

Abstract
The study of normal mammalian cell growth and the defects that contribute to disease pathogenesis constitutes a fundamental avenue of research that links metabolism to cell growth. Here we visit several aspects of this metabolism, emphasizing recent advances in our understanding of how alterations in glucose metabolism affect cytosolic and mitochondrial redox potential and ATP generation. These alterations drive cell growth not only through supporting biosynthesis, energy metabolism, and maintaining redox potential but also through initiating signaling mechanisms that are still poorly characterized. The evolutionary basis of these additional layers of growth control is also discussed.
A renewed interest in research in cellular metabolism has collaborated with advances in analytical technologies and computational methodologies to yield rapid conceptual breakthroughs in our modern understanding of metabolic regulation in cellular systems(McKnight, 2010). One area of rapid progress involves the regulation of mammalian cell growth and the defects in this regulation that can lead to proliferative diseases such as cancer(Deberardinis et al., 2008; Hsu and Sabatini, 2008; Levine and Puzio-Kuter, 2010; Luo et al., 2009; Vander Heiden et al., 2009). It has long been realized that cells undergoing rapid growth and division exhibit changes in metabolism(Warburg, 1956). However, accumulating evidence indicates that alterations in metabolism are necessary, and in some cases sufficient for cell growth. Moreover, the same signal transduction pathways that coordinate the activation of transcription factors and cell cycle progression control and are controlled by changes in cellular metabolism(Jones and Thompson, 2009; Levine and Puzio-Kuter, 2010; Shaw and Cantley, 2006; Vander Heiden et al., 2009). In this review, we will discuss aspects of metabolism in the context of cell growth, emphasizing recent advances in our understanding of glucose metabolism and how they affect redox potential, energy status, biosynthesis, and signal transduction in growing cells.
The Warburg Effect and cell growth
Alterations in cellular glucose metabolism are now recognized to constitute a common feature of the majority of tumor cells (Hanahan and Weinberg, 2011). In a series of papers describing the original experiments, Otto Warburg showed that tumor cells were metabolizing glucose to lactate and that this process, referred to as aerobic glycolysis, was occurring in the presence of oxygen(Warburg, 1925, 1956; Warburg et al., 1924; Weinhouse, 1976). In addition to the preferential utilization of fermentation was the observation that tumor cells also exhibited a substantially enhanced rate of glucose uptake. The quantification obtained from these experiments indicated that tumor cells could metabolize many orders of magnitude larger amounts of glucose than their differentiated, normal counterparts(Warburg et al., 1927). That is, in Warburg’s initial estimation, the amount of glucose being metabolized in a tumor per unit time is on the same order as the amount being metabolized through entire regions of the circulatory system comprising multiple organs. Although this form of metabolism was identified in solid tumors, its features also provide insights into understanding general mechanisms of cell growth.
Why cancer cells and growing cells utilize fermentative metabolism is complicated and a hotly contested topic of considerable debate. One reason for the controversy is that the origins and growth-promoting functions of the Warburg effect are pleiotropic and include multiple, evolutionarily conserved cell autonomous functions observed even in yeast (Brauer et al., 2008). These functions include the support of biosynthetic programs as well as the maintenance of redox and energy potentials. In addition, such a cell-autonomous metabolism functions to initiate signal transduction mechanisms that confer additional layers of growth-regulatory properties to cells. Each of these functions will be discussed below.
In addition to the biological functions of cell-autonomous metabolism, non-cell autonomous effects may result in growth-advantageous tumor-stroma interactions(Gatenby and Gillies, 2004). These non-cell autonomous effects include the ability of lactate, by acidification, to facilitate the disruption of tissue architecture and thus promote tumor cell invasion (Parks et al. 2011). Acidification of the tumor microenvironment may also promote immune evasion(Gatenby and Gillies, 2008). For example, cell migration has a pH dependence and in addition, some chemokines and cytokines may be preferentially degraded under acidic conditions. Nevertheless, despite the complex aspects of the benefits of tumor cell fermentation on non-cell autonomous cell growth, the consequences of enhanced glucose uptake and fermentation have many cell-autonomous growth advantages as well.
High glucose uptake and cell growth
The many orders of magnitude per cell difference in the rate of glucose uptake has been translated to the clinic and Positron Emission Tomography involving 2-deoxy-2(18F)-fluoro-glucose glucose is routinely used to monitor and classify tumors (Engelman et al., 2008; Strauss and Conti, 1991). This difference in glucose metabolism also has far-reaching consequences concerning the rearrangements of metabolic fluxes that occur downstream of glucose capture.
Many of the implications of the high glycolytic rates observed in growing cells can be observed from the chemical equation for glycolysis in which one molecule of glucose is converted into two molecules of pyruvate:
In addition to the generation of pyruvate, two molecules of ATP and two molecules of the reduced form of Nicotinimide Adenine Dinucleotide (NAD) are generated. Thus, for glycolysis to be maintained, a flux balance that consumes its products must be achieved. This mass conservation requires a supply of glucose and inorganic phosphate as well as sufficient consumption of both ATP and NADH.
Alterations in redox potential and cell growth
In terminally differentiated cells or other cell types undergoing low rates of glycolysis, regeneration of NAD+ from NADH resulting from the end product of glycolysis is achieved by redox-coupled mitochondrial shuttles (Lanoue and Williams.Jr, 1971). Such shuttles involve the movement of oxidizable material between subcellular compartments. These processes are accomplished by anti-porter based mechanisms by which the transport of one metabolite from one compartment to another requires the concomitant transport of an additional metabolite from the receiving compartment back into the exiting compartment. The most commonly appreciated shuttle is known as the malate-aspartate shuttle(Lanoue and Williams.Jr, 1971). Other shuttles such as the glycerol phosphate shuttle may also play a role and others may yet be discovered. Additional shuttles may couple redox potential to other organelles such as the peroxisome and lysosome.
In the malate-aspartate shuttle (Figure 1), cytosolic oxaloacetate (OAA) is first reduced to form malate (MAL) and in the process, NAD+ is generated from NADH, eliminating the cytosolic NADH/NAD+ imbalance that results from glycolysis. MAL enters the mitochondria through an antiporter mechanism coupled to mitochondrial alpha-ketoglutarate (αKG) export to the cytosol. Upon entering the mitochondria, MAL is oxidized and converted back to OAA, thereby generating NADH in the mitochondria. By a transamination reaction, OAA is then converted to aspartic acid (ASP) with the nitrogen donor glutamate converted back to αKG. ASP is then transported from the mitochondria to the cytosol in exchange for cytosolic GLU by another antiporter. OAA is then generated in the cytosol from the exported ASP by a transamination reaction that simultaneously converts cytosolic GLU to αKG. This reaction completes the cycle and the net result is that NADH is converted back to NAD+ in the cytosol and NAD+ is converted to NADH in the mitochondria with no net flux in any other metabolite. The NADH to NAD+ conversion in the cytosol satisfies the redox imbalance from glycolysis and the NADH generated in the mitochondria enters the electron transport system to ultimately generate more ATP.
Figure 1. Glucose metabolism during conditions of high and low glucose uptake.
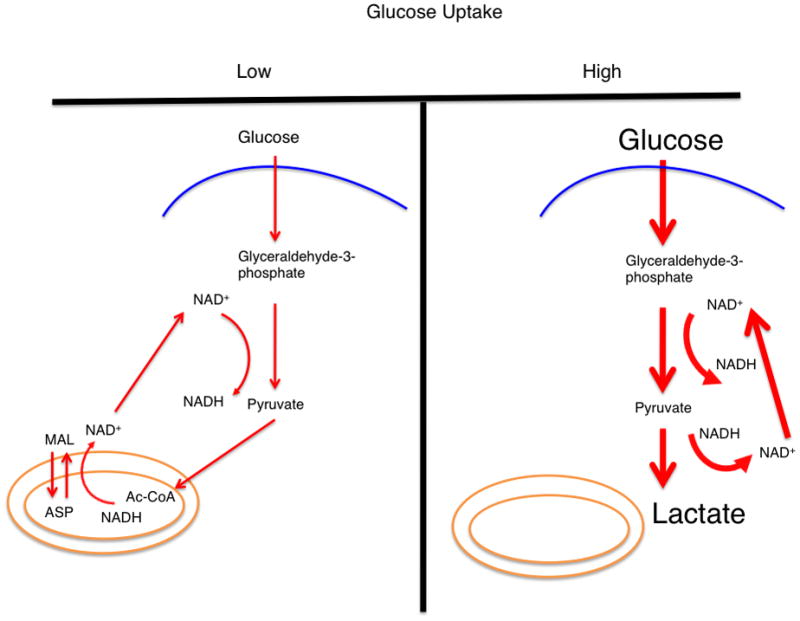
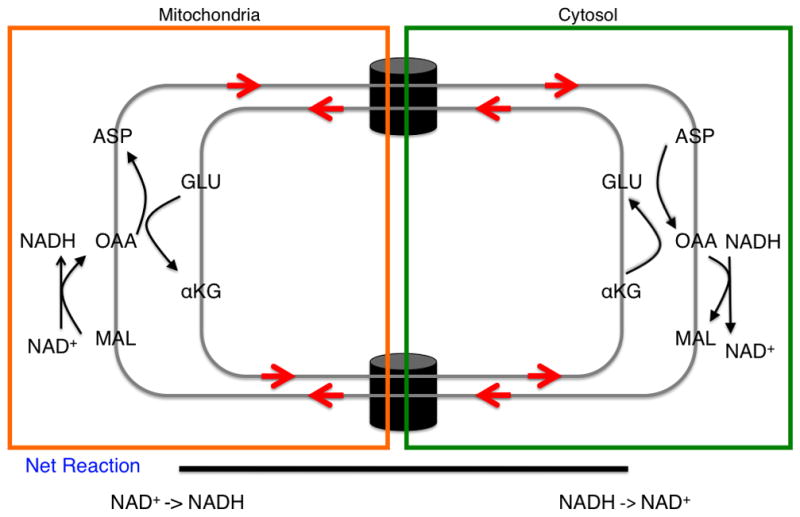
In conditions of low glucose uptake and flux through glycolysis (left), the glucose flux that passes through glycolysis and generates NADH is balanced by a mitochondrial shuttling mechanism. In conditions of high glucose uptake and flux (right), the capacity of the malate-aspartate shuttle to regenerate NAD+ can be saturated and lactate dehydrogenase activity is required to regenerate NAD+. A proposition is that this behavior can explain the Warburg effect. The thickness of the arrows represents relative magnitudes of flux at different points in the glycolytic pathway. (inset) The malate-aspartate shuttle: a series of oxidation-reduction reactions and anti-porter based trafficking of intermediate metabolites that begins with the oxidation of malate and end with reduction of aspartate in a series of six reactions. Directed arrows denote the conversion of substrate to product. The net result is a regeneration of NAD+ in the cytosol and production of NADH in the mitochondria. Abbreviations: MAL – malate, ASP – aspartate, αKG – alpha-ketoglutarate, OAA – oxaloacetate, and GLU – glutamate.
A similar concept is in place with the glycerol phosphate shuttle in which glycerol-3-phosphate is shuttled into the mitochondria and the net result of the processes involves the regeneration of NAD+ from NADH in the cytosol and production of reducing equivalents for oxidative phosphorylation this time in the form of FADH2. Flux through the glycerol phosphate shuttle is thought to occur primarily in brown fat but its generality remains unexplored and it is tempting to speculate that this activity could be upregulated in tumors(Kozak et al., 1991). Although these redox-coupled shuttling pathways were worked out many years ago, much remains to be learned about their regulation and how the fluxes across the compartments are controlled.
For small rates of glycolysis (Figures 1, 2), calculations predict that these shuttles are able to account for the production of NAD+ to balance the rate at which NADH is generated. This behavior is in contrast to the case of high glucose uptake rates when the rate of NADH generation increases by many orders of magnitude compared to the rate observed under normal physiological conditions and thus, typically exceeds the capacity of these relative slow mitochondrial shuttles (Figure 2). It is not likely that these shuttles can regenerate sufficient NAD+ when transport across intracellular membranes approaches its maximum velocity. Therefore, at the rates of glycolysis observed in rapidly dividing cells, these shuttling mechanisms are likely saturated. As a result, other mechanisms are required to regenerate cytosolic NAD+. The major mechanism that is in place to regenerate NAD+ involves the reduction of pyruvate to lactate to generate NAD+ from NADH via lactate dehydrogenase (LDH) (Figure 2). LDH is frequently overexpressed in growing cells and this effect allows for an increased maximum enzyme velocity to account for the enhanced requirement for NAD+ regeneration. As a result, the maintenance of high rates of glycolysis and the concomitant requirement of NAD+ regeneration may, to a large extent, induce the Warburg effect in growing cells (Figure 2).
Figure 2. A mathematical model characterizing the effects of lactate production on changes in glucose uptake and NAD+ regeneration rates.
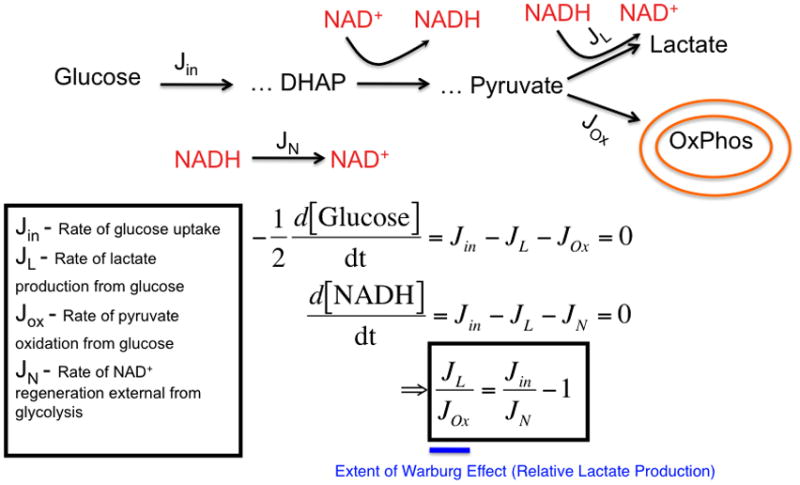
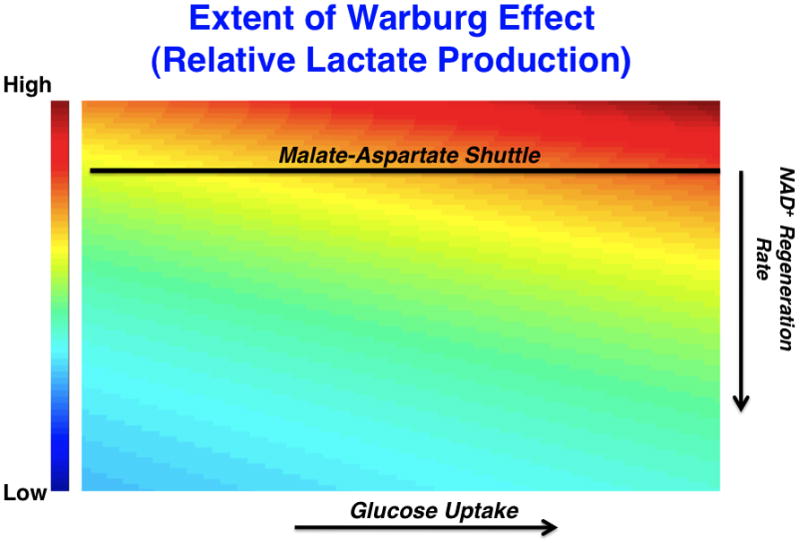
(left) A model that considers the stoichiometric balance of carbon and NAD+/NADH-generating fluxes in glycolysis and peripheral metabolism. In the model, glucose uptake and capture resulting in the generation of 2 glycolytic carbon units occurs at a rate, Jin. Glucose is metabolized through a series of steps eventually reaching an intermediate dihydroxyacetone phosphate (DHAP) at which point it is oxidized to produce NADH from NAD+. Eventually a glycolytic carbon unit reaches pyruvate where it can be reduced to produce lactate and generate NAD+ from NADH at a rate, JL. Alternatively, pyruvate can enter the mitochondria at a rate JOx. External to glycolysis, NAD+ is regenerated NADH by redox shuttles and other mechanisms. As a result, two differential equations can be written describing mass conservation of carbon and redox status and, at steady-state, can be solved for the relative rate of JL over JOx which is a measure of the extent of the Warburg effect or relative glucose-derived lactate production (in blue). (right) A representative plot of the ratio of the rate of lactate production to the rate of pyruvate entry into the mitochondria as a function of glucose uptake and NAD+ regeneration rate is shown. The extent of the Warburg effect is thus proportional to the rate of glucose uptake and inversely proportional to the rate at which NAD+ is regenerated by sources outside of Lactate dehydrogenase. The straight line represents an estimated rate of NAD+ from shuttles such as the malate-aspartate shuttle that is limited by mitochondrial transport rates. As glucose uptake increases (in the direction of the arrow on x-axis), the relative amount of glycolytic flux diverted to lactate production increases. As the rate at which NAD+ is regenerated increases (in the direction of the arrow on the y-axis), the relative amount of glycolytic flux diverted to lactate production decreases. These equations are obtained strictly from mass conservation of a model involving glucose entering glycolysis and leaving as either pyruvate or lactate. If the model allows for carbon units to exit glycolysis, then this would result in lowering the effective Jin in the equation.
The cytosolic ratio of NAD+ to NADH appears greater than one in measured cases and this is observed even in rapidly proliferating transformed cells that undergo high rates of glycolysis(Schwartz et al., 1974). The advantages of a positive oxidative redox potential (high NAD+/NADH ratio) are many(Lin and Guarente, 2003). These effects include signaling functions through for example NAD+-dependent ADP-ribosylation by PARP enzymes that are essential for DNA repair and survival, and NAD+-dependent deacetylases (sirtuins) that play critical roles in regulation of metabolism and transcription. Additionally, such a redox potential promotes biosynthesis and will be discussed later.
ATP requirements for cell growth and the existence of high energy phosphate-consuming futile cycles
Aerobic glycolysis is an inefficient means of generating ATP with nearly twenty-fold less ATP generated per unit of glucose than the amount that can be produced through complete oxidation of glucose to CO2 in the mitochondria. In addition, other materials can be utilized in the mitochondria for ATP generation. These catabolites include amino acids such as glutamine as well as long-chain fatty acids(Deberardinis et al., 2008; Hsu and Sabatini, 2008; Locasale and Cantley, 2010; Schafer et al., 2009). However, the rate of glucose uptake in growing cells is often many orders of magnitude larger than that observed in cells comprising non-growing, differentiated tissue(Warburg et al., 1927). Taking this into account the overall Warburg effect (enhanced glucose uptake accompanied by fermentation) could potentially generate more ATP.
Surprisingly, calculations suggest that the amount of ATP required for mammalian cell growth and division may be far less than that required for basal cellular maintenance(Kilburn et al., 1969). That is, in the time that it takes to undergo growth and cell division, the amount of ATP consumed in processes such as the maintenance of ion gradients via Na+/K+ and Ca2+-ATPases, steady-state protein and RNA synthesis, and intracellular trafficking exceeds the amount used for the de novo synthesis of biomaterials that constitute a new cell.
If it is counterintuitive that ATP is not the limiting reagent for cell growth and division, several illustrative examples are helpful. For example, one of the major energetic costs of biosynthesis is the creation of the peptide bonds that comprise polypeptide chain polymerization leading to protein synthesis. In fact each peptide bond carries an energetic cost of about 3 kcal/mol. However, studies have shown that protein turnover is typically rapid in cells. Proteome-wide studies of global protein turnover rates in exponentially growing mammalian cells have been measured using fluorescence-based photo bleaching techniques(Eden et al., 2011). It was found that the half-lives of most proteins range from minutes to a few hours. Only about 10% of proteins within the proteome had half-lives close to the time scales corresponding to cell cycle turnover. Thus the majority of ATP consumed during protein synthesis occurs to maintain steady-state levels of protein concentrations and not for the production of protein to constitute a new cell.
In the case of the Na+/K+ ATPase pump, one molecule of ATP is consumed for every three Na+ ions that are pumped out of cells to maintain approximately a 20 mM concentration in the cytosol. Measurements of the rate of ion flux have been observed to be around 100nM/minute per milligram of protein(Soltoff et al., 1988). Glucose uptake rates in cultured cells are on that order as well. For example transformed NIH 3T3 fibroblasts have a measured glucose uptake rate to also be around 100nM/minute per milligram of protein(Peterkofsky and Prather, 1982). Thus the amount of ATP consumed by the Na+/K+ ATPase pump is comparable to the amount generated by glycolysis.
Therefore, the generation of ATP by increased glycolysis might far exceed the additional ATP requirements for proliferation. Furthermore this amount of ATP generation could be inhibitory for growth due to many factors. One effect of excess ATP directly on the glycolytic pathway is the inhibition of glycolysis by allosteric inhibition of phosphofructokinase. Other overall effects involve the toxicities associated with a change in the equilibrium of ATP-dependent cellular reactions. In fact, much of the historical discussion of the Warburg Effect concerned how highly glycolytic cells compensate for excess ATP.
Efraim Racker noted this problem of an imbalance of ATP-generating and ATP-consuming processes in glycolytic cells and postulated that large ATP-consuming fluxes must be upregulated to account for the increase in ATP production(Racker, 1976a, b; Scholnick et al., 1973; Soltoff et al., 1988). For example, there was considerable effort spent on searching for a lesion in the Na+/K+ ATPase pump that would decouple ion transport from ATP hydrolysis. Recently, other mechanisms of upregulating ATP hydrolysis have been identified. It has been shown recently that increasing the rates of ATP consumption at the endoplasmic reticulum results in higher rates of aerobic glycolysis(Fang et al., 2010). This phenomenon appears to a large extent to occur in the absence of the PTEN tumor suppressor gene.
One clue for how cells might compensate for enhanced rates of ATP production came from the observation that growing cells express a single isoform of pyruvate kinase, PKM2(Christofk et al., 2008a; Eigenbrodt et al., 1992). This expression is in contrast to the form of pyruvate kinase observed in differentiated cells, PKM1. Pyruvate kinase catalyzes the final step of glycolysis converting phosphoenolpyruvate (PEP) to pyruvate and transferring a phosphate to ADP to regenerate ATP. Biochemically, PKM2 is intrinsically a less active enzyme with a specific activity of about one half that of PKM1. In addition PKM2 is subject to several forms of negative regulation including the inhibition of its activity via phosphotyrosine mediated binding(Christofk et al., 2008b). However, when cells were engineered to express exclusively the PKM1 or PKM2 isoforms, enhanced lactate production was observed in cells expressing PKM2. This observation raised many questions as it was unclear why a less active enzyme would generate more lactate.
A partial resolution to this paradox was the discovery of an alternate enzymatic activity for the pyruvate kinase substrate, PEP (Figure 3) (Locasale et al., 2010; Vander Heiden MG, 2010). In cells expressing PKM2, an enzymatic activity was discovered that converted PEP to pyruvate without generating ATP. This activity also mediated transfer of the phosphate group from PEP to the active site of phosphoglycerate mutase (PGAM). PGAM catalyzes an intermediate step of glycolysis involving a mutase reaction that transfers a phosphate from the three to the two position of the glycerate moiety. This enzymatic activity could be purified away from pyruvate kinase as well as enolase, the enzyme catalyzing the conversion of 2-phosphosphoglycerate to PEP. Quantitatively, it was shown that this mechanism could result in up to one-half of the total pyruvate generated from PEP implying that this mechanism could be an important source of lactate. While the exact enzyme mechanism involved in the phosphotransfer is unclear, it was shown that this reaction occurred at a concentration commensurate with the Michaelis constant of pyruvate kinase. Accompanying the phosphotransfer was the generation of pyruvate as well as the release of inorganic phosphate. The phosphotransfer to the catalytic site of the protein initiates a PEP-dependent feedback loop that may have some additional regulatory properties. The net reaction is a phosphate-burning futile cycle in which pyruvate is generated in the absence of ATP production from glycolysis. This decoupling of glycolysis from ATP synthesis may confer multiple growth-advantages to cells by limiting the availability of ATP. Additional biological functions of this futile cycle remain to be discovered and how this cycle alters glycolytic flux remains to be studied.
Figure 3. Alternative glycolytic pathway that decouples glycolysis from ATP synthesis.
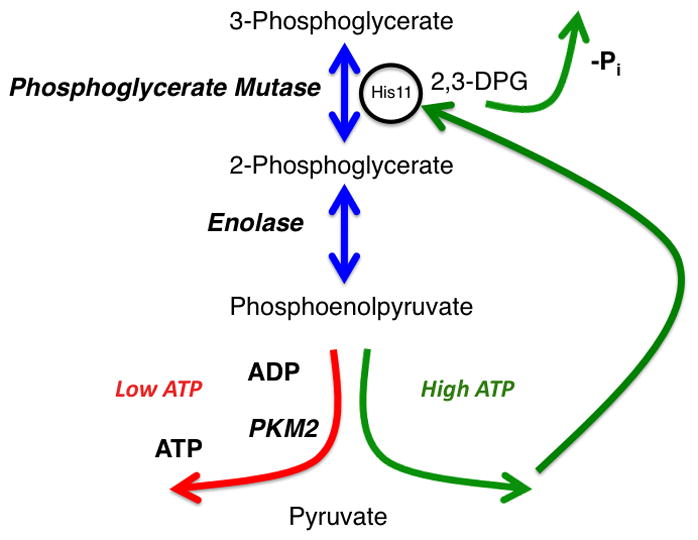
A glycolytic phosphate futile cycle that is initiated when 3-phosphoglycerate is converted to 2-phosphoglycerate by phosphoglycerate mutase. The reaction proceeds through a 2,3-diphosphoglycerate (2,3DPG) intermediate that has a tendency to dissociate from Phosphoglycerate Mutase and release an inorganic phosphate. 2-phosphoglycerate is converted to Phosphoenolpyruvate by Enolase. Two enzyme activities compete for phosphoenolpyruvate as a substrate. One activity (left) involves pyruvate kinase that transfers the phosphate group to ADP to generate pyruvate and ATP. The other involves an enzyme complex that donates the phosphate to Phosphoglycerate Mutase to prime the enzyme active site (right). In turn, this alternate glycolytic pathway results in net zero production of ATP during glycolysis.
Biosynthesis and cell growth
The biosynthetic requirements of growing cells involve de novo synthesis of nucleotide, membrane and protein components. These materials can originate from central carbon metabolism as well as salvage pathways located in peripheral metabolism. Enhanced flux into biosynthetic pathways is achieved in conditions of high glucose uptake. When cells exhibit multiple orders of magnitude of enhanced glucose uptake, fluxes of glucose into biosynthetic pathways are dramatically altered. Under these conditions, even if only 0.1–1% of the glucose that is captured enters biosynthetic pathways such as the pentose phosphate pathway, glycerolipid metabolism, or de novo amino acid biosynthesis, this increase can more than double the basal flux into these pathways compared to the flux in non-transformed cells.
Increases in the rates of NADH generation from glycolysis can drive reductive fluxes through for example, glycerol 3-phosphate dehydrogenase, which utilizes NADH to generate glycerol-3-phosphate from dihydroxy acetone phosphate (DHAP). Glycerol-3-phosphate is required for synthesis of all the glycerol-backbone lipids. Changes in DHAP and NADH concentrations can result in a transition of glycerol phosphate dehydrogenase operating in a kinetic regime in which catalysis is limited by the availability of its substrate up to a regime where the enzyme kinetics are saturated and the enzyme kinetics operate at maximum velocity.
In addition to the high rates of glucose flux that contribute to glycolytic intermediates that could be used to divert glycolytic flux out of glycolysis from any one of the ten glycolytic intermediates, enhanced glucose flux may also function to reorganize mitochondrial flux that contributes to biosynthesis. Within the mitochondria, the tricarboxylic-acid (TCA) based carbon sources that cycle in the mitochondria to contribute electrons to the electron transport chain are also reactive with enzymes that initiate specific biosynthetic programs(DeBerardinis et al., 2007). How enhanced rates of glucose uptake affect mitochondrial biosynthetic functions are unclear but many lines of evidence suggest an essential coupling.
For example, the redox potential in the mitochondria that sets the relative fluxes and steady-state concentrations of TCA cycle intermediates could be regulated by glycolysis and the malate-aspartate and glycerol-phosphate shuttles as well as by the rate of glucose-derived carbon entry into the mitochondria. In principle changes in the redox coupling could direct TCA cycle flux differentially into any of the biosynthetic routes beginning at the TCA cycle. One possibility would be a buildup of acetyl-coA that could be used for enhanced rates of de novo lipid biosynthesis required in growing cells. A change in the mitochondrial redox potential might allow for metabolites such as glutamine to enter the mitochondria and redirect this carbon source into nucleotide and lipid metabolism.
In addition to these biosynthetic functions, alterations in mitochondrial redox potential also affect the generation of reactive oxygen species (ROS)(Finkel and Holbrook, 2000). Changes in NADH and FADH2 levels affect the efficiency of electron transport through electron transport complexes. Leaky electron transfer is implicated in the generation of superoxides that produce hydrogen peroxide. The generation of free radicals has direct biochemical effects on growth signaling. For example, ROS may enhance signal transduction through the inactivation of phosphatases that have catalytic cysteine residues, such as PTEN or protein tyrosine phosphatases, thereby enhancing Phosphoinositide 3-Kinase and protein-Tyr kinase signaling (Salmeen and Barford, 2005). Metabolic enzymes such as Glyceraldehyde-3-phosphate dehydrogenase are also inhibited by ROS. Thus the generation of ROS can result in a multitude of biological consequences.
Metabolism and growth signaling
Cellular decision-making occurs through signal transduction. The signal transduction involved in mammalian cell growth has largely been studied by examining the propagation of signals through networks with information conveyed through modifications in the form of reversible phosphorylation at serine, threonine, tyrosine residues, and inositol lipids. Networks of protein phosphorylation events embedded in cascades with multiple layers of nonlinear regulation such as crosstalk and feedback loops have evolved to control cell growth(Hunter, 1995). These pathways include the Ras-MAPK pathway and the PI3K-mTOR pathways(Shaw and Cantley, 2006).
For signaling flux to propagate at any individual node, both sufficient enzyme activity and metabolic substrate availability are required. In the case of phosphorylation by protein kinases, the metabolic substrate is ATP. Typical concentrations of intercellular ATP are near 5 mM and this value is far above the Michaelis constant for most protein kinases. This implies that enzyme activity of protein kinases is not affected by the availability of its metabolic substrate since cells rarely, if ever see the concentration of ATP that is necessary to affect signaling. Instead, the regulation of signaling responses is generated through the complexity of the networks involving over 500 individual protein kinases and the different ways to activate them(Yaffe et al., 2001).
These networks involve numerous regulatory mechanisms such as the presence of cascades and feedback loops that confer diverse properties to signal transduction networks. For example, phosphorylation at a kinase activation loop by an upstream kinase in the network or binding of a kinase to a scaffold can alter the enzyme activity by varying its kinetic parameters by many orders of magnitude(Locasale et al., 2007). As a result these protein kinase networks can respond to diverse environmental perturbations including alterations in cellular osmotic pressure, salt concentrations, ATP availability, the presence of growth factors, as well as the availability of nutrients. In contrast to mammalian cells, primitive organisms including most prokaryotes lack these evolutionarily advanced protein kinase signaling networks(Parkinson, 1993). Instead these organisms integrate signals to make cellular decisions on the status of cellular metabolism(Kolb et al., 1993; Ptashne, 1988).
In spite of the regulatory power garnered by protein kinase networks, other reversible protein post-translation modifications are increasingly becoming appreciated as having important roles in cellular signal transduction. These modifications include protein acetylation, methylation, O-linked GlcNAC-ylation, ADP-ribosylation, and many others (Figure 4)(D’Amours et al., 1999; Hart, 1997; Johnson, 2004; Roth et al., 2001; Teperino et al., 2010). Many of these modifications are conserved down to the level of prokaryotes. Current estimates from advances in mass spectrometry-based proteomics suggest that many of these modifications may be as prevalent in mammalian systems as the more studied phosphorylation(Choudhary et al., 2009).
Figure 4. Regulation of growth signaling by protein phosphorylation and metabolism.

Modes of mammalian signal transduction. Examples of modifications, enzymes, substrates and the primary form of regulation are shown.
Abbreviations: OGT – O-linked N-acetylglucosamine transferase, GLCNAC – N-acetylglucosamine, PARP – Poly (ADP-ribose) polymerase, SAM – S-adenosylmethionine.
In contrast to reversible protein phosphorylation, each of these modifications is carried out by a relatively small number of enzymes. The number of distinct acetyltransferases, methyltransferases, GlcNAc transferases, ADP-ribosyltransferases, etc are much smaller than the total number of unique protein kinases. Also, in contrast to the case of protein kinases, where physiological concentrations of the substrate ATP far exceed the Michaelis constant, the enzymes that carry out these modifications typically have Michaelis constants for their metabolic substrates that are close to the physiological concentrations. These substrates include Acetyl-CoA, S-Adenosylmethionine, N-Acetylglucosamine, and ADP-ribose and their levels are dynamically regulated by alterations in cellular metabolism (Figure 4).
Changes in these concentrations can result from variations in metabolic flux occurring from countless mechanisms such as changes in nutrient uptake. These changes could affect their concentrations by several orders of magnitude and in turn affect activity of their corresponding enzymes by shifting the substrate concentration to levels well above or below the Michaelis-constant of the enzymes. Therefore, the diversity of signaling responses regulated through these modifications may have evolved from the ability of metabolic networks to adapt metabolism to achieve differing levels of substrate availability. These forms of signal transduction are more ancient than protein kinases and have evolved from more primitive versions of signal transduction observed in lower organisms.
Classic instances of metabolite-regulated signal transduction involve the control of transcription factor activity by small molecules. For example, degradation of hypoxia-inducible factor (HIF1/2) in response to prolyl hydroxylation (Ivan et al., 2001; Jaakkola et al., 2001). In this case, HIF is prolyl-hydroxlyated when sufficient levels of O2 and alpha-ketoglutarate are available. Also, the glucose-responsive transcription factor chREBP activates a program of genes in response to changes in glucose concentration(Yamashita et al., 2001). Finally, the lipid-responsive transcription factor SREBP regulates its activity in response to intracellular cholesterol levels(Brown and Goldstein, 1997). Such behavior is reminiscent of operons in bacteria that control the transcription of genes in response availability of specific nutrients.
Many other examples of metabolic signal transduction of this form in mammalian systems have been described. For example, changes in dietary folate intake have strong associations with changes in methylation status in humans(Christensen et al., 2010). It has also been shown that changes in glucose availability and Acetyl-CoA levels regulate epigenetic programs through direct effects on the modification of acetylation patterns on histones (Cai et al., 2011; Wellen et al., 2009). Another particularly striking example of how altered metabolic flux may affect cell growth through changes in signaling machinery was observed in the hexosamine biosynthetic pathway. In the study by Wellen and colleagues, the authors show that cells when growth-arrested upon deprivation of glucose can glucose, can regain the ability to grow when treated with glucosamine (Wellen et al., 2010). The recovery of cellular GlcNAc modifications that affect diverse cellular processes is implicated in this rescue of cell growth.
Additional layers of feedback control also occur in the context of metabolic regulation of signal transduction. For example, proteomics studies have shown that protein acetylation is overrepresented on metabolic enzymes compared to other proteins(Zhao et al., 2010). This observation creates possibilities that feedback control of NAD+-mediated metabolic fluxes that affect the activity of deacetylases such as the Sirtuin family(Blander and Guarente, 2004). However, the advantages of such feedback controls are unknown. Also, activation of Mammalian Target of Rapamycin (mTOR) is mediated by amino acid availability in addition to protein kinase signaling(Wullschleger et al., 2006). mTOR in turn stimulates a program of metabolic changes manifested in the enhanced transcriptional activity of metabolic genes(Duvel et al., 2010). As a result, enhanced levels of lipids and amino acids could create positive feedback loops that stimulate mTOR signaling and its growth-promoting functions. As has been appreciated for many years, changes in lipid metabolism and the ability of lipids to regulate the generation of secondary messengers in signaling pathways also have essential roles in regulating cell growth(Kim et al., 2011; Nomura et al., 2010; Riese et al., 2011).
Summary and future directions
The ability of cells to sense changes in metabolism to control cell growth is found in the most primitive of organisms. A selection for this behavior may have been one of the first acquired processes in the evolution of cell autonomy. Additional layers of growth control, such as protein kinase networks evolved later and expanded dramatically in the development of multicellular organisms. These networks evolved to institute forms of cellular social control to, for example, sense the presence of growth factors secreted by other cells(Raff, 1992). In the development of cancer, cells universally acquire mutations in these signaling networks that allow them to proliferate outside the social context of normal tissue development. However, the ability of these cancer cells to regulate growth by metabolism and nutrient availability remains. If metabolism then is the primary form of growth control, a fundamental question that remains then is to what extent are these cells now more sensitive to perturbations in metabolism and nutrient availability.
Despite the many recent advances in understanding the metabolism of growing cells in mammalian organisms, much remains to be understood. How the Warburg effect and changes in redox potential might contribute to enhanced or specific anabolic metabolism has not been fully characterized and how enhanced glycolysis may contribute to metabolically-regulated signal transduction is still in its infancy. Also how these behaviors depend on the genetic status within cells is not understood. Nevertheless these areas of study provide a wealth of unanswered questions and addressing them may open up new therapeutic strategies in the confrontation of proliferative diseases.
Highlights.
High rates of glucose uptake alter cytosolic and mitochondrial redox potential
ATP availability is not limiting for cell growth
An alternative glycolytic pathway is initiated in growing cells
Growth control by metabolically-regulated signal transduction is beginning to be explored
Acknowledgments
We thank Matt Vander Heiden, Shyh-Chang Ng, Costas Lyssiotis and the anonymous reviewers for their helpful comments pertaining to the manuscript. This work was supported by grants from the NIH, NCI and the American Cancer Society. LCC is a founder of and JWL is a consultant for Agios pharmaceuticals, a company developing cancer therapies through targeting metabolism.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blander G, Guarente L. The Sir2 family of protein deacetylases. Annual Review of Biochemistry. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, Botstein D. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Cai L, Sutter B, Li B, Tu B. Acetyl-CoA Induces Cell Growth and Proliferation by Promoting the Acetylation of Histones at Growth Genes. Molecular Cell. 2011;42:426–237. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Christensen BC, Kelsey KT, Zheng SC, Houseman EA, Marsit CJ, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Kushi LH, Kwan ML, Wiencke JK. Breast Cancer DNA Methylation Profiles Are Associated with Tumor Size and Alcohol and Folate Intake. Plos Genetics. 2010;6:10. doi: 10.1371/journal.pgen.1001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008a;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008b;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- Deberardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma QC, Gorski R, Cleaver S, Heiden MGV, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Molecular Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Geva-Zatorsky N, Issaeva I, Cohen A, Dekel E, Danon T, Cohen L, Mayo A, Alon U. Proteome Half-Life Dynamics in Living Human Cells. Science. 2011;331:764–768. doi: 10.1126/science.1199784. [DOI] [PubMed] [Google Scholar]
- Eigenbrodt E, Reinacher M, Scheefers-Borchel U, Scheefers H, Friis R. Double role for pyruvate kinase type M2 in the expansion of phosphometabolite pools found in tumor cells. Crit Rev Oncog. 1992;3:91–115. [PubMed] [Google Scholar]
- Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, Garcia-Echeverria C, Weissleder R, Mahmood U, Cantley LC, Wong KK. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Shen ZR, Huang S, Zhao LP, Chen S, Mak TW, Wang XD. The ER UDPase ENTPD5 Promotes Protein N-Glycosylation, the Warburg Effect, and Proliferation in the PTEN Pathway. Cell. 2010;143:711–724. doi: 10.1016/j.cell.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Hypoxia and metabolism - Opinion - A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hart GW. Dynamic O-linked glycosylation of nuclear and cytoskeletal proteins. Annual Review of Biochemistry. 1997;66:315–335. doi: 10.1146/annurev.biochem.66.1.315. [DOI] [PubMed] [Google Scholar]
- Hsu PP, Sabatini DM. Cancer cell metabolism: warburg and beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- Hunter T. PROTEIN-KINASES AND PHOSPHATASES - THE YIN AND YANG OF PROTEIN-PHOSPHORYLATION AND SIGNALING. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang HF, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG. HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O-2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O-2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Johnson ES. Protein modification by SUMO. Annual Review of Biochemistry. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–548. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilburn DG, Lilly MD, Webb FC. ENERGETICS OF MAMMALIAN CELL GROWTH. Journal of Cell Science. 1969;4:645. doi: 10.1242/jcs.4.3.645. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim SF, Maag D, Maxwel MJ, Resnick AC, Juluri KR, Chakraborty A, Koldobskiy MA, Cha SH, Barrow R, Snowman AM, Snyder SH. Amino Acid Signaling to mTOR Mediated by Inositol Polyphosphate Multikinase. Cell Metab. 2011;13:215–221. doi: 10.1016/j.cmet.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb A, Busby S, Buc H, Garges S, Adhya S. TRANSCRIPTIONAL REGULATION BY CAMP AND ITS RECEPTOR PROTEIN. Annu Rev Biochem. 1993;62:749–795. doi: 10.1146/annurev.bi.62.070193.003533. [DOI] [PubMed] [Google Scholar]
- Kozak LP, Kozak UC, Clarke GT. ABNORMAL BROWN AND WHITE FAT DEVELOPMENT IN TRANSGENIC MICE OVEREXPRESSING GLYCEROL 3-PHOSPHATE DEHYDROGENASE. Genes Dev. 1991;5:2256–2264. doi: 10.1101/gad.5.12a.2256. [DOI] [PubMed] [Google Scholar]
- Lanoue KF, Williams INTERRELATIONSHIPS BETWEEN MALATE-ASPARTATE SHUTTLE AND CITRIC ACID CYCLE IN RAT HEART MITOCHONDRIA. Metabolism-Clinical and Experimental. 1971;20:119. doi: 10.1016/0026-0495(71)90087-4. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Puzio-Kuter AM. The Control of the Metabolic Switch in Cancers by Oncogenes and Tumor Suppressor Genes. Science. 2010;330:1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- Lin SJ, Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol. 2003;15:241–246. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- Locasale JW, Cantley LC. Altered metabolism in cancer. BMC Biol. 2010;8:3. doi: 10.1186/1741-7007-8-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Heiden MGV, Cantley LC. Rewiring of glycolysis in cancer cell metabolism. Cell Cycle. 2010;9:4253–4253. doi: 10.4161/cc.9.21.13925. [DOI] [PubMed] [Google Scholar]
- Locasale JW, Shaw AS, Chakraborty AK. Scaffold proteins confer diverse regulatory properties to protein kinase cascades. Proc Natl Acad Sci U S A. 2007;104:13307–13312. doi: 10.1073/pnas.0706311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight SL. On Getting There from Here. Science. 2010;330:1338–1339. doi: 10.1126/science.1199908. [DOI] [PubMed] [Google Scholar]
- Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol Lipase Regulates a Fatty Acid Network that Promotes Cancer Pathogenesis. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson JS. SIGNAL-TRANSDUCTION SCHEMES OF BACTERIA. Cell. 1993;73:857–871. doi: 10.1016/0092-8674(93)90267-t. [DOI] [PubMed] [Google Scholar]
- Parks SK, Chiche J, Pouyssegur J. pH Control Mechanisms of Tumor Survival and Growth. J Cell Physiol. 226:299–308. doi: 10.1002/jcp.22400. [DOI] [PubMed] [Google Scholar]
- Peterkofsky B, Prather W. CORRELATION BETWEEN THE RATES OF AEROBIC GLYCOLYSIS AND GLUCOSE-TRANSPORT, UNRELATED TO NEOPLASTIC TRANSFORMATION, IN A SERIES OF BALB 3T3-DERIVED CELL-LINES. Cancer Res. 1982;42:1809–1816. [PubMed] [Google Scholar]
- Ptashne M. HOW EUKARYOTIC TRANSCRIPTIONAL ACTIVATORS WORK. Nature. 1988;335:683–689. doi: 10.1038/335683a0. [DOI] [PubMed] [Google Scholar]
- Racker E. Why do tumor cells have a high aerobic glycolysis? J Cell Physiol. 1976a;89:697–700. doi: 10.1002/jcp.1040890429. [DOI] [PubMed] [Google Scholar]
- Racker E. WHY DO TUMOR-CELLS HAVE A HIGH AEROBIC GLYCOLYSIS. J Cell Physiol. 1976b;89:697–700. doi: 10.1002/jcp.1040890429. [DOI] [PubMed] [Google Scholar]
- Raff MC. Social controls on cell-survial and cell-death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- Riese MJ, Grewal J, Das J, Zou T, Patil V, Chakraborty AK, Koretzky GA. Decreased Diacylglycerol Metabolism Enhances ERK Activation and Augments CD8(+) T Cell Functional Responses. J Biol Chem. 2011;286:5254–5265. doi: 10.1074/jbc.M110.171884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annual Review of Biochemistry. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Salmeen A, Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxidants & Redox Signaling. 2005;7:560–577. doi: 10.1089/ars.2005.7.560. [DOI] [PubMed] [Google Scholar]
- Schafer ZT, Grassian AR, Song LL, Jiang ZY, Gerhart-Hines Z, Irie HY, Gao SZ, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–U118. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholnick P, Lang D, Racker E. Regulatory mechanisms in carbohydrate metabolism. IX. Stimulation of aerobic glycolysis by energy-linked ion transport and inhibition by dextran sulfate. J Biol Chem. 1973;248:5175. [PubMed] [Google Scholar]
- Schwartz JP, Passonne Jv, Johnson GS, Pastan I. EFFECT OF GROWTH-CONDITIONS ON NAD+ AND NADH CONCENTRATIONS AND NAD+-NADH RATIO IN NORMAL AND TRANSFORMED FIBROBLASTS. J Biol Chem. 1974;249:4138–4143. [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Soltoff S, McMillian M, Cantley L, Talamo B. EXTRACELLULAR ATP ACTIVATES MULTIPLE ION-TRANSPORT SYSTEMS IN THE RAT PAROTID CELL BY A NOVEL MECHANISM. Kidney International. 1988;33:173–173. [Google Scholar]
- Strauss LG, Conti PS. THE APPLICATIONS OF PET IN CLINICAL ONCOLOGY. Journal of Nuclear Medicine. 1991;32:623–648. [PubMed] [Google Scholar]
- Teperino R, Schoonjans K, Auwerx J. Histone Methyl Transferases and Demethylases; Can They Link Metabolism and Transcription? Cell Metab. 2010;12:321–327. doi: 10.1016/j.cmet.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MGLJ, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, Cantley LC. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. Uber den Stoffwechsel der Carcinomzelle. Klin Wochenschr. 1925;4:534–536. [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Warburg O, Posener K, Negelein E. Ueber den Stoffwechsel der Tumoren Biochemische Zeitschrift. 1924;152:319–344. [Google Scholar]
- Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. Journal of General Physiology. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhouse S. The Warburg hypothesis fifty years later. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol. 1976;87:115–126. doi: 10.1007/BF00284370. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Lu C, Mancuso A, Lemons JMS, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, Thompson CB. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010;24:2784–2799. doi: 10.1101/gad.1985910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Leparc GG, Lai J, Obata T, Volinia S, Cantley LC. A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nature Biotechnology. 2001;19:348–353. doi: 10.1038/86737. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W, Arnot D, Uyeda K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci U S A. 2001;98:9116–9121. doi: 10.1073/pnas.161284298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao SM, Xu W, Jiang WQ, Yu W, Lin Y, Zhang TF, Yao J, Zhou L, Zeng YX, Li H, Li YX, Shi J, An WL, Hancock SM, He FC, Qin LX, Chin J, Yang PY, Chen X, Lei QY, Xiong Y, Guan KL. Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]