

Abstract
Morbidity and mortality associated with increased white fat accumulation in visceral fat depots has focused attention on the pathways regulating the development of this tissue during embryogenesis, in adulthood, and while under the influence of obesogenic diets. Adipocytes undergo clonal expansion, differentiation (adipogenesis) and maturation through a complex network of transcriptional factors, most of which are expressed at similar levels in visceral and subcutaneous fat. Rigorous research attempts to unfold the pathways regulating expression and activity of adipogenic transcription factors that act in a fat-depot-specific manner. Peroxisome proliferator-activated receptor-γ (PPARγ) is the master regulator of adipogenesis, and is expressed at higher levels in subcutaneous than in visceral depots. PPARγ expression in adipogenesis is mediated by CCAAT/enhancer binding proteins (C/EBPs) and several transcription factors acting in conjunction with C/EBPs, although alternative pathways through zinc-finger protein-423 (ZFP423) transcription factor are sufficient to induce PPARγ expression and adipogenesis. Vitamin A and its metabolites, retinaldehyde and retinoic acid, are transcriptionally-active molecules that are generated in adipose tissue by the aldehyde dehydrogenase-1 family of enzymes (Aldh1). In this review, we discuss the role of Aldh1 enzymes in the generation of retinoic acid during adipogenesis, in the regulation of the transcriptional network and PPARγ in a fat-depot-specific manner, and suggest the important contribution of this autocrine pathway in the development of visceral obesity.
1) Health risks associated with fat depots
Recent studies show that obesity rates in the United States have reached epidemic proportions along with a dramatic rise in morbidity and mortality [1, 2]. A number of studies attribute such mortalities to central/visceral obesity, characterized by increased fat mass in the abdomen [3, 4]. General obesity and abdominal distribution of fat seem to have two separate entities with different clinical consequences [5, 6]. A population-based study of 1432 Swedish women showed a significant association between waist-hip ratio (WHR) and metabolic risk factors independent of BMI, suggesting the association between visceral fat and metabolic health risks independent of overall obesity [6]. This is one of several epidemiological, pathophysiological and clinical studies that have implicated visceral fat as a major contributor to an adverse metabolic profile (i.e. elevated serum cholesterol, triglycerides, fasting glucose levels) in humans [7]. Moreover, visceral fat accumulation showed a positive correlation with non insulin dependent diabetes mellitus (NIDDM) in the Nurses' Health Study enrolling 43,581 healthy US women followed from 1986 to 1994 [8]. INTERHEART study, involving 30,000 subjects from 52 countries worldwide showed that the proportion of myocardial infarctions attributable directly to visceral obesity was 60% in regions such as Western Europe, North America, Australia and New Zealand and 43.3% overall [9]. These observations serve as a few among many examples that closely associate visceral obesity with the prevalence of insulin resistance, type 2 diabetes, cardiovascular diseases, and even some cancers [3, 7, 8].
In keeping with the notion that visceral fat exerts harmful metabolic effects, liposuction, a common procedure of removing subcutaneous fat in obese patients did not improve metabolic effects [10], whereas omentectomy, i.e. removal of visceral fat, improved glucose metabolism in humans [11] and rodents [12, 13]. Removal of subcutaneous fat in the same Zucker rat model of obesity failed to impart beneficial changes in weight gain, serum glucose, or insulin levels [14]. Thus, both human and mice studies characterize visceral fat as a unique, pathogenic fat depot, associated with an adverse metabolic profile, while subcutaneous fat, constituting peripheral obesity, seems to improve insulin sensitivity and lower the risk of developing metabolic syndrome. Such differences, however, prove not to be due to their different anatomical location. Transplantation of subcutaneous fat into visceral or subcutaneous regions of mice exhibited improved insulin sensitivity, decreased body weight and total fat mass. Visceral fat transplantation into subcutaneous fat had no effects, further emphasizing specific properties of fat from different depots [15].
White adipose depots, apart from being energy storage depots, also function as major endocrine organs secreting many cytokines and adipokines, which potentially affect the function of other tissues [16]. Unlike subcutaneous fat depots, visceral fat formation is associated with elevated production of pro-inflammatory cytokines, i.e. TNF-α, IL-6 and leptin [17-19], which provoke a local and systemic state of chronic low-grade inflammation [20] and induce insulin resistance [21]. Adiponectin, an adipokine specifically secreted at higher levels in subcutaneous than in visceral adipose tissue, is an established marker of insulin sensitivity [22]. Ob/ob mice overexpressing adiponectin showed a massive increase in subcutaneous fat; however, this subcutaneous obesity was associated with improved insulin sensitivity, increased lipid clearance and improved β-cell function [23]. Considering these facts, one could speculate that differences in the degree of adipokine secretion, such as adiponectin, by subcutaneous and visceral fat depots could be causal to their disparate metabolic effects. However, such a conclusion may be an oversimplification of a far more complex depot-specific transcriptome responsible for metabolic responses of subcutaneous and visceral adipose tissue. The development of therapies directed towards reducing depot-specific obesity depends on understanding the congenital and adaptive transcriptional networks and signaling pathways in adipocytes from different depots. Vitamin A derivatives (retinoids) are known resourceful compounds with a broad range of pharmacological action in fat tissues [24] and adipocytes, whereas the autocrine role of retinoids in adipogenesis and fat formation is just beginning to be explored [25]. In this review we will primarily discuss mechanisms for endogenous RA production in adipocytes and its implication for transcription regulation in adipogenesis and regional fat formation.
2) Transcriptional networks defining fat depots
2.1 Developmental genes
Recent efforts to identify differences in human and rodent adipocytes isolated from different white fat depots using gene array profiling yield explicit evidence of participation of developmental genes in the formation of subcutaneous and visceral tissues [26-28]. These genes include numerous transcription factors participating in organogenesis, including members of homeobox (HOX) and nuclear receptor (e.g. COUP-TF1/ NR2F1) families of transcription factors [27], reviewed in [29]. These findings suggest that adipocytes from different white fat depots originate from different precursors and maintain their developmental transcriptional profile. Consistent with the hypothesis that different fat depots have inherited characteristics, multiple propagations and immortalization of human preadipocytes isolated from omental and subcutaneous fat depots of different donors did not alter their depot-specific expression of developmental genes that comprised approximately18% of the adipocyte genes [28] (Figure.1, Group 1). These studies also reveal the heterogeneity of visceral depots in humans, which consist of omental and mesenchymal fat. Mesenchymal fat shares characteristics with subcutaneous fat that is distinct from omental fat [28, 30]. The role of HOX genes in adipocyte biology and the mechanisms of their regulation in response to nutrients deserve further study, especially in light of the discovery by Kahn and colleagues [27] that expression of HOXA5 in subcutaneous and visceral depots correlates with obesity and fat distribution in humans.
Figure 1. Schematic presentation of selected genes that are expressed in a fat-depot-specific manner.

Red box, small font – repressed genes; Green box, large font – induced genes.
Group 1. Developmental genes. Specific developmental genes differ in their expression pattern in subcutaneous and visceral fat depots. Among genes involved in organogenesis, homeobox genes HoxA5, HoxA9, homeobox transcription factor Engrailed 1 (En1), and nuclear receptor COUP-TF1/NR2F1 are expressed at higher levels in the subcutaneous fat depot, whereas in visceral fat, these genes are downregulated. Vice-versa, HoxC9 and homeobox transcription factor Engrailed 1 (En1) have nominal expression in subcutaneous fat, while visceral fat shows their higher expression [27,28].
Group 2. Fundamental adipogenic transcription factors. Among genes regulating adipogenesis, subcutaneous fat expresses higher levels of PPARγ, C/EBPα, and CREBP1 compared to visceral fat [29,30]
Group 3. Proteins involved in vitamin A metabolism. Retinol binding protein RBP4 is preferentially expressed in the visceral fat depot. The expression of the Aldh1 family of enzymes regulating endogenous RA production is also dissimilar in the two fat depots but it differs between humans and mice (indicated by H and M respectively) [65,25].
2.2. PPARγ
In all depots, adipose tissue formation during the development depends on peroxisome proliferator-activated receptor-γ (PPARγ), a master regulator of transcription in adipogenesis [31, 32]. PPARγ is expressed as two isoforms, PPARγ1 and PPARγ2, produced by alternative promoter usage (functions and regulation reviewed in [33]). In the course of development, both human and rodent adipocytes express more PPARγ in subcutaneous than in visceral fat depots [34] and maintain this expression pattern even in cultured adipocytes isolated from these fat depots for several generations, which may highlight their origin from different lineages [28, 30]. Correspondently, PPARγ activity is increased in subcutaneous compared to visceral fat tissues [35], which may account for the high expression of PPARγ target genes that define properties of subcutaneous fat. PPARγ-mediated activation of adiponectin has already been recognized as a major mechanism for insulin sensitivity associated with subcutaneous fat [36, 37]. Moreover, increased adiponectin production by PPARγ is the basis of the insulin sensitizing effects of PPARγ ligands, thiozolidinediones [37]. Differences in PPARγ expression can contribute causally to the formation of specific fat depots. Impaired transcriptional PPARγ2 performance in patients with PPARγ2 Pro12Ala (P12A) polymorphism [38] leads to overweight, prevailing loss of subcutaneous fat, insulin resistance, and other metabolic dysfunctions in children and lean adults [39-42]. In contrast, treatment of rodents and humans with PPARγ agonists increases formation of subcutaneous depots [43], further underscoring the regulatory role of this transcription factor in regional fat formation.
It is currently unknown whether congenital and acquired depot-specific differences in PPARγ expression are regulated by the same transcriptional mechanisms. It is clear; however, that regional obesity in abdominal regions induced by overfeeding in humans is also associated with PPARγ expression [44]. Hormone-dependent fat distribution also proceeds through PPARγ regulation, although PPARγ involvement could be somewhat paradoxical in the context of autocrine mechanisms for glucocorticoid production. Adipose-specific transgenic expression of 11β-hydroxysteroid dehydrogenase, inactivating the hormone corticosterone, is associated with resistance to high-fat diet-induced obesity, but accompanied by increased expression of PPARγ in mouse fat [45]. The factors defining depot-specific PPARγ expression remain poorly understood, whereas numerous potential transcriptional inducers of PPARγ expression have been reported and reviewed in chapters 2.3-2.5 (Figure 2). The pathways downstream of PPARγ are beyond the scope of this review and will not be discussed.
Figure 2. Transcriptional network regulating PPARγ expression.

Preadipocyte differentiation requires the coordinated action of multiple transcription factors induced by hormones or their synthetic analogs. Pathways that are dispensable in vivo are represented by dashed lines. IBMX activates the cAMP signaling cascade resulting in induction of C/EBPβ expression in vitro. C/EBPδ expression is induced by dexamethasone in vitro and mediated through glucocorticoid receptor (GR). C/EBPβ forms homo/heterodimers with C/EBPδ and binds to the C/EBP regulatory region of C/EBPα and PPARγ, activating both transcription factors. C/EBPα, in a positive feedback loop, can further induce PPARγ. Furthermore, C/EBPβ activates the KLF5 promoter, which in turn activates the PPARγ promoter, in conjunction with C/EBPδ. SREBP1c, a transcription factor acting downstream of C/EBPβ, activates PPARγ upon insulin stimulation, possibly through endogenous PPARγ ligand production. Some evidence suggests that fetal bovine serum (FBS) stimulates the STAT5/GR pathway, which induces PPARγ expression, although its role in vivo is yet to be explored. Early B cell factor (EBF1) regulates C/EBPβ expression. Recently, ZFP423 was identified as a potent inducer of PPARγ2 and adipogenesis.
2.3. Transcriptional inducers of PPARγ expression
CCAAT/enhancer binding proteins (C/EBP) were the first identified family of transcription factors contributing to PPARγ expression and activation [46]. The C/EBP family belongs to the large family of basic leucine zipper transcription factors that can both homodimerize and heterodimerize with each other and bind to the same C/EBP consensus sequences (reviewed in [33]). The expression of C/EBPβ in cultured preadipocytes results from activation of the cAMP signaling cascade by β-adrenergic signaling in vivo and 3-isobutyl-1-methylxanthine (IBMX) in vitro. C/EBPδ expression in these cells is mediated by the glucocorticoid pathway, which is stimulated through dexamethasone in vitro. C/EBPβ homodimers or C/EBPβ-C/EBPδ heterodimers bind to the C/EBP regulatory elements in PPARγ and C/EBPα promoters, activating these transcription factors [46-48]. In a positive feedback regulatory loop, C/EBPα can further induce PPARγ expression. Global gene expression studies revealed that both PPARγ and C/EBPα occupy similar regulatory sites of proteins required for terminal adipocyte differentiation [49]. PPARγ, however, plays the chief role and can rescue adipogenesis in C/EBPα−/− adipocytes [50]. Similar to PPARγ, C/EBPα is also preferentially expressed in subcutaneous compared to visceral fat [30], where it is likely to contribute to the increase in PPARγ activation in this tissue (Figure 1, Group 2). The complex relationship between C/EBPβ and C/EBPδ on PPARγ induction was shown in mouse models lacking C/EBPβ and C/EBPδ expression. Whereas isolated mouse embryonic fibroblasts (MEF) obtained from these mice were not capable of PPARγ and C/EBPα expression, poorly developed adipose tissues from these mice expressed both PPARγ and C/EBPα [51]. This observation argues for the presence of an alternative pathway(s) that induces PPARγ in different white adipose depots (reviewed in 2.5).
2.4. Transcription factors acting in conjunction with C/EBP for PPARγ induction
The majority of transcription factors involved directly in the up-regulation of PPARγ expression and activity required interaction with C/EBPs (Figure 2). The comprehensive work of Nagai and colleagues [52], demonstrates that C/EBP participation in the regulation of PPARγ entails another transcription factor, Krueppel-like factor 5 (KLF5). C/EBPβ activates the KLF5 promoter, which, in turn, activates the PPARγ promoter in concert with C/EBPβ or C/EBPδ [52]. Insufficient KLF5 function in KLF5+/- neonate mice results in 50% less PPARγ and C/EBPα in these mice as compared to WT mice, and deficient fat formation, although these differences were not seen in adult mice. Nonetheless, KLF5 expression was not sufficient to rescue PPARγ expression and adipogenesis in NIH-3T3 cells. Insulin stimulation of preadipocytes increases expression of sterol regulatory element binding protein 1 (SREBP1c) [53], another transcription factor that acts downstream of C/EBPβ and facilitates adipogenesis, likely through the production of endogenous PPARγ ligands.
Cell-cycle regulating proteins, including retinoblastoma protein (pRb) have also been proposed to cooperate with C/EBPα for PPARγ2 induction and with transcription factor E2F4 for PPARγ activation [54, 55]. In vivo, however, the marked loss of white fat in adult mice with pRb deficiency in adipose tissue was not associated with alteration of PPARγ expression [56]. A transcription factor early B-cell factor 1 (EBF1) appears to be sufficient to rescue adipogenesis in NIH-3T3 cells after its ectopic expression [57]. It is currently unknown whether EBF1 regulates expression of PPARγ through C/EBPβ, although it appears that EBF1 deficiency in vivo leads to a more profound reduction in C/EBPβ than PPARγ expression [58]. Intriguingly, EBF1 deficiency leads to preferential reduction of visceral fat in this model [58], suggesting that EBF1 participates in the transcriptional regulation of factors defining fat- depot-specific differences (Figure 2).
2.5. Alternative inducers of PPARγ expression
STAT5 and STAT5A/glucocorticoid receptor (GR) complexes have been proposed to induce PPARγ expression and adipogenesis in NIH-3T3 cells [59], but the role of this pathway in fat formation in vivo has not been elucidated yet. Emerging evidence suggests that many growth factors and hormones in fetal bovine serum act through STAT-mediated pathways in adipogenesis [60, 61] (Figure 2). Recent work of Noy and colleagues identifies the RBP-retinol/STRA6/JAK2/STAT5 signaling cascade as an inducer of STAT target genes, such as SOCS3 and PPARγ [62].
The breakthrough in the understanding of PPARγ expression mechanisms came from recent studies by Spiegelman and associates [63], who convincingly demonstrate that ectopic expression of transcription factor ZFP423 (ZNF423 is a human analog) is sufficient to rescue PPARγ2 expression and adipogenesis in NIH-3T3 cells (Figure 2). This pathway appears to bypass C/EBPs, KLFs, and other known PPARγ inducers, in its activation of PPARγ2. Surprisingly, PPARγ1 expression is not influenced in the presence of ZFP423. Although a detailed mechanism for PPARγ2 regulation by ZFP423 remains to be elucidated, it is clear that bone morphogenetic protein activates PPARγ2 in adipogenesis through ZFP423/SMAD interaction. The estimation of ZFP423's contribution to PPARγ regulation in vivo is problematic given the embryonic lethality of ZFP423−/− mice, but the initial observations showed a fewer number of adipocytes in the ZFP423−/− embryo [63]. Thus far, ZFP423 appears to be a powerful alternative pathway for PPARγ regulation. The potent role of ZFP423 in the regulation of PPARγ raises questions about mechanisms controlling the expression of this transcription factor and the contribution of this pathway to PPARγ induction in fat depots. The emerging autocrine mechanisms for ZFP423 regulation by vitamin A are discussed next (Chapter 3.3).
3) Evidence of vitamin A metabolism participation in depot-specific fat formation
3.1 Fat-depot-specific aspects of vitamin A metabolism
Persistent interests in the of role vitamin A in adipogenesis stem from the abundant presence of vitamin A in fat and the numerous studies documenting the significant role of vitamin A metabolism in fat tissue (comprehensively reviewed in this issue). Although liver is a primary source of vitamin A in mammals, several studies have shown that adipose tissue plays an active role in the metabolism and homeostasis of vitamin A by taking up circulatory chylomicron-bound (by lipoprotein lipase) and retinol binding protein 4 (RBP4)-bound (by STRA6) retinol [64, 65]. Interestingly, comparison of RBP4 expression in 66 lean and 130 obese patients revealed higher expression of RBP4 in visceral than in subcutaneous fat [66]. Although some subsequent studies did not find such associations in other patient populations [67], these studies draw attention to possible depot-specific processing of vitamin A (Figure 1, Group 3). Retinol saturase is another example of a potent but still not defined action of retinoids on adipose tissue distribution. Retinol saturase is an enzyme catalyzing the conversion of retinol to dihydroretinoids that is positively regulated by PPARγ in vitro, however, retinol saturase knockout mice exhibit increased adiposity with up-regulation of PPARγ [68-70].
Further studies are needed to determine concentrations of vitamin A (retinol and retinyl esters) and its metabolites (retinaldehyde and RA) in human and rodent visceral and subcutaneous adipose tissues. In an early study in rats, Blaner and colleagues have shown that visceral and subcutaneous adipose depots have comparable amounts of retinol, i.e. 6.4 and 6.9 μg retinol/g tissue [71]. There are substantial differences between hepatic and adipose tissue stores. Hepatic vitamin A accumulation is in the form of retinyl esters. Adipose tissues mostly store the retinol form (75%), while the esterified form accounts for only 25% of total retinol stores [71]. The estimated vitamin A content (both retinol and retinyl esters) in adipose tissues is 15-20% of the total body retinoid stores in rodents [71]. Even less is known about concentrations of retinol metabolites, retinol and retinoic acid, in different depots, although these compounds have been detected in adipose tissue [72] and their concentrations were dependent on retinyl hydrolase activity of hormone sensitive lipase [73,74] and aldehyde dehydrogenase1a1 [75].
Our previous studies [75] showed a 0.1-1.0 μM concentration of retinaldehyde, measured as retinaldehyde oxime in mouse white adipose tissue, that was inversely correlated with adipocyte size. However, no systematic analytical studies are available to show the levels of vitamin A and its metabolites in various white adipose depots and their relevance to adipose tissue differentiation, development, depot- and sex-specific accumulation.
The expression vitamin A metabolizing enzymes was examined and offered insight into possible depot-specificity in RA production [25, 76]. The metabolism of retinol proceeds in two enzymatic steps catalyzed by different families of enzymes encoded in separated genes (reviewed in this issue and in [77]). Alcohol dehydrogenases and retinol dehydrogenases convert retinol to retinaldehyde. RA is produced exclusively from retinaldehyde by the cytosolic aldehyde dehydrogenase-1 family of enzymes (Aldh1, also known as Raldh) [77]. We have found that in female mice, Aldh1 enzymes are expressed at higher levels in subcutaneous than in visceral (perigonadal) fat depots [25]. Aldh1a1 (also known as Raldh1) was the predominately expressed enzyme, followed by Aldh1a3 in both fat depots (Figure 3). In vitro, adipogenesis in 3T3-L1 cells was also accompanied by increased expression of Aldh1a1, which was responsible for approximately 70% of RA production [25] (gene names in italics represent mRNA levels). The causal evidence of Aldh1a1's participation in the formation of depot-specific fat reserves came from observations in Aldh1a1−/− mice. Deficiency in Aldh1a1 leads to impaired accumulation of fat in visceral more than in subcutaneous regions in aged Aldh1a1−/− compared to WT female mice (Figure 3). Interestingly, the regional differences in fat formation in humans are also associated with Aldh1 enzyme expression, although the pattern of Aldh1 expression is dissimilar to that of rodents [25]. In women, Aldh1a2 and a3 (also known as Raldh2, Raldh3) expression was significantly higher in visceral (omental) than in subcutaneous fat, even though Aldh1a1 remained the predominantly expressed enzyme in both tissues. Independently, proteomic comparison of omental and subcutaneous fat in 14 men and women revealed Aldh1a2 as a protein marker of differences between these fat depots [76]. More studies in larger patient cohorts are needed to understand whether Aldh1 expression and concentrations of dependent metabolites, RA and retinaldehyde, are linked to visceral obesity in humans and if these can predict propensity to visceral obesity in response to obesogenic diets in specific ethnic or disease populations. Our previous studies showed that Aldh1a1 deficiency in mice renders them resistant to obesity mediated by a high-fat diet, although the possible regional specificity of these effects has not yet been scrutinized [75].
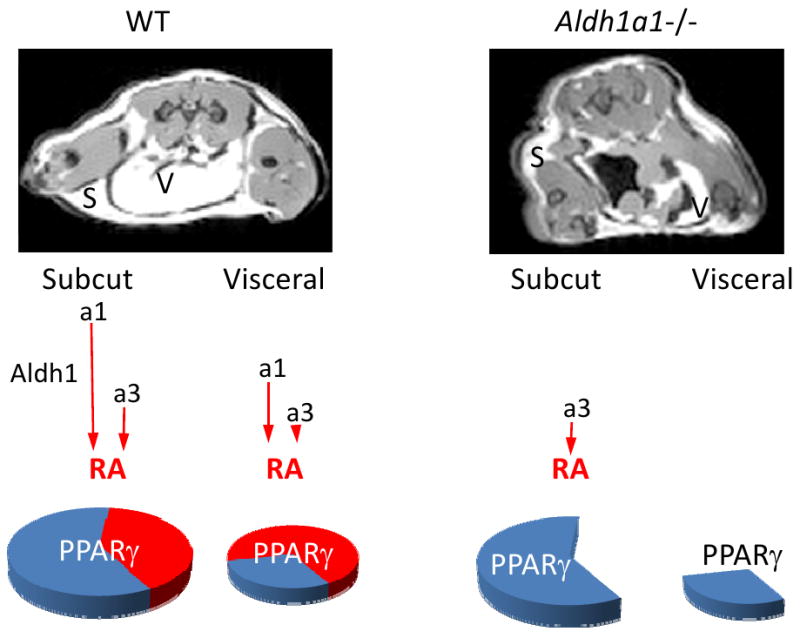
Figure 3. Schematic mechanisms by which autocrine production of retinoic acid (RA) by the aldehyde dehydrogenase-1 family of enzymes regulates depot-specific fat formation and PPARγ expression.

The MRI images of C57/Bl6 (WT) and Aldh1a1−/− female mice on a regular diet showed marked loss of visceral (V) and, to lesser extent, subcutaneous (S) fat. Aldh1 is a family of three cytosolic enzymes (Aldh1a1, a2, a3) that catalyze retinaldehyde oxidation to RA. Our in vitro studies show that Aldh1a1 is responsible for 70% of endogenous RA production (gene names in italics represent mRNA levels). Subcutaneous fat of WT female mice expresses higher levels of Aldh1 enzymes compared to visceral fat (red arrow lengths show relative expression pattern). In both fat depots, Aldh1a1 is the predominantly expressed enzyme, followed by Aldh1a3. Consequently, disruption of RA production was observed in Aldh1a1−/− visceral fat depleted of any Aldh1 enzymes, while subcutaneous fat had marginal Aldh1a3 expression. Such drastic changes in Aldh1 expression in the knockouts result in a 70% decrease in PPARγ expression in visceral fat and a 40% decrease in PPARγ expression in subcutaneous fat, as compared to WT, which corresponds to fat accumulation in visceral and subcutaneous depots [25].
3.2 Distinguished characteristics of autocrine RA production and RA treatments
Autocrine RA production by Aldh1 enzymes plays a role in the regulation of fat formation. These effects are somewhat expected given that many critical transcriptional pathways regulating organogenesis, adipogenesis and fat biology are influenced by RA treatment in vitro and in vivo (Figure 4, reviewed in 3.3). However, the autocrine mechanism for RA generation has distinguished characteristics with potentially divergent molecular responses in cells and tissues, compared to the effects mediated by RA treatments. In fact, Aldh1 controls the concentration of two metabolites, retinaldehyde and RA, regulating transcription through different factors (reviewed in [78]). RA acts principally through retinoic acid receptor (RAR), although other transcriptional factors, including RXR, PPARδ, and redox-sensitive factors, contribute substantially to RA-mediated transcription (reviewed in this issue and in [79, 80]). RA produced in a regulated manner by an autocrine mechanism can exert more specific effects than RA treatments, especially those with high RA (μmol) concentrations. At these pharmacological levels, RA can potentially induce multiple transcription factors and also bind to proteins [81]. Detectable levels of retinoylated proteins in 3T3-L1 adipocytes were found after treatment with 100 nmol RA [81]. Further work needs to examine the contribution of retinoylation reactions to adipogenesis. In contrast to RA, retinaldehyde is a weak agonist of RAR [82], but can inhibit RXR and PPARγ activation in differentiated adipocytes in vitro [75]. This retinaldehyde accumulation and its molecular effects may be primarily responsible for the inhibition of adipogenesis in Aldh1a1−/− fibroblasts that could be only partially rescued by RA treatments [25].
Figure 4. Potential RA effects on the transcriptional network participating in PPARγ regulation.
RA, acting through RAR and Smad3, blocks C/EBPβ and C/EBPα activity, which subsequently inhibits PPARγ expression in vitro. It is suggestive that RA also regulates other transcription factors, i.e. SREBP1c, STAT5, KLF5 and EBF1 in various tissues. Endogenous RA is produced from retinaldehyde by the cytosolic Aldh1 family of enzymes. Our studies showed that autocrine production of RA controls 70% of ZFP423 expression in visceral fat, which in turn induces PPARγ expression.
The Aldh1 family consists of three enzymes catalyzing production of RA [77]. Our studies showed that in adipogenesis all Aldh1 enzymes appear to be redundant in RA production, and forced expression of Aldh1a2 or a3 enzymes can partially rescue adipogenesis in fibroblasts deficient in all Aldh1 enzymes [25]. This concerted autocrine action of the Aldh1 enzymes appears to establish different RA production and transcription factor expression in visceral and subcutaneous depots in mice (Figure 3). In addition to their common feature in RA production, different expression of Aldh1 enzymes appears to exert specific responses correspondent to their expression in different depots [25]. For instance, Aldh1a3-deficient fibroblasts express reduced levels of adipokine visfatin, and expression of Aldh1a1 could not compensate for this effect. These observations could indicate some specific signaling mechanisms activated by Aldh1 enzymes or their products/substrates. In vitro, participation of the Aldh1a1 enzyme in dehydrogenation of other aldehydes including 3-deoxyglucosone [83] and lipid aldehydes [84], has been documented. Retinaldehyde remains the preferential substrate for Aldh1a1 under physiologic conditions; however, consumption of specific diets and oxidative stress can create conditions where alternative aldehydes would be utilized by Aldh1a1 for the production of their respective acids instead of RA. In the context of autocrine RA production, the signaling properties of other aldehydes and their respective acids have not been explored.
Negative feedback is a common regulatory mechanism preventing enzymes from product overproduction. Differentiated 3T3-L1 adipocytes also respond to RA treatments by inhibition of predominantly expressed Aldh1a1 enzyme in a manner dependent on RA concentrations [25]. In consonance, Lobo et.al showed that treatment with 2 μM exogenous RA led to a 12-fold reduction in cellular RA levels in differentiated NIH-3T3 cells [85]. Treatment with RA also decreases expression of its binding protein CRABP-II [86]. Hormone sensitive lipase-null mice exhibiting a lean phenotype could partially recover white adipose tissue mass when fed a RA-supplemented high-fat diet. The validity of RA participation in the obesogenic processes needs to be further examined to shed light on the puzzling similarity in phenotypes of Aldh1a1−/− mice, deficient in the major RA generating enzyme, and mice treated with RA that experience analogous induction in thermogenesis and reduction of white fat depots [24, 80]. Given these considerations, in the next chapter we will review, first, known RA effects on the potential transcriptional regulators of regional fat formation in adipocytes or other cellular systems and, second, conclude with the fat-depot-specific molecular mechanisms induced by autocrine RA production.
3.3 RA effects on transcription factors involved in a depot-specific fat formation
Although HOX genes have only recently been considered as transcriptional markers of subcutaneous and visceral fat depots, their role in organogenesis and their link to RA-dependent RAR regulation has been well documented. HOXA1, HOXB1 HOXB4 and HOXD4 genes are responsive to RA through the RAR response element (RARE) in the 5′ regulatory regions of their enhancers [87,88]. Conversely, HOXB4 and HOXD4 genes can control RAR expression [88]. The complex cross-regulation between HOX and RAR families in embryogenesis has been recently reviewed [89], but not elucidated in adipogenesis.
The pioneering work by Lazar and associates has established a mechanism for the inhibition of adipogenesis in murine 3T3-L1 cells treated with μmolar concentrations of RA, other retinoids, and synthetic RAR ligands during the first 48 hours after initiation of adipogenesis by a hormonal mix [54, 90]. In adipogenesis, the activated RAR receptor can block activity of C/EBP proteins (both alpha and beta), thereby reducing PPARγ expression and adipogenesis. Subsequent studies by the Wiper-Bergeron group showed that RA-mediated occupancy of the C/EBPα promoter by C/EBPβ depends on Smad 3 [91]. RA conveys its inhibitory effects through Smad3 [91]. These interactions could account at least in part for the biphasic action of RA in adipogenesis: 1) inhibitory if added before 48 hours, and 2) stimulatory if added after 48 hours of differentiation [90]. These RA effects in adipogenesis need further investigation, since RAR-dependent pathways can potentially influence all phases of adipogenesis. Indeed, all RAR isoforms, RARα, -β and -γ, were expressed throughout adipogenesis together with RXRβ [92]. RXRα was expressed in the middle phase (after 16h of differentiation) and later, whereas RXRγ was expressed only in mature adipocytes (after 5 days of differentiation) [92]. Some of the RA effects in adipogenesis and fat formation could depend on proteins regulating the cell cycle, such retinoblastoma protein [54, 74]. Interestingly, RA stimulation of adipogenesis in bovine cells induces PPARγ expression [93], suggesting that species' transcriptomes influence RA effects. RA was implicated in the regulation of other transcription factors, participating in the regulation of PPARγ expression and activity, including SREBP1c [94], EBF1 [95], STAT5 [96], and KLF5 [97] in different cell types (Figure 4). The role of these pathways in adipogenesis and fat formation in different depots is still unknown.
The major outcome of disrupted autocrine production of RA in Aldh1a1−/− mice is also a marked decrease in PPARγ expression in adipocytes in vivo [25]. Corresponding to the preferential disruption of visceral compared to subcutaneous fat in Aldh1a1−/− versus WT female mice, PPARγ expression was markedly diminished in visceral compared to subcutaneous fat (70% vs 40%) (Figure 3). Subsequent experiments in adipocytes deficient in Aldh1a1, a3, or all Aldh1 enzymes proved that suppressed PPARγ expression stems at least in part from deficient RA production, hence either treatment with RA concentrations (nanomolar range) or forced expression of any of the Aldh1 enzymes partially rescued PPARγ expression and adipogenesis [25].
Despite recovered PPARγ expression and increased lipid accumulation in Aldh1a-deficient adipocytes treated with RA, expression of fatty acid synthase, downregulation of preadipocyte markers and expression of PPARγ target genes remained reduced [25]. This effect is somewhat expected from the autocrine mechanisms that also control catabolism of retinaldehyde, inhibiting PPARγ activity [75]. The retinaldehyde-dependent mechanisms might play a dominant role in the prevention of diet-induced obesity in Aldh1a1−/− mice, since PPARγ expression was similar (data not shown), but expression of PPARγ target genes was reduced in Aldh1a1−/− compared to WT mice on a high-fat diet [75]. More studies are needed to elucidate endogenous vitamin A metabolism and transcriptional responses affected by vitamin A without or in combination with obesogenic diets. Current studies in rodents describe synergic effects of vitamin A and a high-fat diet in adipose tissue development and PPARγ expression [98], whereas treatment of obese mice on a regular diet with vitamin A resulted in decreased fat mass ([99] and reviewed in this issue).
Autocrine RA production in adipogenesis regulates PPARγ by alternative pathways. Aldh1 loss-of-function experiments revealed that ZFP423 expression was reduced by 99% [25]. Both RA and forced expression of Aldh1 enzymes effectively recovered ZFP423 expression. Although precise mechanisms by which RA regulates ZFP423 remain to be determined, the Aldh1-dependent induction of ZFP423 appears to play a main role in the depot-specific regulation of PPARγ and fat formation (Figure 3). Reduced expression of Aldh1 enzymes in visceral fat of Aldh1a1−/− mice resulted in a marked 70% suppression of ZFP423 expression [25]. In subcutaneous fat of Aldh1a1−/− mice, ZFP423 levels were similar to those seen in WT mice, probably due to the remaining expression of Aldh1a3. Unexpectedly, C/EBPα and C/EBPβ expression could not be conclusively linked to Aldh1 function in RA generation [25]. The mechanisms by which RA induces expression of ZFP423 need to be elucidated; however, ZFP423 has been implicated in the activation of RARE in neuroblastoma cells potentiating RA effects [100]. Thus, important cross-regulation might exist between RAR and ZFP423 pathways that influence differentiation in many tissues. Future studies need to address autocrine mechanisms in human adipose tissues and their relevance in the regulation of the transcriptional network that is ultimately responsible for the endocrine and lipolytic differences between fat depots.
Conclusive remarks
Tissue-specific autocrine pathways are expected to have a broad range of transcriptional and non-genomic responses that can set the transcriptional network in motion and elicit depot-specific fat formation during embryogenesis and in response to nutrients. Autocrine RA production might also influence pathways in mature adipocytes, mitochondrial functions, and lineage commitments that were discovered in the context of treatment with retinoids and were not discussed here. The identification of fat-depot-specific regulatory mechanisms, both autocrine and transcriptional, is undoubtedly a basis for the development of novel approaches in the prevention and treatment of visceral obesity.
Highlights.
Fat accumulation in visceral fat depots increases risks for morbidity and mortality. In this review, we discuss the autocrine role of aldehyde dehydrogenase-1 family of enzymes in the regulation of the transcriptional network in a fat-depot-specific manner and in the development of visceral obesity
Acknowledgments
This research was supported by the EHE SEED grant, Food Innovation Grant (O.Z.); and ICMR-International Fellowship (S.M.J). The project described was supported by Award Number UL1RR025755 (O.Z.) from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deitel M. Overweight and obesity worldwide now estimated to involve 1.7 billion people. Obes Surg. 2003;13:329–330. doi: 10.1381/096089203765887598. [DOI] [PubMed] [Google Scholar]
- 2.Li C, Ford ES, McGuire LC, Mokdad AH. Increasing trends in waist circumference and abdominal obesity among US adults. Obesity (Silver Spring) 2007;15:216–224. doi: 10.1038/oby.2007.505. [DOI] [PubMed] [Google Scholar]
- 3.Zhang C, Rexrode KM, van Dam RM, Li TY, Hu FB. Abdominal obesity and the risk of all-cause, cardiovascular, and cancer mortality: sixteen years of follow-up in US women. Circulation. 2008;117:1658–1667. doi: 10.1161/CIRCULATIONAHA.107.739714. [DOI] [PubMed] [Google Scholar]
- 4.Reis JP, Macera CA, Araneta MR, Lindsay SP, Marshall SJ, Wingard DL. Comparison of overall obesity and body fat distribution in predicting risk of mortality. Obesity (Silver Spring) 2009;17:1232–1239. doi: 10.1038/oby.2008.664. [DOI] [PubMed] [Google Scholar]
- 5.Chan JM, Rimm EB, Colditz GA, Stampfer MJ, Willett WC. Obesity, fat distribution, and weight gain as risk factors for clinical diabetes in men. Diabetes Care. 1994;17:961–969. doi: 10.2337/diacare.17.9.961. [DOI] [PubMed] [Google Scholar]
- 6.Lapidus L, Bengtsson C, Bjorntorp P. The quantitative relationship between “the metabolic syndrome” and abdominal obesity in women. Obes Res. 1994;2:372–377. doi: 10.1002/j.1550-8528.1994.tb00077.x. [DOI] [PubMed] [Google Scholar]
- 7.Kim LJ, Nalls MA, Eiriksdottir G, Sigurdsson S, Launer LJ, Koster A, Chaves PH, Jonsdottir B, Garcia M, Gudnason V, Harris TB. Associations of Visceral and Liver Fat With the Metabolic Syndrome Across the Spectrum of Obesity: The AGES-Reykjavik Study. Obesity (Silver Spring) doi: 10.1038/oby.2010.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carey VJ, Walters EE, Colditz GA, Solomon CG, Willett WC, Rosner BA, Speizer FE, Manson JE. Body fat distribution and risk of non-insulin-dependent diabetes mellitus in women. The Nurses' Health Study. Am J Epidemiol. 1997;145:614–619. doi: 10.1093/oxfordjournals.aje.a009158. [DOI] [PubMed] [Google Scholar]
- 9.Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, Lisheng L. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937–952. doi: 10.1016/S0140-6736(04)17018-9. [DOI] [PubMed] [Google Scholar]
- 10.Klein S, Fontana L, Young VL, Coggan AR, Kilo C, Patterson BW, Mohammed BS. Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N Engl J Med. 2004;350:2549–2557. doi: 10.1056/NEJMoa033179. [DOI] [PubMed] [Google Scholar]
- 11.Thorne A, Lonnqvist F, Apelman J, Hellers G, Arner P. A pilot study of long-term effects of a novel obesity treatment: omentectomy in connection with adjustable gastric banding. Int J Obes Relat Metab Disord. 2002;26:193–199. doi: 10.1038/sj.ijo.0801871. [DOI] [PubMed] [Google Scholar]
- 12.Barzilai N, She L, Liu BQ, Vuguin P, Cohen P, Wang J, Rossetti L. Surgical removal of visceral fat reverses hepatic insulin resistance. Diabetes. 1999;48:94–98. doi: 10.2337/diabetes.48.1.94. [DOI] [PubMed] [Google Scholar]
- 13.Gabriely I, Ma XH, Yang XM, Atzmon G, Rajala MW, Berg AH, Scherer P, Rossetti L, Barzilai N. Removal of visceral fat prevents insulin resistance and glucose intolerance of aging: an adipokine-mediated process? Diabetes. 2002;51:2951–2958. doi: 10.2337/diabetes.51.10.2951. [DOI] [PubMed] [Google Scholar]
- 14.Liszka TG, Dellon AL, Im M, Angel MF, Plotnick L. Effect of lipectomy on growth and development of hyperinsulinemia and hyperlipidemia in the Zucker rat. Plast Reconstr Surg. 1998;102:1122–1127. doi: 10.1097/00006534-199809040-00031. [DOI] [PubMed] [Google Scholar]
- 15.Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 2008;7:410–420. doi: 10.1016/j.cmet.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahima RS. Adipose tissue as an endocrine organ. Obesity (Silver Spring) 2006;14 5:242S–249S. doi: 10.1038/oby.2006.317. [DOI] [PubMed] [Google Scholar]
- 17.Plomgaard P, Nielsen AR, Fischer CP, Mortensen OH, Broholm C, Penkowa M, Krogh-Madsen R, Erikstrup C, Lindegaard B, Petersen AM, Taudorf S, Pedersen BK. Associations between insulin resistance and TNF-alpha in plasma, skeletal muscle and adipose tissue in humans with and without type 2 diabetes. Diabetologia. 2007;50:2562–2571. doi: 10.1007/s00125-007-0834-6. [DOI] [PubMed] [Google Scholar]
- 18.Klover PJ, Zimmers TA, Koniaris LG, Mooney RA. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes. 2003;52:2784–2789. doi: 10.2337/diabetes.52.11.2784. [DOI] [PubMed] [Google Scholar]
- 19.Esteghamati A, Khalilzadeh O, Anvari M, Rashidi A, Mokhtari M, Nakhjavani M. Association of serum leptin levels with homeostasis model assessment-estimated insulin resistance and metabolic syndrome: the key role of central obesity. Metab Syndr Relat Disord. 2009;7:447–452. doi: 10.1089/met.2008.0100. [DOI] [PubMed] [Google Scholar]
- 20.Maury E, Ehala-Aleksejev K, Guiot Y, Detry R, Vandenhooft A, Brichard SM. Adipokines oversecreted by omental adipose tissue in human obesity. Am J Physiol Endocrinol Metab. 2007;293:E656–665. doi: 10.1152/ajpendo.00127.2007. [DOI] [PubMed] [Google Scholar]
- 21.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 22.Trujillo ME, Scherer PE. Adiponectin--journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J Intern Med. 2005;257:167–175. doi: 10.1111/j.1365-2796.2004.01426.x. [DOI] [PubMed] [Google Scholar]
- 23.Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonet ML, Ribot J, Felipe F, Palou A. Vitamin A and the regulation of fat reserves. Cell Mol Life Sci. 2003;60:1311–1321. doi: 10.1007/s00018-003-2290-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reichert B, Yasmeen R, Jeyakumar S, Yang F, Thomou T, Alder H, Duester D, Maiseyeu A, Mihai G, Harrison E, Rajagopalan S, Kirkland J, Ziouzenkova O. Concerted action of aldehyde dehydrogenases influences depot-specific fat formation. Molecular Endocrinology. 2011 doi: 10.1210/me.2010-0465. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vohl MC, Sladek R, Robitaille J, Gurd S, Marceau P, Richard D, Hudson TJ, Tchernof A. A survey of genes differentially expressed in subcutaneous and visceral adipose tissue in men. Obes Res. 2004;12:1217–1222. doi: 10.1038/oby.2004.153. [DOI] [PubMed] [Google Scholar]
- 27.Gesta S, Bluher M, Yamamoto Y, Norris AW, Berndt J, Kralisch S, Boucher J, Lewis C, Kahn CR. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci U S A. 2006;103:6676–6681. doi: 10.1073/pnas.0601752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tchkonia T, Lenburg M, Thomou T, Giorgadze N, Frampton G, Pirtskhalava T, Cartwright A, Cartwright M, Flanagan J, Karagiannides I, Gerry N, Forse RA, Tchoukalova Y, Jensen MD, Pothoulakis C, Kirkland JL. Identification of depot-specific human fat cell progenitors through distinct expression profiles and developmental gene patterns. Am J Physiol Endocrinol Metab. 2007;292:E298–307. doi: 10.1152/ajpendo.00202.2006. [DOI] [PubMed] [Google Scholar]
- 29.Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–256. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Tchkonia T, Giorgadze N, Pirtskhalava T, Thomou T, DePonte M, Koo A, Forse RA, Chinnappan D, Martin-Ruiz C, von Zglinicki T, Kirkland JL. Fat depot-specific characteristics are retained in strains derived from single human preadipocytes. Diabetes. 2006;55:2571–2578. doi: 10.2337/db06-0540. [DOI] [PubMed] [Google Scholar]
- 31.Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- 32.He W, Barak Y, Hevener A, Olson P, Liao D, Le J, Nelson M, Ong E, Olefsky JM, Evans RM. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A. 2003;100:15712–15717. doi: 10.1073/pnas.2536828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006;4:263–273. doi: 10.1016/j.cmet.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lefebvre AM, Laville M, Vega N, Riou JP, van Gaal L, Auwerx J, Vidal H. Depot-specific differences in adipose tissue gene expression in lean and obese subjects. Diabetes. 1998;47:98–103. doi: 10.2337/diab.47.1.98. [DOI] [PubMed] [Google Scholar]
- 35.Sauma L, Franck N, Paulsson JF, Westermark GT, Kjolhede P, Stralfors P, Soderstrom M, Nystrom FH. Peroxisome proliferator activated receptor gamma activity is low in mature primary human visceral adipocytes. Diabetologia. 2007;50:195–201. doi: 10.1007/s00125-006-0515-x. [DOI] [PubMed] [Google Scholar]
- 36.Tsuchida A, Yamauchi T, Takekawa S, Hada Y, Ito Y, Maki T, Kadowaki T. Peroxisome proliferator-activated receptor (PPAR)alpha activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: comparison of activation of PPARalpha, PPARgamma, and their combination. Diabetes. 2005;54:3358–3370. doi: 10.2337/diabetes.54.12.3358. [DOI] [PubMed] [Google Scholar]
- 37.Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, Rossetti L, Scherer PE. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281:2654–2660. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- 38.Heikkinen S, Argmann C, Feige JN, Koutnikova H, Champy MF, Dali-Youcef N, Schadt EE, Laakso M, Auwerx J. The Pro12Ala PPARgamma2 variant determines metabolism at the gene-environment interface. Cell Metab. 2009;9:88–98. doi: 10.1016/j.cmet.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 39.Yen CJ, Beamer BA, Negri C, Silver K, Brown KA, Yarnall DP, Burns DK, Roth J, Shuldiner AR. Molecular scanning of the human peroxisome proliferator activated receptor gamma (hPPAR gamma) gene in diabetic Caucasians: identification of a Pro12Ala PPAR gamma 2 missense mutation. Biochem Biophys Res Commun. 1997;241:270–274. doi: 10.1006/bbrc.1997.7798. [DOI] [PubMed] [Google Scholar]
- 40.Kim KS, Choi SM, Shin SU, Yang HS, Yoon Y. Effects of peroxisome proliferator-activated receptor-gamma 2 Pro12Ala polymorphism on body fat distribution in female Korean subjects. Metabolism. 2004;53:1538–1543. doi: 10.1016/j.metabol.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 41.Herrmann SM, Ringel J, Wang JG, Staessen JA, Brand E. Peroxisome proliferator-activated receptor-gamma2 polymorphism Pro12Ala is associated with nephropathy in type 2 diabetes: The Berlin Diabetes Mellitus (BeDiaM) Study. Diabetes. 2002;51:2653–2657. doi: 10.2337/diabetes.51.8.2653. [DOI] [PubMed] [Google Scholar]
- 42.Buzzetti R, Petrone A, Caiazzo AM, Alemanno I, Zavarella S, Capizzi M, Mein CA, Osborn JA, Vania A, di Mario U. PPAR-gamma2 Pro12Ala variant is associated with greater insulin sensitivity in childhood obesity. Pediatr Res. 2005;57:138–140. doi: 10.1203/01.PDR.0000147728.62185.21. [DOI] [PubMed] [Google Scholar]
- 43.Laplante M, Festuccia WT, Soucy G, Gelinas Y, Lalonde J, Berger JP, Deshaies Y. Mechanisms of the depot specificity of peroxisome proliferator-activated receptor gamma action on adipose tissue metabolism. Diabetes. 2006;55:2771–2778. doi: 10.2337/db06-0551. [DOI] [PubMed] [Google Scholar]
- 44.Tchoukalova YD, Votruba SB, Tchkonia T, Giorgadze N, Kirkland JL, Jensen MD. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc Natl Acad Sci U S A. 107:18226–18231. doi: 10.1073/pnas.1005259107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kershaw EE, Morton NM, Dhillon H, Ramage L, Seckl JR, Flier JS. Adipocyte-specific glucocorticoid inactivation protects against diet-induced obesity. Diabetes. 2005;54:1023–1031. doi: 10.2337/diabetes.54.4.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Z, Xie Y, Bucher NL, Farmer SR. Conditional ectopic expression of C/EBP beta in NIH-3T3 cells induces PPAR gamma and stimulates adipogenesis. Genes Dev. 1995;9:2350–2363. doi: 10.1101/gad.9.19.2350. [DOI] [PubMed] [Google Scholar]
- 47.Christy RJ, Kaestner KH, Geiman DE, Lane MD. CCAAT/enhancer binding protein gene promoter: binding of nuclear factors during differentiation of 3T3-L1 preadipocytes. Proc Natl Acad Sci U S A. 1991;88:2593–2597. doi: 10.1073/pnas.88.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao Z, Umek RM, McKnight SL. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991;5:1538–1552. doi: 10.1101/gad.5.9.1538. [DOI] [PubMed] [Google Scholar]
- 49.Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A, Feng D, Zhuo D, Stoeckert CJ, Jr, Liu XS, Lazar MA. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008;22:2941–2952. doi: 10.1101/gad.1709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosen ED, Hsu CH, Wang X, Sakai S, Freeman MW, Gonzalez FJ, Spiegelman BM. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev. 2002;16:22–26. doi: 10.1101/gad.948702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanaka T, Yoshida N, Kishimoto T, Akira S. Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J. 1997;16:7432–7443. doi: 10.1093/emboj/16.24.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oishi Y, Manabe I, Tobe K, Tsushima K, Shindo T, Fujiu K, Nishimura G, Maemura K, Yamauchi T, Kubota N, Suzuki R, Kitamura T, Akira S, Kadowaki T, Nagai R. Kruppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab. 2005;1:27–39. doi: 10.1016/j.cmet.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 53.Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc Natl Acad Sci U S A. 1998;95:4333–4337. doi: 10.1073/pnas.95.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shao D, Lazar MA. Peroxisome proliferator activated receptor gamma, CCAAT/enhancer-binding protein alpha, and cell cycle status regulate the commitment to adipocyte differentiation. J Biol Chem. 1997;272:21473–21478. doi: 10.1074/jbc.272.34.21473. [DOI] [PubMed] [Google Scholar]
- 55.Classon M, Kennedy BK, Mulloy R, Harlow E. Opposing roles of pRB and p107 in adipocyte differentiation. Proc Natl Acad Sci U S A. 2000;97:10826–10831. doi: 10.1073/pnas.190343597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dali-Youcef N, Mataki C, Coste A, Messaddeq N, Giroud S, Blanc S, Koehl C, Champy MF, Chambon P, Fajas L, Metzger D, Schoonjans K, Auwerx J. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc Natl Acad Sci U S A. 2007;104:10703–10708. doi: 10.1073/pnas.0611568104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akerblad P, Mansson R, Lagergren A, Westerlund S, Basta B, Lind U, Thelin A, Gisler R, Liberg D, Nelander S, Bamberg K, Sigvardsson M. Gene expression analysis suggests that EBF-1 and PPARgamma2 induce adipogenesis of NIH-3T3 cells with similar efficiency and kinetics. Physiol Genomics. 2005;23:206–216. doi: 10.1152/physiolgenomics.00015.2005. [DOI] [PubMed] [Google Scholar]
- 58.Fretz JA, Nelson T, Xi Y, Adams DJ, Rosen CJ, Horowitz MC. Altered metabolism and lipodystrophy in the early B-cell factor 1-deficient mouse. Endocrinology. 151:1611–1621. doi: 10.1210/en.2009-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Floyd ZE, Stephens JM. STAT5A promotes adipogenesis in nonprecursor cells and associates with the glucocorticoid receptor during adipocyte differentiation. Diabetes. 2003;52:308–314. doi: 10.2337/diabetes.52.2.308. [DOI] [PubMed] [Google Scholar]
- 60.Shang CA, Waters MJ. Constitutively active signal transducer and activator of transcription 5 can replace the requirement for growth hormone in adipogenesis of 3T3-F442A preadipocytes. Mol Endocrinol. 2003;17:2494–2508. doi: 10.1210/me.2003-0139. [DOI] [PubMed] [Google Scholar]
- 61.Stewart WC, Baugh JE, Jr, Floyd ZE, Stephens JM. STAT 5 activators can replace the requirement of FBS in the adipogenesis of 3T3-L1 cells. Biochem Biophys Res Commun. 2004;324:355–359. doi: 10.1016/j.bbrc.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 62.Berry DC, Jin H, Majumdar A, Noy N. Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc Natl Acad Sci U S A. 108:4340–4345. doi: 10.1073/pnas.1011115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gupta RK, Arany Z, Seale P, Mepani RJ, Ye L, Conroe HM, Roby YA, Kulaga H, Reed RR, Spiegelman BM. Transcriptional control of preadipocyte determination by Zfp423. Nature. 2010;464:619–623. doi: 10.1038/nature08816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kurlandsky SB, Gamble MV, Ramakrishnan R, Blaner WS. Plasma delivery of retinoic acid to tissues in the rat. J Biol Chem. 1995;270:17850–17857. doi: 10.1074/jbc.270.30.17850. [DOI] [PubMed] [Google Scholar]
- 65.Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, Wiita P, Bok D, Sun H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315:820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
- 66.Kloting N, Graham TE, Berndt J, Kralisch S, Kovacs P, Wason CJ, Fasshauer M, Schon MR, Stumvoll M, Bluher M, Kahn BB. Serum retinol-binding protein is more highly expressed in visceral than in subcutaneous adipose tissue and is a marker of intra-abdominal fat mass. Cell Metab. 2007;6:79–87. doi: 10.1016/j.cmet.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 67.Bajzova M, Kovacikova M, Vitkova M, Klimcakova E, Polak J, Kovacova Z, Viguerie N, Vedral T, Mikulasek L, Sramkova P, Srp A, Hejnova J, Langin D, Stich V. Retinol-binding protein 4 expression in visceral and subcutaneous fat in human obesity. Physiol Res. 2008;57:927–934. doi: 10.33549/physiolres.931379. [DOI] [PubMed] [Google Scholar]
- 68.Moise AR, Lobo GP, Erokwu B, Wilson DL, Peck D, Alvarez S, Dominguez M, Alvarez R, Flask CA, de Lera AR, von Lintig J, Palczewski K. Increased adiposity in the retinol saturase-knockout mouse. FASEB J. 24:1261–1270. doi: 10.1096/fj.09-147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schupp M, Lefterova MI, Janke J, Leitner K, Cristancho AG, Mullican SE, Qatanani M, Szwergold N, Steger DJ, Curtin JC, Kim RJ, Suh MJ, Albert MR, Engeli S, Gudas LJ, Lazar MA. Retinol saturase promotes adipogenesis and is downregulated in obesity. Proc Natl Acad Sci U S A. 2009;106:1105–1110. doi: 10.1073/pnas.0812065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moise AR, Dominguez M, Alvarez S, Alvarez R, Schupp M, Cristancho AG, Kiser PD, de Lera AR, Lazar MA, Palczewski K. Stereospecificity of retinol saturase: absolute configuration, synthesis, and biological evaluation of dihydroretinoids. J Am Chem Soc. 2008;130:1154–1155. doi: 10.1021/ja710487q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsutsumi C, Okuno M, Tannous L, Piantedosi R, Allan M, Goodman DS, Blaner WS. Retinoids and retinoid-binding protein expression in rat adipocytes. J Biol Chem. 1992;267:1805–1810. [PubMed] [Google Scholar]
- 72.Kane MA, Folias AE, Napoli JL. HPLC/UV quantitation of retinal, retinol, and retinyl esters in serum and tissues. Anal Biochem. 2008;378:71–79. doi: 10.1016/j.ab.2008.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wei S, Lai K, Patel S, Piantedosi R, Shen H, Colantuoni V, Kraemer FB, Blaner WS. Retinyl ester hydrolysis and retinol efflux from BFC-1beta adipocytes. J Biol Chem. 1997;272:14159–14165. doi: 10.1074/jbc.272.22.14159. [DOI] [PubMed] [Google Scholar]
- 74.Strom K, Gundersen TE, Hansson O, Lucas S, Fernandez C, Blomhoff R, Holm C. Hormone-sensitive lipase (HSL) is also a retinyl ester hydrolase: evidence from mice lacking HSL. FASEB J. 2009;23:2307–2316. doi: 10.1096/fj.08-120923. [DOI] [PubMed] [Google Scholar]
- 75.Ziouzenkova O, Orasanu G, Sharlach M, Akiyama TE, Berger JP, Viereck J, Hamilton JA, Tang G, Dolnikowski GG, Vogel S, Duester G, Plutzky J. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat Med. 2007;13:695–702. doi: 10.1038/nm1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peinado JR, Jimenez-Gomez Y, Pulido MR, Ortega-Bellido M, Diaz-Lopez C, Padillo FJ, Lopez-Miranda J, Vazquez-Martinez R, Malagon MM. The stromal-vascular fraction of adipose tissue contributes to major differences between subcutaneous and visceral fat depots. Proteomics. 10:3356–3366. doi: 10.1002/pmic.201000350. [DOI] [PubMed] [Google Scholar]
- 77.Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. 2008;134:921–931. doi: 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ziouzenkova O, Plutzky J. Retinoid metabolism and nuclear receptor responses: New insights into coordinated regulation of the PPAR-RXR complex. FEBS Lett. 2008;582:32–38. doi: 10.1016/j.febslet.2007.11.081. [DOI] [PubMed] [Google Scholar]
- 79.Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, De Lera AR, Lotan R, Mangelsdorf DJ, Gronemeyer H. International Union of Pharmacology. LX. Retinoic acid receptors. Pharmacol Rev. 2006;58:712–725. doi: 10.1124/pr.58.4.4. [DOI] [PubMed] [Google Scholar]
- 80.Berry DC, Noy N. All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor beta/delta and retinoic acid receptor. Mol Cell Biol. 2009;29:3286–3296. doi: 10.1128/MCB.01742-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takahashi N, De Luca LM, Breitman TR. Decreased retinoylation in NIH 3T3 cells transformed with activated Ha-ras. Biochem Biophys Res Commun. 1997;239:80–84. doi: 10.1006/bbrc.1997.7434. [DOI] [PubMed] [Google Scholar]
- 82.Repa JJ, Hanson KK, Clagett-Dame M. All-trans-retinol is a ligand for the retinoic acid receptors. Proc Natl Acad Sci U S A. 1993;90:7293–7297. doi: 10.1073/pnas.90.15.7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Collard F, Vertommen D, Fortpied J, Duester G, Van Schaftingen E. Identification of 3-deoxyglucosone dehydrogenase as aldehyde dehydrogenase 1A1 (retinaldehyde dehydrogenase 1) Biochimie. 2007;89:369–373. doi: 10.1016/j.biochi.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiao T, Shoeb M, Siddiqui MS, Zhang M, Ramana KV, Srivastava SK, Vasiliou V, Ansari NH. Molecular cloning and oxidative modification of human lens ALDH1A1: implication in impaired detoxification of lipid aldehydes. J Toxicol Environ Health A. 2009;72:577–584. doi: 10.1080/15287390802706371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lobo GP, Amengual J, Li HN, Golczak M, Bonet ML, Palczewski K, von Lintig J. Beta,beta-carotene decreases peroxisome proliferator receptor gamma activity and reduces lipid storage capacity of adipocytes in a beta,beta-carotene oxygenase 1-dependent manner. J Biol Chem. 285:27891–27899. doi: 10.1074/jbc.M110.132571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Berry DC, Soltanian H, Noy N. Repression of cellular retinoic acid-binding protein II during adipocyte differentiation. J Biol Chem. 285:15324–15332. doi: 10.1074/jbc.M110.110635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Langston AW, Thompson JR, Gudas LJ. Retinoic acid-responsive enhancers located 3′ of the Hox A and Hox B homeobox gene clusters. Functional analysis. J Biol Chem. 1997;272:2167–2175. doi: 10.1074/jbc.272.4.2167. [DOI] [PubMed] [Google Scholar]
- 88.Serpente P, Tumpel S, Ghyselinck NB, Niederreither K, Wiedemann LM, Dolle P, Chambon P, Krumlauf R, Gould AP. Direct crossregulation between retinoic acid receptor {beta} and Hox genes during hindbrain segmentation. Development. 2005;132:503–513. doi: 10.1242/dev.01593. [DOI] [PubMed] [Google Scholar]
- 89.Daftary GS, Taylor HS. Endocrine regulation of HOX genes. Endocr Rev. 2006;27:331–355. doi: 10.1210/er.2005-0018. [DOI] [PubMed] [Google Scholar]
- 90.Schwarz EJ, Reginato MJ, Shao D, Krakow SL, Lazar MA. Retinoic acid blocks adipogenesis by inhibiting C/EBPbeta-mediated transcription. Mol Cell Biol. 1997;17:1552–1561. doi: 10.1128/mcb.17.3.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marchildon F, St-Louis C, Akter R, Roodman V, Wiper-Bergeron NL. Transcription factor Smad3 is required for the inhibition of adipogenesis by retinoic acid. J Biol Chem. 285:13274–13284. doi: 10.1074/jbc.M109.054536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fu M, Sun T, Bookout AL, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. A Nuclear Receptor Atlas: 3T3-L1 adipogenesis. Mol Endocrinol. 2005;19:2437–2450. doi: 10.1210/me.2004-0539. [DOI] [PubMed] [Google Scholar]
- 93.Garcia-Rojas P, Antaramian A, Gonzalez-Davalos L, Villarroya F, Shimada A, Varela-Echavarria A, Mora O. Induction of peroxisomal proliferator-activated receptor gamma and peroxisomal proliferator-activated receptor gamma coactivator 1 by unsaturated fatty acids, retinoic acid, and carotenoids in preadipocytes obtained from bovine white adipose tissue1,2. J Anim Sci. 88:1801–1808. doi: 10.2527/jas.2009-2579. [DOI] [PubMed] [Google Scholar]
- 94.Li R, Chen W, Li Y, Zhang Y, Chen G. Retinoids synergized with insulin to induce Srebp-1c expression and activated its promoter via the two liver X receptor binding sites that mediate insulin action. Biochem Biophys Res Commun. doi: 10.1016/j.bbrc.2011.02.031. [DOI] [PubMed] [Google Scholar]
- 95.Chen X, Esplin BL, Garrett KP, Welner RS, Webb CF, Kincade PW. Retinoids accelerate B lineage lymphoid differentiation. J Immunol. 2008;180:138–145. doi: 10.4049/jimmunol.180.1.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Si J, Collins SJ. IL-3-induced enhancement of retinoic acid receptor activity is mediated through Stat5, which physically associates with retinoic acid receptors in an IL-3-dependent manner. Blood. 2002;100:4401–4409. doi: 10.1182/blood-2001-12-0374. [DOI] [PubMed] [Google Scholar]
- 97.Zhang XH, Zheng B, Han M, Miao SB, Wen JK. Synthetic retinoid Am80 inhibits interaction of KLF5 with RAR alpha through inducing KLF5 dephosphorylation mediated by the PI3K/Akt signaling in vascular smooth muscle cells. FEBS Lett. 2009;583:1231–1236. doi: 10.1016/j.febslet.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 98.Redonnet A, Ferrand C, Bairras C, Higueret P, Noel-Suberville C, Cassand P, Atgie C. Synergic effect of vitamin A and high-fat diet in adipose tissue development and nuclear receptor expression in young rats. Br J Nutr. 2008;100:722–730. doi: 10.1017/S0007114508967568. [DOI] [PubMed] [Google Scholar]
- 99.Jeyakumar SM, Vajreswari A, Giridharan NV. Chronic dietary vitamin A supplementation regulates obesity in an obese mutant WNIN/Ob rat model. Obesity (Silver Spring) 2006;14:52–59. doi: 10.1038/oby.2006.7. [DOI] [PubMed] [Google Scholar]
- 100.Huang S, Laoukili J, Epping MT, Koster J, Holzel M, Westerman BA, Nijkamp W, Hata A, Asgharzadeh S, Seeger RC, Versteeg R, Beijersbergen RL, Bernards R. ZNF423 is critically required for retinoic acid-induced differentiation and is a marker of neuroblastoma outcome. Cancer Cell. 2009;15:328–340. doi: 10.1016/j.ccr.2009.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]