

Abstract
Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1), the principal viral oncoprotein and a member of the tumor necrosis factor receptor superfamily, is a constitutively active membrane signaling protein that regulates multiple signal transduction pathways via its C-terminal-activating region 1 (CTAR1) and CTAR2, and also the less-studied CTAR3. Because protein sumoylation among other posttranslational modifications may regulate many signaling pathways induced by LMP1, we investigated whether during EBV latency LMP1 regulates sumoylation processes that control cellular activation and cellular responses. By immunoprecipitation experiments, we show that LMP1 interacts with Ubc9, the single reported SUMO-conjugating enzyme. Requirements for LMP1-Ubc9 interactions include enzymatically active Ubc9: expression of inactive Ubc9 (Ubc9 C93S) inhibited the LMP1-Ubc9 interaction. LMP1 CTAR3, but not CTAR1 and CTAR2, participated in the LMP1-Ubc9 interaction, and amino acid sequences found in CTAR3, including the JAK-interacting motif, contributed to this interaction. Furthermore, LMP1 expression coincided with increased sumoylation of cellular proteins, and disruption of the Ubc9-LMP1 CTAR3 interaction almost completely abrogated LMP1-induced protein sumoylation, suggesting that this interaction promotes the sumoylation of downstream targets. Additional consequences of the disruption of the LMP1 CTAR3-Ubc9 interaction revealed effects on cellular migration, a hallmark of oncogenesis. Together, these data demonstrate that LMP1 CTAR3 does in fact function in intracellular signaling and leads to biological effects. We propose that LMP1, by interaction with Ubc9, modulates sumoylation processes, which regulate signal transduction pathways that affect phenotypic changes associated with oncogenesis.
INTRODUCTION
Epstein-Barr virus (EBV) is a ubiquitous gammaherpesvirus that establishes life-long latent infection within its hosts. Types I, II, and III latent EBV infections are characteristically associated with distinct lymphoid and epithelial malignancies (36). EBV nuclear antigen 1 (EBNA-1) is the only viral gene consistently expressed in type I latency, and Burkitt's lymphoma exemplifies this latent state. EBNA-1 and latent membrane protein 1 (LMP1), LMP2A, and LMP2B are expressed in type II EBV latency, which characterizes nasopharyngeal carcinoma and EBV-positive Hodgkins lymphoma. EBNA-1, -2, -3A, -3B, and -3C, along with LMP1, LMP2A, and LMP2B, are expressed in type III EBV latency, the viral phenotype in immunoblastic lymphomas, such as posttransplant lymphoproliferative disorder (PTLD) and AIDS-associated central nervous system lymphomas. Type III EBV latency is captured in lymphoblastoid cell lines (LCLs), which can be established following EBV infection of primary B cells and exhibit sustained cellular proliferation and increased cellular survival due to the constitutive activation of cellular signaling pathways. The main viral protein important in regulating these signal transduction events is LMP1.
LMP1 is the principal viral oncoprotein that is essential for immortalization of B cells into proliferating LCLs. It is an integral membrane signaling protein that mimics the tumor necrosis factor (TNF) receptor family members (such as CD40), with the exception that its activation is ligand independent and it is constitutively active (26). LMP1 consists of a short 24-amino-acid cytoplasmic N-terminal domain, six transmembrane domains (required for oligomerization of LMP1 and its constitutive activity), and a 200-amino-acid cytoplasmic C-terminal domain, which contains three C-terminal activating regions (CTARs) (3, 21, 26). Most LMP1-mediated signal transduction events are mediated via the extensively characterized CTAR1 and CTAR2. Function for CTAR3 is less well defined. CTAR1 contains a PXQXT motif through which it interacts with TNF receptor-associated factors (TRAF) 1, 2, 3, and 5. TRAF2 acts as a linker between CTAR1 and TRAF6. CTAR2 contains a YYD motif that binds to TNF receptor-interacting protein (RIP) and the TNF-associated death domains (TRADD), which enable indirect interaction between LMP1, TRAF2, and TRAF6 (15, 26, 35, 39). As a result of the protein-protein interactions at CTAR1 and CTAR2, multiple signal transduction events are initiated (15, 26, 28, 35).
Not much is known about the role of CTAR3, which falls between CTAR1 and CTAR2, in LMP1-induced signaling. CTAR3 has been shown to bind JAK3 via PXXPXP motifs located at amino acids 275 to 280 and 302 to 307 that activate the DNA binding of STAT1 (11). However, Higuchi et al. reported that JAK3 only weakly associated with both LMP1 and LMP1 lacking CTAR3 (13), suggesting that CTAR3 does not bind JAK3, and Brennan et al. showed that CTAR3 was not capable of inducing STAT1 transcriptional activity (5). Others have reported that deletion of the 120 amino acids between LMP1 CTAR1 and CTAR2 (amino acids 231 to 351) did not alter the ability of LMP1 to interact with TRAFs, TRADDs, or RIP (17) or its ability to activate JAK3, STAT2, STAT5 (13), NF-κB, JNK1/2, and Bcl-2 (17), suggesting that deletion of CTAR3 does not affect the protein interactions of CTAR1 and CTAR2. Deletion of amino acids 231 to 351 was also reported not to affect cellular growth, viral growth, or B-cell transformation (7, 17), but it did abolish the ability of LMP1 to induce colony formation (49). Together, these reports raise the question of whether CTAR3 does in fact have a function, which is addressed here.
The C-terminal region of LMP1 induces different types of posttranslational modifications of proteins that regulate their function. The two most common are phosphorylation and ubiquitination. Induction of a third major posttranslational modification, sumoylation, by LMP1 has not been reported, although most of the signaling pathways activated by LMP1, such as phosphatidylinositol 3-kinase (PI3K), NF-κB, and interferon regulatory factor 7 (IRF7), can also be regulated by sumoylation or regulate sumoylation of additional proteins (12, 25, 29, 38).
Protein sumoylation is a posttranslational modification in which the small ubiquitin-like modifier (SUMO), a 12-kDa protein that shares 20% homology with ubiquitin (20), is conjugated to the protein typically at lysine residues found within the conserved ΨKxE motif, where Ψ represents a hydrophobic residue (20). Only 5 to 10% of a given protein is found in a sumoylated form at any given time. Sumoylation results in the regulation of protein function through alteration of intracellular location, affecting the ability of the protein to interact with other proteins and modifying the ability of the protein to interact with DNA (1, 19, 20, 22, 34, 43, 50). In a manner similar to ubiquitination, proteins can be mono- or polysumoylated, but little is known of how these forms differ in regulating protein function (20). Protein sumoylation is a dynamic and reversible process requiring the SUMO-activating enzyme SAE1/SAE2, the SUMO- conjugating enzyme Ubc9, and one of the few identified SUMO-E3 ligases.
Reports on the effects of EBV latency proteins on protein sumoylation are limited and have mostly focused on the EBNAs. SUMO-1 and SUMO-3 interact with EBNA3C (27), allowing it to coactivate the LMP1 promoter with EBNA2 (37). In addition, EBNA3C and EBNA3B are sumoylated (37). Finally, even though it is not sumoylated, interaction of EBNA2 with sumoylated proteins modulates its transactivational function (14). A role for LMP1 in the sumoylation process has not been reported.
Because LMP1 can induce several different posttranslational modifications, most LMP1-regulated pathways can be affected by sumoylation, and preliminary data suggest that LMP1 expression induces sumoylation of IRF7 and STAT1 (data not shown), we hypothesized that EBV LMP1 may also be involved in sumoylation processes during EBV latent infection and control cellular activation and cellular responses. We show here that EBV LMP1 interacts with Ubc9 in a process that requires its enzymatic activity through its CTAR3 region. Neither CTAR1 nor CTAR2 appears to contribute to the LMP1-Ubc9 interaction, which is critical for the viral oncogene to induce sumoylation of cellular proteins. In addition, the LMP1-Ubc9 interaction can bring about phenotypic changes that affect cellular migration, a prominent feature in oncogenesis. Together, these findings demonstrate that LMP1 CTAR3 exerts regulatory functions by enabling sumoylation of cellular proteins, which results in biological effects.
MATERIALS AND METHODS
Cells.
Human embryonic kidney (HEK) 293T cells (293T cells), 293 cells, and U20S cells were maintained in Dulbecco's modified Eagle's medium (DMEM; CellGro) plus 10% fetal bovine serum (FBS; Gemini). Raji cells, EBV-positive cells derived from Burkitt's lymphoma, were maintained in RPMI (Cellgro) plus 10% FBS. EBV-infected MDA-MB–231 cells (16, 41, 47) were maintained in RPMI with 10% FBS and 700 μg/ml G418 (Cellgro).
Plasmids.
FLAG-LMP1, FLAG-LMP1 1–231, FLAG-LMP1 1–187, and FLAG-LMP1 Δ187–351 were gifts from Nancy Raab-Traub (31). FLAG-LMP1 Δ33bpr (base pair repeats), FLAG-LMP1 1xbox1, and FLAG-LMP1 1x33bpr were gifts from Wolfgang Hammerschmidt (11). Hemagglutinin (HA)-Ubc9 and HA-Ubc9 C93S were obtained from Addgene (48).
Construction of FLAG-LMP1 Δ275–307.
A FLAG-tagged LMP1 CTAR3 deletion mutant was constructed with wild-type FLAG-LMP1 and use of a site-directed mutagenesis kit (Stratagene). Using forward primer LMP C3 F (5′-GGA CCC TGA CAA CAC TGA TGA CAA TGG CCA TAG CCC TAG CGA CTC TGC TGG AAA TG-3′) and reverse primer LMP C3 R (5′-CAT TTC CAG CAG AGT CGC TAG GGC TAT GGC CAT TGT CAT CAG TGT TGG CAG GGT CC-3′), PCR was performed. DNA was denatured at 95°C for 30 s followed by 20 cycles of 95°C for 30 s, 60°C for 60 s, and 68°C for 8 min. Final elongation was performed at 68°C for 10 min. The PCR product was subjected to DpnI (Stratagene) digestion, transformed into XL-1 supercompetent cells (Stratagene), and grown on LB-ampicillin plates. Colonies were selected and sequenced to verify the deletion of amino acids 275 to 307 and the correct sequence of the remaining amino acids. Protein expression was verified by Western blot analyses.
Immunoprecipitation.
A total of 1 × 106 293T cells were grown in 60-mm dishes and transfected with a total amount of 2 μg of DNA with use of Effectene transfection reagent (Qiagen). At 48 h posttransfection, cells were lysed in NP-40 lysis buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1% NP-40, 5 mM dithiothreitol, 50 μM Na3VO4, 100 mM NaF, 1 mM phenylmethylsulfonyl fluoride, and Complete protease inhibitors). Supernatant fluids were collected after freezing and thawing the cells to lyse cells. Ten percent of the supernatants fluids were collected to examine protein expression and labeled whole-cell lysates (WCL). The remaining 90% of the fluids were used for immunoprecipitations for which lysates were incubated with 1 μg of antibody (FLAG or HA) for 1 h at 4°C. Washed protein A/G-agarose beads (Santa Cruz Biotechnology) were added to the samples, which were then incubated overnight at 4°C. Beads were washed four times with NP-40 lysis buffer, and sodium dodecyl sulfate (SDS; Sigma) loading buffer was added to the tubes.
Immunoprecipitations for endogenous interactions were performed in a similar manner with LMP1, Ubc9, and isotype control antibodies.
Western blot analysis.
Whole-cell lysates and immunoprecipitates were denatured in SDS loading buffer and boiled for 5 min. Samples were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (PVDF). Membranes were blocked with 5% milk in Tris-buffered saline-Tween 20 (TBST) and incubated overnight at 4°C with primary antibodies. The membranes were washed and incubated with appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Membranes were washed again and bands visualized with enhanced chemiluminescence reagent.
Antibodies.
Mouse anti-FLAG (M2) antibodies were purchased from Sigma. Mouse and rabbit anti-HA (F-7 and Y-11), mouse anti-Ubc9 (67AT1237.95.90), mouse anti-SUMO-1 (D-11), and rabbit anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; FL-335) antibodies were purchased from Santa Cruz Biotechnology. Anti-LMP1 (S12) antibodies were a gift from Nancy Raub-Traab (University of North Carolina at Chapel Hill).
Cellular migration assays.
Modified scratch assays were performed (2, 4). U20S or 293 cells were grown in 12-well plates and transfected with Effectene. At 42 h posttransfection, once cells were confluent, monolayers were scratched with a pipette tip and washed, and fresh DMEM supplemented with 1% FBS was added back to the wells. At 17 h later, medium was removed, and cells were fixed and stained in H2O with 0.1% crystal violet and 20% ethanol. Fifteen images were taken along the scratches (at 0 and 6 h), and scratch widths, percent scratch recovery, and the fold change in migration were calculated (2, 4). Experiments were performed in triplicate.
RESULTS
EBV LMP1 interacts with Ubc9.
We hypothesized that EBV LMP1 regulates sumoylation processes during EBV latent infections, so we first examined if LMP1 interacted with Ubc9. 293T cells transfected with FLAG-LMP1 or control expression constructs showed that when immunoprecipitations were performed with FLAG antibodies (IP:FLAG) for FLAG-LMP1, endogenous Ubc9 could only be detected by immunoblotting when FLAG-LMP1 was expressed (Fig. 1A, lane 2). Control immunoprecipitations were performed with either FLAG antibodies on samples not expressing FLAG-LMP1 (Fig. 1A, lane 1) or with control antibodies, which did not interact with FLAG-LMP1, on samples expressing FLAG-LMP1 (Fig. 1A, lane 3). Endogenous Ubc9 was not pulled down in either sample. To confirm these data, experiments were repeated with overexpression of FLAG-LMP1 and HA-Ubc9. In immunoprecipitations performed with FLAG antibodies (IP:FLAG), HA-Ubc9 was detected only when FLAG-LMP1 was coexpressed (Fig. 1B). Immunoprecipitations with HA antibodies (IP:HA) for HA-Ubc9 further confirmed that Ubc9 interacted with LMP1 (Fig. 1B). When only FLAG-LMP1 or HA-Ubc9 was expressed (Fig. 1B, lanes 1 and 2), neither interactions between the two proteins nor nonspecific pulldown of the proteins was detected. In addition, we verified that nonspecific immunoprecipitation of Ubc9 did not occur when we used control antibodies (Fig. 1C).
Fig. 1.

EBV LMP1 interacts with Ubc9. (A) 293T cells were transfected with FLAG-LMP1 or vector control and cultured for 48 h. Cell lysates were collected and immunoprecipitations (IP) were performed with FLAG or isotype control antibodies. Western blot analyses of the immunoprecipitates, and whole-cell lysates (WCL; 10 μg, or 2% of total lysates) were obtained for detection of FLAG-LMP1 and endogenous Ubc9. (B) 293T cells were transfected with FLAG-LMP1 and/or HA-Ubc9 and cultured for 48 h. Cell lysates were collected, and immunoprecipitations were performed with FLAG and HA antibodies. Western blot analyses were performed to detect FLAG-LMP1 and HA-Ubc9 expression. (C) 293T cells were transfected with FLAG-LMP1 and/or HA-Ubc9 and cultured for 48 h. Cell lysates were collected, and immunoprecipitations were performed with FLAG antibodies or isotype control antibodies. Western blot analyses were performed to detect FLAG-LMP1 and HA-Ubc9 expression. (D) WCL of Raji cells and MDA-MB 231 EBV+ cell clones C3B4 and C4A3 were subjected to Western blot analyses for detection of endogenous LMP1 and Ubc9. (E) Cell lysates were collected from Raji cells and MDA-MB 231 EBV+ cell clones C3B4 and C4A3. One milligram of total protein from each cell lysate was subjected to immunoprecipitations with LMP1 and IgG isotype control antibodies. Western blot analyses were performed to detect LMP1 and Ubc9 expression.
Next we investigated whether LMP1 interacted with Ubc9 endogenously in three different cell lines. Raji cells, EBV-positive B lymphoblastoid cells, express high levels of LMP1 (Fig. 1C, lane 1). MDA-MB 231 EBV+ cells are EBV-infected human breast cancer cell lines (16, 41). Clone C3B4 expresses intermediate levels of LMP1 (Fig. 1D, lane 2). Clone C4A3 does not express detectable levels of LMP1 (Fig. 1D, lane 3). Western blot analyses showed that endogenous Ubc9 expression was similar in all three cell lines (Fig. 1D), suggesting that LMP1 does not regulate Ubc9 expression.
Immunoprecipitations with LMP1 antibodies or isotype control antibodies were performed with equal amounts of proteins from lysates of all three cell lines. Western blot analyses revealed that when immunoprecipitations were performed with LMP1 antibodies, both Ubc9 as well as LMP1 were detected in Raji cells and clone C3B4 (Fig. 1E), but neither protein was detected in clone C4A3 or with isotype control antibodies (IgG) (Fig. 1E). Immunoprecipitations performed with Raji cell extracts, which expressed higher levels of LMP1 than clone C3B4, pulled down larger amounts of Ubc9 than clone C3B4, suggesting a dose-dependent interaction between LMP1 and Ubc9. Taken together, these data demonstrated that LMP1 interacts with Ubc9 endogenously as well as when overexpressed.
EBV LMP1 interacts only with enzymatically active Ubc9.
Cysteine residue 93 of Ubc9 is essential for its SUMO-conjugating activity; mutation of this amino acid (Ubc9 C93S) renders Ubc9 enzymatically inactive (32). However, Ubc9 C93S still interacts with certain proteins and can regulate protein function without mediating sumoylation (18, 23, 24). To test whether LMP1 could interact with Ubc9 C93S, 293T cells were transfected with expression vectors for FLAG-LMP1 and HA-Ubc9 or HA-Ubc9 C93S. Results showed that FLAG-LMP1 only interacted with enzymatically active HA-Ubc9 (Fig. 2A); when HA-Ubc9 C93S was expressed, interaction with FLAG-LMP1 was not detected (Fig. 2A). Thus, Ubc9 activity is necessary for the LMP1-Ubc9 interaction.
Fig. 2.
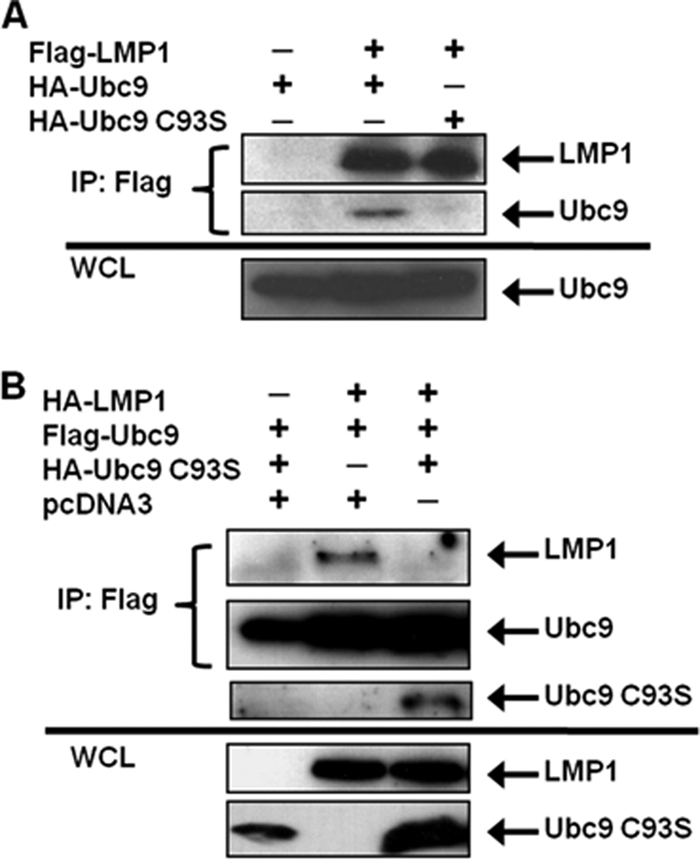
EBV LMP1 interacts only with enzymatically active Ubc9. (A) 293T cells were transfected with FLAG-LMP1 and HA-Ubc9 or HA-Ubc9 C93S. (B) 293T cells were transfected with FLAG-Ubc9, HA-LMP1, and increasing amounts of HA-Ubc9 C93S. At 48 h after transfection, cell lysates were harvested and immunoprecipitations (IP) were performed with FLAG antibodies. Western blot analyses of the immunoprecipitates and WCL were used to detect FLAG and HA expression.
Next, because overexpression of Ubc9 C93S suppresses the function of endogenous Ubc9 (10, 33), and in some cases mutation of cysteine residue 93 of Ubc9 results in loss of protein-protein interaction (9), we tested whether its expression could disrupt the LMP1-Ubc9 interaction by transfecting 293T cells with FLAG-Ubc9, HA-LMP1, and HA-Ubc9 C93S. The results showed that FLAG-Ubc9 only interacted with HA-LMP1 when HA-Ubc9 C93S was not coexpressed (Fig. 2B). With coexpression of HA-Ubc9 C93S, interaction between wild-type Ubc9 and LMP1 was no longer detected. Instead, wild-type FLAG-Ubc9 pulled down HA-Ubc9 C93S, suggesting that the inactive Ubc9 mutant interacted with wild-type FLAG-Ubc9, possibly inhibiting the interaction between Ubc9 and LMP1. These data demonstrate that the LMP1-Ubc9 interaction can be disrupted by the overexpression of the enzymatically inactive Ubc9 mutant.
Ubc9 does not interact with EBV LMP1 CTAR1 or CTAR2.
We next determined the region of LMP1 required for its interaction with Ubc9. The C-terminal region of EBV LMP1 contains two major C-terminal activating regions (CTAR1 at amino acids 204 to 208 and CTAR2 at amino acids 384 to 386), and we selected deletion mutants lacking CTAR1 (FLAG-LMP1 Δ187–351), CTAR2 (FLAG-LMP1 1–231), and CTAR1 and CTAR2 (FLAG-LMP1 1–187) (Fig. 3A) to test in this study (31). 293T cells were transfected with expression constructs for HA-Ubc9 and FLAG-LMP1 or its deletion mutants. The results showed that HA-Ubc9 interacted with wild-type FLAG-LMP1 but not with any of the deletion mutants (Fig. 3B). Thus, neither LMP1 CTAR1 nor LMP1 CTAR2 was sufficient for LMP1 to interact with Ubc9.
Fig. 3.

LMP1 CTAR1 and CTAR2 are not critical for the LMP1-Ubc9 interaction. (A) Diagrammatic alignment of wild-type LMP1 and selected LMP1 mutants. LMP 1–187 is missing both CTAR1 and CTAR2. LMP1 1–231 only contains CTAR1. LMP1 Δ187–351 only contains CTAR2. LMP1 PQAA, YIID, and DM are point mutants with inactive CTAR1 and/or CTAR2. (B and C) 293T cells were transfected with FLAG-LMP1 or the indicated FLAG-LMP1 mutants and HA-Ubc9. Cell lysates were harvested 48 h after transfection, and immunoprecipitations were performed with FLAG antibodies. Western blot analyses of the immunoprecipitates and WCL were used to detect FLAG-LMP1 and HA-Ubc9 expression.
To confirm these results, which suggested that CTAR1 and CTAR2 were not critical for the LMP1-Ubc9 interaction, LMP1 constructs containing point mutations (Fig. 3A) that inactivate CTAR1 (PQAA), CTAR2 (YIID), and both CTAR1 and CTAR2 (DM) were examined for their ability to interact with Ubc9. 293T cells were transfected with expression constructs for HA-Ubc9 and FLAG-LMP1 or its point mutants. The results showed that HA-Ubc9 interacted not only with wild-type FLAG-LMP1 but also the four mutated constructs (Fig. 3), demonstrating that functional CTAR1 and CTAR2 are not required for LMP1 to interact with Ubc9. Taken together, these data suggest that the region of LMP1 between CTAR1 and CTAR2 (amino acids 231 to 351) is critical for the LMP1-Ubc9 interaction.
LMP1 CTAR3 is required for the LMP1-Ubc9 interaction.
Because LMP1 amino acids 231 to 351, which encompass CTAR3, were deleted in all LMP1 mutants that did not interact with Ubc9 and were present in all LMP1 point mutants that did interact with Ubc9, we tested if CTAR3 was necessary by using two CTAR3 deletion mutants. We constructed FLAG-LMP1Δ275–307 and obtained another construct from Wolfgang Hammerschmidt (FLAG-LMP1Δ33bpr, missing amino acids 245 to 307) (Fig. 4A). 293T cells were transfected with HA-Ubc9 and FLAG-LMP1 or either of the CTAR3 deletion mutant expression constructs. Results showed that HA-Ubc9 interacted only with wild-type FLAG-LMP1 but with neither of the deletion mutants (Fig. 4B), suggesting that CTAR3, specifically amino acids 275 to 307, was necessary for the observed LMP1-Ubc9 interaction.
Fig. 4.

LMP1 CTAR3 is required for its interaction with Ubc9. (A) Diagrammatic alignment of LMP1 mutants. LMP1 Δ275–307 was constructed to delete both JAK3-binding motifs. LMP1Δ33bpr is missing the 33-bp repeats, which includes the JAK3-binding motifs. LMP1 1xbox 1 and LMP1 1x33bor are reconstituted LMP1Δ33bpr mutants. (B to D) 293T cells were transfected with HA-Ubc9 and FLAG-LMP1 or the indicated FLAG-LMP1 mutant. Cell lysates were harvested 48 h after transfection, and immunoprecipitations were performed with FLAG antibodies. Western blot analyses of the immunoprecipitates and WCL were performed to determine FLAG and HA expression.
Amino acid sequences found in LMP1 CTAR3 aid the interaction of LMP1 and Ubc9.
Next we examined if reconstitution of one copy of the 11-amino-acid repeats or one copy of the JAK-binding motif found in CTAR3 restored the LMP1-Ubc9 interaction. LMP1 CTAR3 deletion mutants with one copy of the 11-amino-acid repeats (LMP1 1x33bpr) (Fig. 4A) or one copy of the JAK-binding motif (LMP1 1xbox1) (Fig. 4A) were obtained from Wolfgang Hammerschmidt (11). 293T cells were transfected with expression constructs for HA-Ubc9 and FLAG-LMP1 or the LMP1 mutants. The results confirmed that when CTAR3 was missing, the interaction between LMP1 and Ubc9 was not observed (Fig. 4C, lane 3). Reconstitution with one copy of the JAK-binding motif (Fig. 4C, lane 5) restored approximately 10 to 20% of this interaction. Reconstitution with one copy of the 11-amino-acid repeats restored 5 to 10% of the Ubc9-LMP1 interaction (Fig. 4C, lane 4). Thus, while both the JAK-interacting motifs and the 11-amino-acid repeats aid the interaction of LMP1 and Ubc9, the JAK-interacting motif may be somewhat more important for the interaction.
To determine if CTAR3 alone was sufficient to interact with Ubc9, we reconstituted FLAG-LMP1 1–187 with LMP1 amino acids 275 to 288 (LMP1 1–187, 275–288) or 275 to 296 (LMP1 1–187, 275–296). 293T cells were transfected with expression constructs for HA-Ubc9 and FLAG-LMP1 or the LMP1 mutants. The results confirmed that LMP1 1–187 did not interact with Ubc9 (Fig. 4D). Reconstitution of LMP1 amino acids 275 to 288 or 275 to 296 partially restored the interaction between LMP1 and Ubc9 (Fig. 4D, lanes 4 and 5), suggesting CTAR3 is sufficient for the LMP1-Ubc9 interaction.
LMP1 induces the sumoylation of cellular proteins.
Because our findings showed that LMP1 interacted with Ubc9, we next examined if LMP1 regulated sumoylation processes within the cells. 293T cells were transfected with FLAG-SUMO-1, FLAG-SUMO–2, or FLAG-SUMO-3 expression constructs along with HA-LMP1. Results showed that small amounts of cellular proteins were detected as modified by SUMO-1 and SUMO-3 when coexpressed with the vector control (pcDNA3) (Fig. 5A). However, when SUMO-1, SUMO-2, and SUMO-3 were coexpressed with HA-LMP1, increased amounts of cellular proteins appeared to be modified. These data were confirmed by examining effects of endogenous SUMO-1 on modification of cellular proteins. Western blot analyses showed that LMP1 expression coincided with detection of increases in sumoylated cellular proteins (Fig. 5B). Together, these data demonstrate that LMP1 expression increases levels of endogenously sumoylated cellular proteins and suggest that the viral oncoprotein induces sumoylation processes within the cell.
Fig. 5.

LMP1 induces the sumoylation of cellular proteins. (A) 293T cells were transfected with vector control or HA-LMP1 and FLAG-SUMO-1, FLAG-SUMO-2, or FLAG-SUMO-2. (B) 293T cells were transfected with vector control or FLAG-LMP1. (C) 293T cells were transfected with vector control, FLAG-LMP1, or FLAG-LMP1Δ33bpr and HA-SUMO-1. (D) 293T cells were transfected with HA-Ubc9 or HA-Ubc9 C93S along with HA-SUMO-1 and vector control, FLAG-LMP1, or FLAG-LMP1Δ33bpr. At 48 h after transfection, whole-cell lysates were collected and Western blot analyses were performed for SUMO-1, FLAG, HA, Ubc9, and GAPDH expression.
To determine if CTAR3 was required for LMP1-induced sumoylation, 293T cells were transfected with HA-SUMO-1 and FLAG-LMP1 or FLAG-LMP1Δ33bpr expression constructs. Results showed that LMP1 expression coincided with increased amounts of cellular proteins modified by SUMO-1 (Fig. 5C), confirming our earlier data. In contrast, when FLAG-LMP1Δ33bpr was expressed, amounts of sumoylated proteins detected decreased. Repeat experiments revealed that FLAG-LMP1Δ33bpr expression coincided with 80% less sumoylation than when FLAG-LMP1 was expressed. The results suggest that LMP1 CTAR3 accounts for the majority of LMP1-induced protein sumoylation.
The importance of the LMP1-Ubc9 interaction in LMP1-induced protein sumoylation was verified by utilizing overexpression of Ubc9 and its inactive mutant. 293T cells were transfected with HA-Ubc9 or HA-Ubc9 C93S along with HA-SUMO-1 and vector control, FLAG-LMP1, or FLAG-LMP1Δ33bpr expression constructs. Western blot analyses showed that when wild-type Ubc9 was overexpressed, LMP1 expression coincided with increased amounts of cellular proteins modified by SUMO-1, whereas expression of FLAG-LMP1Δ33bpr decreased protein sumoylation. In addition, expression of HA-Ubc9 C93S resulted in decreased modification by SUMO-1 with either the wild-type or mutant LMP1 constructs (Fig. 5D). Together, these data demonstrate the importance of the LMP1-Ubc9 interaction, via CTAR3, in LMP1-induced sumoylation of cellular proteins.
Inhibition of the Ubc9-LMP1 CTAR3 interaction reduces cellular migration.
Increased protein sumoylation has been implicated in tumorigenesis (8, 44). Because our findings suggested that LMP1 CTAR3 could function to induce sumoylation of cellular proteins, we examined whether the LMP1-Ubc9 interaction affected phenotypic changes associated with LMP1-mediated oncogenesis. In a previous report, deletion of CTAR3 abrogated LMP1-induced colony formation (49), suggesting a function for LMP1 CTAR3 in oncogenesis in epithelial cells. We next investigated whether the Ubc9-LMP1 CTAR3 interaction could affect cellular migration, as commonly observed during oncogenesis. HA-LMP1-, HA-LMP1Δ33bpr-, and vector-expressing stable cell lines were generated, starting with MDA-MB 231 EBV+ cells, clone C4A3, which are infected with EBV but do not express detectable levels of LMP1 (16, 41). Scratch assays were performed on confluent cell monolayers, and cellular migration was quantitated (2, 4). Results showed that vector control-expressing cells migrated and filled in approximately 50% of the lesion in the monolayer, whereas HA-LMP1-expressing cells produced a significant (P < 0.05) increase in cellular migration, filling almost 100% of the lesion (Fig. 6A), equivalent to a 2-fold increase in migration. HA-LMP1Δ33bpr-expressing cells exhibited an intermediate phenotype, producing a 1.6-fold increase in migration compared with the vector control, as expected, because CTAR1 and CTAR2 also affect cellular migration (6, 40). These data demonstrated a role for LMP1 CTAR3 in oncoprotein-mediated cellular migration.
Fig. 6.

Inhibition of the Ubc9-LMP1 CTAR3 interaction decreases cellular migration. (A) MDA-MB 231 EBV+ clone C4A3 cells were transfected to stably express vector control, HA-LMP1, or HA-LMP1Δ33bpr. Following selection, cells were plated and grown to confluence. Confluent cell layers were scratched with a pipette tip and washed, and fresh RPMI was added to wells. After 24 h, medium was removed and cells were stained and fixed in a crystal violet and ethanol solution. Multiple images were captured along the scratch, and the wound width was calculated (2, 4). Representative images are shown, and the fold change in migration was determined. (B to D) 293 cells were plated and transfected as indicated. Confluent cell layers were scratched with a pipette tip and washed, and fresh 1% DMEM was added to wells. After 18 h, medium was removed and cells were stained and fixed in crystal violet-ethanol. The wound width was calculated (2, 4), and the fold change in migration was determined. Data are the means ± standard deviations for experiments performed in triplicate.
To confirm the contribution of LMP1 CTAR3 in cellular migration, 293 cells were transfected with FLAG-LMP1, FLAG-LMP1Δ33bpr, or control expression plasmids along with HA-Ubc9 or HA-Ubc9 C93S constructs, and migration was examined by scratch assays (Fig. 6B). The results again showed that FLAG-LMP1-expressing cells exhibited a significant (P < 0.05) increase in cellular migration compared with vector control cells (Fig. 6B). In contrast, FLAG-LMP1Δ33bpr-expressing cells migrated at significantly (P < 0.05) lower levels, with only a 1.6-fold increase, than LMP1-expressing cells, but still at significantly (P < 0.05) higher levels than vector control-expressing cells (Fig. 6B). Coexpression of HA-Ubc9 C93S and FLAG-LMP1 or FLAG-LMP1Δ33bpr significantly (P < 0.05) inhibited LMP1-mediated cellular migration (Fig. 6B). However, levels of migration were still higher than with vector control-expressing cells. Results were similar with U20S cells (data not shown), and cellular proliferation assays yielded similar growth curves for all experimental arms (Fig. 7A and B).
Fig. 7.

The LMP1-Ubc9 interaction does not affect cellular growth. (A) A total of 1 × 104 293 cells were plated and transfected with vector control, FLAG-LMP1, or FLAG-LMP1Δ33bpr. (B) A total of 1 × 104 293 cells were plated and transfected with HA-Ubc9 or HA-Ubc9 C93S and either the vector control, FLAG-LMP1, or FLAG-LMP1Δ33bpr. Cells and supernatant fluids were collected at 24, 48, 72, and 96 h after transfection, and the total numbers of cells were determined.
We next used LMP1 constructs with mutations in CTAR1 and CTAR2 as well as CTAR3. In scratch assays, FLAG-LMP1-expressing cells again migrated at significantly (P < 0.05) higher levels than vector control-expressing cells and at lower levels in FLAG-LMP1Δ33bpr-expressing cells than LMP1-expressing cells, but still higher than vector control cells. Results were similar in cells expressing FLAG-LMP1 1–231 (Fig. 6C), which expressed CTAR1 but not CTAR2 or CTAR3. FLAG-LMP1 Δ187–351 and FLAG-LMP1 1–187, both of which lacked CTAR1 and CTAR3, were not able to induce significant levels of cellular migration (Fig. 6C). These data suggest that the three terminal activating regions are required for LMP1-induced cellular migration, and CTAR3 has an independent effect on this event.
Scratch assays were also performed on cells expressing the CTAR1 and CTAR2 point mutations tested earlier. Again, FLAG-LMP1-expressing cells showed a 2-fold increase in cell migration, whereas FLAG-LMP1Δ33bpr-expressing cells exhibited a 1.6-fold increase compared to vector control cells (Fig. 6D). FLAG-LMP1 YIID-expressing cells yielded similar results as FLAG-LMP1Δ33bpr-expressing cells (Fig. 6D). Both FLAG-LMP1 PQAA and FLAG-LMP1 DM, which bind Ubc9 but lack a functional CTAR1 domain, induced significantly (P < 0.05) lower levels of cellular migration. However, they were both still able to induce significantly (P < 0.05) higher levels of migration than the vector control-expressing cells. These data confirm that regardless of the presence of functional CTAR1 and CTAR2 domains, the ability of LMP1 to interact with Ubc9 contributes to its induction of cellular migration. Furthermore, CTAR3 contributes to the ability of LMP1 to induce cell migration. Together, these data suggest that the LMP1 CTAR3-Ubc9 interaction contributes to phenotypic changes associated with LMP1's oncogenic potential.
DISCUSSION
In this paper, we have shown that EBV LMP1 interacts with the single-known SUMO-conjugating enzyme, Ubc9, suggesting that it may influence sumoylation processes. The interaction between Ubc9 and LMP1 is localized to LMP1's intracellular signaling domain, the CTAR. This interaction does not involve the much-studied CTAR1 or CTAR2, but it is dependent on the much-less-characterized CTAR3. Expression of LMP1 induced sumoylation of cellular proteins, and disruption of its interaction with Ubc9, either by deletion of LMP1 amino acids 275 to 307 or by overexpression of enzymatically inactive Ubc9, significantly reduced sumoylation. These observations confirmed our hypothesis that LMP1 regulates sumoylation processes within the cell. Furthermore, disruption of LMP1's interaction with Ubc9 suppressed cellular migration, a phenotypic change associated with basic oncogenic properties, induced by the viral oncoprotein.
This is one of the few reports on a role for CTAR3 in LMP1-induced cellular signaling and its functional consequences. CTAR3 was first defined by findings that suggested that the domain interacts with JAK3 and activates binding of STAT1 to DNA (11). However, a later report claimed that LMP1 CTAR3 was insufficient for activation of STAT1 transcriptional activity (5). An explanation for these seemingly conflicting results may lie in the sumoylation status of STAT1. STAT1 can be sumoylated (45, 51), and STAT1 sumoylation at K703 depends on its phosphorylation status. Specifically, phosphorylation of STAT1 Y701 inhibits its sumoylation (51), whereas phosphorylation of STAT1 Y727 induces its sumoylation (46). Therefore, it is possible that LMP1 CTAR3 may mediate Y727 phosphorylation of STAT1, induce its sumoylation, and thereby inhibit its transcriptional activity. We have begun studies to test this possibility. Preliminary results have demonstrated that STAT1 can be sumoylated in a CTAR3-dependent manner (data not shown). Our findings not only support the existence of LMP1 CTAR3 as a functional domain, but also they may help to resolve how it activates STAT1 DNA binding yet is not required for STAT1 activation (5, 11).
Others have reported that the 120-amino-acid region between LMP1 CTAR1 and CTAR2 (amino acids 231 to 351) did not influence protein interactions of CTAR1 and CTAR2. Specifically, deletion of this region did not alter the ability of LMP1 to interact with TRAFs, TRADDs, or RIP (17) or its ability to activate JAK3, STAT2, STAT5 (13), NF-κB, JNK1/2, and Bcl-2 (17). Nor did this region of LMP1 influence cellular growth or B-cell transformation (7, 17). However, deletion of these 120 amino acids did abolish the ability of LMP1 to induce colony formation (49). While the LMP1 CTAR3 mutants used in our study (FLAG-LMP1Δ33bpr lacks 65 amino acids and LMP1Δ275–307 lacks 32 amino acids [Fig. 4]) differed slightly from published mutants, we also found that whereas LMP1 CTAR3 did not affect cellular growth (Fig. 7), it did affect another phenotypic response typical of oncogenic processes, specifically, cellular migration (Fig. 6).
Previous reports in which both deletion and point mutants were used documented the importance of CTAR1 and CTAR2 in LMP1-mediated cellular migration (6, 40). Shair et al. tested epithelial cell migration by using LMP1 deletion mutants, while Dawson et al. tested epithelial cell migration using point mutants to inactivate CTAR1 and CTAR2 (6). Both reported that LMP1-induced epithelial cell migration required both CTAR1 and CTAR2 (40). When we used similar mutants, our data confirmed these findings. Additionally, the CTAR3 deletion mutant was still capable of inducing migration, but not to the same extent as full-length LMP1. Therefore, we now propose that all three regulatory regions contribute to LMP1-induced migration. The role of CTAR3 in B-lymphocyte migration needs to be examined.
While the importance of LMP1 CTAR3 in the LMP1-JAK3 interaction has been contentious (11, 13), the data shown here clearly demonstrate the criticalness of CTAR3 in the interaction of LMP1 with Ubc9. The selected deletion mutants (LMP1 1–187, LMP1 1–231, LMP1Δ187–351, LMP1Δ275–307, and LMP1Δ33bpr) all failed to interact with Ubc9. A similarity between these 5 mutants is that they all lack amino acids 275 to 308, suggesting these amino acids are necessary for the LMP1-Ubc9 interaction. Because the two CTAR3 deletion mutants (LMP1Δ275–307 and LMP1Δ33bpr) retain functional CTAR1 and CTAR2 domains, the possibility existed that CTAR1 and CTAR2 could cooperate to mediate Ubc9 association and that deletion of CTAR3 inhibited certain protein-protein interactions needed for CTAR1 or CTAR2. We have now discounted these possibilities by testing point mutants that inactivated either CTAR1 or CTAR2 or both domains. Our data showed that when either region is inactivated, LMP1 still interacted with Ubc9 (Fig. 3C). Thus, neither CTAR1 nor CTAR2 is required for the LMP1-Ubc9 interaction. These findings also rule out the possibility that CTAR1 and CTAR2 are together required for the LMP1-Ubc9 interaction. Instead, the data strongly suggest that CTAR3 is critical for this protein-protein interaction.
Further support for the role of LMP1 CTAR3 in the LMP1-Ubc9 interaction are obtained from the data demonstrating that a JAK-interacting motif and a 33bpr could both partially restore the LMP1-Ubc9 interaction in mutants lacking CTAR3 (LMP1-Δ33bpr). Reconstitution of part of CTAR3 to LMP1 1–187, which lacks the cytoplasmic tail of LMP1, also partially restored the LMP1-Ubc9 interaction. Taken together, these data converge to indicate that CTAR3 is necessary and sufficient, while CTAR1 and CTAR2 are neither required nor sufficient, for LMP1 to interact with Ubc9.
In light of the interaction between LMP1 and enzymatically active Ubc9, we examined if LMP1 was itself sumoylated. However, LMP1 does not contain the conserved sumoylation motif (20), and in the systems tested, sumoylation of the viral oncoprotein was not detected (data not shown).
In any case, sumoylation of cellular proteins was strongly induced by the LMP1-Ubc9 interaction and may regulate LMP1-mediated signaling. Our findings show that expression of LMP1 coincides with increased amounts of cellular proteins modified by SUMO-1, SUMO-2, and SUMO-3. These results were confirmed by examining endogenous sumoylation (by SUMO-1). Together, these findings demonstrate that the effect was LMP1 specific and not due to LMP1-induced plasmid expression. Because LMP1 can induce protein expression, the possibility existed that LMP1-induced sumoylation of cellular proteins was caused by increased expression of Ubc9. However, levels of endogenous Ubc9 were not affected by LMP1 or LMP1Δ33bpr (Fig. 1A and D and 5A and C), thus demonstrating a link between LMP1 expression and the induction of sumoylation of cellular proteins. Additional experiments demonstrated that LMP1 CTAR3 and its ability to interact with Ubc9 was responsible for mediating the majority of LMP1-induced sumoylation. When the LMP1-Ubc9 interaction was inhibited, either by deletion of CTAR3 or by overexpression of Ubc9 C93S, approximately 80% less sumoylation of cellular proteins was observed.
Because LMP1 expression coincided with increased sumoylation of cellular proteins, it is possible that LMP1 acts as a SUMO E3 ligase. However, SUMO E3 ligases are not required for protein sumoylation (19, 20). Therefore, the ability of LMP1 to serve as a ligase will need to be investigated. Additionally, LMP1 may not directly bind to Ubc9 but instead requires another protein for this interaction. It is also probable that the binding of Ubc9 to the CTAR3 region influences signaling via CTAR1 and CTAR2. For instance, our preliminary data suggest that CTAR3 is critical for LMP1-induced sumoylation of IRF7. We recently reported that CTAR2 is necessary for regulating IRF7 activity (35). These findings suggest that LMP1-induced sumoylation is mediated by CTAR3 but may modulate signal transduction pathways induced by CTAR1 and CTAR2. Therefore, examination of how sumoylation processes are involved in cross talk among the different regulatory regions is necessary to clarify the function of CTAR3.
Finally, our data show that overexpression of inactive Ubc9 (Ubc9 C93S) inhibited the LMP1-Ubc9 interaction, resulting in decreased cell motility. Ubc9 is a candidate target for anticancer therapies (7, 30). Means proposed to target Ubc9 include use of small interfering RNA to inhibit Ubc9 expression (21, 26, 42), expression of the avian adenovirus protein Gam1 to induce Ubc9 degradation (4), and expression of a dominant-negative Ubc9 mutant (Ubc9 C93S) to suppress the function of endogenous Ubc9 (8, 31). This last method could be an approach for treatment of LMP1-associated malignancies, because we have shown that expression of Ubc9 C93S inhibits the LMP1-Ubc9 interaction and controls a cellular behavior that is characteristic of the oncogenic effects of the viral protein.
Together, these data disclose a protein-protein interaction and function unique for this region of LMP1. Our findings support the existence of LMP1 CTAR3 as a significant functional domain and suggest that further study of mechanisms by which LMP1 CTAR3 affects cellular behavior will be revealing.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants HL-64851, CA09156, and CA142132.
Footnotes
Published ahead of print on 27 July 2011.
REFERENCES
- 1. Adamson A. L., Kenney S. 2001. Epstein-Barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 75:2388–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Albuquerque M. L., Waters C. M., Savla U., Schnaper H. W., Flozak A. S. 2000. Shear stress enhances human endothelial cell wound closure in vitro. Am. J. Physiol. Heart Circ. Physiol. 279:H293–H302 [DOI] [PubMed] [Google Scholar]
- 3. Aviel S., Winberg G., Massucci M., Ciechanover A. 2000. Degradation of the Epstein-Barr virus latent membrane protein 1 (LMP1) by the ubiquitin-proteasome pathway. Targeting via ubiquitination of the N-terminal residue. J. Biol. Chem. 275:23491–23499 [DOI] [PubMed] [Google Scholar]
- 4. Bentz G. L., Yurochko A. D. 2008. Human CMV infection of endothelial cells induces an angiogenic response through viral binding to EGF receptor and β1 and β3 integrins. Proc. Natl. Acad. Sci. U. S. A. 105:5531–5536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brennan P., Floettmann J. E., Mehl A., Jones M., Rowe M. 2001. Mechanism of action of a novel latent membrane protein-1 dominant negative. J. Biol. Chem. 276:1195–1203 [DOI] [PubMed] [Google Scholar]
- 6. Dawson C. W., Laverick L., Morris M. A., Tramoutanis G., Young L. S. 2008. Epstein-Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. J. Virol. 82:3654–3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dirmeier U., et al. 2003. Latent membrane protein 1 is critical for efficient growth transformation of human B cells by Epstein-Barr virus. Cancer Res. 63:2982–2989 [PubMed] [Google Scholar]
- 8. Duan X., Trent J. O., Ye H. 2009. Targeting the SUMO E2 conjugating enzyme Ubc9 interaction for anti-cancer drug design. Anticancer Agents Med. Chem. 9:51–54 [DOI] [PubMed] [Google Scholar]
- 9. Fan Z., et al. 2006. SARS-CoV nucleocapsid protein binds to hUbc9, a ubiquitin conjugating enzyme of the sumoylation system. J. Med. Virol. 78:1365–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Giorgino F., et al. 2000. The sentrin-conjugating enzyme mUbc9 interacts with GLUT4 and GLUT1 glucose transporters and regulates transporter levels in skeletal muscle cells. Proc. Natl. Acad. Sci. U. S. A. 97:1125–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gires O., et al. 1999. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 18:3064–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo B., Yang S. H., Witty J., Sharrocks A. D. 2007. Signalling pathways and the regulation of SUMO modification. Biochem. Soc. Trans. 35:1414–1418 [DOI] [PubMed] [Google Scholar]
- 13. Higuchi M., Kieff E., Izumi K. M. 2002. The Epstein-Barr virus latent membrane protein 1 putative Janus kinase 3 (JAK3) binding domain does not mediate JAK3 association or activation in B-lymphoma or lymphoblastoid cell lines. J. Virol. 76:455–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hille A., Badu-Antwi A., Holzer D., Grasser F. A. 2002. Lysine residues of Epstein-Barr virus-encoded nuclear antigen 2 do not confer secondary modifications via ubiquitin or SUMO-like proteins but modulate transcriptional activation. J. Gen. Virol. 83:1037–1042 [DOI] [PubMed] [Google Scholar]
- 15. Huye L. E., Ning S., Kelliher M., Pagano J. S. 2007. Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol. Cell. Biol. 27:2910–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Imai S., Nishikawa J., Takada K. 1998. Cell-to-cell contact as an efficient mode of Epstein-Barr virus infection of diverse human epithelial cells. J. Virol. 72:4371–4378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Izumi K. M., et al. 1999. The residues between the two transformation effector sites of Epstein-Barr virus latent membrane protein 1 are not critical for B-lymphocyte growth transformation. J. Virol. 73:9908–9916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaul S., Blackford J. A., Jr., Cho S., Simons S. S., Jr 2002. Ubc9 is a novel modulator of the induction properties of glucocorticoid receptors. J. Biol. Chem. 277:12541–12549 [DOI] [PubMed] [Google Scholar]
- 19. Kerscher O. 2007. SUMO junction—what's your function? New insights through SUMO-interacting motifs. EMBO Rep. 8:550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kerscher O., Felberbaum R., Hochstrasser M. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22:159–180 [DOI] [PubMed] [Google Scholar]
- 21. Kieser A. 2008. 13th Bienn. Conf. Int. Assoc. Res. Epstein-Barr Virus Assoc. Dis., Guangzhou, China, abstr. 27. International Association for Research on Epstein-Barr Virus and Associated Diseases, Houston, TX [Google Scholar]
- 22. Kroetz M. B. 2005. SUMO: a ubiquitin-like protein modifier. Yale J. Biol. Med. 78:197–201 [PMC free article] [PubMed] [Google Scholar]
- 23. Kurihara I., et al. 2005. Ubc9 and protein inhibitor of activated STAT 1 activate chicken ovalbumin upstream promoter-transcription factor I-mediated human CYP11B2 gene transcription. J. Biol. Chem. 280:6721–6730 [DOI] [PubMed] [Google Scholar]
- 24. Kurtzman A. L., Schechter N. 2001. Ubc9 interacts with a nuclear localization signal and mediates nuclear localization of the paired-like homeobox protein Vsx-1 independent of SUMO-1 modification. Proc. Natl. Acad. Sci. U. S. A. 98:5602–5607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lang V., Rodriguez M. S. 2008. Innate link between NF-κB activity and ubiquitin-like modifiers. Biochem. Soc. Trans. 36:853–857 [DOI] [PubMed] [Google Scholar]
- 26. Li H. P., Chang Y. S. 2003. Epstein-Barr virus latent membrane protein 1: structure and functions. J. Biomed. Sci. 10:490–504 [DOI] [PubMed] [Google Scholar]
- 27. Lin J., Johannsen E., Robertson E., Kieff E. 2002. Epstein-Barr virus nuclear antigen 3C putative repression domain mediates coactivation of the LMP1 promoter with EBNA-2. J. Virol. 76:232–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luftig M., et al. 2003. Epstein-Barr virus latent membrane protein 1 activation of NF-κB through IRAK1 and TRAF6. Proc. Natl. Acad. Sci. U. S. A. 100:15595–15600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mabb A. M., Miyamoto S. 2007. SUMO and NF-κB ties. Cell. Mol. Life Sci. 64:1979–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maul G. G., Negorev D., Bell P., Ishov A. M. 2000. Review: properties and assembly mechanisms of ND10, PML bodies, or PODs. J. Struct. Biol. 129:278–287 [DOI] [PubMed] [Google Scholar]
- 31. Miller W. E., Mosialos G., Kieff E., Raab-Traub N. 1997. Epstein-Barr virus LMP1 induction of the epidermal growth factor receptor is mediated through a TRAF signaling pathway distinct from NF-κB activation. J. Virol. 71:586–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mo Y. Y., Moschos S. J. 2005. Targeting Ubc9 for cancer therapy. Expert Opin. Ther. Targets 9:1203–1216 [DOI] [PubMed] [Google Scholar]
- 33. Mo Y. Y., Yu Y., Shen Z., Beck W. T. 2002. Nucleolar delocalization of human topoisomerase I in response to topotecan correlates with sumoylation of the protein. J. Biol. Chem. 277:2958–2964 [DOI] [PubMed] [Google Scholar]
- 34. Muller S., Matunis M. J., Dejean A. 1998. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J. 17:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ning S., Campos A. D., Darnay B. G., Bentz G. L., Pagano J. S. 2008. TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol. Cell. Biol. 28:6536–6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pagano J. S. 2009. EBV diseases, p. 217–240 In Damania B., Pipas J. M. (ed.), DNA tumor viruses. Springer Science and Business Media, New York, NY [Google Scholar]
- 37. Rosendorff A., et al. 2004. EBNA3C coactivation with EBNA2 requires a SUMO homology domain. J. Virol. 78:367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rui H. L., et al. 2002. SUMO-1 modification of the C-terminal KVEKVD of Axin is required for JNK activation but has no effect on Wnt signaling. J. Biol. Chem. 277:42981–42986 [DOI] [PubMed] [Google Scholar]
- 39. Schultheiss U., et al. 2001. TRAF6 is a critical mediator of signal transduction by the viral oncogene latent membrane protein 1. EMBO J. 20:5678–5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shair K. H., Schnegg C. I., Raab-Traub N. 2008. EBV latent membrane protein 1 effects on plakoglobin, cell growth, and migration. Cancer Res. 68:6997–7005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shimizu N., Yoshiyama H., Takada K. 1996. Clonal propagation of Epstein-Barr virus (EBV) recombinants in EBV-negative Akata cells. J. Virol. 70:7260–7263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sides M. D., et al. 2011. Arsenic mediated disruption of promyelocytic leukemia protein nuclear bodies induces ganciclovir susceptibility in Epstein-Barr positive epithelial cells. Virology 416:86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Song J., Durrin L. K., Wilkinson T. A., Krontiris T. G., Chen Y. 2004. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. U. S. A. 101:14373–14348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stewart M. J., Smoak K., Blum M. A., Sherry B. 2005. Basal and reovirus-induced beta interferon (IFN-beta) and IFN-beta-stimulated gene expression are cell type specific in the cardiac protective response. J. Virol. 79:2979–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ungureanu D., Vanhatupa S., Gronholm J., Palvimo J. J., Silvennoinen O. 2005. SUMO-1 conjugation selectively modulates STAT1-mediated gene responses. Blood 106:224–226 [DOI] [PubMed] [Google Scholar]
- 46. Vanhatupa S., Ungureanu D., Paakkunainen M., Silvennoinen O. 2008. MAPK-induced Ser727 phosphorylation promotes SUMOylation of STAT1. Biochem. J. 409:179–185 [DOI] [PubMed] [Google Scholar]
- 47. Wakisaka N., et al. 2004. Epstein-Barr virus latent membrane protein 1 induces synthesis of hypoxia-inducible factor 1 alpha. Mol. Cell. Biol. 24:5223–5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yasugi T., Howley P. M. 1996. Identification of the structural and functional human homolog of the yeast ubiquitin conjugating enzyme UBC9. Nucleic Acids Res. 24:2005–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Z., Zhang Q., Yu Y., Ouyang Y., He Z. 2008. Construction and function analysis of the Epstein-Barr virus-encoded latent membrane protein-1 of CTAR3 region. Wei Sheng Wu Xue Bao 48:1308–1313 (In Chinese.) [PubMed] [Google Scholar]
- 50. Zhong S., et al. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 95:2748–2752 [PubMed] [Google Scholar]
- 51. Zimnik S., Gaestel M., Niedenthal R. 2009. Mutually exclusive STAT1 modifications identified by Ubc9/substrate dimerization-dependent SUMOylation. Nucleic Acids Res. 37:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]