

Abstract
Highly pathogenic avian influenza (HPAI) H7 virus infection in humans frequently results in conjunctivitis as a major symptom. However, our understanding of what properties govern virus subtype-specific tropism, and of the host responses responsible for eliciting ocular inflammation and pathogenicity following influenza virus infection, are not well understood. To study virus-host interactions in ocular tissue, we infected primary human corneal and conjunctival epithelial cells with H7, H5, and H1 subtype viruses. We found that numerous virus subtypes were capable of infecting and replicating in multiple human ocular cell types, with the highest titers observed with highly pathogenic H7N7 and H5N1 viruses. Similar patterns of proinflammatory cytokine and chemokine production following influenza virus infection were observed in ocular and respiratory cells. However, primary ocular cells infected with HPAI H7N7 viruses were found to have elevated levels of interleukin-1β (IL-1β), a cytokine previously implicated in ocular disease pathology. Furthermore, H7N7 virus infection of corneal epithelial cells resulted in enhanced and significant increases in the expression of genes related to NF-κB signal transduction compared with that after H5N1 or H1N1 virus infection. The differential induction of cytokines and signaling pathways in human ocular cells following H7 virus infection marks the first association of H7 subtype-specific host responses with ocular tropism and pathogenicity. In particular, heightened expression of genes related to NF-κB-mediated signaling transduction following HPAI H7N7 virus infection in primary corneal epithelial cells, but not respiratory cells, identifies activation of a signaling pathway that correlates with the ocular tropism of influenza viruses within this subtype.
INTRODUCTION
Avian influenza A viruses of the H7 subtype have resulted in over 100 cases of human infection since 2002 (5). Highly pathogenic avian influenza (HPAI) H7 viruses frequently cause conjunctivitis in infected individuals but also possess the ability to cause severe respiratory disease and even death (21). While rare, sporadic reports of ocular-related symptoms following H5N1, seasonal, and 2009 H1N1 virus infection have also been documented (1, 15, 19, 33, 44, 45). The properties which govern the ocular tropism of influenza viruses, and of H7 viruses in particular, are poorly understood. It has been proposed that the predominance of α2-3-linked sialic acids on ocular epithelial cells facilitates the ability of avian influenza viruses which exhibit this binding preference to infect the ocular surface (38). However, studies using a murine model demonstrated that the ability of influenza viruses to bind to or replicate in ocular tissue cannot be explained by sialic acid binding preference alone (8). Understanding the properties which govern the ability of influenza viruses to preferentially replicate in ocular tissue (such as H7 viruses) or potentially use the eye as a portal of entry to establish a respiratory infection (such as H5 viruses) is important for public health preparedness and the response to emerging influenza viruses (30).
Further hindering our understanding of H7 subtype-specific tropism is limited knowledge of the host immune responses elicited following H7 subtype infection. We recently showed that infection with HPAI H7 viruses from both Eurasian and North American lineages resulted in a delayed and weakened induction of innate immune responses compared with that after infection with other HPAI H5N1, low-pathogenic H7, and human influenza A virus subtypes in human respiratory cells (9). Human ocular cells have been shown to elicit proinflammatory mediators following infection with numerous viruses, including respiratory syncytial virus (RSV), herpes simplex virus, and adenovirus (10, 31, 42). However, characterization of host immune responses following influenza virus infection in ocular cells has been limited, and responses to H7 subtype infection in this tissue have not been reported to date (36).
Given the diversity of documented laboratory and occupational ocular exposures to influenza virus, numerous ocular cell types could play a role in influenza-related ocular infection and pathology observed in humans. Independent studies have evaluated the permissiveness of human corneal epithelial cells, ex vivo conjunctival biopsy specimens, and retinal pigment epithelial cells to influenza virus infection, demonstrating the ability of select influenza viruses to replicate in these cell types (8, 13, 36). Despite this, side-by-side comparisons of virus infection in multiple ocular cell types have not been performed, making it difficult to compare the magnitudes of host responses between ocular cell types or virus subtypes.
Here, we investigated the induction of the innate immune response to human and avian influenza virus infection in both corneal and conjunctival epithelial cells to characterize host responses in ocular tissue. This information allowed us to then examine H7 subtype-specific host responses in both human corneal and bronchial epithelial cells to more accurately delineate ocular tropism determinants of viruses within this subtype. H7N7 virus infection of corneal epithelial cells resulted in enhanced and significant increases in the expression of genes related to NF-κB signal transduction compared with H5N1 or H1N1 virus infection. In contrast, H5N1 virus infection resulted in heightened NF-κB signal transduction in respiratory and not ocular cells. Identification of tissue-specific and subtype-specific host responses following infection with human and avian influenza viruses is crucial for achieving a more precise understanding of properties governing virus tropism in the human host.
MATERIALS AND METHODS
Viruses.
Influenza A viruses were grown in the allantoic cavities of 10-day-old embryonated hens' eggs (H7, H5, and seasonal H1 viruses) or in Madin-Darby canine kidney (MDCK) cells (2009 H1N1 virus) and then clarified by centrifugation and stored at −80°C until use, as previously described (6, 34, 35). Stocks were titrated by standard plaque assay for determination of PFU titer (51). All experiments were conducted under biosafety level 3 containment, including enhancements required by the U.S. Department of Agriculture and the Select Agent Program (17).
Cell culture and viral infection.
Primary human corneal epithelial cells (HCEpiC), primary human conjunctival epithelial cells (HConEC), and primary human trabecular meshwork cells isolated from juxtacanalicular and corneoscleral regions of the human eye (HTMC) were obtained from ScienCell (San Diego, CA). Primary cells were cryopreserved at passage 1 and then grown on poly-l-lysine-coated flasks per the manufacturer's instructions. Cells were grown to confluence in 6-well (for replication kinetics), 12-well (for PCR array analysis), or 24-well (for cytokine quantification and real-time PCR) poly-l-lysine coated plates in serum-free keratinocyte medium (epithelial cells) or complete fibroblast medium (trabecular meshwork cells) (ScienCell). All primary cells were infected with virus within 10 population doublings. The bronchial epithelial cell line Calu-3 (ATCC, Manassas, VA) was cultured on membrane inserts and infected apically as previously described (9, 51).
Virus was added to cells at a multiplicity of infection (MOI) of 0.01 (for replication kinetics) or 2 (for cytokine quantification and real-time PCR) for 1 h before washing. Cell type-specific serum-free medium was added to all wells, with the addition of 300 μg/liter N–p-tosyl-l-phenylalanine chloromethyl ketone (TPCK)-treated trypsin (Sigma-Aldrich, St. Louis, MO) in cultures using primary human cells. Aliquots of culture supernatant taken postinfection (p.i.) were immediately frozen at −80°C until use. Medium with 10% normal allantoic fluid was used as a mock control. Samples collected for replication kinetics were titrated for the presence of infectious virus by standard plaque assay.
Cell viability assays.
Quantification of nucleosomes at 24 h following virus infection of HCEpiC at an MOI of 2 was performed with the ELISAPlus cell death detection assay (Roche Applied Science, Indianapolis, IN), according to manufacturer's instructions. A minimum of three independent samples were collected and tested for each condition. Quantification of cell proliferation at 24, 48, and 72 h following virus infection of HCEpiC and Calu-3 cells at an MOI of 2 was performed using the WST-1 cell proliferation reagent (Roche Applied Science), according to manufacturer's instructions. Four independent samples were tested for each condition.
Cytokine and chemokine quantification.
Culture supernatants were analyzed using an enzyme linked immunosorbent assay (ELISA) for the presence of human IP-10, tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), and IL-10 (BD Biosciences, San Jose, CA) and with the BioPlex protein array system (Bio-Rad, Hercules, CA), according to manufacturer's instructions. A minimum of two independent samples were collected and tested for each condition.
Real-time qRT-PCR.
Total RNA was extracted from mock-infected or virus-infected cells (Qiagen, Valencia, CA). The DyNAmo SYBR green two-step quantitative reverse transcription-PCR (qRT-PCR) kit was used for cDNA synthesis and SYBR green quantitative PCR (New England BioLabs, Ipswich, MA) according to manufacturer's instructions, with all reactions performed in duplicate or triplicate. All quantifications (threshold cycle [CT] values) were normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and analyzed as described previously to determine the relative level of gene expression (51). Primer sequences were designed with PrimerExpress (Applied Biosystems, Carlsbad, CA) and have been published previously (51), with the exception of IL-1β (5′, CCA CGG CCA CAT TTG GTT; 3′, TAG GGA AGC GGT TGC TCA TC).
RT2 Profiler PCR array analysis (SABiosciences, Frederick, MD) for the human NF-κB signaling pathway was performed with total RNA extracted at 24 h p.i. and analyzed per manufacturer's instructions.
SDS-PAGE immunoblot analysis.
Cell lysates from virus- or mock-infected HCEpiC and Calu-3 cells were resuspended in Laemmli buffer (Bio-Rad) and boiled for 10 min. Protein content was determined using a NanoDrop spectrophotometer at 280 nm (Thermo Scientific, Wilmington, DE), and equal amounts of protein were loaded in a sodium dodecyl sulfate-polyacrylamide gel. Western blot analysis was conducted as described previously using antibodies to IκBα (Santa Cruz Biotechnology, Santa Cruz, CA) or β-actin (Sigma, St. Louis, MO), with detection using a horseradish peroxidase-conjugated secondary antibody (Cell Signaling, Beverly, MA) (51). Densitometry quantification was performed using ImageJ software.
Statistics.
Significance in all experiments presented in this study was assessed by Student's t test.
RESULTS
Multiple human ocular cell types support replication of avian influenza A viruses.
The diverse range of potential ocular exposures to influenza virus in humans indicates that numerous ocular cell types may be exposed to infectious virus and support productive virus replication. We examined two ocular epithelial cell types, human corneal epithelial cells (HCEpiC) and human conjunctival epithelial cells (HConEC), which are likely exposed to virus during human ocular exposure. The influenza viruses used in these infections are listed in Table 1. The permissiveness of primary human corneal and conjunctival cells to influenza virus infection has been previously shown, and expression of the influenza virus M1 transcript at 2 h p.i. in these cell types was found to be similar for all viruses examined (data not shown) (8, 13). To examine the possibility of intraocular influenza virus replication, we additionally infected human trabecular meshwork cells (HTMC), fibroblasts located in the anterior chamber angle of the eye which are exposed to draining aqueous fluid (32). The replication kinetics for each virus were generally found to be similar in all ocular cell types examined. Primary cells from all ocular anatomical locations tested supported productive replication of all HPAI H5 and H7 viruses, whereas titers from low-pathogenic avian influenza (LPAI), seasonal, and 2009 H1N1 viruses were reduced (Fig. 1). While reaching similar titers by 72 h p.i., the HPAI viruses NL/219, Thai/16, and VN/1203, all of which possess a lysine at position 627 of PB2, replicated to significantly (P < 0.001) higher titers by 24 h p.i. than the HPAI viruses NL/230 and Can/504, which lack this residue in PB2. As epithelial cells are the first barrier of the ocular surface to encounter influenza virus, HCEpiC and HConEC cultures were utilized in subsequent experiments.
Table 1.
Influenza A viruses used in this study
| Virus | Name in this study | Subtype | Pathogenicitya | PB2 627b | Patient symptomsc |
|---|---|---|---|---|---|
| A/Netherlands/219/03 | NL/219 | H7N7 | HPAI | K | Respiratory, fatal |
| A/Netherlands/230/03 | NL/230 | H7N7 | HPAI | E | Conjunctivitis |
| A/Canada/504/04 | Can/504 | H7N3 | HPAI | E | Conjunctivitis, respiratory |
| A/Canada/444/04 | Can/444 | H7N3 | LPAId | E | Conjunctivitis, respiratory |
| A/New York/107/03 | NY/107 | H7N2 | LPAI | E | Respiratory |
| A/Vietnam/1203/04 | VN/1203 | H5N1 | HPAI | K | Respiratory, fatal |
| A/Thailand/16/04 | Thai/16 | H5N1 | HPAI | K | Respiratory, fatal |
| A/Brisbane/59/07 | Brisbane | H1N1 | NAe | E | Respiratory |
| A/Mexico/4482/09 | Mex/4482 | H1N1 | NA | E | Respiratory |
Fig. 1.

Replication kinetics of influenza viruses in primary human ocular cells. HCEpiC (A), HConEC (B), and HTMC (C) were infected at an MOI of 0.01 with the indicated viruses. Culture supernatants were collected at the indicated times p.i., and titers were determined by plaque assay to quantify infectious virus. HP, highly pathogenic; LP, low pathogenic. The means from duplicate independent cultures per virus ± standard deviations are shown.
HPAI H5 and H7 viruses reduce cell viability following infection of human ocular cells.
High levels of virus replication in respiratory tissues have been associated with impaired cell function and increased cell death (3, 25, 47). However, the viability of human ocular cells following influenza virus infection has not been extensively studied. To determine whether influenza virus replication in ocular cells resulted in a decrease in cell proliferation, we measured the ability of mitochondrial dehydrogenases in HCEpiC cultures infected with influenza virus to cleave tetrazolium salt added to the culture medium, as a marker for cellular metabolic activity (Fig. 2A). HCEpiC cultures infected with viruses which replicated to the highest titers in HCEpiC cultures (NL/219, VN/1203, and Thai/16 viruses) were found to possess fewer metabolically active cells than mock-infected cultures at 24 and 48 h p.i., resulting in less mitochondrial dehydrogenase activity in culture supernatant, indicative of attenuated cell proliferation. In contrast, HCEpiC cultures infected with seasonal and 2009 H1N1 viruses retained levels of cell proliferation comparable to those in mock-infected cultures through 72 h p.i. (Fig. 2A and data not shown). To examine whether the decrease in cell proliferation observed following HPAI virus infection was associated with increased cell death, we quantified levels of cytoplasmic histone-associated DNA fragments (nucleosomes) in cell lysates (indicative of apoptosis) and culture supernatants (indicative of necrosis) at 24 h p.i. in HCEpiC cultures (Fig. 2B and C). While infection with all influenza viruses tested resulted in the significantly elevated (P < 0.005) enrichment of nucleosomes in cell lysates (>15-fold over that in mock-infected cells), NL/219, VN/1203, and Thai/16 virus infection resulted in nucleosome levels among the highest observed (Fig. 2B). Levels of nucleosomes in culture supernatant followed a similar trend but were within 3-fold of the level for mock-infected cells at 24 h p.i. (Fig. 2C). Interestingly, two viruses which did not replicate efficiently in corneal epithelial cells, Can/444 and NY/107, were still capable of eliciting elevated levels of nucleosomes in both cell lysate and culture supernatant. Significantly reduced cell proliferation compared with mock-infected cells was further observed in NY/107 virus-infected HCEpiC cultures (P < 0.005). In summary, while viruses of multiple subtypes were capable of inducing cell death in human corneal epithelial cells, viruses which replicated to high titers were generally associated with the greatest corresponding decrease in cell proliferation.
Fig. 2.

Viability of HCEpiC cells following influenza virus infection. HCEpiC cells were infected at an MOI of 2 with the indicated viruses. (A) Quantification of cell proliferation following the addition of the reagent WST-1 was performed by measuring the absorbance of formazan dye produced by metabolically active cells at 450 nm. The data are presented as the mean absorbance for each virus subtracted from the absorbance for mock-infected cells for each time point plus standard deviation and represent quadruple cultures. (B and C) Cell lysates (B) or supernatants (C) were collected at 24 h p.i., and the presence of nucleosomes was detected by ELISA to measure apoptosis and necrosis, respectively. HP, highly pathogenic; LP, low pathogenic. *, P < 0.05; **, P < 0.005 (compared with mock-infected cells). The data are presented as the relative fold above mock-infected cells and results for represent triplicate samples plus standard deviations.
HPAI H7N7 viruses exhibit differential cytokine production in HCEpiC and HConEC compared with H5N1 and H1N1 viruses.
Human corneal and conjunctival epithelial cells elicit elevated levels of proinflammatory cytokines and chemokines following exposure to toxins or other stimuli (20, 23). Cytokine dysregulation has been reported following HPAI H5N1 virus infection in numerous human respiratory cell types, and levels of selected proinflammatory cytokines were elevated following H5N1 virus infection in retinal pigment epithelial cells (16, 36, 51). However, the induction of innate immune responses in ocular surface epithelial cells following influenza virus infection was not known. Therefore, we infected HCEpiC and HConEC with avian and human influenza viruses and measured the production of key inflammatory mediators early after infection (Fig. 3).
Fig. 3.

Cytokine production following influenza virus infection in human ocular epithelial cells. HCEpiC (A) or HConEC (B) were infected with influenza virus at an MOI of 2. Supernatants were collected at the indicated times p.i., and levels of each analyte were quantified by ELISA or BioPlex. The means from duplicate independent cultures per virus ± standard deviations are shown.
Similar trends in proinflammatory cytokine (TNF-α, IL-6, and IL-1β) and chemokine (IP-10) production were generally observed in both ocular cell types (Fig. 3). HPAI H5N1 viruses consistently resulted in the highest levels of TNF-α, IL-6, and IP-10 production in ocular epithelial cells, with the LPAI H7N7 virus NY/107 also eliciting high levels, a property of this virus that was previously shown in respiratory cell types (9). Infection with HPAI H7N7 viruses, notably NL/219 virus, produced significantly lower levels of these cytokines and chemokines at 24 h p.i. in human ocular epithelial cells than were induced by H5N1, H7N3 and H7N2 viruses (P < 0.05). However, the cytokine IL-1β was produced by HCEpiC at 24 h following NL/219 virus infection at higher levels than after infection with H5N1 or H1N1 virus subtypes (P < 0.02) (Fig. 3B). Interestingly, while the trends in cytokine production were similar in both cell types examined, the magnitudes of IP-10 and IL-1β production were approximately 10-fold lower in HConEC than in HCEpiC. Despite pronounced differences in cytokine production in these cultures, comparable levels of mRNAs for key cytokines and chemokines were detected with HPAI H7 and H5 viruses, indicating that the diminished cytokine production observed in H7 virus-infected ocular cells was not a function of reduced transcript levels in infected HCEpiC or HConEC cultures (see Fig. S1 in the supplemental material).
Endogenous levels of IL-8 and granulocyte colony-stimulating factor (G-CSF) (800 to 2,000 pg/ml) were detected in cultures of both cell types by 24 h p.i. but did not rise >5-fold over those in mock-infected cells following virus infection (data not shown). IL-2, IL-4, IL-5, IL-7, IL-10, IL-12 (p70), IL-13, IL-17, monocyte chemoattractant protein 1 (MCP-1), MIP-1β, gamma interferon (IFN-γ), and granulocyte-macrophage CSF (GM-CSF) were detected at only nominal levels (<30 pg/ml) through 24 h p.i. and were not significantly elevated above levels in mock-infected cells following influenza virus infection in either cell type (data not shown). These results indicate that, generally, proinflammatory cytokine production in human ocular epithelial cells closely resembles that observed in respiratory cell types, with the exception of IL-1β (9). The greater magnitude of cytokine production in HCEpiC than in HConEC cultures suggests that corneal epithelial cells are an important contributor to ocular immune responses following influenza virus infection. These results further indicate that cytokine transcript is present in infected cells, but detectable protein is not being secreted following HPAI H7 virus infection.
HPAI H7N7 virus infection leads to upregulation of NF-κB signaling in human ocular but not respiratory cells.
IL-1β has known roles in ocular pathology and inflammation in ocular cells and has previously been shown to disrupt the barrier function of human corneal epithelial cells in an NF-κB-dependent manner (27). The detection of elevated levels of IL-1β following NL/219 virus infection in human ocular but not respiratory cells prompted us to examine the induction of NF-κB activation in both cell types to explore both tissue-specific and subtype-specific responses. HCEpiC and Calu-3 cells were infected with representative H7N7 (NL/219), H5N1 (Thai/16), and H1N1 (Brisbane) viruses, and cell lysates were collected and examined for the degradation of IκBα to detect activation of the NF-κB pathway (Fig. 4). IκBα belongs to a family of proteins which function to prevent NF-κB nuclear translocation and inhibit the DNA binding activity of NF-κB; NF-κB activation is associated with the degradation of IκBα (50). Degradation of IκBα was detected by 12 h p.i. following infection with all virus subtypes in HCEpiC cultures, with cells infected with NL/219 virus possessing undetectable levels of IκBα by densitometry by 24 h p.i. Similar results were observed following virus infection in Calu-3 cells, indicating that this early step of NF-κB activation occurred independent of cell type. As levels of IκBα degradation were similar in ocular and respiratory cell types, we next examined a panel of genes related to NF-κB-mediated signal transduction by real-time PCR array analysis in both corneal and respiratory epithelial cells to determine if differential downstream NF-κB responses play a role in the ocular tropism of influenza viruses.
Fig. 4.
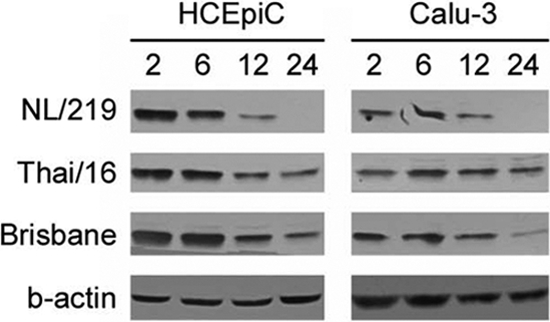
Degradation of IκBα following influenza virus infection in human ocular and respiratory cells. HCEpiC or Calu-3 cells were infected with the indicated viruses at an MOI of 2. Protein samples were harvested at 2, 6, 12, or 24 h p.i. and examined by SDS-PAGE for the presence of IκBα. Equal loading was achieved by loading 200 μg of sample per well and was confirmed by β-actin (a representative blot is shown).
Genes related to the NF-κB signal transduction pathway that were significantly (P < 0.05) upregulated following virus infection of HCEpiC cultures are presented in Fig. 5 and in Table S1 in the supplemental material; the fold change in expression over that in mock-infected controls is shown. As predicted by the heightened production of IL-1β in H7N7 virus-infected ocular cells, NL/219 virus infection resulted in the highest upregulation of numerous genes related to NF-κB signaling in HCEpiC cells, notably, those for transcription factors such as FOS, JUN, and RELB, and NF-κB responsive genes, including those for CSF2, ICAM,1, and LTA (Fig. 5). In contrast, infection with the North American lineage H7 virus Can/504 did not broadly elicit NF-κB gene activation. Thai/16 virus infection of HCEpiC cultures did elicit elevated levels of select genes involved in NF-κB signal transduction, including those for IL-6, IL-8, and TNF, but fewer genes overall than with NL/219 virus infection. Infection with the H1N1 viruses Brisbane and Mex/4482 resulted in an overall reduced activation of genes related to NF-κB signal transduction pathways compared with H7 and H5 virus subtypes (Fig. 5).
Fig. 5.

Fold regulation of selected genes related to NF-κB signaling in ocular cells following influenza virus infection. HCEpiC were infected with influenza viruses at an MOI of 2. Total RNA was isolated from cells at 24 h p.i. and examined by real-time RT-PCR array analysis. Genes which exhibited a significant (P < 0.05) >3-fold change in expression over that in mock-infected cells are shown. A full list of genes upregulated and downregulated following infection of HCEpiC cultures with these influenza viruses is shown in Table S1 in the supplemental material.
To determine if the heightened NF-κB activity observed in HCEpiC cultures following H7N7 virus infection was tissue specific, we compared genes related to the NF-κB signal transduction pathway that were either significantly (P < 0.05) upregulated or downregulated compared with those in mock-infected cells following infection of HCEpiC or Calu-3 cells with NL/219, Thai/16, or Brisbane virus (Fig. 6; see Table S1 in the supplemental material). The upregulation of numerous genes related to NF-κB signaling in HCEpiC following H7N7 virus infection was found to be both virus subtype specific and tissue specific; unlike H7N7 virus infection, Thai/16 or Brisbane virus infection in HCEpiC cultures did not result in substantial upregulation of NF-κB genes specifically in ocular cells (Fig. 6A to C). H5N1 virus infection upregulated numerous genes pertaining to NF-κB signaling; however, the genes exhibiting upregulation were found solely in Calu-3 cells or were similarly upregulated in both respiratory and ocular cell types (Fig. 6B). H1N1 virus infection resulted in a similar profile, though with a lesser total number of genes (Fig. 6C). In comparison to the observed upregulation of NF-κB-related genes in ocular cells following NL/219 virus infection, H7N7 virus infection in respiratory cells resulted in a broad downregulation of NF-κB-related genes (Fig. 6D). This downregulation was also found to be virus subtype specific and tissue specific, as only 6% of the genes downregulated following H7N7 virus infection were specific to HCEpiC cultures only (Fig. 6D). In contrast, the majority of NF-κB-related genes which were downregulated following H5N1 virus infection were detected in ocular cells only (Fig. 5E). H1N1 virus infection resulted in the downregulation of numerous genes, but many of these genes behaved similarly in both ocular and respiratory cells (Fig. 6F).
Fig. 6.

Comparison of fold regulation of NF-κB signaling in human respiratory and ocular cells relative to virus subtype. HCEpiC or Calu-3 cells were infected with influenza viruses at an MOI of 2. Total RNA was isolated from cells at 24 h p.i. and examined by real-time RT-PCR array analysis. Genes included in this analysis possessed the highest (A to C) or lowest (D to F) fold expression over that in mock-infected cells relative to virus subtype in Calu-3 cells (black), HCEpiC cultures (dark gray), or both cell types (light gray). Genes where virus infection did not result in a ≥3-fold change in expression over that in mock-infected cells within that cell type or where this fold change was not significant were excluded from this analysis.
In summary, while early IκBα degradation of NF-κB activation was not affected by cell type origin, we found that the H7N7 virus NL/219 resulted in the upregulation of numerous genes related to NF-κB signaling following infection of ocular cells and in the broad downregulation of these genes following infection of respiratory cells. In contrast, the H5N1 virus Thai/16 resulted in the upregulation of numerous genes related to NF-κB signaling in respiratory cells and in the downregulation of these genes following infection of ocular cells. H1N1 virus infection resulted in high upregulation of relatively few NF-κB-related genes, and most of these genes were similarly upregulated in respiratory cells.
DISCUSSION
Despite the frequency of ocular symptoms following human infection with H7 viruses, the properties which confer the ability of this influenza virus subtype to use the eye as a portal of entry are not known. As previous studies have shown that viruses of multiple subtypes are capable of binding to and replicating in ocular tissue, we chose to examine the early induction of innate immune responses elicited by influenza viruses of multiple subtypes in human ocular cells to determine whether H7 subtype-specific host responses contribute to the ocular tropism of this virus subtype. Unlike viruses of the H5N1 or H1N1 subtype, we identified an HPAI H7N7 virus which elicited elevated levels of IL-1β and upregulated genes associated with NF-κB signal transduction following infection of ocular but not respiratory cells, representing the first in vitro evidence of an H7 subtype-specific and tissue-specific response in ocular tissue.
The development of conjunctival symptoms following influenza virus exposure in humans has been documented following both laboratory exposures (e.g., by liquid containing influenza virus being splashed on a laboratorian's face or infected animals sneezing in the face and eye of a laboratorian) and occupational exposures (e.g., by direct ocular exposure to infected poultry or eye abrasions from fomites during culling operations) (5). Both corneal and conjunctival epithelial cells express glycoconjugates containing terminal sialic acids on their surfaces, and it is credible that virus comes into contact with both tissue surfaces during an ocular exposure (8, 13, 40, 46). Furthermore, both corneal and conjunctival epithelial cells are exposed to the lacrimal drainage system in the human eye, which governs drainage from tear ducts to nasal passages (40). As such, both cell types are likely candidates to be exposed to virus and/or support virus replication during the course of an ocular infection. While cell types located in the interior of the eye would be expected to be less accessible to influenza virus during initial environmental exposure than surface epithelial cells, their ability to support high-titer influenza virus replication as shown for retinal pigment epithelial cells (36) and trabecular meshwork cells (Fig. 1) underscores the diversity of cell types in ocular tissue which are potential sources of infection.
While similar kinetics of virus infection and replication were observed for both ocular epithelial cell types, HCEpiC cultures secreted 10-fold-higher levels of select cytokines and chemokines following virus infection than HConEC cultures (Fig. 3), indicating that virus infection of these cell types can elicit differential host responses. This finding is of particular importance when assessing the permissiveness of ocular cells to 2009 pandemic viruses. Documented instances of ocular symptoms following exposure to seasonal or 2009 H1N1 viruses are rare, suggesting that these viruses are poorly suited to use the eye as a portal of entry (19, 33, 44). In support of this, we demonstrated previously that seasonal and 2009 H1N1 viruses do not readily infect mice when inoculated by the ocular route and do not replicate to high titer in human corneal cells (7, 8). However, a recent publication demonstrated that a 2009 H1N1 pandemic virus, but not a seasonal H1N1 virus, was capable of high-titer replication in human conjunctival tissue (13). Interestingly, our study found that the 2009 H1N1 virus Mex/4482 replicated to titers up to 100-fold higher than those of the seasonal virus Brisbane by 72 h p.i. in HConEC cultures but not in other ocular cell types examined (Fig. 1). Additionally, infection with Mex/4482 virus was capable of producing elevated levels of TNF-α and IP-10 in human conjunctival but not corneal cell cultures (Fig. 3). The finding of differences in the magnitudes of certain host responses following influenza virus infection in general and the potential preference of particular viruses for select ocular cells underscore the necessity of studying numerous ocular cell types when evaluating the ocular tropism of influenza viruses. Further study is needed to ascertain what properties of 2009 H1N1 viruses facilitate this apparent enhanced replication and proinflammatory cytokine and chemokine production in conjunctival but not corneal cells.
The efficient replication in all ocular cell types of HPAI viruses bearing a lysine at position 627 in PB2 suggests that this residue confers a replication advantage in both respiratory and ocular cells, as similar patterns of replication with HPAI H7N7 viruses bearing a lysine at this position have been observed in both murine corneal epithelial sheets and human bronchial and lung cells (8, 18, 22). The abundant expression of α2-3-linked sialic acids on the surfaces of the primary ocular cell types tested here may have further facilitated high-titer replication of viruses with an avian receptor binding preference, including subtypes such as H5N1, which are only infrequently associated with ocular disease (4, 5). Despite their high sequence homology, the HPAI H7N3 virus Can/504 replicated to titers >1,000-fold higher than those of the LPAI Can/444 virus in HCEpiC and HTMC cultures, and >100-fold higher in HConEC cultures, by 48 h p.i., indicating the presence of H7 virus molecular determinants of ocular replication that are independent of position 627 (Fig. 1) (24, 39). Heightened levels of apoptosis and necrosis following infection of HCEpiC cultures with the LPAI viruses Can/444 and NY/107 compared with the HPAI viruses Can/504 and NL/230 highlight a distinct heterogeneity in the behavior of H7 subtype viruses in ocular cells and demonstrate that influenza viruses can elicit pronounced host responses in this tissue in the absence of high-titer virus replication. Interestingly, the attenuated proliferation and induction of nucleosomes detected following infection of ocular cell types with NY/107 virus were also observed in human respiratory cell types; this H7N2 virus possesses receptor binding and transmissibility properties distinct from those of other North American H7 viruses, and further study is warranted to better understand the atypical behavior of this strain (4, 9).
Direct comparisons between virus infection of respiratory and ocular cells allow for greater identification of features specific to one tissue type and can provide clues regarding the tropism of individual viruses for one cell type over another. Despite different epidemiologic profiles of human infection, the infectivity and replication kinetics of HPAI H5N1 and H7N7 viruses are consistent between both ocular and respiratory cells (9, 13). The attenuated production of proinflammatory cytokines and chemokines following HPAI H7N7 virus infection is further maintained in both tissue types. While HPAI H7 and H5 viruses are detrimental to cell viability in both ocular and respiratory cells, we have shown that subtype-specific differences affect levels of necrosis but not apoptosis in Calu-3 cells, while more pronounced subtype-specific differences in apoptosis but not necrosis predominate in HCEpiC cultures (Fig. 2B and C) (9, 52). Notably, the production of IL-1β in HCEpiC cultures following NL/219 virus infection is not observed in Calu-3 cells; this respiratory cell type does not produce substantial levels of IL-1β following influenza virus infection (data not shown). Previous studies have similarly found increased mRNA expression of IL-1β in alveolar and monocyte-derived macrophages, but not primary human bronchial epithelial cells, following lung injury or influenza virus infection (14, 16, 41). IL-1β, a cytokine involved in inflammatory responses, cell growth, and tissue repair, is frequently associated with ocular disease processes (10, 20, 26, 27, 43). Similar to our findings, a previous study found that RSV infection of corneal epithelial cells resulted in an upregulation of secreted IL-1β cytokine without a substantial change in cellular IL-1β mRNA levels (10).
Previous studies have demonstrated an association between dysregulated host innate immune responses and ocular surface inflammation; in particular, many of the documented effects of IL-1β in ocular inflammation and pathology occur in an NF-κB-dependent manner (2, 10, 27, 49). The heightened expression of genes associated with NF-κB signal transduction following NL/219 virus infection (relative to other virus subtypes) in HCEpiC and the corresponding decreased expression of these genes following infection of Calu-3 cells mirror the tropism for ocular and not respiratory disease observed following H7 virus infection in humans. In addition to heightened expression of NF-κB-related genes, HCEpiC cultures infected with NL/219 virus possessed levels of cytokine mRNA transcript comparable to those in H5N1 virus-infected cells, in contrast to the delayed and attenuated levels of cytokine mRNA transcript detected following HPAI H7N7 virus infection in respiratory Calu-3 cells, with reduced expression of NF-κB-related genes (9). However, divergent patterns of NF-κB-related gene activation following infection of HCEpiC cultures with NL/219 and Can/504 viruses indicate that other, yet-unknown host factors likely contribute toward ocular tropism. Ongoing research on potential virus- and strain-specific posttranscriptional regulation of cytokine production to ascertain the disassociation of mRNA and cytokine secretion into the supernatant (notably with regard to TNF-α) will allow for a better understanding of innate immune responses in ocular tissue following influenza virus infection. Furthermore, additional study is needed to explore the potential correlation and implications of heightened transcriptional activity of cytokine and NF-κB-related genes in ocular cells following H7 virus infection. Augmented NF-κB signal transduction in human corneal cells following H7 virus infection identifies a prospective target for immunomodulatory agents to mitigate conjunctival symptoms following H7 virus infection in humans (10, 11, 27, 29).
Understanding the properties which govern the ocular tropism of influenza A viruses has implications which reach beyond H7 subtype viruses. Reports of conjunctivitis concurrent with influenza-like illness have been reported for both H5 and H7 subtype viruses (15, 28, 45, 48). The presence of both α2-3- and α2-6-linked sialic acids on epithelial cells of the human eye suggests that a broad range of influenza viruses could bind to this tissue, including virus subtypes (such as H5N1) which may use the eye as an initial site of virus replication before establishment of a respiratory infection (30). Continued study of the interplay between influenza viruses, the human eye, and the link between ocular infection and development of respiratory disease will provide much-needed information to guide prevention, treatment, and control strategies for all influenza virus subtypes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Yan Li (Canadian Center for Human and Animal Health) and the Vietnam and Thailand Ministries of Health for providing viruses used in this study, Akiko Wilson for graphical assistance, and Shivaprakash Gangappa for helpful discussions.
The findings and conclusions in this report are those of the authors and do not necessarily reflect the views of the funding agency.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 20 July 2011.
REFERENCES
- 1. Abdel-Ghafar A. N., et al. 2008. Update on avian influenza A (H5N1) virus infection in humans. N. Engl. J. Med. 358:261–273 [DOI] [PubMed] [Google Scholar]
- 2. Auron P. E. 1998. The interleukin 1 receptor: ligand interactions and signal transduction. Cytokine Growth Factor Rev. 9:221–237 [DOI] [PubMed] [Google Scholar]
- 3. Baskin C. R., et al. 2009. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl. Acad. Sci. U. S. A. 106:3455–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Belser J. A., et al. 2008. Contemporary North American influenza H7 viruses possess human receptor specificity: implications for virus transmissibility. Proc. Natl. Acad. Sci. U. S. A. 105:7558–7563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Belser J. A., Bridges C. B., Katz J. M., Tumpey T. M. 2009. Past, present, and possible future human infection with influenza virus A subtype H7. Emerg. Infect. Dis. 15:859–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belser J. A., et al. 2007. Pathogenesis of avian influenza (H7) virus infection in mice and ferrets: enhanced virulence of Eurasian H7N7 viruses isolated from humans. J. Virol. 81:11139–11147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Belser J. A., et al. 2010. Pathogenesis of pandemic influenza A (H1N1) and triple-reassortant swine influenza A (H1) viruses in mice. J. Virol. 84:4194–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Belser J. A., Wadford D. A., Xu J., Katz J. M., Tumpey T. M. 2009. Ocular infection of mice with influenza A (H7) viruses: a site of primary replication and spread to the respiratory tract. J. Virol. 83:7075–7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belser J. A., Zeng H., Katz J. M., Tumpey T. M. 2011. Infection with highly pathogenic H7 influenza viruses results in an attenuated proinflammatory cytokine and chemokine response early after infection. J. Infect. Dis. 203:40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bitko V., et al. 2004. Activation of cytokines and NF-kappa B in corneal epithelial cells infected by respiratory syncytial virus: potential relevance in ocular inflammation and respiratory infection. BMC Microbiol. 4:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bitko V., Velazquez A., Yang L., Yang Y. C., Barik S. 1997. Transcriptional induction of multiple cytokines by human respiratory syncytial virus requires activation of NF-kappa B and is inhibited by sodium salicylate and aspirin. Virology 232:369–378 [DOI] [PubMed] [Google Scholar]
- 12. CDC 2004. Update: influenza activity—United States and worldwide, 2003-04 season, and composition of the 2004-05 influenza vaccine. MMWR Morb. Mortal. Wkly. Rep. 53:547–552 [PubMed] [Google Scholar]
- 13. Chan M. C., et al. 2010. Tropism and innate host responses of the 2009 pandemic H1N1 influenza virus in ex vivo and in vitro cultures of human conjunctiva and respiratory tract. Am. J. Pathol. 176:1828–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chan M. C., et al. 2005. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir. Res. 6:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chan P. K. 2002. Outbreak of avian influenza A(H5N1) virus infection in Hong Kong in 1997. Clin. Infect. Dis. 34 (Suppl. 2):S58–S64 [DOI] [PubMed] [Google Scholar]
- 16. Cheung C. Y., et al. 2002. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360:1831–1837 [DOI] [PubMed] [Google Scholar]
- 17. Chosewood L. C., Wilson D. E. 2009. Biosafety in microbiological and biomedical laboratories, 5th ed. U.S. Dept. of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institutes of Health, Washington, DC [Google Scholar]
- 18. de Wit E., et al. 2010. Molecular determinants of adaptation of highly pathogenic avian influenza H7N7 viruses to efficient replication in the human host. J. Virol. 84:1597–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dubnov-Raz G., Somech R., Warschawski Y., Eisenberg G., Bujanover Y. 12 October 2010. Clinical characteristics of children with 2009 pandemic H1N1 influenza virus infections. Pediatr. Int. doi:10.1111/j.1442-200X.2010.03271.x [DOI] [PubMed] [Google Scholar]
- 20. Epstein S. P., Chen D., Asbell P. A. 2009. Evaluation of biomarkers of inflammation in response to benzalkonium chloride on corneal and conjunctival epithelial cells. J. Ocul. Pharmacol. Ther. 25:415–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fouchier R. A., et al. 2004. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc. Natl. Acad. Sci. U. S. A. 101:1356–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gabriel G., et al. 2005. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl. Acad. Sci. U. S. A. 102:18590–18595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gamache D. A., et al. 1997. Secretion of proinflammatory cytokines by human conjunctival epithelial cells. Ocul. Immunol. Inflamm. 5:117–128 [DOI] [PubMed] [Google Scholar]
- 24. Hirst M., et al. 2004. Novel avian influenza H7N3 strain outbreak, British Columbia. Emerg. Infect. Dis. 10:2192–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kash J. C., et al. 2006. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443:578–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim S. H., Mok J. W., Kim H. S., Joo C. K. 2008. Association of −31T>C and −511 C>T polymorphisms in the interleukin 1 beta (IL1B) promoter in Korean keratoconus patients. Mol. Vis. 14:2109–2116 [PMC free article] [PubMed] [Google Scholar]
- 27. Kimura K., Teranishi S., Nishida T. 2009. Interleukin-1beta-induced disruption of barrier function in cultured human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 50:597–603 [DOI] [PubMed] [Google Scholar]
- 28. Koopmans M., et al. 2004. Transmission of H7N7 avian influenza A virus to human beings during a large outbreak in commercial poultry farms in the Netherlands. Lancet 363:587–593 [DOI] [PubMed] [Google Scholar]
- 29. Kopp E., Ghosh S. 1994. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 265:956–959 [DOI] [PubMed] [Google Scholar]
- 30. Kumlin U., Olofsson S., Dimock K., Arnberg N. 2008. Sialic acid tissue distribution and influenza virus tropism. Influenza Other Respir. Viruses 2:147–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li H., et al. 2006. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 117:167–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Llobet A., Gasull X., Gual A. 2003. Understanding trabecular meshwork physiology: a key to the control of intraocular pressure? News Physiol. Sci. 18:205–209 [DOI] [PubMed] [Google Scholar]
- 33. Lopez-Prats M. J., Sanz Marco E., Hidalgo-Mora J. J., Garcia-Delpech S., Diaz-Llopis M. 2010. Bleeding follicular conjunctivitis due to influenza H1N1 virus. J. Ophthalmol. 2010:423672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maines T. R., et al. 2009. Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science 325:484–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maines T. R., et al. 2005. Avian influenza (H5N1) viruses isolated from humans in Asia in 2004 exhibit increased virulence in mammals. J. Virol. 79:11788–11800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Michaelis M., Geiler J., Klassert D., Doerr H. W., Cinatl J., Jr 2009. Infection of human retinal pigment epithelial cells with influenza A viruses. Invest. Ophthalmol. Vis Sci. 50:5419–5425 [DOI] [PubMed] [Google Scholar]
- 37. OIE 1996. Highly pathogenic avian influenza (fowl plague), p. 155–160 In Cullen G. A., Linnance S. (ed.), Manual of standards for diagnostic tests and vaccines. OIE, Paris, France [Google Scholar]
- 38. Olofsson S., Kumlin U., Dimock K., Arnberg N. 2005. Avian influenza and sialic acid receptors: more than meets the eye? Lancet Infect. Dis. 5:184–188 [DOI] [PubMed] [Google Scholar]
- 39. Pasick J., et al. 2005. Intersegmental recombination between the haemagglutinin and matrix genes was responsible for the emergence of a highly pathogenic H7N3 avian influenza virus in British Columbia. J. Gen. Virol. 86:727–731 [DOI] [PubMed] [Google Scholar]
- 40. Paulsen F., et al. 1998. Functional anatomy of human lacrimal duct epithelium. Anat. Embryol. (Berl.) 198:1–12 [DOI] [PubMed] [Google Scholar]
- 41. Puneet P., Moochhala S., Bhatia M. 2005. Chemokines in acute respiratory distress syndrome. Am. J. Physiol. Lung Cell. Mol. Physiol. 288:L3–L15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rajaiya J., Xiao J., Rajala R. V., Chodosh J. 2008. Human adenovirus type 19 infection of corneal cells induces p38 MAPK-dependent interleukin-8 expression. Virol. J. 5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Solomon A., et al. 2000. Doxycycline inhibition of interleukin-1 in the corneal epithelium. Am. J. Ophthalmol. 130:688. [DOI] [PubMed] [Google Scholar]
- 44. Stuart-Harris C. H. 1961. Twenty years of influenza epidemics. Am. Rev. Respir. Dis. 83:54–75 [Google Scholar]
- 45. Tam J. S. 2002. Influenza A (H5N1) in Hong Kong: an overview. Vaccine 20(Suppl. 2):S77–S81 [DOI] [PubMed] [Google Scholar]
- 46. Terraciano A. J., et al. 1999. Sialyl Lewis X, Lewis X, and N-acetyllactosamine expression on normal and glaucomatous eyes. Curr. Eye Res. 18:73–78 [DOI] [PubMed] [Google Scholar]
- 47. Tumpey T. M., Lu X., Morken T., Zaki S. R., Katz J. M. 2000. Depletion of lymphocytes and diminished cytokine production in mice infected with a highly virulent influenza A (H5N1) virus isolated from humans. J. Virol. 74:6105–6116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tweed S. A., et al. 2004. Human illness from avian influenza H7N3, British Columbia. Emerg. Infect. Dis. 10:2196–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ueta M., Kinoshita S. 2010. Innate immunity of the ocular surface. Brain Res. Bull. 81:219–228 [DOI] [PubMed] [Google Scholar]
- 50. Verma I. M., Stevenson J. K., Schwarz E. M., Van Antwerp D., Miyamoto S. 1995. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 9:2723–2735 [DOI] [PubMed] [Google Scholar]
- 51. Zeng H., et al. 2007. Highly pathogenic avian influenza H5N1 viruses elicit an attenuated type I interferon response in polarized human bronchial epithelial cells. J. Virol. 81:12439–12449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zeng H., Pappas C., Katz J. M., Tumpey T. M. 2011. The 2009 pandemic H1N1 and triple-reassortant swine H1N1 influenza viruses replicate efficiently but elicit an attenuated inflammatory response in polarized human bronchial epithelial cells. J. Virol. 85:686–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.