

Abstract
Doxorubicin (DXR), among the most widely used cancer chemotherapy agents, promotes cancer cell death via activation of ATM and the resultant up-regulation of tumor suppressor p53. The exact mechanism by which DXR activates ATM is not fully understood. Here we discovered a novel role for Regulator of G protein Signaling 6 (RGS6) in mediating activation of ATM and p53 by DXR. RGS6 was robustly induced by DXR, and genetic loss of RGS6 dramatically impaired DXR-induced activation of ATM and p53, as well as its in vivo apoptotic actions in heart. The ability of RGS6 to promote p53 activation in response to DXR was independent of RGS6 interaction with G proteins but required ATM. RGS6 mediated activation of ATM and p53 by DXR via a ROS-dependent and DNA damage-independent mechanism. This mechanism represents the primary means by which DXR promotes activation of the ATM-p53-apoptosis pathway that underlies its cytotoxic activity. Our findings contradict the canonical theories that DXR activates ATM primarily by promoting DNA damage either directly or indirectly (via ROS) and that RGS6 function is mediated by its interactions with G proteins. These findings reveal a new mechanism for the chemotherapeutic actions of DXR and identify RGS6 as a novel target for cancer chemotherapy.
Keywords: RGS6, doxorubicin, ATM, p53, reactive oxygen species
Introduction
DXR belongs to the anthracycline class of anti-tumor agents that are among the most effective and widely used chemotherapeutic drugs for treatment of human cancers (1). DXR interferes with DNA topoisomerase II (2, 3) and also generates reactive oxygen species (ROS) (4). Both of these mechanisms can induce double-stranded DNA breaks (DSBs) that promote death of cancer cells by activating the DNA damage response (DDR), a complex signaling cascade that carries out DNA repair or induces apoptosis in cells with severe DNA damage (5, 6). The ATM protein kinase plays a critical role in sensing such DSBs and in orchestrating the signaling output including induction of the tumor suppressor p53 by mechanisms involving ATM-dependent phosphorylation of p53 and Mdm2 (7–9). p53 plays a primary role in mediating growth arrest and apoptosis during DNA damage as evidenced by the finding that thymocytes from p53−/− mice do not undergo apoptosis in response to radiation-induced DNA damage (10). This role of p53 appears to represent its major tumor suppressor function in view of the fact that over half of sporadic tumors have mutations in p53 that are exclusive of the nearly 15% of sporadic tumors with ATM mutations (11).
RGS6 is a member of the RGS family of proteins that function as negative regulators of G protein signaling by virtue of their GTPase activating protein activity toward Gα subunits (12–14). A link between RGS6 and cancer was suggested by the finding that a SNP in the RGS6 gene, which increases its translational efficiency about three-fold, is associated with an overall reduction in the risk of bladder tumor formation (15). This risk reduction was more pronounced in smokers (40%), especially those who began smoking at a young age (58%), suggesting a gene-environment interaction. Cigarette smoking is responsible for up to two thirds of bladder cancer and represents the most common mechanism of self-inflicted DNA damage (16). Thus, we hypothesized that RGS6 might somehow function to prevent proliferation of abnormal cells arising from DNA damage. Recently, we discovered pro-apoptotic actions of RGS6 mediated by its ability to induce reactive oxygen species (ROS) and found that MEFs lacking RGS6 exhibited an impaired apoptotic response to DXR (17). Thus it is of particular interest that Guo et al. (18) recently demonstrated direct activation of ATM by ROS. Given the evidence that RGS6 seems to protect against bladder cancers arising from DNA damage, induces ROS that might activate ATM, and is required for the apoptotic response to DXR, we undertook an investigation of whether RGS6 functioned to modulate the DDR.
Material and Methods
Cell Culture and Transfection
MEFs were isolated from E14.5 wild-type (WT), RGS6+/− and RGS6−/− mouse embryos using standard protocols. MEFs, U-87 and MCF-7 cells were grown in DMEM supplemented with 10% FBS and penicillin/streptomycin (10 U/ml). Cells were grown in a 5% CO2-humidified atmosphere at 37 °C. Cells were transiently transfected with LipofecTAMINE 2000 (Invitrogen) using the manufacturer’s protocol. RGS6 cDNAs were prepared as described previously (19). Wild type and kinase-deficient ATM cDNAs were kindly provided by Dr. Michael Kastan.
Luciferase Assays
We amplified and cloned the human RGS6 promoter (− 1450 to +52 bp) into pGL3 basic vector (pGL3-RGS6). U-87 and MCF-7 cells were transfected with pGL3-RGS6 together with a Renilla luciferase control reporter pRL-SV40 (Promega), which was used to normalize transfection efficiency. Thirty-six h after transfection, cells were treated with DXR (1 μM) for 6 h before harvesting and assay of firefly and renilla luciferase activities using the dual luciferase assay kit (Dual-Luciferase® Reporter Assay System 10-Pack) according to the manufacturer’s protocol (Promega).
Western Blotting
Western blotting was performed as described previously (19). Antibodies used were GFP (Santa Cruz, 1:2000), actin (Sigma, 1:2000), p53 (Santa Cruz, 1:2000), pp53-S15 (Cell Signaling, 1:2000), pATM-S1981 (Cell Signaling 1:500), and flag (Sigma, 1:500). Mdm2 monoclonal antibody 2A10 (1:10) was kindly provided by Dr. Arnold Levine. RGS6 antiserum was made previously in the lab (19) and the RGS6 antibody used in Western blotting was affinity purified (20). Western blot signals were recorded and measured using the Odyssey infrared imaging system (LI-COR Biosciences).
Apoptosis Analysis
MEFs plated in 8-well glass coverslip chamber slides at a density of 2000 cells/well were grown overnight before treatment with 0.5 μM DXR for 6 h. Growth medium was then removed and cells were stained by addition of 0.5 ml of HEPES buffer (10 mM HEPES/NaOH, ph7.4, 150 mM NaCl, 1 mM MgCl2, 5 mM KCl, 1.8 mM CaCl2) containing Annexin V (FITC) (Sigma, 0.5 μg/ml final concentration), propidium iodide (Sigma, 0.5 μg/ml final concentration) and Hoechst 33342 (Sigma, 2 μg/ml final concentration). After incubation at room temperature for 15 min, annexin-positive cells were counted using a Leica DMI6000B fluorescence microscope. We did not observe propidium iodide-positive cells indicating that the condition we used in this experiment did not induce necrosis.
Comet Assay and Measurement of Intracellular ROS Levels
MEFs of different genotypes were treated with vehicle or DXR (0.5 μM, 6 h). Comet assay (alkaline single cell gel assay, pH>13) was performed using a standard protocol to determine DNA damage. Comet tail length was measured with Comet Assay IV software (Perceptive Instruments Ltd, UK). Measurement of intracellular ROS levels in WT and RGS6−/− MEFs with or without DXR treatment (0.5 μM, 6 h) was done as we recently described (17).
H&E staining and TUNEL staining
Two month-old mice were treated with saline (4 WT mice and 4 RGS6−/− mice) or DXR (10 mg/kg i.p. 4 WT mice and 5 RGS6−/− mice) on days 1 and 4 and sacrificed on day 8. Five micrometer paraffin-embedded slices of ventricle tissues were prepared in the Central Microscopy Research Facility (CMRF) of the University of Iowa according to standard protocol. H&E staining following standard procedures was also performed in the CMRF. TUNEL staining was performed using the TADS® 2 TdT-DAB in Situ Apoptosis Detection Kit from TREVIGEN, according to the manufacturer’s protocol.
Statistical analysis
Data were analyzed by Student’s t-test. Results were considered significantly different at p < 0.05. Values are expressed as means ± S.E.
Results
DXR induces up-regulation of RGS6 in cancer cells
We reasoned that expression of RGS6 may be regulated during genotoxic stress if it functions as a positive modulator of DDR, because RGS6 expression is low in most mitotically active cells. Hence we examined effects of DXR on RGS6 protein levels in human tumor cell lines. Because DXR is used to treat breast cancer, we performed these experiments in MCF-7 breast cancer cells, in addition to U-87 glioma cells where DXR also promotes apoptosis (21). MCF-7 and U-87 cells expressed low levels of RGS6 that were increased greatly by DXR treatment (Fig. 1A). The apparent molecular mass of the induced RGS6 protein corresponded to that of the long (RGS6L) splice forms of RGS6 we described (19). Up-regulation of RGS6 by DXR appeared due, at least in part, to induction of RGS6 gene transcription as shown by the ability of DXR to dose-dependently promote activation of the human RGS6 gene promoter in MCF-7 and U-87 cells (Fig. 1B). The finding that DXR treatment of cancer cells transcriptionally activates the RGS6 gene and dramatically up-regulates RGS6 expression is consistent with a possible role for RGS6 in the chemotherapeutic actions of DXR.
Figure 1. DXR induces up-regulation of RGS6 in human cancer cell lines.

A. DXR treatment (1 μM, 24 h) induces up-regulation of endogenous RGS6 protein levels in U-87 and MCF-7 human cancer cells. A representative Western blot of three independent experiments is shown. B. DXR dose-dependently activates transcription from the RGS6 gene promoter in U-87 and MCF-7 cells. The human RGS6 promoter (bp −1450 to +52) in pGL3 basic vector was transfected into U-87 and MCF-7 cells. Thirty-six h after transfection, cells were then treated with indicated concentrations of DXR for 6 h. Luciferase activity was determined as described under “Material and Methods.” The luciferase activity of untreated (0 μM DXR) cells was set as 1. Results represent means ± S.E. of three independent experiments (*p < 0.001).
RGS6 is required for DXR-induced phosphorylation and up-regulation of p53 in MEFs and MCF-7 breast cancer cells
We next examined the role of endogenous RGS6 in DXR-induced p53 activation by employing mouse embryonic fibroblasts (MEFs) from wild-type (WT), RGS6+/− and RGS6−/− mice that we recently developed (20). ATM is activated in response to DSBs and ROS by its autophosphorylation on S1981 (22) and, once activated, ATM phosphorylates p53 on S15 which contributes to its stabilization and up-regulation (7, 8). DXR treatment induced a significant up-regulation of RGS6 in MEFs from WT and heterozygote mice, but not in those from RGS6−/− mice (Fig 2A), confirming our evidence that RGS6 is a key gene target of DXR. RGS6 protein levels were stringently regulated by DXR, as RGS6 expression was undetectable in untreated MEFs of all RGS6 genotypes. Likewise, basal levels of p53 and phospho-p53(pp53)-S15 were very low in MEFS of all RGS6 genotypes and were dramatically increased in response to treatment with DXR at a concentration as low as 0.25 μM for 24 h in WT and heterozygote MEFs. Importantly this effect was severely impaired in RGS6−/− MEFs (Fig. 2A), with DXR-induced levels of pp53-S15 and total p53 reduced by 90% or more compared to those observed in WT and heterozygote MEFs. We then tested whether this loss of DXR-induced p53 activation in RGS6−/− MEFs could be rescued by overexpression of RGS6L to levels comparable to those induced by DXR in WT MEFs. Fig. 2B shows that expression of RGS6L in RGS6−/− MEFs restored the ability of DXR to induce phosphorylation and up-regulate p53 to that observed in WT MEFs, which is also illustrated in Supplemental Figure 1 summarizing the results of multiple rescue experiments. No significant activation of p53 was observed due to RGS6L-GFP expression in the absence of DXR. Thus, in the absence of RGS6, there is a severe impairment in the ability of DXR to activate and up-regulate p53. These findings also demonstrate that RGS6 as opposed to other members of the RGS protein family are essential in this process.
Figure 2. RGS6 is required for DXR-induced activation of p53.

A. Marked impairment of DXR-induced activation and stabilization of p53 in MEFs from RGS6−/− mice. MEFs isolated from different genotypes of mice were treated with the indicated concentrations of DXR for the indicated time periods. Levels of RGS6, phosphorylated p53 (pp53-S15) and total p53 were analyzed by Western blotting. B. Overexpression of RGS6 in MEFs from RGS6−/− mice rescues the ability of DXR to induce phosphorylation of p53 and up-regulation of p53. MEFs (RGS6−/− and WT) were transfected with RGS6L-GFP or GFP. Thirty-six h after transfection, cells were treated with 0.5 μM DXR for 6 h. Levels of pp53-S15 and total p53 were determined. C. Knockdown of RGS6 via shRNAi attenuates DXR-induced activation of p53 in MCF-7 cells. MCF-7 cells were transfected with GFP or different shRNAi constructs. Sixty h after transfection, cells were treated with DXR (1 μM) for 6 h. Levels of RGS6, pp53-S15 and total p53 were determined by Western blotting. Each panel is representative of at least three independent experiments.
We next investigated whether RGS6 played a similar role in cancer cells. For these studies, we evaluated effects of knock-down of RGS6 in MCF-7 cells on DXR-induced responses. Using an RGS6-specific shRNAi (shRNAi-RGS6) that efficiently ablated ectopic expression of RGS6 and one that did not (shRNAi-control), we investigated DXR-induction of RGS6 protein expression and p53 activation in MCF-7 cells. When expressed in MCF-7 cells, shRNAi-RGS6 completely blocked DXR-induced increases in RGS6 and largely prevented DXR-induced increases in pp53-S15 (Fig. 2C). In contrast, there were no effects of the shRNAi-control or GFP transfection control on DXR-mediated increases in RGS6 and pp53-S15. Thus, induced expression of RGS6 by DXR in MCF-7 cells, as in MEFs, is required for DXR-induced S15 phosphorylation of p53. The effect of DXR on total p53 levels in MCF-7 cells is either small or undetectable probably due to high basal expression of p53 in these cells.
RGS6 promotes DXR-induced p53 phosphorylation by mechanisms requiring ATM but independent of its canonical interactions with G proteins
Subsequent studies revealed that expression of RGS6 in MCF-7 cells was sufficient to induce p53 activation to an otherwise ineffective dose of DXR, and that RGS6 promoted this response by mechanisms independent of G proteins. RGS6 possesses, in addition to its G protein-interacting RGS domain, DEP (Dishevelled, Egl-10, Pleckstrin) and GGL (G gamma-like) domains that mediate binding to R7BP and Gβ5 to control localization and stability of RGS6, respectively (23, 24). Previously, we identified numerous splice variant forms of RGS6, some of which lack DEP and/or GGL domains (19). Thus, we evaluated the ability of these RGS6 splice forms and a G protein-interaction deficient mutant of RGS6 to promote p53-S15 phosphorylation in response to a dose of DXR (0.2 μM) insufficient to both induce RGS6 and promote DDR in MCF-7 cells (Fig. 3A, GFP control). As shown in Fig. 3A, expression of RGS6L, the long form of RGS6 with intact DEP and GGL domains, potentiated robust increases in pp53-S15 in response to this dose of DXR. In contrast, RGS6 splice variants lacking GGL (RGS6L-GGL) or DEP (RGS6S) or both of these domains (RGS6S-GGL) minimally enhanced DXR-induced p53 phosphorylation. However, RGS6LN401V, a mutant form of RGS6L that does not interact with or accelerate the GTPase activity of Gα subunits (25), was as capable as its wild-type counterpart in promoting p53 activation. The ability of RGS6 to sensitize cells to DXR by mechanisms dependent upon its DEP and GGL domains and independent of its interaction with G proteins identifies an entirely novel signaling mechanism for RGS6.
Figure 3. RGS6 promotes DXR-induced activation of p53 by mechanisms dependent upon ATM and independent of interactions with G proteins.

A. Comparison of the ability of RGS6 protein isoforms and a non-G protein-regulating mutant (RGS6LN401V) to promote activation of p53 by an ineffective dose of DXR. MCF-7 cells were transfected with GFP, GFP-tagged RGS6 isoforms or RGS6LN401V. Thirty-six h after transfection, cells were treated with vehicle or 0.2 μM DXR, a concentration insufficient to induce p53 phosphorylation without overexpression of RGS6L, for 6 h. A representative Western blotting result of three independent experiments is shown. B. RGS6L enhancement of DXR-induced p53 activation is blocked by a dominant negative ATM lacking its kinase activity (FLAG-ATM(KD)). MCF-7 cells were co-transfected with GFP or RGS6L-GFP and FLAG-ATM(KD) or pcDNA3. Thirty-six h after transfection, cells were treated with DXR as in A. A representative Western blot of three independent experiments is shown. C. Loss of RGS6 significantly blocks DXR-induced ATM activation as well as ATM-mediated phosphorylation of Mdm2. WT, RGS6+/−, and RGS6−/− MEFs were treated with 0.5μM DXR for 6 h. Upper panel representative results of three independent experiments. Lower panel quantification of Mdm2 immunoreactivity. Mdm2 level relative to actin in WT MEFs without DXR treatment was set as 1. Results represent means ± S.E. of three independent experiments (*p<0.005).
Although RGS6 is clearly required for DXR-induced phosphorylation of S15 of p53, a direct target of ATM, there are several other kinases downstream of ATM that can phosphorylate this site (26, 27). Therefore, we determined whether RGS6 functioned upstream of ATM to promote activation of both ATM and p53. Expression of RGS6 in MCF-7 cells induced autophosphorylation of ATM (S1981) and S15 of p53 in response to a low dose of DXR (0.2 μM), which alone did not produce any effects on ATM or p53 phosphorylation (Fig. 3B). Co-expression of dominant-negative (kinase-deficient, KD) ATM nearly completely blocked these actions of RGS6, demonstrating that RGS6 functions upstream of ATM to promote its activation and downstream signaling. Stabilization of p53 in response to DNA damage requires ATM-mediated inactivation of the oncoprotein Mdm2, a ubiquitin ligase that acts as a major negative regulator of p53 (9). ATM phosphorylates Mdm2 on S395, which significantly reduces its reactivity with the monoclonal Mdm2 antibody 2A10 (28) and its ability to cause p53 degradation (9). Figure 3C shows that DXR-induced activation of ATM was greatly impaired in RGS6−/− MEFs compared to WT and heterozygote MEFs, demonstrating that RGS6 is required for DXR-induced activation of ATM. Consistent with a decrease in ATM activity in RGS6−/− MEFs, DXR-induced loss of Mdm2 (2A10) reactivity was greatly impaired in RGS6−/− MEFs (Fig. 3C). Together, these results show that RGS6 is required for DXR-induced ATM activation, which leads to phosphorylation of p53 and Mdm2 and the ensuing stabilization and up-regulation of p53.
RGS6 promotes DXR-induced ATM activation by enhancing ROS generation
We undertook several studies to elucidate the mechanism by which RGS6 promotes activation of ATM by DXR. First, we investigated whether RGS6 somehow increased the extent of DNA damage induced by DXR and thereby promoted ATM activation. For these experiments, we assessed DXR-induced DNA damage in WT and RGS6−/− MEFs using COMET assays, a sensitive method for determining DNA damage in cells in which COMET tail length correlates directly to extent of DNA damage (29). However, as shown in Fig. 4A, DXR produced significantly less, rather than more, DNA damage in WT MEFs compared to RGS6−/− MEFs. Thus, RGS6 does not promote ATM activation by increasing the level of DNA damage from DXR. The higher level of DXR-induced DNA damage in RGS6−/− MEFs is likely due to impaired repair of DNA from loss of ATM activation in these cells (Fig. 3C). Second, we examined whether RGS6 might activate ATM by mechanisms involving ROS. This possibility was suggested by recent evidence that RGS6 induces apoptosis by generation of ROS (17) and that ROS can directly activate ATM (18). Moreover, it has long been known that DXR induces ROS (4), and Kurz et al. (30) demonstrated that ATM-dependent phosphorylation of p53 (S15) and p53 binding to its cognate DNA binding site in response to DXR is ROS-dependent. Fig. 4B shows that brief treatment of MEFs with NAC (N-acetyl-cysteine), a ROS scavenger that significantly inhibits RGS6-induced ROS (17) and ROS-induced ATM activation (18), significantly blocked DXR-induced increases in pATM-S1981, pp53-S15 and total p53 in WT MEFs as well as in RGS6−/− MEFs in which RGS6 was expressed to rescue the DDR. Similarly, NAC blocked RGS6-dependent phosphorylation of ATM and p53 in MCF-7 cells using a DXR dose that alone is ineffective in promoting these responses (Fig. 4C). DXR-induced ROS generation is significantly lower in RGS6−/− MEFs compared to WT MEFs (Fig. 4D), indicating that DXR-induced ROS generation is mediated in part by RGS6. Together, these findings provide new evidence that RGS6 promotes DXR-induced activation of ATM by enhancing ROS generation.
Figure 4. RGS6 promotes DXR-induced ATM activation by enhancing ROS generation.

A. DXR treatment (as in Fig. 3C) induces more DNA damage (DSBs) in RGS6−/− MEFs compared to WT MEFs. Left pane representative images of single cells treated with or without DXR. Right panel quantification of comet tail length. Results represent means ± S.E. of six independent experiments (*p < 0.001). B. ROS scavenger NAC significantly blocks DXR-induced ATM and p53 activation in WT MEFs and RGS6−/− MEFs overexpressing RGS6. Cells were pre-treated with NAC (30 mM) for 1 h followed by DXR (0.5 μM) for 3 h. C. RGS6-dependent ATM activation in MCF-7 cells is blocked by pre-treatment of NAC (30 mM, 1 h). MCF-7 cells were incubated with an ineffective dose of DXR (0.2 μM) for 3 h following NAC treatment. D. DXR-induced ROS generation is markedly reduced in RGS6−/− MEFs compared to WT MEFs. MEFs of both genotypes were treated with DXR as in Fig. 3C. Results represent means ± S.E. of three independent experiments (*p < 0.01)
RGS6 is required for DXR-induced apoptosis both in isolated cells and in vivo
Activation of the DDR can induce either cell cycle arrest, which allows for higher fidelity repair of DSBs by homologous recombination, or apoptosis if damage is insurmountable. The therapeutic efficacy of DXR relies largely upon its ability to kill severely damaged cells. Since RGS6 is required for DXR-induced DDR (ATM and p53) activation, we next assessed the role of RGS6 in DXR-induced apoptosis in WT and RGS6−/− MEFs. MEFs of both genotypes were treated with DXR as in Fig. 3C. Figure 5A shows that loss of RGS6 in MEFs dramatically impairs the early apoptotic response to DXR assessed by Annexin V staining. These results are consistent with our previous finding that RGS6 enhances apoptotic signaling initiated by DXR (17)
Figure 5. RGS6 is required for DXR-induced apoptosis in MEFs and in vivo.

A. RGS6−/− MEFs show significantly impaired apoptosis in response to DXR (0.5 μM, 6 h) treatment. The percentage of annexin V positive cells was normalized to non-treated control. Results represent means ± S.E. of three independent experiments (*p < 0.005). B. and C. Loss of RGS6 dramatically reduces DXR-induced ventricular cell death. Cell death in ventricles from mice of different genotypes treated with saline or DXR (10 mg/kg) were analyzed by TUNEL staining as described in “Material and Methods”. B. Representative images from 8 WT (4 for saline and 4 for DXR treatment) and 9 RGS6−/− mice (4 for saline and 5 for DXR treatment) of TUNEL staining of ventricular tissue slides are shown. TUNEL-positive cells were marked with arrows. C. Quantification of TUNEL-positive cells. Results represent means ± S.E. of 8 WT and 9 RGS6−/− mice (*p < 0.001). D. Loss of RGS6 prevents DXR-induced cardiac toxicity. Tissue morphology of ventricles from mice of different genotypes treated with saline or DXR (10 mg/kg) were examined using H&E staining as described in “Material and Methods.” Representative images from 8 WT (4 for saline and 4 for DXR treatment) and 9 RGS6−/− mice (4 for saline and 5 for DXR treatment) are shown. Damage of ventricular tissue in WT mice are marked with arrows.
To confirm the significance of RGS6 to DXR-induced cell death in vivo, we compared DXR-induced cardiotoxicity in WT and RGS6−/− mice. The major limitation to DXR use is its induction of life-threatening cardiotoxicity, mediated by the same ATM-p53 DNA damage pathway responsible for its tumor-killing actions (31). Reduction of p53 activity by generation of dominant-negative p53 transgenic mice, by genetic disruption of the gene, or via chemical inhibition protects against DXR-induced heart injuries in mice (32–34). Based upon our finding that RGS6 is essential for DXR-induced p53 activation and the pivotal role of p53 in DXR-induced cardiotoxicity, we hypothesized that loss of RGS6 will prevent or dramatically reduce DXR-induced heart injury. DXR treatment of WT mice induced marked apoptosis of ventricular cells and ventricular damage, which were dramatically reduced (Figs. 5B and 5C) and absent (Fig. 5D), respectively, in RGS6−/− mice. Together our results demonstrate the requirement of RGS6 for the ability of DXR to induce cell death both in isolated cells and in vivo.
Discussion
This study reveals a novel and significant role of RGS6 in DXR-induced activation of the ATM-p53-apoptosis pathway that underlies the chemotherapeutic actions of DXR. Loss of RGS6 severely impaired ATM and p53 activation in response to DXR, as well as DXR-induced apoptosis in cells and in the heart in vivo. Importantly, the ability of RGS6 to promote DXR-induced ATM activation and p53 up-regulation is independent of its canonical actions as a G protein regulator. Instead, RGS6 promoted DXR-induced ATM activation by enhancing DXR-induced ROS generation, in keeping with the recent discovery that ATM can be directly activated by ROS (18) and our finding that RGS6 promotes apoptosis by mechanisms involving ROS generation (17). Importantly this mechanism appears to represent the major means by which DXR promotes ATM and p53 activation as most of this response is dependent upon RGS6 and ROS (Fig.4B). Although RGS6 enhancement of ROS generation could also lead to DSBs to activate ATM, we found less DNA damage in response to DXR in WT vs RGS6−/− MEFs (Fig. 4A). Moreover, increased DNA damage in RGS6−/− MEFs did not enhance ATM activation (Fig. 3C) indicating that DNA damage is not the major mechanism for DXR-induced ATM activation in these cells. Therefore, our findings are consistent with RGS6-induced ROS activating ATM directly. We cannot exclude the possibility that RGS6 might also enhance ROS-mediated ATM activation because loss of RGS6 led to larger decreases in DXR-induced ATM activation than it did on DXR-induced ROS generation (Figs. 4B, 4D). Our results further suggest that RGS6-dependent activation of ATM leads to phosphorylation of p53 and Mdm2, which promotes stabilization and up-regulation of p53 by impairing its degradation. Collectively these data suggest a model in which RGS6 plays an essential role in DXR-induced activation of ATM and p53 as illustrated in Figure 6.
Figure 6. Schematic illustration of the role of RGS6 in DXR-induced DDR.
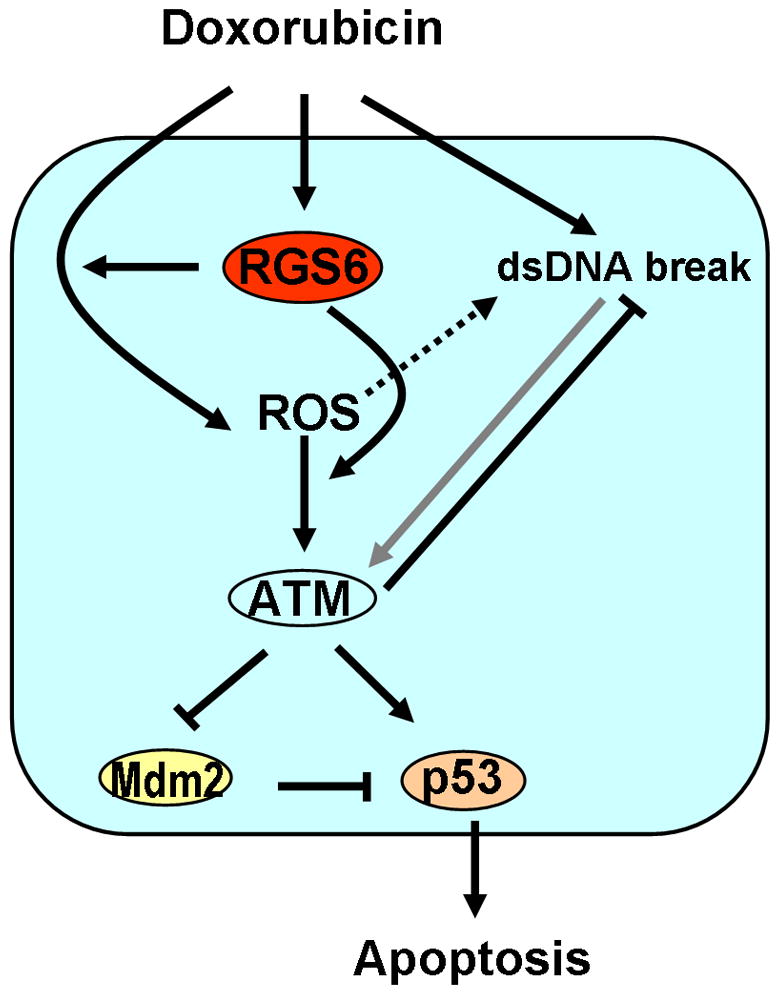
The anti-tumor activity of DXR is mediated by its ability to activate DDR, especially the ATM-p53 pathway, which ultimately leads to apoptosis of severely damaged cells. Our study reveals an essential role of RGS6 in DXR-induced activation of the ATM-p53 pathway. RGS6 promotes DXR-induced generation of ROS, which directly activates the ATM-p53 pathway. RGS6 might also promote ROS-induced activation of ATM as loss of RGS6 in MEFs had a greater effect on DXR-induced ATM activation than DXR-induced ROS generation. In contradiction to the canonical theory that DXR activates ATM primarily by promoting DNA damage (DSBs) either directly or indirectly (via ROS), our study shows that the major mechanism by which DXR activates ATM DXR is via ROS (black arrows) and that DNA damage has only a minor effect (grey arrow) on ATM activation.
The possibility that RGS6 might somehow modulate DNA damage signaling to either kill badly damaged cells or stimulate DNA repair was suggested by the finding that a SNP in the RGS6 gene, which increased its expression, protected against the risk of bladder cancer, particularly in smokers (15). Here we provide emphatic confirmation that RGS6 has a crucial role in modulating both of these responses that hinges on its mediation of ATM activation. While it has long been known that ATM is the primary responder to DNA damage and facilitates DNA repair or cell death in the case of insurmountable DNA damage, our results show that the recently described activation of ATM by ROS (18) is also critical for these downstream actions of ATM in response to a widely used chemotherapeutic agent. The key finding that RGS6 is a dramatic modulator of this response is entirely consistent with the findings suggesting that increased expression of RGS6 protects against bladder cancer risk from smoking, which induces ROS generation and DNA damage. Further work will be required to determine whether RGS6 functions to activate ATM to prevent genetic instability and cellular transformation in situations other than during DXR treatment. It is well known that ROS is generated during normal metabolism as well as during exposure to environmental agents including DXR that can induce oxidation of DNA bases and cause DNA breaks (4, 11). The present findings raise the possibility that RGS6 has a role in ROS-induced ATM activation in such situations. In this regard, we recently provided new evidence for a tumor suppressor role of RGS6 in breast cancer (17). RGS6 had powerful apoptotic actions in breast cancer cells, and its loss in ductal epithelial cells correlates with cancer progression (17). RGS6 expression is also dramatically lost in both human glioma and bladder cancer (Stewart & Assem, unpublished data). Together, these findings and the present results suggest that loss or mutations in RGS6 may have severe consequences including tumor formation and their resistance to treatment with DXR.
The rapid induction of RGS6 expression by DXR and its G protein-independent mechanism of mediating DXR-induce p53 activation are unexpected for a member of the RGS family of proteins. Expression of RGS proteins is usually under tight regulation since these proteins are strong negative regulators of G protein signaling. RGS6 indeed possesses G protein regulating actions in vivo as shown by its essential role as a negative regulator of G protein activation of GIRK channels mediating bradycardic responses to acetylcholine in heart (20). Here we found that DXR induced robust increases in RGS6 protein expression in both cancer cells and MEFs. Although DXR increased transcription of the human RGS6 promoter, it seems likely that post-transcriptional mechanisms are also involved in RGS6 up-regulation because DXR promoted maximal increases in RGS6 protein levels at the earliest times we have measured (2–3 h) (data not shown). The fact that RGS6 expression is so dramatically increased actually points to a novel function for RGS6 as such increases are not at all consistent or expected for its role in regulating G protein signaling. Thus, it is especially intriguing that RGS6 mediates DXR-induced p53 activation by mechanisms dependent upon its GGL and DEP domains but not its G protein-interacting RGS domain. RGS6 is a member of the R7 subfamily of RGS proteins that includes RGS7, RGS9 and RGS11 (35). The crystal structure of the RGS9-Gβ5 complex recently was elucidated (36). In this structure, the RGS9 GGL domain bound to Gβ5 is sandwiched between its DEP and RGS domains creating two distinct surfaces, one for interaction with Gα subunits and one important for interactions with Gβ5 and the DEP domain. RGS6 likely shares an overall similar structural organization with RGS9. If so, our findings suggest that the important face of RGS6 for promoting DDR is on the opposite side of its Gα subunit binding RGS domain. Thus our findings define a novel functional role of RGS6 that depends upon structural elements quite separate from those employed for its canonical action as a G protein regulator.
In summary, this work identifies a novel signaling action of RGS6 that is required for the ability of one of the most effective and widely employed chemotherapeutic agents to promote cellular responses that kill cancer cells. Importantly RGS6 functions as a key modulator of DXR-induced ATM activation by mechanisms unrelated to its canonical function as a G protein regulator, broadening our understanding of RGS protein function. Our findings provide new evidence that the recently described activation of ATM by ROS (18) plays a major role in how DXR activates p53, and we found that RGS6 has a central role in this new ATM activation mechanism. In view of these findings and the ability of RGS6 to sensitize cancer cells to otherwise ineffective doses of DXR, the present findings identify RGS6 as a prominent therapeutic target for treatment of cancer.
Supplementary Material
Supplemental Figure 1. Rescue of the severe impairment in DXR-induced increases in p53 (A) and pp53-S15 (B) in RGS6−/− MEFs by expression of RGS6L-GFP. Results show quantification of western blot signals of p53 and pp53-S15 from three independent experiments like that shown in Fig 2B. Cells were transfected with GFP or RGS6L-GFP and treated with DXR as described in Fig 2B, and western blot signals were quantified by densitometry. The level of p53 and pp53-S15 in DXR treated WT MEFs (lane 6 in Fig. 2B) were set as 1. Because pp53-S15 levels are undetectable in the absence of DXR, only DXR-stimulated increases in pp53-S15 are shown in panel B. (*p<0.001, **p<0.0001)
Acknowledgments
Financial support: This project was supported by NIH (GM075033, ARRA GM075033- 03S1)
We thank Dr. John Koland for his careful reading of and useful suggestions for this manuscript.
References
- 1.Carter SK. Adriamycin-a review. J Natl Cancer Inst. 1975;55(6):1265–74. doi: 10.1093/jnci/55.6.1265. [DOI] [PubMed] [Google Scholar]
- 2.Capranico G, Zunino F. DNA topoisomerase-trapping antitumour drugs. Eur J Cancer. 1992;28A(12):2055–60. doi: 10.1016/0959-8049(92)90255-z. [DOI] [PubMed] [Google Scholar]
- 3.Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226(4673):466–8. doi: 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- 4.Lown JW, Sim SK, Majumdar KC, Chang RY. Strand scission of DNA by bound adriamycin and daunorubicin in the presence of reducing agents. Biochem Biophys Res Commun. 1977;76(3):705–10. doi: 10.1016/0006-291x(77)91557-1. [DOI] [PubMed] [Google Scholar]
- 5.Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281(5381):1317–22. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- 6.Iliakis G, Wang Y, Guan J, Wang H. DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene. 2003;22(37):5834–47. doi: 10.1038/sj.onc.1206682. [DOI] [PubMed] [Google Scholar]
- 7.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281(5383):1677–9. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 8.Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, et al. ATM associates with and phosphorylates p53: mapping the region of interaction. Nat Genet. 1998;20(4):398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- 9.Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15(9):1067–77. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362(6423):847–9. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 11.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Molecular cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dohlman HG, Thorner J. RGS proteins and signaling by heterotrimeric G proteins. J Biol Chem. 1997;272(7):3871–4. doi: 10.1074/jbc.272.7.3871. [DOI] [PubMed] [Google Scholar]
- 13.Berman DM, Gilman AG. Mammalian RGS proteins: barbarians at the gate. J Biol Chem. 1998;273(3):1269–72. doi: 10.1074/jbc.273.3.1269. [DOI] [PubMed] [Google Scholar]
- 14.Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem. 2000;69:795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 15.Berman DM, Wang Y, Liu Z, Dong Q, Burke LA, Liotta LA, et al. A functional polymorphism in RGS6 modulates the risk of bladder cancer. Cancer Res. 2004;64(18):6820–6. doi: 10.1158/0008-5472.CAN-04-1916. [DOI] [PubMed] [Google Scholar]
- 16.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maity B, Yang J, Huang J, Askeland RW, Bera S, Fisher RA. Regulator of G protein signaling 6 (RGS6) induces apoptosis via a mitochondrial-dependent pathway not involving its GTPase-activating protein activity. J Biol Chem. 2010;286(2):1409–19. doi: 10.1074/jbc.M110.186700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330(6003):517–21. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 19.Chatterjee TK, Liu Z, Fisher RA. Human RGS6 gene structure, complex alternative splicing, and role of N terminus and G protein gamma-subunit-like (GGL) domain in subcellular localization of RGS6 splice variants. J Biol Chem. 2003;278(32):30261–71. doi: 10.1074/jbc.M212687200. [DOI] [PubMed] [Google Scholar]
- 20.Yang J, Huang J, Maity B, Gao Z, Lorca RA, Gudmundsson H, et al. RGS6, a modulator of parasympathetic activation in heart. Circ Res. 2010;107(11):1345–9. doi: 10.1161/CIRCRESAHA.110.224220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gopinath S, Vanamala SK, Gujrati M, Klopfenstein JD, Dinh DH, Rao JS. Doxorubicin-mediated apoptosis in glioma cells requires NFAT3. Cell Mol Life Sci. 2009;66(24):3967–78. doi: 10.1007/s00018-009-0157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 23.Martemyanov KA, Yoo PJ, Skiba NP, Arshavsky VY. R7BP, a novel neuronal protein interacting with RGS proteins of the R7 family. J Biol Chem. 2005;280(7):5133–6. doi: 10.1074/jbc.C400596200. [DOI] [PubMed] [Google Scholar]
- 24.Cabrera JL, de Freitas F, Satpaev DK, Slepak VZ. Identification of the Gbeta5-RGS7 protein complex in the retina. Biochem Biophys Res Commun. 1998;249(3):898–902. doi: 10.1006/bbrc.1998.9218. [DOI] [PubMed] [Google Scholar]
- 25.Liu Z, Chatterjee TK, Fisher RA. RGS6 interacts with SCG10 and promotes neuronal differentiation. Role of the G gamma subunit-like (GGL) domain of RGS6. J Biol Chem. 2002;277(40):37832–9. doi: 10.1074/jbc.M205908200. [DOI] [PubMed] [Google Scholar]
- 26.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91(3):325–34. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 27.Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997;11(24):3471–81. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, Shkedy D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc Natl Acad Sci U S A. 1999;96(26):14973–7. doi: 10.1073/pnas.96.26.14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rojas E, Lopez MC, Valverde M. Single cell gel electrophoresis assay: methodology and applications. J Chromatogr B Biomed Sci Appl. 1999;722(1–2):225–54. doi: 10.1016/s0378-4347(98)00313-2. [DOI] [PubMed] [Google Scholar]
- 30.Kurz EU, Douglas P, Lees-Miller SP. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J Biol Chem. 2004;279(51):53272–81. doi: 10.1074/jbc.M406879200. [DOI] [PubMed] [Google Scholar]
- 31.Yoshida M, Shiojima I, Ikeda H, Komuro I. Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J Mol Cell Cardiol. 2009;47(5):698–705. doi: 10.1016/j.yjmcc.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 32.Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, et al. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation. 2009;119(1):99–106. doi: 10.1161/CIRCULATIONAHA.108.799700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shizukuda Y, Matoba S, Mian OY, Nguyen T, Hwang PM. Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice. Mol Cell Biochem. 2005;273(1–2):25–32. doi: 10.1007/s11010-005-5905-8. [DOI] [PubMed] [Google Scholar]
- 34.Liu X, Chua CC, Gao J, Chen Z, Landy CL, Hamdy R, et al. Pifithrin-alpha protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am J Physiol Heart Circ Physiol. 2004;286(3):H933–9. doi: 10.1152/ajpheart.00759.2003. [DOI] [PubMed] [Google Scholar]
- 35.Zheng B, De Vries L, Gist Farquhar M. Divergence of RGS proteins: evidence for the existence of six mammalian RGS subfamilies. Trends Biochem Sci. 1999;24(11):411–4. doi: 10.1016/s0968-0004(99)01474-7. [DOI] [PubMed] [Google Scholar]
- 36.Cheever ML, Snyder JT, Gershburg S, Siderovski DP, Harden TK, Sondek J. Crystal structure of the multifunctional Gbeta5-RGS9 complex. Nat Struct Mol Biol. 2008;15(2):155–62. doi: 10.1038/nsmb.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Rescue of the severe impairment in DXR-induced increases in p53 (A) and pp53-S15 (B) in RGS6−/− MEFs by expression of RGS6L-GFP. Results show quantification of western blot signals of p53 and pp53-S15 from three independent experiments like that shown in Fig 2B. Cells were transfected with GFP or RGS6L-GFP and treated with DXR as described in Fig 2B, and western blot signals were quantified by densitometry. The level of p53 and pp53-S15 in DXR treated WT MEFs (lane 6 in Fig. 2B) were set as 1. Because pp53-S15 levels are undetectable in the absence of DXR, only DXR-stimulated increases in pp53-S15 are shown in panel B. (*p<0.001, **p<0.0001)