

Abstract
The cellular requirements for activation of herpesvirus fusion and entry remain poorly understood. Low pH triggers change in the antigenic reactivity of the prefusion form of the herpes simplex virus (HSV) fusion protein gB in virions, both in vitro and during viral entry via endocytosis (S. Dollery et al., J. Virol. 84:3759–3766, 2010). However, the mechanism and magnitude of gB conformational change are not clear. Here we show that the conformation and oligomeric state of gB with mutations in the bipartite fusion loops were similarly altered despite the fusion-inactivating mutations. Together with previous studies, this suggests that fusion loop mutants undergo conformational changes but are defective for fusion because they fail to make productive contact with the outer leaflet of the host target membrane. A direct, reversible effect of low pH on the structure of gB was detected by fluorescence spectroscopy. A soluble form of gB containing cytoplasmic tail sequences (s-gB) was triggered by mildly acidic pH to undergo changes in tryptophan fluorescence emission, hydrophobicity, antigenic conformation, and oligomeric structure and thus resembled the prefusion form of gB in the virion. In contrast, soluble gB730, for which the postfusion crystal structure is known, was only marginally affected by pH using these measures. The results underscore the importance of using a prefusion form of gB to assess the activation and extent of conformation change. Further, acidic pH had little to no effect on the conformation or hydrophobicity of gD or on gD's ability to bind nectin-1 or HVEM receptors. Our results support a model in which endosomal low pH serves as a cellular trigger of fusion by activating conformational changes in the fusion protein gB.
INTRODUCTION
Membrane fusion during enveloped virus entry is mediated by conformational change in viral fusion proteins triggered by cellular factors such as endosomal low pH, receptor binding, or proteolytic cleavage. Herpesviruses are a paradigm for viral entry mediated by a multicomponent fusion machinery. Herpes simplex virus (HSV) glycoproteins gB, gD, and gH-gL are necessary for entry and membrane fusion (12, 35, 55). Herpesviral fusion and entry are further complicated by the likely requirement of multiple cellular cues. There is mounting evidence for the critical, direct role of endosomal pH during HSV entry by endocytosis, which is the predominant entry pathway for HSV in many cell types, including human epithelial cells (40, 41).
We recently demonstrated that the prefusion conformation of the HSV fusion protein gB is altered in direct response to low pH (20), providing in part, a possible molecular explanation for why herpesviruses require endosomal pH for entry. Specifically, a mildly acidic pH of ∼<6.2 causes specific changes in the antigenic structure of the functional region of gB containing the hydrophobic, bipartite fusion loops. Impor-tantly, this conformational change is also detected during viral entry by endocytosis, when the incoming virus arrives in an acidic compartment. Three independent approaches were used to demonstrate that a similar range of pHs causes a change in the oligomeric structure of gB. Low pH triggers gB to become more hydrophobic, suggesting that membrane-interacting regions are revealed. A highly fusogenic form of gB has antigenic changes similar to those induced in wild-type gB by acidic pH (49), suggesting that gB conformation change correlates with fusion activity. All conformational changes in gB detected to date are reversible, which is a hallmark of class III fusion proteins.
gB is conserved among all herpesviruses. The crystal structure of gB730, an ectodomain fragment of HSV type 1 (HSV-1) gB bears striking architectural homology to the low-pH, postfusion form of G glycoprotein from vesicular stomatitis virus (33, 47). In this report, the extent and nature of gB conformation changes were investigated. We demonstrate that reversible, pH-triggered changes in gB occur regardless of fusion loop activity and that low pH has little to no effect on the conformation or function of gD. We also investigate the capacity of two recombinant forms of soluble gB to undergo changes in response to low pH. The results suggest that the soluble ectodomain of gB directly fused to the cytoplasmic tail undergoes conformational changes and may reflect the prefusion structure of gB present in the virion. Further, gB730, for which the crystal structure is known, resembles gB that is locked in a postfusion conformation which undergoes limited conformational change.
MATERIALS AND METHODS
Cells and viruses.
Vero cells (American Type Culture Collection; ATCC; Rockville, MD) were propagated in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-Products, West Sacramento, CA). HSV-1 strain KOS (provided by Priscilla Schaffer, Harvard University) and HSV-2 strain 333 (provided by Stephen Straus, National Institutes of Health) were propagated and titers were determined on Vero cells.
Recombinant baculoviruses expressing gC(457t) (60), gD(Δ290-299t) (44), HVEMt (64), or nectin-1t (36) were obtained from Gary Cohen and Roselyn Eisenberg, University of Pennsylvania. Baculoviruses were grown and titers determined as described previously (66).
Purified proteins.
Recombinant s-gB is HSV-2 strain 333 gB with its transmembrane domain deleted and the cytoplasmic tail fused directly to the ectodomain. Specifically, ectodomain residues Ala23 to Asn725 are fused to tail residues Gln799 to Leu904 (58, 65). s-gD is a similarly constructed soluble form of HSV-2 333 gD (58). s-gB and s-gD were produced in CHO cells and obtained from Stephen Straus, NIAID. gC(457t) (60) is HSV-1 strain KOS gC truncated at residue 457 just prior to the transmembrane region (TMR). gB730 (a gift from R. Eisenberg and G. Cohen [10]) is truncated at residue 730 and lacks TMR and cytoplasmic tail regions. It is expressed in Sf9 insect cells. gD(Δ290-299t) (44) is HSV-1 strain KOS gD truncated at residue 306 with a 4-amino-acid linker inserted in place of amino acids 290 to 299 and has a higher affinity for receptors than its wild-type counterpart (44). gD(Δ290-299t) lacks TMR and cytoplasmic tail sequences. HVEMt (64) and nectin-1t (36) are truncated at residues 200 and 346, respectively. gC(457t), gD(Δ290-299t), HVEMt, and nectin-1t each have a 6-histidine C-terminal tag and were purified from recombinant baculovirus-infected Sf9 cell supernatants by metal-chelate affinity chromatography essentially as described previously (66).
Antibodies.
Anti-gB monoclonal antibodies (MAbs) H126 (domain I), H1359 (domain III), and H1817 (domain VI) were purchased from Virusys. Anti-gB monoclonal antibodies DL16 (trimer-specific), SS10 (domain IV), SS55 (domain I) (9), SS106 (domain V), and SS144 (domain V) (8) and R69 polyclonal antibody to gB were provided by G. Cohen and R. Eisenberg, University of Pennsylvania. MAb 1C8 (25) to gC and anti-gD MAbs DL11 (group Ib) (15), DL6 (group IIb) (21), and DL2 (group VI) (15) were provided by R. Eisenberg and G. Cohen. MAb 1103 (group VII) (45) to gD was purchased from Rumbaugh-Goodwin Institute, Plantation, FL.
Intrinsic fluorescence spectroscopy.
A 10- to 20-μg/ml portion of s-gB (7 Trp residues), gB730 (7 Trp residues), gC(457t) (8 Trp residues), s-gD (6 Trp residues), gD(Δ290-299t) (4 Trp residues), or ovalbumin (3 Trp residues) was prepared in 137 mM NaCl and 5 mM (each) HEPES (Life Technologies), 2-(N-morpholino)ethanesulfonic acid (MES) (Sigma), and sodium succinate (Sigma) to achieve final pHs ranging from 7.3 to 5.1. Proteins were incubated at 37°C for 2 min. Using a Shimadzu RF-5301PC spectrofluorophotometer, samples were excited at 280 nm. A 10-nm spectral bandwidth with a 0.5-s response time was used. Emission was recorded over 310 to 400 nm. To test the reversibility of the pH-induced spectral transition, samples were prepared at pH 5.1, incubated for 2 min at 37°C, and then neutralized with a predetermined amount of NaOH. For control samples, water was added in place of NaOH, which did not alter the shift in peak emission.
Dot blot analysis.
Purified HSV glycoproteins, cell-free virion preparations, or infected cell lysates were diluted in serum-free, bicarbonate-free DMEM with 0.2% bovine serum albumin (BSA) and 5 mM (each) HEPES, MES, and sodium succinate to achieve final pHs ranging from 7.4 to 4.8. Samples were incubated at 37°C for 10 min. Samples either were blotted directly to nitrocellulose with a Mini Fold dot blot system (Whatman) or were first neutralized by addition of pretitrated amounts of 0.05 N NaOH. Membranes were blocked and incubated with antibodies to gB or gD. After incubation with horseradish peroxidase-conjugated secondary antibodies, enhanced chemiluminescent substrate (Pierce) was added, and blots were exposed to X-ray film (Kodak).
Antigenic analysis of gD by ELISA.
Portions (8 μg/ml) of gD(Δ290-299t) in phosphate-buffered saline (PBS) were bound to microtiter plates overnight at 4°C. Plates were incubated with 1% BSA and 1% chicken egg ovalbumin in PBS (blocking buffer) for 1 h at room temperature. Dilutions (2-fold) of MAb DL11, DL6, 1103, or 1C8 were added for 1 h at room temperature. Protein A-peroxidase (Roche) in blocking buffer was added for 1 h at room temperature. Next, 2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid (Pierce) substrate was added. The A410 was recorded with a microtiter plate reader (Biotek).
gD binding to cognate receptors measured by ELISA.
The methods for gD binding to cognate receptors were modified from references 36 and 64. Soluble receptors HVEMt (15 μg/ml) or nectin-1t (10 μg/ml) in PBS were bound to microtiter plates overnight at 4°C. Plates were blocked with blocking buffer for 1 h at room temperature. Dilutions of gD(Δ290-299t) that had been adjusted to pH 4.7 for 10 min at 37°C, heated to 80°C for 10 min, or mock treated were prepared in blocking buffer and were added for 3 h at room temperature. Anti-gD MAb DL6 in blocking buffer was added for 1 h at room temperature. Detection with protein A-peroxidase and substrate was as indicated above for the antigenic enzyme-linked immunosorbent assay (ELISA). Nonspecific binding of gD in the absence of receptor was subtracted as background.
Triton X-114 partitioning.
As described in reference 20, 200-ng portions of soluble forms of gB, gC, or gD were incubated with 2% Triton X-114 in fusion medium buffered to various pHs on ice for 10 min. Samples were incubated at 37°C for 10 min and then centrifuged at 300 × g for 3 min at 25°C. Aqueous and detergent phases were collected and immunoprecipitated with MAb H1359 to gB or MAb DL6 to gD. Precipitates were analyzed by SDS-PAGE followed by Western blotting.
Analysis of gB oligomeric structure by PAGE.
As described in reference 20, infected cell lysates were diluted in the same medium used for dot blot analysis. Samples were adjusted to the indicated pHs with pretitrated amounts of 0.05 N HCl and incubated at 37°C for 10 min. SDS (1%) was then added or samples remained untreated. Polyacrylamide gel electrophoresis (PAGE) sample buffer containing 0.2% sodium dodecyl sulfate (SDS) and no reducing agent was added (“native” conditions), and proteins were resolved by PAGE. After transfer to nitrocellulose, membranes were blocked and incubated with rabbit polyclonal antibody specific for gB (provided by G. Cohen and R. Eisenberg). After incubation with horseradish peroxidase-conjugated secondary antibodies, enhanced chemiluminescent substrate (Pierce) was added and membranes were exposed to X-ray film (Kodak).
Preparation of lysates of cells infected with recombinant HSVs expressing gB with fusion loop mutations.
The construction of mutant HSVs bearing gB with a Y179K mutation in fusion loop 1 or A261D in fusion loop 2 was described previously (67). F6 cells (24) (provided by Tony Minson, Cambridge University) were derived from Vero cells, express HSV-1 gH, and were maintained in DMEM with 8% FBS and 150 μg/ml G418 (Mediatech, Herndon, VA). F6 cells were infected with the following recombinant HSVs at a multiplicity of infection (MOI) of 5 for 18 to 24 h: F-BACgH−, which expresses wild-type gB but no gH (w.t.gB/gH−); F-BAC-gBgalK/gH−, which expresses no gB and no gH; F-BAC-gB(Y179K); or F-BAC-gB(A261D)/gH−. Vero cells were infected with HSV-1 KOS under similar conditions. Infected cells were scraped and collected by centrifugation at 500 × g. Cells were resuspended in DMEM with 1% FBS and then sonicated on ice. Samples were clarified of large debris by centrifugation at 2,000 × g for 5 min.
RESULTS
Low-pH-induced changes in gB detected by intrinsic tryptophan fluorescence spectroscopy.
Conformational change in HSV glycoproteins was measured by direct spectroscopic analysis. Fluorescence from the aromatic amino acid tryptophan is sensitive to the nature of its local environment. Following excitation at 280 nm, tryptophan has emission spectra centered between 330 and 340 nm. A shift in the emission wavelength maximum is an indicator of conformation change (7). The intrinsic fluorescence emission spectra of purified forms of HSV gB, gC, or gD were recorded at pH 5.1 or 7.3 (Fig. 1).
Fig. 1.

Effect of acid pH on tryptophan fluorescence of HSV glycoproteins. The indicated protein (10 to 20 μg/ml) was adjusted to a pH of 7.3 or 5.1. Indicated gB samples were acidified to pH 5.1 and then reneutralized to 7.4. Excitation was performed at 280 nm, and emission was recorded over 310 to 400 nm. Peak fluorescence is shown. Representative spectra from at least three independent experiments are shown.
At the neutral pH of 7.3, s-gB had a peak emission at 330 nm. Upon treatment at a pH of 5.1, the emission maximum of s-gB shifted >2 nm toward longer wavelength (Fig. 1). This indicates change in the microenvironment of one or more tryptophan residues and suggests surface exposure of tryptophan as a result of low-pH-induced conformational changes. Interestingly, when gB730 was treated at low pH, a slight shift toward shorter wavelength in peak emission occurred. The seven tryptophans in s-gB and gB730 are conserved. While it is not clear which tryptophan(s) undergo a change in microenvironment, the results suggest that s-gB undergoes a conformational change that is different from gB730 and perhaps greater.
Reversibility of conformational change is a characteristic of class III fusion proteins. Changes in prefusion gB antigenic reactivity and oligomeric structure are reversible (20). When samples were first treated at pH 5.1 and then neutralized back to 7.4, there was little change in emission wavelength maximum relative to either form of gB at neutral pH (Fig. 1). This suggests that the low-pH-induced spectral transition of gB is reversible. The tryptophan fluorescence analysis is antibody-independent, biophysical evidence that herpesvirus gB undergoes reversible, low-pH-induced conformational changes.
The emission wavelength maxima of gC(457t), s-gD or gD(Δ290-299t) were very similar at pHs of 7.3 or 5.1 (Fig. 1). Thus, pH-induced conformational changes were not detected under these conditions. As an additional control, treatment with pH 5.1 had no detectable effect on the peak fluorescence emission spectra of the control protein ovalbumin. The absence of changes in the spectra of the other proteins further indicates that changes in gB are specific.
Effect of low pH on the antigenic conformation of gD.
During endocytic entry it is unclear whether the essential role of gD occurs at the plasma membrane or inside an acidic endosome. There was no detectable change in the local environment of tryptophan residues in gD upon exposure to low pH (Fig. 1). To extend this analysis, we tested the effect of pH on the antigenic conformation of gD using a panel of MAbs to different sites across the molecule.
The conformation of gD is important for HSV entry (22). Soluble gD blocks viral entry (34, 44). However, reduced or heat-denatured gD neither binds to receptors nor blocks HSV penetration (36, 44, 64). An increase in temperature destroys discontinuous epitopes of gD that are recognized by conformation-dependent monoclonal antibodies (MAbs) such as those belonging to groups Ib and VI (44). The group Ib MAb DL11 blocks HSV binding to HVEM and nectin-1 and neutralizes virus entry (36, 42). Group VII MAbs block both gD binding and virion binding to HVEM (42). The group VII epitope overlaps the N-terminal hairpin of gD which contains contact residues for HVEM binding (13).
HSV-1 KOS virions were exposed to pHs ranging from 7.4 to 5.2 and blotted directly to nitrocellulose, and then antibody binding was measured at neutral pH. Control polyclonal antibody R7 to gD detected virion gD to an equivalent extent under all conditions tested (Fig. 2A). The four representative MAbs to gD, DL11 (group Ib), DL6 (group IIb), DL2 (group VI), and 1103 (group VII), displayed unaltered binding to acid-treated virions, suggesting that pH does not induce changes in virion gD conformation. Control MAbs to gB were included. Four MAbs that recognize distinct epitopes in functional region 1 of prefusion gB have diminished reactivity with pH-treated gB (20). As expected, the MAb to gB domain I, H126, displayed diminished binding to gB from virions that had been treated at pH <6.2, and MAb H1817 (to domain VI) had unaltered binding to low-pH-treated virions (Fig. 2A).
Fig. 2.

Effect of low pH on the antigenic conformation of virion gD or soluble gD. (A) HSV-1 KOS virions were treated for 10 min at 37°C with medium buffered to pHs ranging from 7.4 to 5.2. Next, 105 PFU were blotted immediately to a nitrocellulose membrane. Blots were probed at neutral pH with gD-specific polyclonal antibody R7 or MAbs specific for gD (DL2, DL6, 1103, or DL11) or for gB (H1817 or H126), followed by horseradish peroxidase-conjugated goat secondary antibody. (B) Microtiter plates were coated with soluble gD(Δ290-299t) that had been treated with pH 7.4 or 4.7. MAb DL2, DL6, DL11, or 1103 to gD or control MAb to gC was added, followed by horseradish peroxidase-conjugated protein A and substrate. Values shown are the means of duplicate determinations.
Antigenic ELISA (44) was performed to assess the effect of pH on soluble gD structure. Acid-treated or untreated gD(Δ290-299t) (referred to as gDt) had similar reactivity with a range of dilutions of MAb DL6 and 1103 (Fig. 2B), indicating that these epitopes are not obscured following exposure to low pH. Compared with that of untreated gDt, low-pH-treated gDt had similar reactivity overall with MAbs DL11 and DL2, although slightly less reactivity was observed with intermediate dilutions. This suggests that these discontinuous epitopes are not destroyed following acid treatment. As expected, control anti-gC MAb 1C8 failed to recognize either treated or untreated gDt (Fig. 2B). By the measures used here, acidic pH has little detectable effect on gD conformation, including regions that are important for receptor binding.
Effect of low pH on the receptor-binding function of gD.
Upon binding to the HVEM receptor, HSV gD undergoes pH-independent conformational change, resulting in movement of the C terminus to reveal receptor contact sites (13, 26, 37). The effect of low pH on gD function has not been reported. The ability of acid-treated, soluble gD to bind to HVEM or nectin-1 was evaluated by a binding ELISA. Soluble gD that was treated at pH 4.7 bound to immobilized HVEMt in a concentration-dependent manner similar to that of untreated gDt (Fig. 3A). Control, heat-treated gDt, however, had severely diminished binding activity (Fig. 3A). Acid-treated and untreated gDt also bound comparably to immobilized nectin-1t (Fig. 3B). Taken together, the results indicate that low-pH pretreatment had no effect on the interaction of gD with its receptors, supporting the notion that endosomal acidic pH affects a required, post-gD binding step in entry. By the methods used here, pH-induced changes in the structure or function of gD were not detected (Fig. 1, 2, and 3).
Fig. 3.

Effect of low-pH pretreatment of gD on binding to HSV entry receptors HVEM or nectin-1. gD (Δ290-299t) was treated at pH 4.7, heated to 80°C, or left untreated. Increasing concentrations of gD were added to microtiter plates coated with HVEMt (A) or nectin-1t (B). Bound gD was detected with MAb DL6 followed by peroxidase-conjugated protein A and substrate. Absorbance was read at 410 nm. Each value represents the average of results for duplicate wells.
The antigenic structure of s-gB is more susceptible to pH than gB730.
A soluble form of gB with its transmembrane domain deleted and the cytoplasmic tail fused directly to the ectodomain (s-gB) undergoes conformational changes reminiscent of that observed with prefusion virion gB (20). gB730 lacks the TMR and cytoplasmic tail regions, and its crystal structure is a hairpin that is believed to reflect the postfusion state (33). We tested the effect of low pH on the antigenic reactivity of these two forms of gB.
s-gB or gB730 was exposed to pHs ranging from 7.4 to 5.1 and were then immediately blotted to nitrocellulose membrane. Antibody binding was then assessed at pH 7.4. MAb H126 displayed diminished binding to s-gB that had been treated at pH <6.6 (Fig. 4A). Similar results were obtained with a MAb to domain V, SS106 (Fig. 4A). This suggests a specific change in the antigenic structure of the functional region of s-gB responsible for fusion. Notably, H126 and SS106 both bound well to acid-treated gB730. Importantly, the trimer-specific MAb DL16 displayed decreased binding to low-pH-treated s-gB, but acid treatment appeared to have less effect on this epitope in gB730. As a control, SS10, directed to domain IV and gB-specific polyclonal Ab R69, bound well to both low-pH-treated forms of gB (Fig. 4A). This also confirms that the structure of gB is not globally misfolded and that the detected changes in reactivity are specific. Together, current and previous results suggest that the antigenic and oligomeric structure of purified recombinant s-gB is altered by low pH in a manner similar to prefusion gB found in virions. In contrast, gB730 is less susceptible to pH, consistent with it resembling a postfusion, terminally activated form of gB.
Fig. 4.
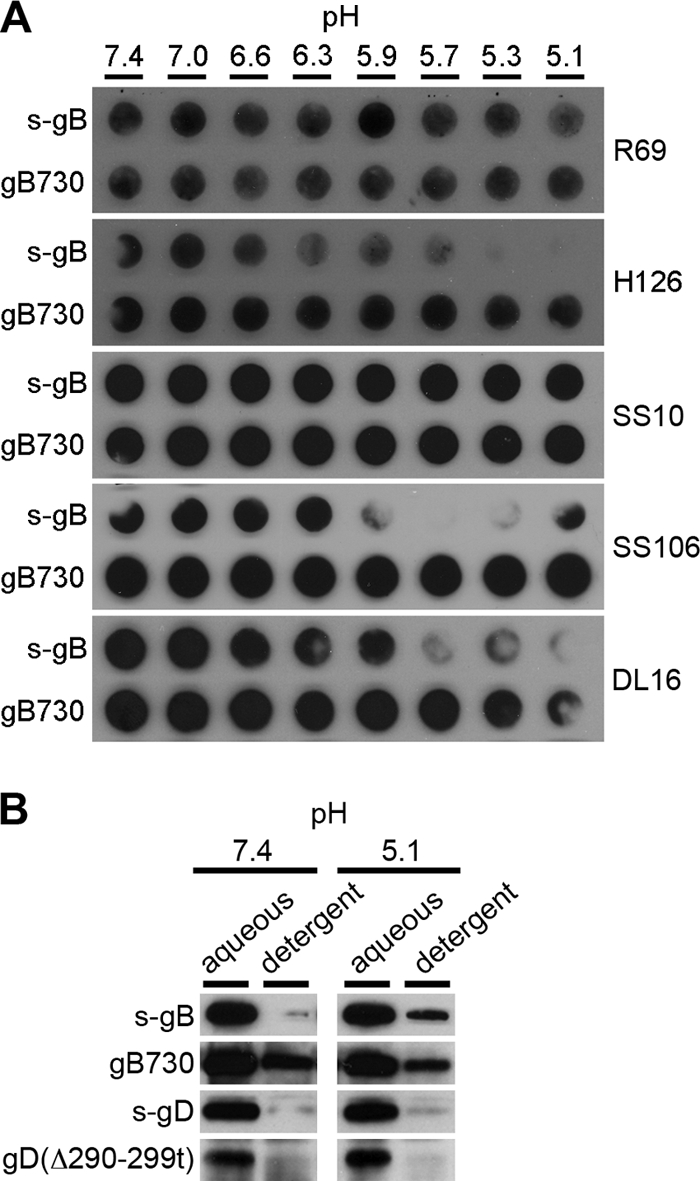
Effect of low pH on antigenic reactivity and hydrophobicity of different forms of soluble gB. (A) s-gB or gB730 was treated for 10 min at 37°C with medium buffered to pHs ranging from 7.4 to 5.1 and was blotted immediately to a nitrocellulose membrane. An equivalent amount of R69-reactive gB (50 to 100 μg) was blotted in each case. Blots were probed at neutral pH with gB-specific antibodies R69 (polyclonal) or H126, SS10, SS106, or DL16 (monoclonal), followed by horseradish peroxidase-conjugated goat secondary antibody. The exposures shown highlight the pH threshold of conformational change. (B) Proteins were added to 2% Triton X-114 that had been adjusted to the indicated pH. Samples were incubated for 10 min at 37°C and were centrifuged at 300 × g for 3 min. The aqueous supernatant phase or detergent phase was collected and diluted 20-fold in PBS. Samples were subjected to immunoprecipitation (H1817 for gB, DL6 for gD) followed by SDS-PAGE and immunoblotting (H1359 for gB, DL6 for gD).
gB730 is hydrophobic at physiological and acidic pHs.
Conformational changes associated with fusion are often accompanied by transient exposure of hydrophobic regions, namely, fusion peptides or fusion loops. We used the nonionic detergent Triton X-114 to assess the hydrophobicity of soluble forms of HSV glycoproteins. Upon centrifugation at room temperature, Triton X-114 can be separated into hydrophilic (aqueous, top) and amphiphilic (detergent, bottom) phases (11). Protein forms that are more hydrophobic associate with hydrophobic Triton X-114 molecules and partition with the amphiphilic phase. We used this approach previously to demonstrate the effect of pH on the structure of s-gB (20).
Purified s-gB that was treated at pH 7.4 was mixed with Triton X-114 at 30°C. The mixture was centrifuged, and gB was recovered primarily from the aqueous phase (Fig. 4B) (20), where hydrophilic proteins are expected to partition. At pH 7.4, gB730 was detected in the aqueous phase but also in the detergent phase, suggesting that gB730 is already hydrophobic at neutral pH. This is consistent with surface exposure of the hydrophobic fusion loops in the structure of gB730 (33). In contrast, little s-gB was detected in the detergent phase. Similarly, the majority of s-gD and gD(Δ290-299t) partitioned with the aqueous phase, suggesting limited hydrophobic character of gD at physiological pH.
Of the glycoproteins tested, s-gB was the only one affected by treatment with pH 5.1. A fraction of the total s-gB partitioned with the detergent phase (Fig. 4B), suggesting that low pH caused s-gB to become more hydrophobic. It is not clear whether gB exists as a mixture of more than one conformation at a given pH. This increase in hydrophobicity is consistent with the notion that gB is a pH-activated fusion protein. By this approach, low pH caused little detectable increase in the hydrophobic nature of the other glycoprotein constructs, gB730, s-gD, and gD(Δ290-299t). The results from three independent approaches (Fig. 1, Fig. 4A and B) suggest that s-gB is sensitive to pH and gB730 is less so. The acid-induced increase in hydrophobicity of s-gB correlates with changes in conformation measured by a battery of approaches, including fluorescence spectroscopy, changes in reactivity with antibodies to the fusion domain, and changes in oligomeric structure (20, 53; this work). The effects of pH on s-gB are similar to what is expected of the prefusion form of gB that is found in the virion as it functions during viral entry. We propose that mildly acidic pH triggers exposure of hydrophobic, membrane-interacting regions in gB.
Fusion-defective mutants of gB undergo conformational changes.
To probe the relationship between conformational change and fusion, we determined the effect of pH on fusion loop mutants of gB. Substituting polar residues for hydrophobic residues in either of the internal fusion loops diminishes cell-cell fusion activity (32, 59) and fusion of newly formed virions with the outer nuclear membrane during viral egress (67). The bipartite hydrophobic loops of class III fusion proteins are thought to insert into the host cell membrane during the fusion process. We analyzed low-pH-induced changes in the antigenic conformation of gB bearing fusion-defective mutations, Y179K in fusion loop 1 or A261D in fusion loop 2.
Lysates of infected cells were examined because high titers of these mutant viruses are difficult to obtain. Cell-expressed gB is competent for fusion, as evidenced during cell-to-cell spread, cell-cell fusion assays, and syncytium formation. Thus, it reflects a prefusion conformation. Lysates of cells infected with mutant viruses were exposed to pHs ranging from 7.4 to 5.1. Samples were immediately blotted to nitrocellulose membranes, and antibody binding was assessed at pH 7.4. For these experiments, the relatively large amount of cell lysate necessary to detect gB often resulted in blots that were of lower quality than those in analyses of purified virions or proteins. Monoclonal antibodies H126 (Fig. 5A) and SS106 (Fig. 6A) displayed diminished binding to Y179K and A61D lysates that had been treated at lower pHs, whereas MAbs SS10 (Fig. 5A) displayed unaltered binding. A similar pattern was observed with lysates of cells infected with matched wild-type (w.t.gB/gH−) or KOS viruses (Fig. 5A), suggesting that changes in antigenic structure occur regardless of whether fusion loops are active. Control lysates lacking gB (gBgalK/gH−) failed to react with gB-specific antibody (Fig. 5A).
Fig. 5.

Effect of low pH on the antigenic and oligomeric conformation of mutant gBs that are defective for fusion. (A) The indicated infected cell lysates were diluted in medium buffered to pHs ranging from 7.4 to 5.1. Samples were incubated at 37°C for 5 min and then blotted directly to nitrocellulose. Amounts of cell-associated virus that reacted equally to MAbs prior to pH treatment were blotted. Membranes were blocked and then incubated at neutral pH with anti-gB MAbs H126 (domain I) or SS10 (domain III) or (B, left panel) DL16 (trimer-specific). Similar results were obtained in at least three independent experiments per antibody. (B, right panel) Assay for sensitivity of oligomeric gB to detergent. The indicated infected cell lysates were treated with medium adjusted to pHs ranging from 7.4 to 5.0. Samples were adjusted to 1% SDS and were added to PAGE sample buffer containing 0.2% SDS and no reducing agent (“native” conditions). Unheated samples were resolved by PAGE. After transfer to nitrocellulose, membranes were blocked and incubated with gB-specific rabbit polyclonal antibody (R69). After incubation with horseradish peroxidase-conjugated goat-anti-rabbit antibody, enhanced chemiluminescent substrate was added and membranes were exposed to X-ray film. Molecular size markers in kilodaltons are indicated at the left. Positions of gB oligomer and monomer are indicated at the right. Each exposure (A and B) highlights the pH threshold of conformational change.
Fig. 6.

Reversibility of conformational changes in fusion loop mutants of gB. (A) The indicated infected cell lysates were treated with medium buffered to pH 7.4 or 5.3. For the indicated samples, the pH of 5.3 was neutralized to 7.4 for 10 min at 37°C and then the sample was blotted immediately to nitrocellulose. Membranes were probed at neutral pH with MAbs H126, SS10, SS106, or (B, left panel) DL16 followed by horseradish peroxidase-conjugated secondary antibody. Similar results were obtained in at least three independent experiments per antibody. (B, right panel) Cell-associated gB preparations were treated with the indicated pH conditions, treated with 1% SDS, and then analyzed by PAGE and immunoblotting with gB-specific polyclonal antibody R69. Images show gB of >181 kDa. Exposures shown highlight the reversibility of conformational change.
Two approaches were used to examine the effect of pH on the oligomeric structure of the fusion loop mutants. First, the trimer-specific MAb DL16 had diminished reactivity with gB from lysates of cells infected with the Y179K or A261D viruses (Fig. 5B). Second, we used an assay established in our laboratory that takes advantage of an experimentally useful characteristic of HSV gB (20, 53). When gB is first exposed to low pH, its oligomeric structure becomes susceptible to 1% SDS as measured by the migration of oligomeric species on native PAGE. Treatment of each of the infected cell lysates with pH 7.4 followed by 1% SDS yielded oligomeric forms of >181 kDa (Fig. 5B). However, upon pretreatment with pH <6.3 or 6.0 followed by 1% SDS, the slowest-migrating species of mutant gB seemed to disappear, leaving a stable oligomeric species of higher mobility (Fig. 5B). This suggests that low pH alters the oligomeric structure of gB containing mutations in its hydrophobic fusion loops, making it more sensitive to SDS. Mildly acidic pH had a similar effect on gB oligomers from lysates of cells infected with matched wild-type or KOS viruses (Fig. 5B). The precise identity of the two major high-molecular-weight species is of great interest. These species do not comigrate with gC, gD, gH, or gL (20). They are clearly not monomeric gB, but whether they represent dimers, trimers, or still higher-order oligomers is not clear. Another possibility is that both forms may be trimeric gB but have altered migration due to differences in conformation. With decreasing pH, there was an apparent decrease in detection of gB-reactive species by Western blotting. One explanation is that monomers are detected only weakly relative to oligomers under standard native PAGE analysis (data not shown). Alternately, upon activation by pH, gB may become part of a larger complex that does not enter the native gel. Notably, the total amount of gB detected by gB-specific polyclonal antibody via dot blot does not change upon exposure to mildly acidic pH (20, 53). The two sets of results (Fig. 5A and B) suggest that the oligomeric forms of fusion-defective and wild-type gBs undergo similar changes upon exposure to acidic pH.
Low-pH-triggered changes in the prefusion form of virion gB have been detected and characterized (20, 53; Fig. 2). Importantly, the current results indicate that cell-associated gB present in infected cells is subject to similar pH-induced changes in domains I and V and oligomeric state. The pH threshold is also similar. Thus, gB from infected cells resembles prefusion gB. Taken together, results here and elsewhere are consistent with a model in which the fusion loop mutants of gB fail to insert into the target membrane despite undergoing structural changes similar to those of the wild type.
pH-induced changes in fusion loop mutants of gB are reversible.
Low-pH-dependent conformational changes in class III fusion proteins, including HSV gB, are reversible (20, 27, 68). During viral assembly and egress, reversibility may allow fusion glycoproteins to avoid nonproductive activation during transport through the low-pH environment of the secretory pathway. To test whether a defect in reversibility correlates with the diminished fusion function of the loop mutants, the reversal of conformational changes in cell-associated gB and the mutants was analyzed. Y179K- and A261D-infected cell lysates were treated at pH 5.3 to trigger conformational change in gB, and were then adjusted back to pH 7.4 prior to blotting to nitrocellulose. Reactivity to MAbs H126 and SS106 was partially recovered relative to samples that received pH 5.3 treatment only (Fig. 6A), suggesting that pH-triggered alterations in the antigenic structure of the loop mutants are reversible. MAb SS10 reacted similarly with lysates that had been subjected to each of the different pH conditions (Fig. 6A). To address the reversibility of changes in the oligomeric state of the loop mutants, we tested the reversibility of DL16 reactivity. There was partial restoration of reactivity of the oligomer-specific MAb DL16 with the fusion loop mutants (Fig. 6B), similar to what was observed with wild-type forms of gB (Fig. 6B). To extend these findings, Y179K and A261D lysates were incubated at pH 5.3 and reneutralized to pH 7.4, and then 1% SDS was added (Fig. 6B). High-molecular-weight, oligomeric forms of gB were detected that were similar to those that had been kept at pH 7.4. The results in Fig. 6 suggest that low-pH-induced changes in the oligomeric structure of gB bearing mutations in the hydrophobic fusion loops are reversible. Results from this study and previous reports (20, 53) indicate that pH-triggered changes in s-gB and wild-type gB from virions or infected cells are reversible. Thus, pH-dependent changes in the antigenic and oligomeric structures are similar for wild-type and fusion-defective mutant forms of gB. This suggests that pH-triggered changes occur independently of fusion loop function.
DISCUSSION
The full complement of cellular factors necessary for herpes simplex virus membrane fusion has remained elusive. In contrast, the viral proteins required for fusion and entry, gB, gD, and gH-gL, have been known for some time and are well studied. An emerging theme in herpesviral entry is that cellular endosomal pH is required, often in a cell-specific manner (23, 30, 40, 41, 46, 50, 63). gB conformation change is central to the molecular understanding of why herpesviruses require low pH to initiate infection. Conformational changes in gB likely provide the energy to drive the viral and host membranes together during fusion. We propose that the combination of cell-induced changes in gB structure and functional hydrophobic fusion loops is needed for herpesviral fusion and entry. Different cellular triggers, namely, endosomal pH or a gB-binding receptor (2), may be necessary for entry via pH-dependent or pH-independent entry mechanisms, respectively.
Cellular factors and HSV entry.
Initially, cell surface heparan sulfate proteoglycans serve as attachment sites for viral envelope gB or gC (52). Then gD binds to a required cell receptor, such as nectin-1, nectin-2, HVEM, or a modified form of heparan sulfate (12, 35, 55). Upon interaction with HVEM, gD undergoes a pH-independent conformational change, which is thought to initiate fusion (37). Low pH has no effect on HSV binding to cell surface HVEM or nectin-1 (unpublished data). HSV utilizes cellular endocytosis machinery to enter epithelial tissues, the major target of infection in vivo (40, 41). Nectin-1 or αVβ3 integrin may mediate endocytic internalization of HSV into some cell types but not others (29, 39, 43, 57). Host cell kinases, such as phosphatidylinositol 3-kinase, facilitate endocytic trafficking of HSV (43, 61).
Low endosomal pH is required for penetration following endocytosis in several cell types, and triggers conformational change in gB (20, 41). Cellular receptors for gB include PILRalpha (51) and nonmuscle myosin IIA (1). The molecular ramifications of gB-receptor interactions are not known. Nectin-2 or PILRalpha can direct HSV to a pH-independent entry pathway (2, 18). Following fusion, which requires gB, gD, and gH-gL, the host 26S proteasome facilitates incoming capsid transport, which may be regulated by capsid-associated tegument ICP0 (17, 19). The capsid utilizes microtubules and associated motor proteins for targeting to the nucleus, which involves tegument UL36 protein (6, 16, 38, 54).
Soluble forms of gB respond differently to low pH.
Using three independent approaches, mildly acidic pH triggered conformational changes in s-gB that appeared to be greater than those in gB730. As measured by tryptophan fluorescence spectroscopy (Fig. 1) and antigenic analysis (Fig. 4A), the conformation of s-gB was altered by pH, but gB730 was only marginally affected. Upon activation, fusion glycoproteins become hydrophobic. Notably, low pH caused s-gB to become hydrophobic, whereas gB730 was already hydrophobic at neutral pH. The hydrophobicity of gB730 was not increased by low pH (Fig. 4B). These results provide insight into the functional relevance of using gB730 to measure the extent of conformational change in gB and the mechanism of fusion activation. The differences between s-gB and gB730 are not likely explained by type-specific effects. Prefusion gB from both HSV-1 and HSV-2 strains undergo acid-induced alterations (20, 53).
While this paper was in preparation, it was reported that low pH caused small changes in the structure of gB730, including alteration of fusion loop 2 (56). From this analysis, the authors propose that gB does not undergo the large-scale conformational changes expected of a viral fusion protein and that low pH is not a trigger of fusion. However, gB730 is not an ideal form of gB on which to base conclusions about the extent of conformational change or the activation of gB's fusion function. Results presented here and elsewhere indicate that gB730 has characteristics of postfusion gB that has already been activated. The hydrophobic nature of gB730 (Fig. 4B) and its ability to bind to lipid membranes at neutral pH (31) further suggest that it does not reflect a functional prefusion state.
By definition, gB that is present in the virion envelope is the prefusion form. The quaternary structure and domains I and V of prefusion gB are specifically altered following exposure to low pH (20). s-gB resembles prefusion gB in these regards (Fig. 4A). gD or gH-gL in the virion or the gB transmembrane and tail regions may be required to capture gB in its truest prefusion form. However, our data with s-gB suggest that gB sequences alone may result in a conformation that resembles prefusion gB. The specific contribution of the cytoplasmic tail to gB conformation change is under investigation.
Given the multiple cellular and viral cues for herpesviral fusion, low pH is likely required but not sufficient to induce the full range of gB conformational changes that occur during HSV endocytosis. Thus, the low-pH-treated form of s-gB may represent an intermediate conformation distinct from the postfusion form of gB represented by gB730. This would explain why the prefusion epitopes on gB that are diminished upon low-pH treatment are intact in gB730. A highly fusogenic form of gB is antigenically altered in domain I and in the trimer-specific DL16 epitope (49, 53). Since similar alterations are detected in acid-treated wild-type gB, changes in these antigenic sites correlate with fusion activity. The current work puts fusion loop activity in context with conformational change.
Cellular activation of gB versus activation by a virion component.
How the fusogenic potential of gB is triggered is an open question. The idea that a cellular factor, namely, endosomal pH, induces structural change in gB has mounting experimental support. Current models of fusion based on bimolecular complementation assays invoke lateral interactions among gD, gB, and gH-gL prior to fusion (3–5, 28). It has been proposed that a gH-gB interaction activates gB for fusion (14). While gH clearly plays a critical, yet-to-be defined functional role in assisting gB in the execution of fusion, there is currently no direct evidence that HSV gH binds to gB or that gH induces a change in gB. In contrast, acid pH directly affects gB (20, 53, 56; this report). Interestingly, human cytomegalovirus gB can be coimmunoprecipitated with gH (62). Data indicating that the structure or function of gB is directly altered or activated by a viral component are not currently available. We posit that cellular endosomal pH triggers a direct change in gB that allows the fusion loops to make initial contact with the outer leaflet of the target membrane, which is a critical destabilizing step in the fusion reaction. During fusion, there may be a large-scale rearrangement of folded domains such as that proposed for the class III fusion protein VSV G (48). In addition to low pH, the full range of conformation change in gB during entry into epithelial cells may require gD and gH-gL and additional cellular factors, including a gD receptor. In the case of the gB fusion loop mutants, wild-type-like conformational changes may occur but productive interaction with the host membrane does not, derailing the fusion reaction.
ACKNOWLEDGMENTS
This investigation was supported by Public Health Service grant AI-083850 from the National Institute of Allergy and Infectious Diseases and grant SAA4832 from the Japan Health Sciences Foundation.
We are grateful to Gary Cohen, Roselyn Eisenberg, Tony Minson, and Stephen Straus for generous gifts of reagents. We thank John Hackett for use of the fluorometer, Kayla Pfab for technical assistance, and Hector C. Aguilar for critical reading of the manuscript.
Footnotes
Published ahead of print on 3 August 2011.
REFERENCES
- 1. Arii J., et al. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862 [DOI] [PubMed] [Google Scholar]
- 2. Arii J., et al. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor alpha. J. Virol. 83:4520–4527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atanasiu D., Saw W. T., Cohen G. H., Eisenberg R. J. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Atanasiu D., et al. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J. Virol. 84:3825–3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Avitabile E., Forghieri C., Campadelli-Fiume G. 2009. Cross talk among the glycoproteins involved in herpes simplex virus entry and fusion: the interaction between gB and gH/gL does not necessarily require gD. J. Virol. 83:10752–10760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Batterson W., Furlong D., Roizman B. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J. Virol. 45:397–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beechem J. M., Brand L. 1985. Time-resolved fluorescence of proteins. Annu. Rev. Biochem. 54:43–71 [DOI] [PubMed] [Google Scholar]
- 8. Bender F. C., et al. 2007. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J. Virol. 81:3827–3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bender F. C., Whitbeck J. C., Lou H., Cohen G. H., Eisenberg R. J. 2005. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol. 79:11588–11597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bender F. C., et al. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 77:9542–9552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bordier C. 1981. Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 256:1604–1607 [PubMed] [Google Scholar]
- 12. Campadelli-Fiume G., et al. 2007. The multipartite system that mediates entry of herpes simplex virus into the cell. Rev. Med. Virol. 17:313–326 [DOI] [PubMed] [Google Scholar]
- 13. Carfi A., et al. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8:169–179 [DOI] [PubMed] [Google Scholar]
- 14. Chowdary T. K., et al. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 17:882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cohen G. H., Isola V. J., Kuhns J., Berman P. W., Eisenberg R. J. 1986. Localization of discontinuous epitopes of herpes simplex virus glycoprotein D: use of a nondenaturing (“native” gel) system of polyacrylamide gel electrophoresis coupled with Western blotting. J. Virol. 60:157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Copeland A. M., Newcomb W. W., Brown J. C. 2009. Herpes simplex virus replication: roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 83:1660–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Delboy M. G., Nicola A. V. 2011. A pre-immediate early role for tegument ICP0 in the proteasome-dependent entry of herpes simplex virus. J. Virol. 85:5910–5918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Delboy M. G., Patterson J. L., Hollander A. M., Nicola A. V. 2006. Nectin-2-mediated entry of a syncytial strain of herpes simplex virus via pH-independent fusion with the plasma membrane of Chinese hamster ovary cells. Virol. J. 3:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Delboy M. G., Roller D. G., Nicola A. V. 2008. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J. Virol. 82:3381–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dollery S. J., Delboy M. G., Nicola A. V. 2010. Low pH-induced conformational change in herpes simplex virus glycoprotein B. J. Virol. 84:3759–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eisenberg R. J., et al. 1985. Localization of epitopes of herpes simplex virus type 1 glycoprotein D. J. Virol. 53:634–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eisenberg R. J., et al. 1994. Structure and function of glycoprotein D of herpes simplex virus, p. 43–65.In Becker Y., Darai G. (ed.), Frontiers in virology, vol. 3 Springer-Verlag, Heidelberg, Germany [Google Scholar]
- 23. Finnen R. L., et al. 2006. Postentry events are responsible for restriction of productive varicella-zoster virus infection in Chinese hamster ovary cells. J. Virol. 80:10325–10334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Forrester A., et al. 1992. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J. Virol. 66:341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Friedman H. M., Cohen G. H., Eisenberg R. J., Seidel C. A., Cines D. B. 1984. Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells. Nature 309:633–635 [DOI] [PubMed] [Google Scholar]
- 26. Fusco D., Forghieri C., Campadelli-Fiume G. 2005. The pro-fusion domain of herpes simplex virus glycoprotein D (gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc. Natl. Acad. Sci. U. S. A. 102:9323–9328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gaudin Y. 2000. Reversibility in fusion protein conformational changes. The intriguing case of rhabdovirus-induced membrane fusion. Subcell. Biochem. 34:379–408 [DOI] [PubMed] [Google Scholar]
- 28. Gianni T., Amasio M., Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J. Biol. Chem. 284:17370–17382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gianni T., Gatta V., Campadelli-Fiume G. 2010. αVβ3-integrin routes herpes simplex virus to an entry pathway dependent on cholesterol-rich lipid rafts and dynamin2. Proc. Natl. Acad. Sci. U. S. A. 107:22260–22265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gillet L., Colaco S., Stevenson P. G. 2008. Glycoprotein B switches conformation during murid herpesvirus 4 entry. J. Gen. Virol. 89:1352–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hannah B. P., et al. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 83:6825–6836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hannah B. P., Heldwein E. E., Bender F. C., Cohen G. H., Eisenberg R. J. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 81:4858–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heldwein E. E., et al. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220 [DOI] [PubMed] [Google Scholar]
- 34. Johnson D. C., Burke R. L., Gregory T. 1990. Soluble forms of herpes simplex virus glycoprotein D bind to a limited number of cell surface receptors and inhibit virus entry into cells. J. Virol. 64:2569–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krummenacher C., Carfi A., Eisenberg R. J., Cohen G. H. 2007. Herpesvirus entry into cells: the enigma variations. In Poehlmann S., Simmons G. (ed.), Viral entry into host cells. Landes Bioscience, Austin, TX. [Google Scholar]
- 36. Krummenacher C., et al. 1998. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J. Virol. 72:7064–7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krummenacher C., et al. 2005. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 24:4144–4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luxton G. W., et al. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. U. S. A. 102:5832–5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Milne R. S., Nicola A. V., Whitbeck J. C., Eisenberg R. J., Cohen G. H. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 79:6655–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nicola A. V., Hou J., Major E. O., Straus S. E. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 79:7609–7616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nicola A. V., McEvoy A. M., Straus S. E. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nicola A. V., et al. 1998. Monoclonal antibodies to distinct sites on herpes simplex virus (HSV) glycoprotein D block HSV binding to HVEM. J. Virol. 72:3595–3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nicola A. V., Straus S. E. 2004. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 78:7508–7517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nicola A. V., Willis S. H., Naidoo N. N., Eisenberg R. J., Cohen G. H. 1996. Structure-function analysis of soluble forms of herpes simplex virus glycoprotein D. J. Virol. 70:3815–3822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pereira L., Dondero D. V., Gallo D., Devlin V., Woodie J. D. 1982. Serological analysis of herpes simplex virus types 1 and 2 with monoclonal antibodies. Infect. Immun. 35:363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raghu H., Sharma-Walia N., Veettil M. V., Sadagopan S., Chandran B. 2009. Kaposi's sarcoma-associated herpesvirus utilizes an actin polymerization-dependent macropinocytic pathway to enter human dermal microvascular endothelial and human umbilical vein endothelial cells. J. Virol. 83:4895–4911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roche S., Bressanelli S., Rey F. A., Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191 [DOI] [PubMed] [Google Scholar]
- 48. Roche S., Rey F. A., Gaudin Y., Bressanelli S. 2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315:843–848 [DOI] [PubMed] [Google Scholar]
- 49. Roller D. G., Dollery S. J., Doyle J. L., Nicola A. V. 2008. Structure-function analysis of herpes simplex virus glycoprotein B with fusion-from-without activity. Virology 382:207–216 [DOI] [PubMed] [Google Scholar]
- 50. Ryckman B. J., Jarvis M. A., Drummond D. D., Nelson J. A., Johnson D. C. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 80:710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Satoh T., et al. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shukla D., Spear P. G. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Invest. 108:503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Siekavizza-Robles C. R., Dollery S. J., Nicola A. V. 2010. Reversible conformational change in herpes simplex virus glycoprotein B with fusion-from-without activity is triggered by mildly acidic pH. Virol. J. 7:352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sodeik B., Ebersold M. W., Helenius A. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Spear P. G., et al. 2006. Different receptors binding to distinct interfaces on herpes simplex virus gD can trigger events leading to cell fusion and viral entry. Virology 344:17–24 [DOI] [PubMed] [Google Scholar]
- 56. Stampfer S. D., Lou H., Cohen G. H., Eisenberg R. J., Heldwein E. E. 2010. Structural basis of local, pH-dependent conformational changes in glycoprotein B from herpes simplex virus type 1. J. Virol. 84:12924–12933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stiles K. M., Milne R. S., Cohen G. H., Eisenberg R. J., Krummenacher C. 2008. The herpes simplex virus receptor nectin-1 is down-regulated after trans-interaction with glycoprotein D. Virology 373:98–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stuve L. L., et al. 1987. Structure and expression of the herpes simplex virus type 2 glycoprotein gB gene. J. Virol. 61:326–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sun X., Belouzard S., Whittaker G. R. 2008. Molecular architecture of the bipartite fusion loops of vesicular stomatitis virus glycoprotein G, a class III viral fusion protein. J. Biol. Chem. 283:6418–6427 [DOI] [PubMed] [Google Scholar]
- 60. Tal-Singer R., et al. 1995. Interaction of herpes simplex virus glycoprotein gC with mammalian cell surface molecules. J. Virol. 69:4471–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tiwari V., Shukla D. 2010. Phosphoinositide 3 kinase signalling may affect multiple steps during herpes simplex virus type-1 entry. J. Gen. Virol. 91:3002–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vanarsdall A. L., Ryckman B. J., Chase M. C., Johnson D. C. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 82:11837–11850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang D., Yu Q. C., Schroer J., Murphy E., Shenk T. 2007. Human cytomegalovirus uses two distinct pathways to enter retinal pigmented epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 104:20037–20042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Whitbeck J. C., et al. 1997. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the tumor necrosis factor receptor superfamily and a mediator of HSV entry. J. Virol. 71:6083–6093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Williams R. K., Straus S. E. 1997. Specificity and affinity of binding of herpes simplex virus type 2 glycoprotein B to glycosaminoglycans. J. Virol. 71:1375–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Willis S. H., et al. 1997. Expression and purification of secreted forms of herpes simplex virus glycoproteins from baculovirus-infected insect cells. Methods Mol. Med. 10:131–156 [DOI] [PubMed] [Google Scholar]
- 67. Wright C. C., et al. 2009. Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J. Virol. 83:11847–11856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou J., Blissard G. W. 2006. Mapping the conformational epitope of a neutralizing antibody (AcV1) directed against the AcMNPV GP64 protein. Virology 352:427–437 [DOI] [PMC free article] [PubMed] [Google Scholar]