

Abstract
A series of C1, C2, C3 and N6 analogs of nantenine (2) was synthesized and evaluated in 5-HT2A and α1A receptor functional assays. Alkyl substitution of the C1 and N6 methyl groups of nantenine provided selective 5-HT2A and α1A antagonists respectively. The C2 alkyloxy analogs studied were generally selective for α1A vs 5-HT2A. The C3 bromo analog 15 is one of the most potent aporphinoid 5-HT2A antagonists known presently.

Keywords: aporphine, nantenine, MDMA, 5-HT2A, α 1A
1. Introduction
Aporphines are a group of tetracyclic alkaloids that are a subset of the ubiquitous tetrahydroisoquinoline family. The aporphine template is known to be associated with a range of biological activities. For example, aporphines have been explored as antituberculosis and cytotoxic agents.1, 2 In the realm of central nervous system (CNS) activity, members of the aporphine class are known to inhibit the enzyme acetylcholinesterase - an important therapeutic target in current treatment modalities for Alzheimers disease.3-5 Behavioral and physiological effects of the designer drug MDMA (“Ecstasy”, 1) are antagonized by nantenine (2), an isolate of a number of Lauraceous plants. With regards to clinically available aporphine drugs, apomorphine (3) is a potent dopamine D1/D2 agonist that is used to treat symptoms of Parkinson’s disease.6
Naturally occurring aporphines and their synthetic derivatives are known to have affinity for dopaminergic, adrenergic and serotonergic receptors.7-14 The aporphine chemotype may be regarded as a “privileged” CNS receptor template. Indeed, this scaffold represents an attractive opportunity to identify and develop selective mono-potent as well as multi-potent CNS receptor ligands for dopaminergic, adrenergic and serotonergic receptors. Such molecules will be useful as interrogative chemical tools and potential therapeutic interventions for a variety of neuropsychiatric conditions such as stress, depression, anxiety, psychosis and psychostimulant abuse.
As indicated earlier, a number of groups have investigated the structure-activity properties of aporphines (particularly apomorphine derivatives) as dopamine D1 and D2 receptor ligands. In contrast, the structure-activity relationships (SAR) of aporphines as 5-HT2A receptor ligands have been little explored. We are particularly interested in “tooling” aporphines as 5-HT2A and α1A dual antagonists and as probes to study antagonism of MDMA’s behavioral and physiological effects. Our interest is propelled by abundant literature studies that implicate 5-HT2A and α1 adrenergic receptors in mediating physiological and psychobehavioral effects of MDMA in animals and humans. 15-21 Currently, no medications are specifically approved to treat adverse effects of “Ecstasy” overdose although the muscle relaxant, dantrolene, has been effectively used as an antidote in cases of hospitalization due to “Ecstasy”- induced hyperthermia.22
Prior to our forays in this area, only one SAR study on aporphines at the 5-HT2A receptor was reported. In that study, conducted with (±)-nantenine (2) as the lead, it was revealed that replacement of the C1 methoxy group with a hydroxyl group gave decreased 5-HT2A affinity.23 Replacement of the N6 methyl group with a hydrogen or ethyl group also gave reduced affinity. We have recently reported further investigations at the C1 position and have identified new 5-HT2A antagonists up to 12 times more potent than nantenine.24, 25
In addition, we have screened (±)-nantenine in a CNS panel of over 30 receptors, ion-channels and transporters and determined that the molecule is a potent and selective α1A ligand.26 Others have documented the affinity trends of aporphines at the α1A receptor, but no study has done so with nantenine as the lead.12 One prior study has investigated the structure-affinity relationships of nantenine at α1 receptors.27 Here it was reported that replacement of the C1 methoxy group with a hydroxyl group gave improved α1 affinity. Substitution of the N6 methyl group of nantenine with a hydrogen or ethyl group gave reduced affinities however.
Taken together, the results of previous investigations of (±)-nantenine suggest that the substituents on the aporphine core are important determinants of its 5-HT2A and α1A activity. While the ultimate goal of optimizing aporphines as 5-HT2A/α1A dual antagonist probes seems achievable, more extensive SAR studies are necessary. In this manuscript, we describe an SAR study to further probe the effects of structural manipulations in the ring A and N6 regions of the molecule on antagonism of 5-HT2A and α1A receptors. For comparison to our prior work, all evaluations in the following discussion were conducted with racemic aporphine derivatives.
2. Results and Discussion
2.1 Chemistry
The ring A C1 modified racemic analogs were prepared using methods analogous to that previously described via the key phenolic precursor 5.24, 28 Standard Williamson ether O-alkylation of 5 followed by reduction of the carbamate group gave the C1 analogs 6a-i. Synthesis of the ring A C2 analogs is shown in Scheme 1. The key intermediate 12 was synthesized from readily available 7 and used to prepare C2 analogs as shown. Thus, coupling of 7 with readily available bromomethylenedioxyphenylacetic acid gave amide 8.29 Bischler-Napieralski cyclization of 8 gave an imine which was subsequently reduced to secondary racemic amine 9 30 (without purification of the imine). Reaction of 9 with Boc anhydride gave the carbamate 10, which was then cyclized via a palladium-mediated microwave-assisted procedure to afford the aporphine 11. Removal of the benzyl protecting group of 11 then gave phenol 12. Alkylation of 12 with respective alkyl bromides afforded the boc protected aporphines 13a-13f. Removal of the Boc group with ZnBr2 and standard reductive amination gave the target C2 derivatives 14a-14f. (We attempted to reduce the Boc group directly to the N-methyl group with lithium aluminium hydride but found that this gave competing cleavage of the Boc group).
Scheme 1.

Synthesis of C2 ring A analogs
Bromination of (±)-nantenine with bromine/acetic acid (Figure 3) gave the C3-bromo analog 15. This reaction also produced a substantial amount of tribromo derivative 16.
Figure 3.

Bromination of nantenine
The preparation of N6 analogs was achieved by N-alklyation of secondary (±)-amine 17 (see ref 4) under reductive amination conditions (Figure 4)
Figure 4.
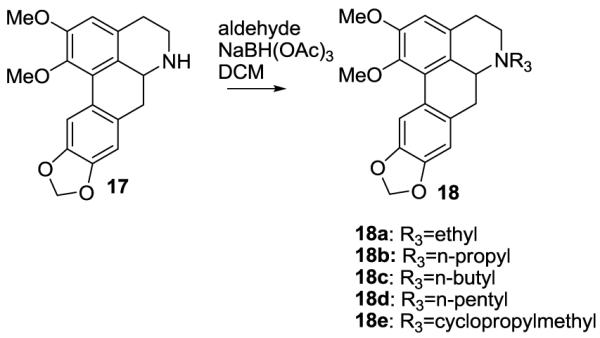
Synthesis of N-alkyl analogs
2.2 Pharmacology
All analogs were screened at 10 μM in multi-well format for intrinsic (agonist) and antagonist activity at the human 5-HT2A receptor using FLIPR Tetra (Molecular Devices, Sunnydale, CA) functional assays that detect receptor-mediated mobilization of internal calcium with a calcium sensitive fluorescent dye. Compounds that showed no intrinsic activity in the functional assay and inhibited the increase in basal fluorescence elicited by the EC80 of 5-HT by at least 50%, had their Ke (apparent affinity in a functional assay) determined. Ke values were determined by running an 8-point half-log 5-HT concentration response curve in the presence and absence of a single concentration of antagonist. EC50 values were calculated for 5-HT (A) and 5-HT + test compound (A’), and these used to calculate the test compound Ke using the formula: Ke = [L]/(DR-1), where [L] equals the concentration of test compound in the assay and DR equals the dose ratio or A’/A. A similar set of assays was performed for the α1A-adrenergic receptor.
In our previous SAR study, we found that successive alkyl homologation of the C1 methyl group of nantenine resulted in a progressive increase in 5-HT2A antagonist activity (ethyl to n-pentyl: 890 to 171 nM). 24 Additionally, the C1 cyclopropylmethyloxy analog was the most potent 5-HT2A antagonist (68 nM) identified in that study. Therefore, we continued our exploration of this position by investigating other alkyloxy, and cycloalkyloxy analogs. n-Hexyloxy analog 6a (Ke=71 nM, Table 1), showed activity comparable to the cyclopropylmethyloxy compound and 2-fold improved activity as compared to the previously reported n-pentyloxy analog. Compound 6b was prepared as a ring-opened analog for comparison to the cyclopropylmethyloxy compound but this had 5-fold lower 5-HT2A antagonist activity. Nevertheless, we anticipated that homologation of 6b would cause a rebound in antagonist activity as seen with the C1 n-alkyloxy series and so we prepared and evaluated isopentyloxy analog 6c and 2-ethylbutyloxy analog 6d. However, homologation did not improve the antagonist activity. In fact in the case of 6c, this compound had weak agonist activity (19% of 5-HT Emax). Next we decided to investigate other cycloalkyloxy analogs. The cyclohexylmethyloxy analog (6g) showed moderate activity, albeit 25-fold lower than the cyclopropyloxy analog. Other cycloalkyloxy derivatives tested showed weak agonist activity (25% and 35% of 5-HT Emax for 6e and 6f respectively). However, we were pleased to observe that the allyloxy analog (6h) had activity comparable to the cyclopropylmethyloxy analog. Since the alkene functionality of 6h is electronically similar to a cyclopropyloxy group, this may account for the similar activities seen; this is in need of further investigation. We then decided to test the tolerance for a polar group in this region. Addition of a hydroxypropyloxy moiety (6i) was associated with a reduction in 5-HT2A antagonist activity. Compounds 6a-6i were evaluated in α1A assays but were devoid of activity.
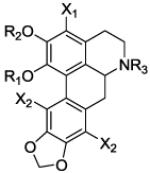
Table 1.
Ke values for ring A and N6 analogs at 5-HT2A and α1A receptors
 |
|||||||
|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | X1 | X2 | Ke ± SEM (nM)a |
|
| 5-HT2A | α 1A | ||||||
| 6a | n-Hex | Me | Me | H | H | 71±19 | >10,000 |
| 6b | Isobu | Me | Me | H | H | 367±82 | >10,000 |
| 6c | Isopen | Me | Me | H | H | NDb | >10,000 |
| 6d | 2-EthylBu | Me | Me | H | H | 806±152 | >10,000 |
| 6e | CyclobutylMe | Me | Me | H | H | NDb | >10,000 |
| 6f | CyclopentylMe | Me | Me | H | H | NDb | >10,000 |
| 6g | CyclohexylMe | Me | Me | H | H | 1722±258 | >10,000 |
| 6h | Allyl | Me | Me | H | H | 70±15 | >10,000 |
| 6i | HydroxyPr | Me | Me | H | H | 708±316 | >10,000 |
| 14a | Me | Et | Me | H | H | 378±92 | 52±13 |
| 14b | Me | n-Pr | Me | H | H | 389±93 | 133±45 |
| 14c | Me | n-Bu | Me | H | H | 943±283 | 234±52 |
| 14d | Me | n-Pen | Me | H | H | >10,000 | 449±169 |
| 14e | Me | CyclopropylMe | Me | H | H | 484±123 | 195±86 |
| 14f | Me | Benzyl | Me | H | H | 154±76 | 1917±226 |
| 15 | Me | Me | Me | Br | H | 53±14 | >10,000 |
| 16 | Me | Me | Me | Br | Br | 43±11 | >10,000 |
| 18a | Me | Me | Et | H | H | >10,000 | 26±6 |
| 18b | Me | Me | n-Pr | H | H | >10,000 | 38±3 |
| 18c | Me | Me | n-Bu | H | H | >10,000 | 210±50 |
| 18d | Me | Me | n-Pen | H | H | >10,000 | 720±204 |
| 18e | Me | Me | CyclopropylMe | H | H | >10,000 | 319±60 |
| 2 | Me | Me | Me | H | H | 850±6c | 36±7 |
| prazosin | 1.1 ± 0.4 | ||||||
| ketanserin | 32c,d | ||||||
Values represent mean ± SEM for three independent experiments.
ND = Ke not determined (compounds were weak agonists).
Data from reference 25
IC50 determined in the presence of 5-HT EC80
We then progressed to examine C2 nantenine analogs. With regards to 5-HT2A activity, the ethyloxy and n-propyloxy analogs (14a and 14b respectively) were equipotent. Further extension of the alkyl chain resulted in decreased antagonist activity with a plummet in activity in going from the n-butyloxy analog to the n-pentyloxy homolog (14c to 14d). The C2 cyclopropylmethyloxy analog was found to have moderate activity that was comparable to 14a and 14b. Benzyloxy analog (14f) was the most potent C2 analog tested. In the α1A assay a clear SAR trend was observed. The C2 ethyloxy analog had the highest activity though this was slightly lower than that obtained for nantenine in this assay. Increasing the alkyl chain length at this position led to a progressive decrease in α1A antagonist activity. The C2 alkyloxy analogs (14a-14d) are generally more selective for α1A vs 5-HT2A. Interestingly, this selectivity is reversed with the benzyloxy analog 14e.
The C3 bromo analog (15) had high 5-HT2A antagonist activity (Ke=53 nM). Polybromination of ring D (16) did not affect this activity. Compounds 15 and 16 are the most potent 5-HT2A aporphine antagonists identified up to now. Both of these halogenated analogs were found to be highly selective for the 5-HT2A receptor vs the α1A receptor.
None of the N6 analogs tested had activity at the 5-HT2A receptor. The N6 ethyl (18a) and N6 n-propyl (18b) congeners had activities that were comparable to nantenine in the α1A assay. Further lengthening of the alkyl chain gave reduced α1A antagonist activity.
The preceding evaluations were conducted with the compounds in racemic form. It will be interesting to evaluate enantiomers of these molecules to ascertain whether there is any enantio-preference with respect to selectivity and activity. This is especially necessary in light of the fact that series of aporphine enantiomers have been shown to have opposing effects (agonism and antagonism) at the related 5-HT1A receptor.11, 31, 32
3. Conclusions
In summary, we have identified a number of new aporphine 5-HT2A and α1A antagonists via manipulation of the ring A and N6 substituents of nantenine. The results of our SAR study suggest that C1 and C3 substituents may be modified to develop potent 5-HT2A antagonists that are highly selective vs the α1A receptor. In contrast, the C2 alkyloxy analogs are generally more selective for the α1A receptor vs. 5-HT2A. However, it would appear that only relatively small alkyl groups are tolerated at this position for optimal activity at either receptor. Similarly, at N6 the relatively small methyl group is best tolerated. Compounds 15 and 16 are the most potent aporphine 5-HT2A antagonists known. It is clear that the aporphine skeleton is sensitive to structural changes and that this template is amenable to the development of selective 5-HT2A vs α1A and selective α1A vs 5-HT2A antagonists.
Altogether, these results suggest that it is possible to synthesize potent dual-acting 5-HT2A/α1A aporphine ligands because the receptor selectivity can be directed through modifications of different substituents. Evaluation of the aporphine enantiomers is an important direction for future work. Our research team is actively investigating these avenues.
4. Experimental section
4.1 General experimental procedures
Reagent grade chemicals and solvents were purchased from Sigma-Aldrich Inc. or Fisher Scientific Inc. Reactions were monitored by TLC with Analtech Uniplate silica gel G/UV 254 precoated plates (0.2 mm). TLC plates were visualized by UV (254 nm), by iodine vapour or by staining with phosphomolybdic acid reagent followed by heating. Microwave reactions were conducted on a CEM Discover microwave reactor. Flash column chromatography was performed with Silicagel 60 (EMD Chemicals, 230-400 mesh, 0.04-0.063 μm particle size). High Resolution Electrospray Mass Spectra (HRESIMS) were obtained using an Agilent 6520 Q-TOF instrument. NMR data were collected on a Bruker 500 MHz machine with TMS as internal standard. Chemical shift (δ) values are reported in ppm and coupling constants in Hertz (Hz). Melting points were obtained on a Mel-Temp capillary electrothermal melting point apparatus.
4.2 Synthesis of C1 analogs (6)
C1 analogs were prepared from 4 using methods as described in reference 25.
4.2.1 1-(Hexyloxy)domesticine (6a)
Off white solid; mp: 68-70 °C; 1H NMR (500 MHz, CDCl3) δ 7.95 (s, 1H), 6.74 (s, 1H), 6.57 (s, 1H), 5.97 (d, J = 1.5 Hz, 1H), 5.95 (d, J = 1.5 Hz, 1H), 3.84 (s, 3H), 3.83.3.81 (m, 1H), 3.61-3.59 (m, 1H), 3.16-3.11 (m, 1H), 3.03 (dd, J=11.3, 6.0 Hz, 1H), 2.98-2.95 (m, 2H), 2.68-2.64 (br d, J = 16 Hz, 1H), 2.54-2.49 (m, 5H), 1.74-1.60 (m, 2H), 1.45-1.26 (m, 6H), 0.88-0.85 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 152.2, 146.48, 143.9, 130.8, 129.9, 128.8, 128.5, 127.5, 126.0, 110.8, 109.5, 108.3, 101.0, 73.4, 64.5, 56.0, 53.4, 44.1, 35.3, 31.8, 30.3, 29.3, 26.0, 22.8, 14.2 ; HRMS (ESI) m/z calcd. for C25H31NO4 ([M+H]+), 410.2326, found 410.2330
4.2.2 1-(Isobutyloxy)domesticine (6b)
Brown Oil; 1H NMR (500 MHz, CDCl3) δ 7.95 (s, 1H), 6.74 (s, 1H), 6.57 (s, 1H), 5.97 (s, 1H), 5.94 (s, 1H), 3.84 (s, 3H), 3.61 (t, J=7.6 Hz, 1H), 3.36 (t, J = 7.6 Hz, 1H), 3.16-3.10 (m, 1H), 3.04-2.95 (m, 2H), 2.67-2.64 (br. d, J=15.7 Hz, 1H), 2.54-2.48 (m, 5H), 2.04-1.99 (m, 1H), 1.01-0.95 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 152.2, 146.4 (×2), 144.1, 130.7, 128.4, 127.4, 125.8, 110.9, 109.6, 108.2, 101.0, 100.84, 79.8, 62.8, 56.1, 53.5, 44.2, 35.3, 29.3, 29.2, 19.7, 19.5; HRMS (ESI) m/z calcd for C23H27NO4 ([M+H]+), 382.2013, found 382.2017.
4.2.3 1-(Isopentyloxy)domesticine (6c)
Brown Oil; 1H NMR (500 MHz, CDCl3) δ 7.95 (s, 1H), 6.74 (s, 1H), 6.57 (s, 1H), 5.96 (s, 1H), 5.95 (s, 1H), 3.86-3.82 (m, 4H), 3.66-3.61 (m, 1H), 3.17-3.10 (m, 1H), 3.04-3.01 (m, 1H), 2.98-2.94 (m, 2H), 2.68-2.64 (m, 1H), 2.55-2.48 (m, 5H), 1.84-1.76 (m, 1H), 1.63-1.52 (m, 2H), 0.88-0.87 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 152.1, 146.4, 144.1, 143.7, 130.7, 129.7, 128.5, 127.3, 125.7, 110.5, 109.2, 108.1, 100.8, 71.5, 62.5, 55.8, 53.2, 40.3, 39.1, 35.1, 24.7, 22.6, 22.4; HRMS (ESI) m/z calcd for C24H29NO4 ([M+H]+), 396.4914, found 396.4914
4.2.4 1-(2-Ethylbutyloxy)domesticine (6d)
Brown solid; mp: 62-64 °C; 1H NMR (500 MHz, CDCl3) δ 7.91 (s, 1H), 6.74 (s, 1H), 6.57 (s, 1H), 5.95 (d, J= 1.4 Hz, 1H), 5.94 (d, J= 1.4 Hz, 1H), 3.84 (s, 3H), 3.74 (dd, J= 9.1, 5.3 Hz, 1H), 3.49 (dd, J= 9.0, 5.3 Hz, 1H), 3.17-3.10 (m, 1H), 3.03 (dd, J=11.3, 5.1 Hz, 1H), 2.98-2.94 (m, 2H), 2.66 (dd, J=16.3, 3.4 Hz, 1H), 2.54-2.48 (m, 5H), 1.56-1.34 (m, 5H), 0.86 (d, J= 7.2 Hz, 3H), 0.83 (d, J=7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 152.1, 146.4, 146.3, 144.1, 130.6, 128.3, 127.5, 127.4, 125.8, 110.8, 109.6, 108.2, 100.9, 75.2, 62.7, 55.9, 53.4, 44.1, 42.1, 35.2, 29.3, 23.4, 23.2, 11.3, 11.1; HRMS (ESI) m/z calcd for C25H31NO4 ([M+H]+), 410.2326, found 410.2329.
4.2.5 1-(cyclobutylmethoxy)domesticine (6e)
Brown Oil; 1H NMR (500 MHz, CDCl3) δ 7.93 (s, 1H), 6.74 (s, 1H), 6.56 (s, 1H), 5.98 (d, J=1.5 Hz 1H), 5.94 (d, J=1.5 Hz, 1H), 3.85 (s, 3H), 3.83 (dd, J=9.4, 6.7 Hz, 1H), 3.59 (dd, J=9.4, 7.4 Hz, 1H), 3.18-3.11 (m, 1H), 3.05 (dd, J=6.0, 5.5 Hz, 1H), 3.0-2.95 (m, 2H), 2.69-2.64 (m, 2H), 2.56-2.50 (m, 5H), 2.06-1.95 (m, 2H), 1.92-1.85 (m, 1H), 1.83-1.66 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 152.1, 146.4, 146.7, 143.7, 130.7, 128.4, 127.5, 127.2, 125.9, 110.7, 109.7, 108.2, 101.0, 62.6, 56.1, 53.3, 44.0, 35.5, 35.2, 29.8, 29.1, 25.5, 25.0, 18.6; HRMS (ESI) m/z calcd for C24H27NO4 ([M+H]+), 394.2013, found 394.2017.
4.2.6 1-(cyclopentylmethoxy)domesticine (6f)
Brown Oil; 1H NMR (500 MHz, CDCl3) δ 7.97 (s, 1H), 6.74 (s, 1H), 6.57 (s, 1H), 5.97 (d, J=1.3 Hz, 1H), 5.94 (d, J=1.3 Hz, 1H), 3.85 (s, 3H), 3.73 (dd, J = 7.0, 7.0 Hz, 1H), 3.47 (dd, J = 7.6, 7.6 Hz, 1H), 3.16-3.10 (m, 1H), 3.03 (dd, J = 11.6, 5.5 Hz, 1H), 2.98-2.94 (m, 2H), 2.66 (br. d, J = 16.1 Hz, 1H), 2.54-2.47 (m, 4H), 2.31-2.25 (m, 1H), 1.92-1.85 (m, 1H), 1.80-1.71 (m, 2H), 1.55-1.49 (m, 4H), 1.33-1.22 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 152.1, 146.4, 146.3, 144.0, 130.7, 128.4, 127.4, 127.2, 125.9, 110.8, 109.7, 108.2, 101.0, 62.7, 56.0, 53.4, 44.1, 40.2, 35.2, 29.9, 29.4, 29.3, 25.6, 25.0, 25.5; HRMS (ESI) m/z calcd for C25H29NO4 ([M+H]+), 408.2169, found 408.2172.
4.2.7 1-(cyclohexylmethoxy)domesticine (6g)
Yellow Oil; 1H NMR (500 MHz, CDCl3) δ 7.94 (s, 1H), 6.74 (s, 1H), 6.57 (s, 1H), 5.97 (s, 1H), 5.94 (s, 1H), 3.84 (s, 3H), 3.60 (dd, J = 8.7, 6.1 Hz, 1H), 3.40 (t, J = 7.6 Hz, 1H), 3.17-3.10 (m, 1H), 3.05-2.94 (m, 3H), 2.66 (br. d. J = 16.0 Hz, 1H), 2.53-2.49 (m, 5H), 1.90-1.63 (m, 10H), 1.17-1.10 (m,1H); 13C NMR (125 MHz, CDCl3) δ 152.2, 146.49, 146.45, 144.2, 130.7, 128.4, 127.4 (× 2), 125.8, 110.9, 109.7, 108.2, 101.0, 78.7, 62.7, 56.1, 53.4, 44.1, 38.7, 35.2, 30.2, 29.9, 29.1, 26.7, 26.1, 26.0 ; HRMS (ESI) m/z calcd for C26H31NO4 ([M+H]+), 422.2326, found 422.2329.
4.2.8 1-(allyloxy)domesticine (6h)
Brown solid; mp: 63-65 °C; 1H NMR (500 MHz, CDCl3) δ 7.95 (s, 1H), 6.74 (s, 1H), 6.58 (s, 1H), 6.01-5.93 (m, 3H), 5.27 (ddd, J= 17.3, 3.0, 1.4 Hz, 1H), 5.14 (dd, J=10.4, 1.4 Hz, 1H), 4.37-4.33 (m, 1H), 4.22-4.21 (m, 1H), 3.86 (s, 3H), 3.17-3.10 (m, 1H), 3.04-2.94 (m, 3H), 2.66 (dd, J = 16.3, 3.35 Hz, 1H), 2.54-2.48 (m, 2H), 2.53 (s, 3H) ; 13C NMR (125 MHz, CDCl3) δ 152.2, 146.5, 146.4, 143.1, 134.4, 130.8, 128.8, 127.6, 127.3,125.8, 117.6, 110.7, 109.4, 108.3, 101.0, 73.8, 62.6, 56.0, 53.3, 44.1, 35.2, 29.3; HRMS (ESI) m/z calcd for C22H23NO4 ([M+Na]+), 388.1519, found 388.1520.
4.2.9 1-(3-hydroxy-propanyloxy)domesticine (6i)
Brown Oil; 1H NMR (500 MHz, CDCl3) δ 7.95 (s, 1H), 6.74 (s, 1H), 6.59 (s, 1H), 5.98 (s, 1H), 5.96 (s, 1H), 3.97-3.92 (m, 2H), 3.90-3.84 (m, 5H), 3.82-3.77 (m, 1H), 3.19-3.13 (m, 1H), 3.07-2.95 (m, 3H), 2.68 (dd, J=16.2, 2.8 Hz, 1H), 2.54 (m, 5H), 2.05-1.97 (s, 1H), 1.85-1.78 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 151.8, 146.7, 146.6, 143.4, 130.8, 128.9, 127.4, 125.6, 110.6, 108.7, 108.5, 101.1, 71.0, 62.6, 61.2, 56.0, 53.3, 44.1, 44.0, 35.1, 29.9; HRMS (ESI) m/z calcd for C22H25NO5 ([M+H]+), 384.4376, found 384.4376
4.3 3-(6-bromobenzo[d][1,3] dioxol-5-yl)-N-(3,4-dimethoxyphenethyl) ethanamide (8)
A solution of bromomethylenedioxyphenylacetic acid (4.76 g, 17.5 mmol) and 1,10-carbonyldiimidazole (2.84 g, 17.5 mmol) in anhydrous THF (40 mL) was stirred at 0 °C for 1.5 h and then at room temperature for 1 h. The mixture was cooled in an ice-bath and stirred for 1 h. Then 3-benzyloxy-4-methoxyphenethylamine (7) (4.50 g, 17.5 mmol) was added and the solution was stirred at 0 °C for 4 h and left to stir overnight at room temperature. The reaction mixture was evaporated under reduced pressure and the residue was dissolved in EtOAc and washed sequentially with 1 N HCl (25 mL), water (50 mL), satd. NaHCO3 solution (25 mL) and finally with brine (50 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was crystallized from EtOAc/diethylether to furnish amide 8 (6.65 g, 76.5%).
White solid; mp: 163-165 °C; 1H NMR (500 MHz, CDCl3) δ 7.45 (m, 2H), 7.39 (m, 2H), 7.30 (m, 1H), 7.00 (s, 1H), 6.80 (d, J=8.2 Hz, 1H), 6.75 (s, 1H), 6.70 (br. s., 1H), 6.66 (d, J=8.2 Hz, 1H), 6.00 (s, 2H), 5.38 (br. s, 1H), 5.12 (s, 2H), 3.89 (s, 3H), 3.55 (s, 2H), 3.44 (dd, J=13.0, 6.5 Hz, 2H), 2.69 (dd, J=6.9, 6.9 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 169.5, 148.4, 148.3, 147.9, 147.7, 137.1, 131.0, 128.5, 127.9, 127.5, 127.4, 121.4, 115.3, 114.6, 112.9, 112.0, 110.9, 102.0, 71.1, 56.1, 43.8, 40.6, 34.9; HRMS (ESI) m/z calcd for C25H24BrNO5 ([M+H]+), 498.0838, found 498.0840.
4.4 6-(benzyloxy)-1-((6-bromobenzo[d][1,3]dioxol-5-yl)methyl)-7-methoxy-1,2,3,4-tetrahydroisoquinoline (9)
To a magnetically stirred ice-cooled solution of 8 (5.00 g, 10.05 mmol) in dry DCM (40 mL) was added solid phosphorus pentachloride (4.19 g, 20.1 mmol) in portions over 10 min. The reaction mixture was stirred at 0 °C for 1 h and then left to stir at room temperature for 20 h. The reaction mixture was then poured onto a saturated solution of aqueous NaHCO3 (100 mL) and the contents of the flask were stirred for 1 h. The aqueous layer was extracted with dichloromethane (3 × 20 mL). The combined organic layer was washed with saturated NaHCO3 solution (100 mL) and brine (40 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. To a magnetically stirred ice-cooled solution of the crude imine thus obtained in a mixture of dry MeOH (40 mL) and dry DCM (20 mL), was added powdered NaBH4 (3.60 g, 98 mmol) in three portions over 10 min. The reaction mixture was stirred at 0 °C for 2 h. The reaction mixture was diluted with water and extracted with dichloromethane (3 × 10 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified by flash column chromatography over deactivated silica gel using 0.7% MeOH/DCM as eluant to furnish pure 9 (3.92 g, 81 % yield from 8)
White solid; mp: 115-117 °C; 1H NMR (500 MHz, CDCl3) δ 7.44 (m, 2H), 7.36 (m, 2H), 7.30 (m, 1H), 7.05 (s, 1H), 6.79 (s, 1H), 6.76 (s, 1H), 6.63 (s, 1H), 6.00 (s, 2H), 5.12 (s, 2H), 4.21 (t, J=6.5 Hz, 1H), 3.84 (s, 3H), 3.28-3.20 (m, 2H), 2.99-2.89 (m, 2H), 2.71 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 147.9, 147.3, 147.2, 147.0, 137.3, 131.6, 130.8, 128.5, 127.8, 127.3, 127.1, 115.0, 114.5, 112.9, 111.4, 110.4, 101.7, 71.1, 56.2, 55.2, 42.8, 40.0, 29.1; HRMS (ESI) m/z calcd for C25H24BrNO4 ([M+H]+), 482.0889, found 482.0886.
4.5 tert-butyl-6-(benzyloxy)-1-((6-bromobenzo[d][1,3]dioxol-5-yl)methyl)-7-methoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (10)
To a stirred solution of 9 (3.67 g, 7.63 mmol) in DCM (30 mL) was added DIPEA (2.66 mL, 15.3 mmol), a catalytic amount of DMAP (0.01 g) followed by BOC anhydride (3.26 mL, 15.3 mmol) at room temperature and the solution stirred under inert atmosphere for 18 h. The reaction mixture was washed with saturated NH4Cl solution and water, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel using 30% EtOAc/hexanes as eluant to furnish pure 10 (5.10 g, 65 %).
Off-white solid; mp: 128-130 °C; 1H NMR (500 MHz, CDCl3) δ 7.43 (m, 2H), 7.36 (m, 2H), 7.29 (m, 1H), 7.02 (s, 1H), 6.96 (s,1H), 6.63 (s, 1H), 6.65 (s, 1H), 5.97 (s, 1H), 5.93 (s, 1H), 5.27 (m, 1H), 5.10-5.09 (s, 2H), 4.35 (dd, J = 13.2, 5.3 Hz, 1H), 3.85 (s, 3H), 3.21 (m, 2H), 2.89 (m, 2H), 1.40-1.23 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 154.9, 148.3, 147.2, 137.8, 131.3, 128.6, 127.9, 127.8, 127.3, 126.6, 115.3, 114.2, 112.5, 111.4, 111.0, 110.6, 101.6, 79.5, 71.1, 56.2, 54.0, 42.6, 36.4, 28.1; HRMS (ESI) m/z calcd for C30H32BrNO6 ([M]+), 581.1413, found 581.1415.
4.6 2-(benzyloxy)N-boc-nor-isodomesticine (11)
In a microwave reaction vial, compound 10 (0.12 g, 0.21 mmol), Pd(OAc)2 (0.009 g, 0.04 mmol), triphenylphosphine (0.02 g, 0.08 mmol) and Cs2CO3 (0.13 g, 0.4 mmol),) were added and dissolved in degassed DMF (2 mL). The mixture was irradiated in the CEM microwave reactor for 15 min at 175 °C with the power level at 300W. After cooling to room temperature, the reaction mixture was loaded directly onto a deactivated silica gel column and eluted in gradient fashion with 15-30% EtOAc/hexanes to furnish compound 11 (0.05 g, 52%).
Off-white solid; mp: 170-172 °C; 1H NMR (500 MHz, CDCl3) δ 8.07 (s, 1H), 7.52-7.29 (m, 5H), 6.76 (s, 1H), 6.68 (s, 1H), 6.01 (s, 2H), 5.15 (m, 2H), 4.65 (m, 1H), 4.42 (m, 1H), 3.75 (s, 3H), 2.86-2.64 (m, 5H), 1.52 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 151.1, 146.6, 146.5, 145.5, 137.1, 129.7, 128.6, 127.9, 127.3, 125.2, 112.8, 109.1, 100.9, 79.9, 71.0, 60.1, 51.8, 30.3, 28.6; HRMS (ESI) m/z calcd for C30H31NO6 ([M]+), 501.2151, found 501.2154.
4.7 N-boc-nor-isodomesticine (12)
To a solution of 11 (0.50 g, 1 mmol) in dry methanol (10 mL) was added 10% Pd/C (25 mg) under inert atmosphere. The reaction was purged with H2 and stirred under a balloon of H2 for 6 h. Pd/C was filtered through a Celite pad and the filtrate was evaporated to give the corresponding phenol 12 (0.38 g, 92%).
White solid; mp: 218-220 °C; 1H NMR (500 MHz, CDCl3) δ 7.90 (s, 1H), 6.79 (s, 1H), 6.73 (s, 1H), 6.02 (s, 2H), 5.86 (s, 1H), 4.70-4.68 (m, 1H), 4.61 (m, 1H), 4.40 (m, 1H), 3.61 (s, 3H), 2.92-2.76 (m, 4H), 2.66-2.62 (m, 1H), 1.57-1.52 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 148.1, 146.9, 146.8, 142.6, 131.5, 131.1, 126.5, 125.4, 124.9, 124.8, 113.4, 108.6, 107.8, 101.0, 79.9, 60.2, 51.8, 30.2, 28.6; HRMS (ESI) m/z calcd for C23H25NO6 ([M]+), 411.1682, found 411.1684.
4.8 General procedure for preparation of C2 analogs (14)
O-methylation of 12
To a stirred solution of phenol (1eq) in acetone (10 mL) was added solid K2CO3 (2 eq) and potassium iodide (2 eq) at r.t. The resulting mixture was stirred for 5 min and then alkyl halide (1.5 eq) was added and refluxed for 7h. The solvent was evaporated and the resulting solid dissolved in water and extracted with dichloromethane twice (20 mL each) . The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by flash column chromatography using 12.5% EtOAc/hexanes.
N-Boc deprotection and reductive amination of 13
To a solution of the boc-protected amine (1 eq) in dichloromethane (15 mL) was added dry ZnBr2 (2 eq) and the reaction allowed to stir at rt overnight under N2. The mixture was diluted with dichloromethane (20 mL) and wahed with saturated aqueous NaHCO3 twice (10 mL each). The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The crude amine product was purified by comlumn chromatography on water-deactivated silica gel in 5% methanol/dichloromethane.
To a solution of this amine (1 eq) in dichloromethane (10 mL) was added the aldehyde (3 eq) and sodium triacetoxyborohydride (3 eq) under N2. The reaction mixture was allowed to stir at r.t. overnight. The reaction was quenched with water and extracted with dichloromethane twice (20 mL each) . The combined organic layer was washed with 10% aq. NaHCO3, then with brine, dried over anhydrous Na2SO4, and concentrated to give crude product. The crude product was subjected to column chromatography over water-deactivated silica gel using methanol/dichloromethane (2:98) as eluant, to furnish N-alkylated final product.
4.8.1 2-(ethoxy)isodomesticine (14a)
Yellow solid; mp: 101-104 °C; 1H NMR (500 MHz, CDCl3) δ 7.92 (s, 1H), 6.74 (s, 1H), 6.57 (s, 1H), 5.96 (d, J = 1.4 Hz, 1H), 5.95 (d, J = 1.4 Hz, 1H), 4.12-4.02 (m, 2H), 3.67 (s, 3H), 3.15-3.08 (m, 1H), 3.02-2.94 (m, 3H), 2.66-2.62 (m, 1H), 2.54-2.45 (m, 5H), 1.47 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.5, 146.5, 146.4, 144.7, 130.5, 128.9, 127.3, 127.1, 125.6, 111.6, 109.1, 108.4, 100.9, 64.2, 62.4, 60.2, 53.4, 44.2, 35.3, 29.3, 15.1; HRMS (ESI) m/z calcd for C21H23NO4 ([M+H]+), 354.4116, found 354.4120.
4.8.2 2-(propyloxy)isodomesticine (14b)
Yellow solid; mp: 76-77 °C; 1H NMR (500 MHz, CDCl3) δ 7.95 (s, 1H), 6.76 (s, 1H), 6.59 (s, 1H), 5.98 (d, J=1.4 Hz, 1H) 5.95 (d, J= 1.4 Hz, 1H), 4.04-3.90 (m, 2H), 3.69 (s, 3H), 3.17-3.10 (m, 1H), 3.04-2.96 (m, 3H), 2.65 (m, 1H), 2.55-2.48 (m, 5H), 1.93-1.85 (m, 3H), 1.09 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.6, 146.5, 146.4, 144.7, 131.0, 128.6, 127.3, 127.1, 125.8, 111.8, 109.1, 108.4, 101.0, 70.3, 62.6, 60.3, 53.4, 44.2, 35.3, 29.4, 22.9, 10.9; HRMS (ESI) m/z calcd for C22H25NO4 ([M+H]+), 368.1882, found 368.1888
4.8.3 2-(butyloxy)isodomesticine (14c)
Yellow solid; mp: 95-97 °C; 1H NMR (500 MHz, CDCl3) δ 7.95 (s, 1H), 6.76 (s, 1H), 6.60 (s, 1H), 5.98 (d, J = 1.4 Hz, 1H), 5.97 (d, J = 1.4 Hz, 1H), 4.05-3.94 (m, 2H), 3.68 (s, 3H), 3.17-3.10 (m, 1H), 3.04-2.95 (m, 3H), 2.68-2.64 (m, 1H), 2.55-2.46 (m, 5H), 1.92-1.78 (m, 2H), 1.63-1.47 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.6, 146.5, 146.4, 144.7, 130.9, 128.6, 127.3, 127.0, 125.8, 111.7, 109.1, 108.3, 100.9, 68.4, 62.6, 60.3, 53.4, 44.2, 35.3, 31.6, 27.4, 19.5, 14.0; HRMS (ESI) m/z calcd for C23H27NO4 ([M+H]+), 382.2016 found 382.2013
4.8.4 2-(pentyloxy)isodomesticine (14d)
Yellow oil; 1H NMR (500 MHz, CDCl3) δ 7.94 (s, 1H), 6.75 (s, 1H), 6.59 (s, 1H), 5.97 (d, J = 1.4 Hz, 1H), 5.95 (d, J = 1.4 Hz, 1H), 4.05-3.94 (m, 2H), 3.68 (s, 3H), 3.15-3.09 (m, 1H), 3.03-2.95 (m, 3H), 2.67-2.63 (m, 1H), 2.54-2.46 (m, 5H), 1.92-1.81 (m, 2H), 1.55-1.37 (m, 4H), 0.96 (t, J=7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.6, 146.5, 146.4, 144.7, 130.9, 128.6, 127.3, 127.1, 125.8, 111.8, 109.1, 108.4, 100.9, 68.7, 62.6, 60.3, 53.4, 44.2, 35.3, 29.4, 29.2, 28.4, 22.6, 14.2; HRMS (ESI) m/z calcd for C24H29NO4 ([M+H]+), 396.2194, found 396.2192
4.8.5 2-(cyclopropylmethyloxy)isodomesticine (14e)
Yellow solid; mp: 91-92 °C; 1H NMR (500 MHz, CDCl3) δ 7.93 (s, 1H), 6.74 (s, 1H), 6.56 (s, 1H), 5.97 (d, J=1.4 Hz, 1H), 5.95 (d, J=1.4 Hz, 1H), 3.94-3.90 (m, 1H), 3.79-3.75 (m, 1H), 3.70 (s, 3H), 3.14-3.07 (m, 1H), 3.02-2.94 (m, 3H), 2.65-2.61 (m, 1H), 2.54-2.45 (m, 5H), 1.37-1.24 (m, 1H), 0.64 (m, 2H), 0.37 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 151.6, 146.5, 146.4, 144.7, 130.9, 128.6, 127.3, 127.1, 125.8, 111.8, 109.1, 108.4, 100.9, 68.7, 62.6, 60.3, 53.4, 44.2, 35.3, 29.4, 29.2, 28.4, 22.6, 14.2; HRMS (ESI) m/z calcd for C23H25NO4 ([M+H]+), 380.1872 found 380.1874.
4.8.6 2-(benzyloxy)isodomesticine (14f)
Brown solid; mp: 112-114 °C; 1H NMR (500 MHz, CDCl3) δ 7.95 (s, 1H), 7.50-7.32 (m, 5H), 6.77 (s, 1H), 6.67 (s, 1H), 5.98 (d, J=1.4 Hz, 1H), 5.97 (d, J=1.4 Hz, 1H), 5.12 (d, J=11.8 Hz, 1H), 5.11 (d, J=11.8 Hz, 1H), 3.72 (s, 3H), 3.11 (m, 1H), 3.09-2.97 (m, 3H), 2.64 (m, 1H), 2.56-2.46 (m, 5H); 13C NMR (125 MHz, CDCl3) δ 151.2, 146.5, 146.4, 145.0, 137.2, 130.8, 128.6, 128.5, 127.9, 127.8, 127.4, 127.3, 127.2, 125.6, 112.7, 109.0, 108.3, 100.9, 70.9, 62.5, 60.3, 53.2, 44.0, 35.1, 29.1; HRMS (ESI) m/z calcd for C26H25NO4 ([M+H]+), 416.1784, found 416.1788.
4.9 Synthesis of 3-bromonantenine (15) and 3,8,11-tribromonantenine (16)
To a solution of nantenine (2) (25 mg, 1 eq) in acetic acid (5 mL) was slowly added a solution of bromine (3 eq) dissolved in acetic acid (2 mL) at room temperature. The mixture was stirred overnight at room temperature and the solvent evaporated in vacuo. The residue was dissolved in toluene and the solvent again removed by rotary evaporation. The residue was purified by column chromatography with elution in 2% MeOH/DCM to give compounds 15 (5 mg, 16%) and 16 (22 mg, 52%).
4.9.1 3-bromonantenine (15)
Off white solid; mp: 142-144 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (s, 1H), 6.75 (s, 1H), 5.98 (d, J=1.2 Hz, 1H), 5.97 (d, J=1.2 Hz, 1H), 3.93 (s, 3H), 3.73 (s, 3H), 3.10-3.07 (dd, J=10.7, 5.2 Hz, 1H), 2.98-2.89 (m, 3H), 2.83-2.79 (m, 1H), 2.52 (s, 3H), 2.49-2.41 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 150.1, 149.3, 146.77, 146.76, 132.6, 130.7, 129.4, 127.0, 124.9, 118.5, 108.8, 108.3, 101.1, 62.9, 60.8, 60.6, 53.2, 44.0, 34.8, 30.5; HRMS (ESI) m/z calcd for C20H20BrNO4 ([M+H]+), 418.0648, found 418.0651.
4.9.2 3,8,11-tribromonantenine (16)
White solid; mp: 193-195 °C; 1H NMR (500 MHz, CDCl3) δ 6.18 (s, 1H), 6.14 (s, 1H), 3.95 (s, 3H), 3.54 (s, 3H), 3.08 (dd, J=11.5, 5.2 Hz, 1H), 2.96-2.90 (m, 1H), 2.85-2.77 (m, 2H), 2.54 (s, 3H), 2.49 (dt, J=11.8, 4.1 Hz, 1H), 2.18 (dd, J=14.5, 12.9 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 149.6, 145.9, 145.0, 133.8, 132.6, 128.7, 127.5, 127.1, 125.7, 119.7, 101.7, 101.5, 101.4, 63.0, 61.5, 61.1, 53.0, 44.1, 34.9, 30.3; HRMS (ESI) m/z calcd for C20H18 Br3NO4 ([M+H]+), 572.8859, found 572.8859.
4.10 Synthesis of N6 analogs (18)
These compounds were prepared by reductive amination on amine 17 using the procedure as described above for synthesis of the C2 analogs.
4.10.1 N-ethyl-nornantenine (18a)
Brown solid; mp: 220-222 °C; 1H NMR (500 MHz, CDCl3) δ 7.92 (s, 1H), 6.76 (s, 1H), 6.60 (s, 1H), 5.98 (d, J=1.4 Hz, 1H), 5.95 (d, J=1.4 Hz, 1H), 3.88 (s, 3H), 3.65 (s, 3H), 3.27 (d, J=8 Hz, 1H), 3.19-3.17 (m, 1H), 3.12-3.08 (m, 2H), 2.99-2.96 (m, 1H), 2.71-2.48 (m, 4H), 1.15 (t, J=7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 152.0, 146.6, 146.4, 144.6, 131.0, 129.0, 127.3, 125.7, 110.7, 109.1, 109.0, 108.4, 101.0, 60.4, 59.2, 55.9, 48.4, 47.9, 35.0, 29.3, 10.8; HRMS (ESI) m/z calcd for C21H23NO4 ([M+H]+), 354.4116, found, 354.4115.
4.10.2 N-propyl-nornantenine (18b)
Yellow Oil; 1H NMR (500 MHz, CDCl3) δ 7.91 (s, 1H), 6.76 (s, 1H), 6.59 (s, 1H), 5.98 (d, J =1.1 Hz, 1H), 5.97 (d, J =1.1 Hz, 1H), 3.90 (s, 3H), 3.87 (s, 3H), 3.23-3.20 (d, 1H), 3.17-3.14 (m, 2H), 2.98-2.95 (m, 1H), 2.69-2.66 (d, 1H), 2.51-2.41 (m, 4H), 1.60-1.57 (m, 2H), 0.97 (t, J=7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ; 151.9, 146.5, 146.4, 144.5, 131.2, 129.2, 128.2, 128.1, 127.3, 125.7, 110.8, 109.0, 108.3, 101.0, 60.3, 60.0, 56.4, 55.9, 49.3, 35.3, 29.8, 29.5, 19.7,12.2; HRMS (ESI) m/z calcd for C22H25NO4 ([M+H]+), 368.4382, found, 368.4380
4.10.3 N-butyl-nornantenine (18c)
Yellow Oil; 1H NMR (500 MHz, CDCl3) δ 7.92 (s, 1H), 6.77 (s, 1H), 6.60 (s, 1H), 5.99 (d, J = 1.5 Hz, 1H), 5.97 (d, J = 1.5 Hz, 1H), 3.88 (s, 3H), 3.65 (s, 3H), 3.17-3.15 (m, 2H), 2.99-2.95 (m, 3H), 2.70 (m, 1H), 2.54-2.43 (m, 3H), 1.59-1.54 (m, 2H), 1.41-1.37 (m, 2H), 0.97 (t, J=7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.9, 146.6, 146.4, 144.5, 131.3, 129.3, 128.6, 127.3, 125.8, 110.7, 109.0, 108.4, 101.0, 60.4, 60.0, 56.0, 54.2, 49.3, 35.3, 29.6, 28.7, 21.0, 14.3; HRMS (ESI) m/z calcd for C23H27NO4 ([M+H]+), 382.4648, found 382.4645
4.10.4 N-pentyl-nornantenine (18d)
Yellow Oil; 1H NMR (500 MHz, CDCl3) δ 7.91 (s, 1H), 6.77 (s, 1H), 6.59 (s, 1H), 5.99 (d, J =1.4 Hz, 1H), 5.97 (d, J=1.4 Hz, 1H), 3.87 (s, 3H), 3.65 (s, 3H), 3.20-3.15 (m, 1H), 2.98-2.94 (m, 4H), 2.69 (m, 1H), 2.54 (m, 1H), 2.45-2.42 (m, 2H), 1.57 (m, 2H), 1.39-1.33 (m, 4H), 0.93 (t, J=6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.9, 146.6, 146.4, 144.54, 131.3, 129.2, 128.1, 127.3, 125.8, 110.7, 109.0, 108.4, 101.0, 60.4, 60.0, 56.0, 54.5, 49.3, 35.3, 30.1, 29.6, 26.2, 22.9, 14.3; HRMS (ESI) m/z calcd for C24H29NO4 ([M+H]+), 396.4914, found 396.4918.
4.10.5 N-cyclopropylmethyl-nornantenine (18e)
Yellow Oil; 1H NMR (500 MHz, CDCl3) δ 7.92 (s, 1H), 6.76 (s, 1H), 6.60 (s, 1H), 5.98 (d, 1.5 Hz, 1H), 5.97 (d, 1.5 Hz, 1H), 3.88 (s, 3H), 3.65 (s, 3H) 3.38-3.37 (m, 1H), 2.99-2.92 (m, 4H), 2.72-2.71 (m, 1H), 2.57-2.56 (m, 1H), 2.53-2.47 (m, 1H), 2.40-2.36 (m, 1H), 0.96 (m, 1H), 0.59 (dd, J=8.0, 8.0 Hz, 1H), 0.54 (dd, J=8.0, 8.0 Hz, 1H), 0.19 (d, J=4.8 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 151.9, 146.6, 146.4, 144.5, 131.2, 129.2, 128.0, 127.2, 125.8, 110.7, 109.0, 108.3, 101.0, 60.4, 59.5, 59.2, 56.0, 49.5, 35.2, 29.5, 7.7, 5.2, 3.1; HRMS (ESI) m/z calcd for C23H25NO4 ([M+H]+), 380.4489, found 380.4490
Supplementary Material
Figure 1.

Structures of MDMA (1), ( ±)-Nantenine (2) and Apomorphine (3)
Figure 2.

Preparation of C1 analogs
Acknowledgements
This publication was made possible by Grant Number RR03037 and R03DA025910 from the National Center for Research Resources (NCRR) and National Institute of Drug Abuse (NIDA) respectively, components of the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or its divisions.
Footnotes
Supplementary data Supplementary data associated with this article are available in the online version
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guzman JD, Gupta A, Evangelopoulos D, Basavannacharya C, Pabon LC, Plazas EA, Munoz DR, Delgado WA, Cuca LE, Ribon W, Gibbons S, Bhakta SJ. Antimicrob. Chemother. 2010;65:2101. doi: 10.1093/jac/dkq313. [DOI] [PubMed] [Google Scholar]
- 2.Stevigny C, Bailly C, Quetin-Leclercq J. Curr. Med. Chem. Anticancer Agents. 2005;5:173. doi: 10.2174/1568011053174864. [DOI] [PubMed] [Google Scholar]
- 3.Lane RM, Potkin SG, Enz A. Int. J. Neuropsychopharmacol. 2006;9:101. doi: 10.1017/S1461145705005833. [DOI] [PubMed] [Google Scholar]
- 4.Pecic S, McAnuff MA, Harding WW. J. Enzyme Inhib. Med. Chem. 2011;26:46. doi: 10.3109/14756361003671078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang H, Wei YB, Zhang C, Ning FX, Qiao W, Huang SL, Ma L, Huang ZS, Gu LQ. Eur J. Med. Chem. 2009;44:2523. doi: 10.1016/j.ejmech.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 6.Stocchi F. Neurol. Sci. 2008;29(Suppl 5):S383. doi: 10.1007/s10072-008-1053-8. [DOI] [PubMed] [Google Scholar]
- 7.Ye N, Wu Q, Zhu L, Zheng L, Gao B, Zhen X, Zhang A. Bioorg. Med. Chem. 2011;19:1999. doi: 10.1016/j.bmc.2011.01.053. [DOI] [PubMed] [Google Scholar]
- 8.Liu Z, Zhang H, Ye N, Zhang J, Wu Q, Sun P, Li L, Zhen X, Zhang A. J. Med. Chem. 2010;53:1319. doi: 10.1021/jm9015763. [DOI] [PubMed] [Google Scholar]
- 9.Zhang A, Zhang Y, Branfman AR, Baldessarini RJ, Neumeyer JL. J. Med. Chem. 2007;50:171. doi: 10.1021/jm060959i. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z, Chen X, Sun P, Yu L, Zhen X, Zhang A. Bioorg. Med. Chem. 2008;16:8335. doi: 10.1016/j.bmc.2008.08.056. [DOI] [PubMed] [Google Scholar]
- 11.Cannon JG, Flaherty PT, Ozkutlu U, Long JP. J. Med .Chem. 1995;38:1841. doi: 10.1021/jm00011a002. [DOI] [PubMed] [Google Scholar]
- 12.Ivorra MD, Valiente M, Martinez S, Madrero Y, Noguera MA, Cassels BK, Sobarzo EM, D’Ocon P. Planta Med. 2005;71:897. doi: 10.1055/s-2005-871281. [DOI] [PubMed] [Google Scholar]
- 13.Indra B, Matsunaga K, Hoshino O, Suzuki M, Ogasawara H, Muramatsu I, Taniguchi T, Ohizumi Y. Eur. J. Pharmacol. 2002;445:21. doi: 10.1016/s0014-2999(02)01601-1. [DOI] [PubMed] [Google Scholar]
- 14.Eltze M, Grebe T, Michel MC, Czyborra P, Ullrich B. Eur. J. Pharmacol. 2002;443:151. doi: 10.1016/s0014-2999(02)01591-1. [DOI] [PubMed] [Google Scholar]
- 15.Orejarena MJ, Lanfumey L, Maldonado R, Robledo P. Int. J. Neuropsychopharmacol. 2010:1. doi: 10.1017/S1461145710001215. [DOI] [PubMed] [Google Scholar]
- 16.Herin DV, Liu S, Ullrich T, Rice KC, Cunningham KA. Psychopharmacology (Berl) 2005;178:505. doi: 10.1007/s00213-004-2030-4. [DOI] [PubMed] [Google Scholar]
- 17.Liechti ME, Saur MR, Gamma A, Hell D, Vollenweider FX. Neuropsychopharmacology. 2000;23:396. doi: 10.1016/S0893-133X(00)00126-3. [DOI] [PubMed] [Google Scholar]
- 18.Selken J, Nichols DE. Pharmacol. Biochem. Behav. 2007;86:622. doi: 10.1016/j.pbb.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fantegrossi WE, Kiessel CL, Leach PT, Van Martin C, Karabenick RL, Chen X, Ohizumi Y, Ullrich T, Rice KC, Woods JH. Psychopharmacology (Berl) 2004;173:270. doi: 10.1007/s00213-003-1741-2. [DOI] [PubMed] [Google Scholar]
- 20.Sprague JE, Moze P, Caden D, Rusyniak DE, Holmes C, Goldstein DS, Mills EM. Crit. Care Med. 2005;33:1311. doi: 10.1097/01.ccm.0000165969.29002.70. [DOI] [PubMed] [Google Scholar]
- 21.Bexis S, Docherty JR. Br. J. Pharmacol. 2006;147:926. doi: 10.1038/sj.bjp.0706688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grunau BE, Wiens MO, Brubacher JR. CJEM. 2010;12:435. doi: 10.1017/s1481803500012598. [DOI] [PubMed] [Google Scholar]
- 23.Indra B, Matsunaga K, Hoshino O, Suzuki M, Ogasawara H, Ishiguro M, Ohizumi Y. Can. J. Physiol. Pharmacol. 2002;80:198. doi: 10.1139/y02-019. [DOI] [PubMed] [Google Scholar]
- 24.Chaudhary S, Pecic S, Legendre O, Navarro HA, Harding WW. Bioorg. Med. Chem. Lett. 2009;19:2530. doi: 10.1016/j.bmcl.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pecic S, Makkar P, Chaudhary S, Reddy BV, Navarro HA, Harding WW. Bioorg. Med. Chem. 2010;18:5562. doi: 10.1016/j.bmc.2010.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Legendre O, Pecic S, Chaudhary S, Zimmerman SM, Fantegrossi WE, Harding WW. Bioorg. Med. Chem. Lett. 2010;20:628. doi: 10.1016/j.bmcl.2009.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Indra B, Matsunaga K, Hoshino O, Suzuki M, Ogasawara H, Ohizumi Y. Eur. J. Pharmacol. 2002;437:173. doi: 10.1016/s0014-2999(02)01303-1. [DOI] [PubMed] [Google Scholar]
- 28.Chaudhary S, Pecic S, Legendre O, Harding WW. Tetrahedron Lett. 2009;50:2437. doi: 10.1016/j.tetlet.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kano S, Yokomatsu T, Ebata T, Shibuya S. Chem.Pharm. Bull. 1977;25:1456. [Google Scholar]
- 30.Chen L-J, Xi Y, Pang D-W, Zhou Q-T, Jin G-Z. Zhongguo Yaoli Xuebao. 1996;17:185. [PubMed] [Google Scholar]
- 31.Cannon JG, Raghupathi R, Moe ST, Johnson AK, Long JP. J. Med. Chem. 1993;36:1316. doi: 10.1021/jm00062a002. [DOI] [PubMed] [Google Scholar]
- 32.Cannon JG, Moe ST, Long JP. Chirality. 1991;3:19. doi: 10.1002/chir.530030105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.