthesis of fulvenes that are not readily accessible by the Thiele procedure.5–6 Alkylation of sodium cyclopentadienide with 1-chloroalkyl acetates followed by elimination of acetic acid from acetoxyalkyl cyclopentadienes with base gives fulvenes in moderate to low yields. However, this method allows the synthesis and isolation of volatile fulvenes such as the unsubstituted parent fulvene, 6-vinylfulvene and 6-ethynylfulvene since no aqueous work-up is necessary. This three-step method requires the synthesis of 1-chloroalkyl acetates and is also less suited for the synthesis of 6,6-dialkyl or diarylfulvenes.
Hafner’s synthesis7 of 6-monosubstituted fulvenes is based on alkyl or aryl lithium reagents onto 6-dimethylaminofulvene; since the pyrrolidine variant (Little and Stone, vide infra) with lower MW aldehydes such as ethanal and propanal is unsatisfactory, Hafner’s two-step method appears to be the method of choice for the synthesis of 6-methylfulvene, a substrate most frequently used in organometallic chemistry for fulvene-based metal complexes as well as materials chemistry.8–12 In this full report we also describe an exceptionally simple and practical synthesis of the aforementioned 6-methyl and 6-ethylfulvenes that allows their isolation in pure form without extensive work-up and chromatography (separation of these volatile fulvenes from chromatography solvents without significant losses is extremely difficult).
A method for the synthesis of volatile and sensitive fulvenes such as the parent fulvene, 6-methyl and 6-ethylfulvene employing basic ion exchange resin has been reported, however, the authors were not able to isolate the aforementioned fulvenes from the reaction mixtures and their formation was inferred from the UV absorption of possible fulvene contents.13–14
Stone and Little reported a procedure whereby excess pyrrolidine was used as base with methanol as solvent and achieved good yields in the case of aliphatic aldehydes and ketones and aromatic aldehydes.15 The Stone and Little variant has been widely used in the last 25 years (over 275 citations), and deservedly attained the status as ’the method of choice for fulvene synthesis’ from simple aldehydes and non-aromatic ketones. Yet it has some limitations mostly brought on by the fact that a) at least 1.5 equivalents of the highly toxic and malodorous pyrrolidine and excess cyclopentadiene need to be used. Some of the other limitations include b) conjugate addition of pyrrolidine onto the enone when α,β-unsaturated ketones or aldehydes are used although this side reaction may be overcome in some instances by using diethylamine16 c) sterically hindered ketones, in particular aromatic ketones cannot be used; d) sterically hindered enones as well as β-aryl enones undergo Michael attack by the cyclopentadienide ion.17
The only method utilizing catalytic amount of amine base was reported by Freiesleben in 1963.3 The author claimed having achieved near quantitative yields with catalytic HNEt2 (or any other base, such as methyl-and phenylhydrazine) without solvent. Among the systems that gave exceptionally high yields were also aromatic ketones, α,β-unsaturated carbonyl compounds, including methyl vinyl ketone. In the two reports by Freiesleben, there was no spectral characterization of the products. These claims were later refuted by a careful study by Neuenschwander, Kyburz and Iseli5 who found that the best yield that was achieved by the Swiss group under Freiesleben’s conditions was 25% for 6,6-dimethylfulvene (instead of 99.8%), while in other cases the yields were 0–10%. Even methylvinyl ketone was reported by Freiesleben in a patent to give the corresponding fulvene, however, Griesbeck found that this reaction leads to a Diels-Alder adduct rather than the fulvene.17
In 1995 we reported a survey of fulvene syntheses and implemented existing condensation methods (either Thiele or Little & Stone) in the syntheses of functionalized fulvenes as well as several 6-vinylfulvenes and explored the scope and limitations of each method.18 More recently we disclosed a convenient and high yield synthesis of 6-methyl-6-vinylfulvene by a three step protocol in an overall yield of 67% (previously available in only 0.5% yield by the Thiele procedure).19
In the course of our studies on the synthesis of new functionalized fulvenes, their implementation in organic synthesis and the chemistry of the endoperoxide derived from fulvenes20–24 we needed an improved and environmentally friendly method for fulvene synthesis. Herein we report new efficient procedures for the preparation of a wide variety of fulvenes using catalytic pyrrolidine in MeOH-H2O that not only give high yields but also minimize product losses during extractive work-up and avoid the use of excess cyclopentadiene whose dimer presents challenging separation problems from the desired fulvenes. Under our conditions, even low molecular-weight fulvenes such as 6-methyl and 6-ethylfulvene are prepared and isolated in high yields and for which the Little procedure proves unsatisfactory. In this report the scope and limitations of our catalytic variant are also discussed and alternative routes suggested. The optimization of the method is based on some mechanistic studies involving effect of base, solvent and substituent effects.
2. Results and Discussion
2.1. Effect of base
The use of cyclopentadiene in excess causes the formation of dicyclopentadiene which is troublesome in the purification process as well. Moreover, we found that the use of excess pyrrolidine prevents the crystallization of solid fulvenes in the aromatic aldehyde series. Thus, we decided to test whether less than stoichiometric amounts of pyrrolidine might be enough for the formation of the iminium ion intermediate and the use of NEt3 would help the condensation/dehydration step in acceptable yields and rates. The reaction of 1a was first conducted only in the presence of triethylamine, however no fulvene formation was observed after 3 h stirring at room temperature. Pyrrolidine (10 mol%) was then added and the reaction was followed by 1H NMR; after 1h the conversion was only 15%. However, performing the reaction in the presence of pyrrolidine and molecular sieves accelerated the reaction significantly (47% after 1h). The reaction was quantitative after stirring overnight at room temperature. Moreover, the isolation of the product consisted of only vacuum-filtration of the crude mixture and evaporation of the solvent to give pure fulvene.
A series of aromatic aldehydes (Table 1, entries a–j, Method A) were subjected to condensation with cyclopentadiene at room temperature under the conditions mentioned above and the initial rates were determined at the 0.83rd h. The correlation of the initial rates (Rx) with Hammett substituent constants σ provided a linear plot with ρ = −2.19 (Figure 1). As the plot clearly indicates, electron-donating groups accelerate the reaction while electron withdrawing substituents decelerate it. In the cases of strong electron withdrawing groups such as p-, and m-NO2 groups the reactions do not proceed at all. The observed reactivity order of the aldehyde series was surprising since results from a previous study revealed that the reactivity order with nitrogen nucleophiles in THF is opposite to the one observed here.25 Therefore, we followed the equilibria for hemiacetal formations of aromatic aldehydes 2a–b, g–i in MeOD at room temperature in order to understand this reversal of reactivity (Table 3). The data revealed that in the cases of aromatic aldehydes with strong electron-withdrawing groups hemiacetal formation equilibria are shifted toward right which explains the reversed reactivity order in the fulvene formations. The Hammett type plot (Figure 1) of logK vs. σ constants produced ρ constants with the same magnitude as for the fulvene formations but with opposite sign.
Table 1.
Method Development Study on Effects of NEt3 and Molecular Sieves on Pyrrolidine-catalyzed Fulvene Formation Rates
| Entry | R | R1 | Method Aa: % 0.83 h (after completion) |
Method Bb: % 0.83 h (after completion) |
|---|---|---|---|---|
| a | H | 4-MeC6H4 | 47(98) | 71(93) |
| b | H | 4-MeOC6H4 | 70(97) | 99(94) |
| c | H | 4-Me2NC6H4 | 50(100) | 67(97) |
| d | H | 4-HOC6H4 | 9(85) | f |
| e | H | 3-MeOC6H4 | f | 35(96) |
| f | H | 3,4-(MeO)2C6H3 | 47(98) | 79(99) |
| g | H | C6H5 | 23(96) | 50(95) |
| h | H | 4-ClC6H4 | 6(96) | 18(96) |
| i | H | 4-NO2C6H4 | 025 | 39c(30)h |
| j | H | 3-NO2C6H4 | 0 | 45c(31) |
| k | Me | Me | 69 | 86 (89) |
| l | Me | 4-MeC6H4 | 0d | (82)e |
| m | Ph | Ph | 0d | 0 |
| n | H | i-propyl | 35 | 64(88) |
| o | R=R1= cyclobutyl | 55 | 80(90) | |
| p | R=R1=cyclopentyl | 51 | 77(85) | |
| t | Me | (CH2)2CO2H | 74g | |
| u | Me | (CH2)3CO2H | 87g | |
| w | H | 2-BrC6H4 | (50) | |
| x | H | 4-CF3C6H4 | (45) | |
The reactions completed within 24 h except entry d where the conversion was 88%;.
The reactions completed within 5 h except in the case of 1h which was stirred overnight for completion.
The reactions were performed in acetonitrile with 15 mol% pyrrolidine and 0.3 g/mmol molecular sieves and the data are for the 3rd h.
Addition of 10 mol% of DBU did not result in fulvene formation after 24 h stirring at room temperature.
Method B with 3 equiv of added pyrrolidine as catalyst; reaction time 48 h. The yield of the product when no MS was used is 79%.
Reactions not performed; Method A: 5 mmol carbonyl compound, 2.5 equiv cyclopentadiene (CP), 1.5g 3A molecular sieves, 10 mol% pyrrolidine, 1.5 equiv. NEt3, 5 mL MeOH; Method B: same as A, no NEt3, 1.2 equiv CP.
Method A without molecular sieves with 20 mol% of added pyrrolidine as catalyst; isolated yield after overnight stirring and regular aqueous work-up was performed (see Experimental Section).
The reaction according to method B gave 10% fulvene. The main product cyclopenta-1,3-dien-1-yl(4-nitrophenyl)methanol and cyclopenta-1,4-dien-1-yl(4-nitrophenyl)methanol (1:1) was isolated in 40% yield after 18h stirring at room temperature.
Figure 1.


Hammett plot of initial rate log(RX) of fulvene 3a–b,f–h (top graph) and hemiacetal formation equlibrium constants (logKX) of 4a–b, g–i versus σ constants (The correlation equations are log(RX) = −2.19σ-log(RX=H); r2 = 0.99 and logKX= 2.16σ−logKX=H; r2 = 0.98 respectively).26
Table 3.
Substituent effect on the hemiacetal formation equilibria of aromatic aldehydes with MeOD
| 2 | σ | log Ka | ||
|---|---|---|---|---|
| a | −0.17 | −1.510 | −1.510 | −1.495 |
| b | −0.27 | −1.440 | −1.996 | −1.880 |
| g | 0 | −1.227 | −1.061 | −0.974 |
| h | 0.23 | −0.989 | −0.630 | −0.568 |
| i | 0.78 | −0.288 | 0.477 | 0.477 |
K=[4]/[2]; K for 1h, 3.5h and 24h
The correlations discussed above impelled us to take a closer look at the pyrrolidine-catalyzed fulvene formation reaction mechanism.
The Hammett correlations performed confirm the assumption that the rate determining step of the reaction must involve the formation of a positively charged species. The question was whether it is solely the iminium salt C or the C–N bond heterolysis in the transformation of D to 3. The other possibility was that both steps may affect the reaction rate. Our observations with other C–H acids and the aromatic aldehyde series where the addition products are detectable clearly revealed that the addition step of the reaction is faster in the cases of electron-withdrawing substituents. However the substituent effect is reversed for the elimination step of the reaction. The same effect of strong electron-withdrawing substituents was observed under the conditions of Methods B and C. The competing formations of cyclopenta-1,3-dien-1-yl(4-aryl)methanol and cyclopenta-1,4-dien-1-yl(4-aryl)methanols 5 were monitored by TLC as well as 1H NMR. To exclude the possibility of formation of 4 by water addition to the corresponding fulvenes, 3x was stirred in MeOD in the presence of 20% water and 50 mol% pyrrolidine. No hydration was observed after 24h. This implies that in the cases of strong electron-withdrawing groups amine displacement competes with amine elimination (Scheme 3). Similarly, the reaction of 4-nitrobenzaldehyde with cyclopentadiene in the presence of only NEt3 in MeOH-d4 did not give compounds 4i after 24 h stirring at room temperature. The experiments summarized in Table support a second order reaction with rate = k[2]a[1]b where a and b are ca. 1.1 and 0.73, respectively. In the present case, the observed substituent effect actually confirms the fact that the rate-limiting step for the overall fulvene formation is the elimination reaction of D to give 3 and the catalyst.
Scheme 3.

Equilibria in pyrrolidine-catalyzed condensation of carbonyl compounds with cyclopentadiene
If this conclusion is correct, the reaction without NEt3 should be much faster because the presence of base may shift the equilibrium from E to C by deprotonating E and/or engaging cyclopentadiene in a hydrogen bond (for solvent effect on the catalyst elimination, see Scheme 4).
Scheme 4.

Solvent role in catalytic cycle
To understand the role of NEt3 on the reaction rate we set up a series of experiments reacting the carbonyl compounds with slight excesses of cyclopentadiene in the presence of only 10 mol% pyrrolidine and molecular sieves (Table 1, Method B). To our surprise the absence of NEt3 enhanced the fulvene formation rates. Again the correlation of the initial rates for the aromatic aldehyde series with Hammett σ constants provided a linear plot with slope (ρ) with a lower absolute value (Figure 2). The comparison of the reaction susceptibility constants implies that the presence of excess amine decelerates the reaction. This may arise from the direct interaction of intermediate D with the amine or an effect on the iminium salt formation. Therefore the use of molecular sieves makes an important difference in the method with a base. In the absence of NEt3 the rate-limiting step is probably the elimination reaction of D to 3. If so, the use of molecular sieves should not have an effect on the reaction rate (Table 5).
Figure 2.

Hammett plot of initial rate according to method B (log(RX) of fulvene 3a–b, g–i versus σ constants. The correlation equation is log(RX) = −1.34σ− log(RX=H) r2 = 0.9727
Table 5.
Effect of molecular sieves amount and the pyrrolidine concentration on the reaction rate
| Entrya | MS (g/mm ol) |
Cat. (mol%) |
Solvent | %Conversioni | ||
|---|---|---|---|---|---|---|
| 1 | 0 | 10 catb | MeOH | 80 | 100 | |
| 2 | 0.1 | 10 catb | MeOH | 74 | 100 | |
| 3 | 0.2 | 10 catb | MeOH | 78 | 100 | |
| 4 | 0.3 | 10 catb | MeOH | 85 | 100 | |
| 5 | 0.4 | 10 catb | MeOH | 84 | 100 | |
| 6 | 0.5 | 10 catb | MeOH | 84 | 100 | |
| 7 | 0.7 | 10 catb | MeOH | 84 | 100 | |
| 8 | 0 | 5 catb | MeOH | 54 | 84 | 100 |
| 9 | 0 | 1 catb | MeOH | 11 | 28 | 58 |
| 10 | 0 | 0.1 catb | MeOH | 0 | 0 | 0 |
| 11 | 0 | 0.01 catb | MeOH | 0 | 0 | 0 |
| 12 | 0 | 40 catc | MeOH | 0.5e | ||
| 13 | 0 | 80 catc | MeOH | 5f | ||
| 14 | 0 | 10 catb | CH3CN | 23 | 55 | 89g |
| 15 | 0 | 10 catb | DMSO | 19 | 48 | 88g |
| 16 | 0 | 10 catb | Ether | 0 | 0 | 0 |
| 17 | 0 | 10 catb | THF | 0 | 0 | 0 |
| 18 | 0 | 10 catb,d | Ether | 0 | 1 | 4h |
| 19 | 0 | 10 catb,d | THF | 0 | 1 | 4h |
All entries are for the reactions of 2b with cyclopentadiene in 1:1.2 ratios at room temperature.
Catalyst pyrrolidine.
Catalyst DEA.
200mol% MeOH.
The yield after 24 h is 39.
66% after 24 h.
100% after 24 h.
20% after 24 h.
Conversion at 0.5h, 1.7h and 5h, respectively.
Diethylamine was also tested for its catalytic activity (entries 12–13) and found that it is less effective than pyrrolidine. However, in spite of the higher concentrations required in some cases, especially in the synthesis of more volatile fulvenes, diethylamine could be preferred due to its lower boiling point.
2.2. Solvent effects
Finally, we thought that a study of the solvent effect on the reaction rate would shed some light on the mechanism of the reaction. The solvent series was chosen among those having polarity above and below that of MeOH (entries 1 and 14–17). The reaction rate in acetonitrile was only 1/4th of the rate in MeOH though the polarity of the former is very close to that of MeOH. The rate in much more polar DMSO was again similar to that in acetonitrile. In aprotic solvents as ether and THF no reaction was detected. However, addition of small amounts of MeOH to the latter mixtures provided the formation of 3b (Table 5, entries 18–19). This clearly proves the catalytic activity of protic solvents in the fulvene formation reactions. Having determined the importance of the solvent to be necessarily protic we have postulated a role for it as depicted in Scheme 4. Intermediate D is in equilibrium with E which probably undergoes concerted or near concerted elimination to 3 and the catalyst.28
If this postulate is correct, the presence of the more acidic and sterically less demanding water should accelerate the reaction. To prove this we reacted 2a with cyclopentadiene in water containing MeOH (Table 6). The result was dramatic: water did not lower the rate. Rather, dramatic rate acceleration was observed at 20% water concentration in MeOH. This clearly proves that the rate-determining step in the reactions without additional base involves the elimination step. However, it seems that this is not the case when sterically encumbered ketones and aldehydes are used (Tables 1–2, Entries l,w) where the presence of water decelerates the reactions (e.g., the conversions according to method C are 11 and 25% while according to B they are 82 and 50%, respectively, for entries l and w). This is most probably due to the fact that the iminium ion formation equilibrium is being shifted to the left.
Table 6.
Effect of water concentration on the reaction rate in the absence of NEt3.
| H2O (conc. %) |
Cata (mol%) | %Conversion(0.25 h) |
|---|---|---|
| 0 | 10 | 35b |
| 2.5 | 10 | 35 |
| 5 | 10 | 37 |
| 10 | 10 | 39 |
| 20 | 10 | 62 |
| 2.5 | 200 | 81 |
Pyrrolidine.
Product 3a crystallized in the reaction medium in all cases except the last entry at the 0.5 h and the reactions were completed when checked at 4.5 h by 1H NMR.
Table 2.
Method Ca: optimal conditions (% isolated yields)
| Entry | Carbonyl | Fulvene | (%) |
|---|---|---|---|
| a | 98 | ||
| b | 97 | ||
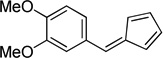
| c | 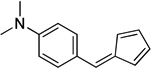 |
98 | |
| eb | 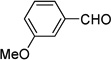 |
 |
95 |
| f | 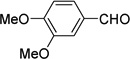 |
 |
98 |
| gb | 97 | ||
| hb | 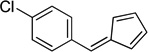 |
86 | |
| kb | 77 | ||
| le | 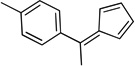 |
11 | |
| nb | 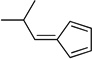 |
90 | |
| ob | CH3CHO | 90 | |
| pb | 85 | ||
| qb | 92 | ||
| rb | 95 | ||
| sb,c | 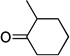 |
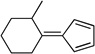 |
44 |
| vb,d | 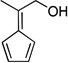 |
60 | |
| w | 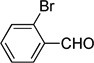 |
 |
25 |
| x | 30 |
Method C: same as B, no molecular sieves, 5 mL MeOH-H2O (4:1).
To a solution of 1o (20 mmol) and CP (10.0 mmol), in MeOH-H2O (10 mL 7/3) pyrrolidine (10 mol%,) was added, and the reaction mixture stirred for 2.5 h at 15 °C. The mixture was poured into an ice-cold mixture of brine solution (10 mL) containing AcOH (1 mmol). The upper oil phase was separated and dried over molecular sieves.
To a solution of 2s (5 mmol) and CP (10 mmol) in 5 mL of methanol-water (4:1), pyrrolidine (10 mmol, 2 equiv) was added, and the mixture stirred at room temperature overnight. The mixture was subjected to flash chromatography on silica gel to give 3s in 44% yield.
Due to anticipated greater water solubility of product, extractive workup was used.
3equiv of pyrrolidine was used: reaction time 24 h.
In addition to the accelerating effect discussed above, the use of MeOH-H2O proved to be of crucial importance in achieving excellent yields in amine-catalyzed synthesis of fulvenes derived from unbranched aliphatic aldehydes by suppressing substrate decomposition due to aldol condensation.
2.3. Effect of carbonyl structure and limitations of the amine base promoted fulvene syntheses
The structure of the carbonyl component appears to dictate the success of our amine catalyzed variants of fulvene syntheses. With aldehydes and sterically unencumbered aliphatic ketones pyrrolidine catalysis (Method C) appears to be the ideal method. As the steric hindrance increases in the ketone, the use of larger quantities of pyrrolidine (transition to Little-Stone procedure) becomes necessary. As a test substrate for a sterically hindered ketone we used 2-methylcyclohexanone (Table 1, entry s). Whereas methods A and B failed to affect fulvene formation, finally the use of 2 equivalents of pyrrolidine (as compared to 5 equiv. in Little’s procedure) gave the corresponding fulvene in 44% isolated yield. Benzophenone is unreactive under amine base catalysis as well as stoichiometric (or excess base) conditions. However, the use of excess (e.g., 3 equivalents) pyrrolidine provided the synthesis of 6-methyl-6-aryl fulvene from the corresponding acetophenone in good yield according to Method B and also according to Little’s conditions, providing that the reaction times are prolonged (Table 1, entry l). In the cases of diaryl ketones the classical Thiele procedure or a variant thereof with alkoxide base is the method of choice. In the case of keto acids (entries t and u) as well as phenolic substrates (e.g., Table 1, entry d) the best results were obtained with method A (without molecular sieves), still avoiding the use of excess pyrrolidine.
Another important outcome of our studies is the fact that our catalytic method is ideally suited for the synthesis and isolation of fulvenes derived from unbranched aldehydes, in particular those derived from acetaldehyde and propionaldehyde.29 The stoichiometric (or multiple equivalents) pyrrolidine method using unbranched aldehydes fails to give satisfactory results is presumably due to the fact that excess pyrrolidine in methanol solution results in considerable enamine formation which induces intermolecular reactions (i.e. aldol condensations) thus rendering the method unsuitable for these systems. We found that this difficulty can be ameliorated by working in a methanol-H2O mixture. It is therefore not surprising to note that particularly 6-alkylfulvenes with short alkyl chains (C1 and 2) that are popular starting materials used in organometallic chemistry are still prepared by the three-step Hafner procedure7 by way of alkyllithium addition to 6-N,N-dimethylaminofulvene instead of the Little procedure. Our catalytic method now offers an excellent direct alternative to Hafner’s multistep procedure for the synthesis of the aforementioned 6-alkylfulvenes.
3. Conclusions
A systematic investigation of the initial rates of sec-amine catalyzed fulvene formations with or without additional NEt3 and correlations with substituent constants allowed elucidating the mechanism of these reactions. Use of excess secondary amine as well as additional NEt3 suppressed the reaction rates. In the presence of added NEt3, the use of molecular sieves as water capturing agent was shown to be necessary. Our studies have established the following facts: a) protic solvents such as MeOH and water accelerate the reaction of cyclopentadiene with carbonyl compounds; b) the presence of tertiary amines retards reaction rates; c) electron-donating substituents on the phenyl ring accelerate rates. Based on our mechanistic studies a mechanism involving the interaction of cyclopentadiene with the preformed iminium salt appears reasonable. Based on the studies on the effects of base, MS and solvent we have developed an efficient, catalytic method for the synthesis of a variety of fulvenes. For crystalline products method C is exceptionally simple and practical. The use of MeOH-H2O provides the formation of products directly isolable in excellent purity and nearly quantitative yields by vacuum filtration. For liquid fulvenes the procedure is almost as simple. The isolation of the products involves the addition of a specified amount of brine solution containing an equivalent of AcOH at 0 °C resulting in the separation of the organic phase containing practically pure (>95% by 1H NMR) fulvene that can be pipetted out. Our amine-catalyzed procedure for fulvene synthesis also boasts the added advantage of providing a green alternative to the existing base-promoted methods. Moreover, our alternative methods A and B may be employed in certain cases with success when a phenolic OH or carboxylic acid functionality is present without sacrificing the catalytic use of pyrrolidine. Thus, a wider range of fulvenes, including 6-alkylfulvenes have now become accessible by our catalytic method.
4. Experimental Section
4.1. General
Melting points were determined in open class capillary tubes and are uncorrected. 1H and 13C NMR spectra were recorded on 300 MHz and 500 MHz NMR spectrometers, using CDCl3 as solvent and TMS as internal standard, unless specified otherwise. Most column chromatographic separations were carried out on a flash chromatography system using 40–60 µm silica gel columns using ethyl acetate/n-hexane solvent mixtures. For preparative TLC, Merck silica gel (grade 60 PF254) was used. All reactions were conducted under an atmosphere of dry nitrogen or argon. Non-deuterated solvents were dried and distilled prior to use. Fresh commercial samples of ethanal (free of trimer) and propanal and 2-methylpropanal were used for fulvene synthesis. Pyrrolidine is an exceptionally foul-smelling and toxic compound and should be handled with care in a well-ventilated hood. Fulvenes are oxygen- and heat-sensitive compounds; all reactions should be carried out under a nitrogen or argon atmosphere. Though all fulvenes prepared in this study are known compounds, their high resolution NMR spectra are included in the Supporting Information since they are not available in literature in several cases. The initial rates of the reactions used in Hammett30 correlations were measured at 23 (±1) °C. The change of fulvene or hemiacetal concentrations within the specified periods of time were monitored by 1H NMR by integrating the appropriate signals of the products and the starting carbonyl compounds.
4.2. Synthesis of (2-ylidene)cyclopenta-1,3-dienes 3. Method A. General Procedure
To a solution of aldehyde or ketone (5 mmol) and cyclopentadiene (12.5 mmol, 0.8262 g, 1.025 mL) in reagent grade methanol (5 mL), molecular sieve (3A, 1.5 g) was added; to this solution pyrrolidine (10 mol%, 0.0355 g) in NEt3 (7.24 mmol, 0.733 g) was added dropwise within 10 min. The mixture was stirred under nitrogen overnight. The mixture was filtered through sintered glass funnel and the solvent evaporated under vacuum to produce the corresponding fulvene in quantitative yield.
4.3. Synthesis of (2-ylidene)cyclopenta-1,3-dienes 3. Method B. General Procedure
To a solution of aldehyde or ketone (5 mmol) and cyclopentadiene (6.0 mmol, 0.3966 g, 0.492 mL) in reagent grade methanol (5 mL) molecular sieve (1.5 g, 3Å) was added, followed by dropwise addition of pyrrolidine (0.0355 g, 10 mol%). The mixture was filtered through sintered glass funnel and the solvent evaporated under vacuum to yield the corresponding fulvene in quantitative yield.
4.4. Synthesis of (2-ylidene)cyclopenta-1,3-dienes 3. Method C. General Procedure
To a solution of aldehyde or ketone (5 mmol) and cyclopentadiene (0.3966 g, 6.0 mmol/0.492 mL for the crystalline fulvenes or 0.4 mL/5 mmol for the aliphatic substrates) in MeOH-H2O (5 mL 4/1) pyrrolidine (0.0355 g, 10 mol%) was added. The formed crystals were filtered, washed with small amounts of cold MeOH-H2O mixture and dried in air. For the isolation of oily products the reaction mixture was transferred to an ice-cold mixture of brine solution and 10 mol% AcOH in a narrow graduated cylinder. The organic phase pure was pipetted out. Drying over molecular sieves produced practically pure (>95%) fulvene.
4.5. 1-(Cyclopenta-2,4-dienylidenemethyl)-4-methylbenzene (3a)
(Method C). Orange crystals, mp 60–61°C. Lit31 mp 70.5 °C. 1H NMR (500 MHz, CDCl3): δ 7.54 (d, J = 8.1 Hz, 2H), 7.26 (d, J = 8.0 Hz, 2H), 7.23 (s, 1H), 6.76 (d, J = 5.3 Hz, 1H), 6.71 – 6.68 (m, 1H), 6.55 – 6.52 (m, 1H), 6.36 (dt, J = 5.1, 1.7 Hz, 1H), 2.43 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 145.2, 140.2, 139.2, 135.9, 134.8, 131.5, 131.1, 130.2, 128.0, 120.9, 22.1.
4.6. 1-(Cyclopenta-2,4-dienylidenemethyl)-4-methoxybenzene (3b)
(Method C). Orange crystals, mp 68–70 °C. 1H NMR (500 MHz, CDCl3): δ 7.61 (d, J = 8.6 Hz, 2H), 7.19 (s, 1H), 6.97 (d, J = 8.8 Hz, 2H), 6.76 (d, J = 5.3 Hz, 1H), 6.69 (ddd, J = 5.2, 3.5, 1.6 Hz, 1H), 6.53 – 6.49 (m, 1H), 6.35 (dt, J = 5.1, 1.8 Hz, 1H), 3.88 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 161.4, 144.0, 139.0, 135.7, 133.2, 130.52, 130.3, 128.1, 120.6, 115.0, 56.1.
4.7. 4-(Cyclopenta-2,4-dienylidenemethyl)-N,N-dimethylbenzenamine (3c)
(Method C). Orange crystals, mp 104–105 °C. Lit31 mp 106.5 °C. 1H NMR (500 MHz, CDCl3): δ 7.62 (d, J = 8.7 Hz, 2H), 7.18 (s, 1H), 6.85 (d, J = 5.2 Hz, 1H), 6.74 (d, J = 9.0 Hz, 2H), 6.69 (ddd, J = 5.2, 3.6, 1.6 Hz, 1H), 6.52 – 6.45 (m, 1H), 6.37 (dt, J = 5.0, 1.8 Hz, 1H), 6.37 (dt, J = 5.0, 1.8 Hz, 1H), 3.07 (s, 6H). 13C NMR (126 MHz, CDCl3): δ 151.8, 141.5, 140.7, 134.4, 133.6, 128.9, 128.3, 125.4, 120.2, 112.7, 40.8.
4.8. 4-(Cyclopenta-2,4-dienylidenemethyl)phenol (3d)
(Method A). Solid, does not melt below 260 °C. 1H NMR (300 MHz, CDCl3): δ 7.54 (d, J = 8.6 Hz, 2H), 7.16 (s, 1H), 6.88 (d, J = 8.6 Hz, 2H), 6.72 (d, J = 5.2 Hz, 1H), 6.67 (dd, J = 5.2, 1.6 Hz, 1H), 6.50 (d, J = 4.9 Hz, 1H), 6.33 (dd, J = 3.3, 1.7 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ 156.6, 143.2, 138.1, 134.9, 132.6, 129.8, 129.6, 127.3, 119.8, 115.7. ESI-HRMS calculated for C12H11O+ (M+H+) 171.0804, found 171.0895
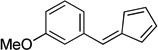
4.9. 1-(Cyclopenta-2,4-dienylidenemethyl)-3-methoxybenzene (3e)
(Method C). 1H NMR (300 MHz, CDCl3): δ 7.34 (t, J = 7.9 Hz, 1H), 7.19 (d, J = 8.7 Hz, 2H), 7.13 (s, 1H), 6.93 (dd, J = 8.2, 2.1 Hz, 1H), 6.69 (dt, J = 5.3, 3.5 Hz, 2H), 6.53 (d, J = 5.0 Hz, 1H), 6.34 (dd, J = 3.4, 1.7 Hz, 1H), 3.86 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 159.6, 145.3, 138.0, 135.4, 130.9, 129.6, 127.1, 123.2, 120.3, 115.56, 114.9, 55.2.
4.10. 4-(Cyclopenta-2,4-dienylidenemethyl)-1,2-dimethoxybenzene (3f)
(Method C). Orange crystals, mp 72–73 °C. Lit32 mp 72 °C. 1H NMR (500 MHz, CDCl3): δ 7.23 – 7.19 (m, 2H), 7.18 (s, 1H), 6.93 (d, J = 8.2 Hz, 1H), 6.75 (d, J = 5.3 Hz, 1H), 6.70 (ddd, J = 5.2, 3.5, 1.6 Hz, 1H), 6.51 (dd, J = 3.7, 1.4 Hz, 1H), 6.35 (dt, J = 5.1, 1.8 Hz, 1H), 3.95 (s, 3H), 3.95 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 151.1, 149.8, 144.2, 139.2, 135.8, 130.6, 130.5, 128.2, 125.5, 120.5, 113.9, 111.9, 56.7, 56.6.
4.11. 1-(Cyclopenta-2,4-dienylidenemethyl)benzene (3g)
(Method C). 1H NMR (300 MHz, CDCl3): δ 7.61 (d, J = 6.9 Hz, 2H), 7.51 – 7.32 (m, 3H), 7.24 (s, 1H), 6.76 – 6.64 (m, 2H), 6.54 (d, J = 5.1 Hz, 1H), 6.35 (dd, J = 3.4, 1.7 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ 145.2, 138.1, 136.7, 135.4, 130.8, 130.6, 128.9, 128.6, 127.1, 120.3.
4.12. 1-Chloro-4-(cyclopenta-2,4-dienylidenemethyl)benzene (3h)
(Method C). Red crystals, mp 43–45 °C. 1H NMR (300 MHz, CDCl3): δ 7.52 (d, J = 8.5 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H), 7.16 (s, 1H), 6.68 (dt, J = 13.4, 3.3 Hz, 2H), 6.54 (d, J = 4.8 Hz, 1H), 6.33 (dd, J = 3.6, 1.6 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ 145.6, 136.4, 135.8, 135.1, 135.1, 131.7, 131.1, 128.9, 127.1, 119.9.
4.13. 1-(Cyclopenta-2,4-dien-1-ylidenemethyl)-4-nitrobenzene (3i)
To a solution of 4-nitrobenzaldehyde (5 mmol) and cyclopentadiene (6.0 mmol, 0.3966 g, 0.492 mL) in reagent grade acetonitrile (5 mL) molecular sieve (1.5 g, 3Å) was added, followed by dropwise addition of pyrrolidine (0.0532 g, 15 mol%). The mixture was stirred overnight and filtered through sintered glass funnel and the solvent evaporated under vacuum. The mixture was separated by flash chromatography using silica packed column and hexane-ethyl acetate as eluent. For further purification preparative TLC was used. Reddish oil. 1H NMR (500 MHz, CDCl3): δ 8.27 (1H, d, J=9 Hz), 7.70 (1H, d, J=9 Hz), 7.20 (1H, s), 6.81 – 6.63 (1H, m), 6.57 (1H, dd, J=5, 2 Hz), 6.38 – 6.26 (1H, m). 13C NMR (126 MHz, CDCl3): δ 148.6, 147.8, 143.3, 137.5, 134.6, 132.9, 131.2, 127.2, 124.1, 120.1.
4.14. 1-(Cyclopenta-2,4-dien-1-ylidenemethyl)-3-nitrobenzene (3j)
Prepared and purified as 3i. Reddish oil. 1H NMR (300 MHz, CDCl3): δ 8.42 (1H, s), 8.21 (1H, dd, J=8, 1 Hz), 7.86 (1H, d, J=8 Hz), 7.60 (1H, t, J=8 Hz), 7.20 (1H, s), 6.76 – 6.67 (1H, m), 6.61 (1H, d, J=5 Hz), 6.56 (1H, d, J=5 Hz), 6.36 – 6.28 (1H, m). 13C NMR (75 MHz, CDCl3): δ 148.3, 147.4, 138.2, 136.9, 135.9, 134.2, 132.2, 129.5, 126.9, 124.8, 123.2, 119.6.
4.15. 5-(Propan-2-ylidene)cyclopenta-1,3-diene (3k)
(Method C). 1H NMR (500 MHz, CDCl3): δ 6.58 – 6.56 (m, 2H), 6.52 (dd, J = 4.0, 2.3 Hz, 2H), 2.25 (s, 6H). 13C NMR (126 MHz, CDCl3): δ 150.8, 143.3, 131.4, 121.3, 23.8.
4.16. 1-(1-(Cyclopenta-2,4-dien-1-ylidene)ethyl)-4-methylbenzene (3l)
Prepared according to Method B by using 3 equiv of pyrrolidine. Reddish oil. 1H NMR (300 MHz, CDCl3): δ 7.31 (2H, d, J=8 Hz), 7.21 (2H, d, J=8 Hz), 6.67 – 6.62 (1H, m), 6.57 (1H, d, J=5 Hz), 6.49 (1H, d, J=5 Hz), 6.24 – 6.20 (1H, m), 2.54 (3H, s), 2.40 (3H, s).13C NMR (75 MHz, CDCl3): δ 150.0, 143.0, 139.0, 138.2, 131.5, 131.2, 129.2, 128.5, 123.7, 121.0, 22.5, 21.2.
4.17. 5-(2-Methylpropylidene)cyclopenta-1,3-diene (3n)
(Method C). Yellow oil. 1H NMR (300 MHz, CDCl3): δ 6.54 (t, J = 3.6 Hz, 2H), 6.48 (d, J = 5.0 Hz, 1H), 6.26 (d, J = 10.1 Hz, 1H), 6.23 – 6.19 (m, 1H), 3.11 – 2.94 (m, 1H), 1.15 (d, J = 6.6 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 149.7, 143.4, 132.8, 130.6, 125.8, 119.1, 30.3, 23.0.
4.18. 5-Ethylidenecyclopenta-1,3-diene (3o)
(Method C). Yellow oil. 1H NMR (300 MHz, CDCl3): δ 6.61 (t, J = 1.6 Hz, 2H), 6.58 – 6.48 (m, 2H), 6.26 (dt, J = 5.1, 1.7 Hz, 1H), 2.21 (d, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 146.9, 137.8, 132.9, 130.6, 125.4, 118.8, 16.7.
4.19. 5-Propylidenecyclopenta-1,3-diene (3p)
(Method C). Yellow oil. Yield 85 %. 1H NMR (300 MHz, CDCl3): δ 6.55 (dd, J = 4.7, 3.5 Hz, 2H), 6.45 (dd, J = 18.2, 6.5 Hz, 2H), 6.23 (dd, J = 3.5, 1.6 Hz, 1H), 2.57 (p, J = 7.6 Hz, 2H), 1.17 (t, J = 7.5 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 145.2, 144.6, 132.9, 130.7, 125.6, 118.9, 24.3, 14.0.
4.20. 5-Cyclobutylidenecyclopenta-1,3-diene (3q)
(Method C). Yellow oil. 1H NMR (300 MHz, CDCl3): δ 6.45 (d, J = 5.2 Hz, 2H), 6.31 (dd, J = 4.4, 2.0 Hz, 2H), 3.14 (t, J = 7.8 Hz, 4H), 2.18 (p, J = 7.9 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 160.3, 138.7, 130.2, 119.8, 32.2, 16.9.
4.21. 5-Cyclopentylidenecyclopenta-1,3-diene (3r)
(Method C). Yellow oil. 1H NMR (300 MHz, CDCl3): δ 6.44 (dt, J = 6.2, 2.9 Hz, 4H), 2.84 (t, J = 7.2 Hz, 4H), 1.91 – 1.73 (m, 4H). 13C NMR (75 MHz, CDCl3): δ 162.5, 138.3, 130.0, 121.3, 33.0, 26.0.
4.22. 1-(Cyclopenta-2,4-dien-1-ylidene)-2-methylcyclohexane (3s)
Yellow oil. (Method C). 1H NMR (300 MHz, CDCl3): δ 6.70 – 6.45 (m, 4H), 3.48 – 3.32 (m, 1H), 2.94 (d, J = 14.0 Hz, 1H), 2.49 (td, J = 13.7, 4.9 Hz, 1H), 2.06 – 1.93 (m, 1H), 1.86 – 1.43 (m, 5H), 1.29 (d, J = 7.2 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 162.4, 139.2, 130.6, 120.1, 119.5, 35.1, 33.7, 28.9, 28.4, 20.5, 19.5.
4.23. 4-(Cyclopenta-2,4-dien-1-ylidene)pentanoic acid (3t)
(Method A without MS). Yellow solid mp 65–67 °C. Lit12 mp 66–67 °C. 1H NMR (300 MHz, CDCl3): δ 10.9 (br. s, 1H), 6.52 (d, J = 5.7 Hz, 4H), 2.95 – 2.82 (m, 2H), 2.62 (dd, J = 8.8, 7.0 Hz, 2H), 2.22 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 178.9, 149.6, 143.4, 131.5, 131.3, 120.7, 120.1, 33.4, 31.4, 20.5.
4.24. 5-(Cyclopenta-2,4-dien-1-ylidene)hexanoic acid (3u)
(Method A without MS). Yellow oil which solidifies on standing in the fridge. 1H NMR (300 MHz, CDCl3): δ 10.01 (br. s, 1H), 6.58 – 6.41 (m, 4H), 2.60 (t, J = 7.7 Hz, 2H), 2.41 (t, J = 7.4 Hz, 2H), 2.22 (s, 3H), 1.93 (p, J = 7.5 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 179.5, 151.6, 143.2, 131.1, 131.0, 120.6, 120.2, 35.7, 33.35, 23.6, 20.6. ESI-HRMS calculated for C11H15O2+ (M+H+) 179.1067, found 179.1086.
4.25. 2-(Cyclopenta-2,4-dienylidene)propan-1-ol (3v)
(Method C) Yellow oil which slowly solidifies in the fridge. mp 52–53 °C. Lit33 mp 54–54.5 °C. 1H NMR (300 MHz, CDCl3): δ 6.52 (dd, J = 6.2, 4.0 Hz, 4H), 4.54 (s, 2H), 2.28 (s, 3H), 1.91 (br.s, 1H). 13C NMR (75 MHz, CDCl3): δ 149.0, 142.5, 132.3, 131.7, 121.2, 119.5, 64.6, 18.3.
4.26. 1-Bromo-2-(cyclopenta-2,4-dien-1-ylidenemethyl)benzene (3w)
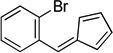
Reddish oil. 1H NMR (300 MHz, CDCl3): δ 7.62 (2H, ddd, J=14, 8, 1 Hz), 7.41 (1H, s), 7.38 – 7.30 (1H, m), 7.22 (1H, td, J=8, 2 Hz), 6.67 (1H, ddd, J=5, 3, 2 Hz), 6.57 (1H, d, J=5 Hz), 6.49 (1H, d, J=5 Hz), 6.43 – 6.36 (1H, m) 13C NMR (75 MHz, CDCl3): δ 146.4, 136.4, 135.5, 132.9, 132.8, 131.8, 129.9, 127.2, 126.4, 120.5. ESI-HRMS calculated for C12H10Br+ (M+H+) 232.9960, not observed.
4.27. 1-(Cyclopenta-2,4-dien-1-ylidenemethyl)-4-(trifluoromethyl)benzene (3x)
Red crystals mp 60–62 °C.1H NMR (300 MHz, CDCl3): δ 7.67 (4H, s), 7.21 (1H, s), 6.73 – 6.66 (1H, m), 6.61 (1H, d, J=5 Hz), 6.56 (1H, d, J=5 Hz), 6.33 (1H, dd, J=4, 2 Hz) 13C NMR (75 MHz, CDCl3): δ 147.0, 140.0, 136.4, 135.6, 131.9, 130.5, 126.9, 125.42 (q, J=3.75Hz), 120.0.
4.28. Cyclopenta-1,3-dien-1-yl(4-nitrophenyl)methanol compound with cyclopenta-1,4-dien-1-yl(4-nitrophenyl)methanol (1:1) (4i)
Yellowish oil. 1H NMR (300 MHz, CDCl3): δ 8.15 (2H, d, J=9 Hz), 7.54 (2H, dd, J=8, 7 Hz), 6.49 – 6.20(3H, m), 5.73 (1H, d, J=12 Hz), 3.01 (1H, t, J=3 Hz), 3.11 – 2.59 (2H, m) 13C NMR (75 MHz, CDCl3): δ 150.6, 149.9, 149.0, 148.0, 147.0, 147.0, 135.3, 133.8, 131.6, 130.9, 130.0, 128.9, 127.0, 126.8, 123.5, 72.0, 71.4, 41.3, 40.0. ESI-HRMS calculated for C12H10Br+ (M+H+) 232.9960, not observed.
Supplementary Material
Scheme 1.

Synthesis of Fulvene derivatives
Scheme 2.

Hemiacetal formation equilibria of aromatic aldehydes
Table 4.
Determination of rate law for the pyrrolidine (5 mol%) -catalyzed fulvene formation between 2b and 1 in MeOH
Initial concentration.
The initial rates (mole L−1 h−1) were determined at the 10th min of the reactions at room temperature
Acknowledgment
This work was supported by funds from the National Institutes of Health, (Grant No. 1SC1GM082340). Dr. Coskun aknowledges a BIDEP-2219 fellowship from the Turkish Scientific and Technological Research Council (TUBİTAK). We also acknowledge funding from the National Science Foundation (Grants No. DBI-0521342and DUE-9451624) for the purchase of 500 MHz and 300 MHz NMR spectrometers.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
1H and 13C NMR spectra of compounds 3a–h, k, n–s and v. Supplementary data associated with this article can be found, in the online version, at
References and notes
- 1.For reviews, see: Yates P. Advanced Alicyclic Chemistry. Vol. 2. New York: Academic Press; 1968. pp. 59–184. Bergman ED. Chem. Rev. 1968;68:41–84. Day JH. Chem. Rev. 1963;53:167–189. Zeller KP. Pentafulvenes. Methoden der Organischen Chemie. 1985;Vo1.5/2c:504–684. Neuenschwander M. Fulvenes. In: Patai S, editor. The Chemistry of Double-Bonded Functional Groups. New York: John Wiley; 1989. pp. 1131–1268..
- 2.(a) Thiele J. Chem. Ber. 1900;33:666. [Google Scholar]; (b) Thiele J. Justus Liebigs Ann. Chem. 1906;348:1. [Google Scholar]
- 3.(a) Freiesleben W. Angew. Chem. 1963;75:576. [Google Scholar]; (b) Freiesleben W. 3192275. US Patent. 1965
- 4.(a) Kerber RC, Linde HG., Jr J. Org. Chem. 1966;31:4321. [Google Scholar]; (b) Olah GA, Prakash GKS, Liang G. J. Org. Chem. 1977;42:661. [Google Scholar]; (c) Hanack M, Eggensperger H. Justus Liebigs Ann. Chem. 1963;31:663. [Google Scholar]
- 5.Neuenschwander M, Kyburz R, Iseli R. Chimia. 1970;24:342. [Google Scholar]
- 6.Kyburz R, Schaltegger H, Neuenschwander M. Helv. Chim. Acta. 1971;54:1037–1046. [Google Scholar]
- 7.Hafner K, Sturm E. Angew. Chem. Int. Ed. Engl. 1964;3:749. [Google Scholar]
- 8.Macomber DW, Spink WC, Rausch MD. J. Organomet. Chem. 1983;250:311–318. [Google Scholar]
- 9.Alves NP, Maia AdS, de Oliveira WJ. Alloys Compd. 2000:3003–3304. 178–181. and references cited therein. [Google Scholar]
- 10.Greger I, Kehr G, Frohlich R, Erker G. Organometallics. 2010;29:3210–3221. [Google Scholar]
- 11.Boul PJ, Reutenauer P, Lehn J-M. Org. Lett. 2005;7:15–18. doi: 10.1021/ol048065k. [DOI] [PubMed] [Google Scholar]
- 12.Reutenauer P, Buhler E, Boul PJ, Candau SJ, Lehn J-M. Chem. Eur. J. 2009;15:1893–1900. doi: 10.1002/chem.200802145. [DOI] [PubMed] [Google Scholar]
- 13.McCain GH. J. Org. Chem. 1958;23:632–633. [Google Scholar]
- 14.McCain GH. 3051765. US Patent. 1962
- 15.Stone KJ, Little RD. J. Org. Chem. 1984;49:1849–1853. [Google Scholar]
- 16.Little RD. private communication. [Google Scholar]
- 17.Griesbeck AG. J. Org. Chem. 1989;107:5308–5309. [Google Scholar]
- 18.Erden I, Xu F-P, Sadoun A, Smith W, Sheff G, Ossun M. J. Org. Chem. 1995;60:813–820. [Google Scholar]
- 19.Erden I, Gärtner C. Tetrahedron Lett. 2009;50:2381–2383. doi: 10.1016/j.tetlet.2009.02.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erden I, Amputch M. Tetrahedron Lett. 1987;28:3779–3782. [Google Scholar]
- 21.Erden I, Drummond J, Alstad R, Xu F. Tetrahedron Lett. 1993;34:1255–1258. [Google Scholar]
- 22.Erden I, Drummond J, Alstad R, Xu F. J. Org. Chem. 1993;58:3611–3612. [Google Scholar]
- 23.Erden I, Xu F, Cao W. Angew. Chem., Int. Ed. 1997;36:1516–1518. [Google Scholar]
- 24.Erden I, Öcal N, Song J, Gleason C, Gärtner C. Tetrahedron. 2006;62:10676–10682. doi: 10.1016/j.tet.2006.07.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The powerful electron-withdrawing NO2 group, as expected, prevented fulvene formation, instead, aldehyde 2i rapidly converted to a polymeric material. To explore the mechanism of this unusual conversion we reacted equimolar amounts of 2i with cyclopentadiene in methanol at room temperature. The 1H NMR spectrum of the mixture revealed the formation of the hemiacetal of 2i in 23% yield within 15 min. The addition of NEt3 to the mixture only led to the polymerization of 2i; cyclopentadiene remained unchanged.
- 26.Coşkun N, Aksoy C. Tetrahedron Lett. 2009;50:3008–3012. [Google Scholar]
- 27.The 4-NMe2 group somewhat deviates from linearity presumably due to direct interaction with the reaction center. In the case of 2d the reaction rate is lowered because of the engagement of the catalyst with the acidic phenol function.
- 28.Patil MP, Sunoj RB. J. Org. Chem. 2007;72:8202–8215. doi: 10.1021/jo071004q. [DOI] [PubMed] [Google Scholar]
- 29.In our hands, Little’s procedure did not produce 6-methylfulvene from ethanal while with propionaldehyde under the same conditions a complex mixture was obtained containing 6-ethylfulvene in 17% along with dicyclopentadiene, as determined by integration of the relevant signals in the crude 1H NMR spectrum.
- 30.(a) Jaffe HH. Chem. Rev. 1953;53:191–261. [Google Scholar]; b) Hansch C, Leo A, Taft RW. Chem. Rev. 1991;91:165–195. [Google Scholar]; c) March J. Advanced Organic Chemistry Reactions Mechanism and Structure. 3rd ed. Wiley & Sons; 1985. p. 244. [Google Scholar]
- 31.Kresze G, Goetz H. Chem. Ber. 1957;90:2161–2176. [Google Scholar]
- 32.Öcal N, Bagdatli E, Arslan M. Turk. J. Chem. 2005;29:7–16. [Google Scholar]
- 33.Knight DB, Burnette JC, Hall RW, Lambeth GH. J. Chem. Eng. Data. 1980;25:184–186. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.