

Abstract
Objective
Septic shock heterogeneity has important implications for clinical trial implementation and patient management. We previously addressed this heterogeneity by identifying 3 putative subclasses of children with septic shock based exclusively on a 100-gene expression signature. Here we attempted to prospectively validate the existence of these gene expression-based subclasses in a validation cohort.
Design
Prospective observational study involving microarray-based bioinformatics.
Setting
Multiple pediatric intensive care units in the United States.
Patients
Separate derivation (n=98) and validation (n=82) cohorts of children with septic shock.
Interventions
None other than standard care.
Measurements and Main Results
Gene expression mosaics of the 100 class-defining genes were generated for 82 individual patients in the validation cohort. Using computer-based image analysis, patients were classified into 1 of 3 subclasses (“A”, “B”, or “C”) based on color and pattern similarity relative to reference mosaics generated from the original derivation cohort. After subclassification, the clinical database was mined for phenotyping. Subclass A patients had higher illness severity relative to subclasses B and C, as measured by maximal organ failure, fewer ICU-free days, and a higher PRISM score. Patients in subclass A were characterized by repression of genes corresponding to adaptive immunity and glucocorticoid receptor signaling. Separate subclass assignments were conducted by 21 individual clinicians, using visual inspection. The consensus classification of the clinicians had modest agreement with the computer algorithm.
Conclusions
We have validated the existence of subclasses of children with septic shock based on a biologically relevant, 100-gene expression signature. The subclasses have relevant clinical differences.
Keywords: microarray, gene expression, stratification, staging, septic shock, pediatrics
INTRODUCTION
Septic shock is a heterogeneous syndrome with variable physiological and biological manifestations across patient groups [1, 2]. A major challenge in critical care medicine is the development of clinically feasible stratification strategies to manage this heterogeneity for the design of more effective clinical trials and individualized patient management [3, 4]. We are addressing the challenge of septic shock heterogeneity by leveraging the discovery potential of whole-genome expression profiling [5].
Viewing septic shock as a syndrome implies the existence of septic shock subclasses predicated on distinct biological processes leading to distinct clinical phenotypes. We previously identified three putative subclasses of children with septic shock based exclusively on differential gene expression patterns representing the first 24 hours of admission to the pediatric intensive care unit (PICU) [6]. Further analyses of the subclasses revealed important phenotypic differences, thus suggesting that the gene expression-based subclasses are clinically relevant. In a subsequent cross validation study, we demonstrated that clinicians could reliably assign patients to the putative subclasses based on 100 class-defining genes depicted as visually intuitive gene expression mosaics [7].
We now seek to prospectively validate our subclassification strategy in a new cohort of children with septic shock. We test the hypothesis that the differential expression patterns of 100 class-defining genes, depicted using visually intuitive expression mosaics, can be used to allocate a validation cohort into septic shock subclasses having relevant clinical differences and biologically relevant differential gene expression.
METHODS
Patients and data collection
The study protocol was approved by the Institutional Review Boards of each institution (n = 11) and is identical for both the derivation and validation cohorts. Children ≤10 years of age admitted to the PICU and meeting pediatric-specific criteria for septic shock were eligible [8]. Controls were recruited from the ambulatory departments of participating institutions using published inclusion and exclusion criteria [9, 10]. The derivation cohort of 98 patients with septic shock and 32 normal controls was previously published [6, 7]. The validation cohort consists of 82 new patients with septic shock and 21 new controls (control median age 2.9 years (1.3–5.6); 11 males and 10 females). The microarray data for both cohorts have been deposited in the NCBI Gene Expression Omnibus (Accession numbers: GSE26440 and GSE26378).
After informed consent from parents or legal guardians, blood samples were obtained within 24 hours of initial presentation to the PICU with septic shock. Clinical and laboratory data were collected daily while in the PICU and stored using a web-based database. The following variables were tracked as indicators of illness severity: organ failure, PRISM score [11], PICU free days, and mortality. Organ failure was defined using pediatric-specific criteria and tracked up to the first 7 days of PICU admission [8]. The calculation of PICU free days was based on the difference between a maximum PICU admission of 28 days and the actual PICU days. Patients who died during the 28 day study period and patients that remained in the PICU for ≥28 days were assigned zero PICU free days. Mortality was tracked for 28 days after enrollment.
RNA extraction and microarray hybridization
Total RNA was isolated from whole blood using the PaxGene™Blood RNA System (PreAnalytiX, Qiagen/Becton Dickson, Valencia, CA). Microarray hybridization was performed as previously described using the Human Genome U133 Plus 2.0 GeneChip (Affymetrix, Santa Clara, CA) [6, 9, 10, 12–14]. CEL files were preprocessed using Robust Multiple-Array Average (RMA) normalization and GeneSpring GX 7.3 software (Agilent Technologies, Palo Alto, CA). All signal intensity-based data were used after RMA normalization [15]. All chips were normalized to the respective median values of controls.
Gene expression mosaics
In the aforementioned study (the derivation cohort), we used discovery-oriented expression and statistical gene filters, and unsupervised hierarchical clustering, to identify 3 putative subclasses of children with septic shock (subclasses “A”, “B”, and “C”) [6]. We subsequently refined the subclass-defining signature to 100 genes using a 2 stage approach. In stage 1 we used K-means clustering to identify coordinately regulated gene clusters corresponding to signaling pathways related to inflammation and immunity. In stage 2 we tested the genes identified in stage 1 for the ability to predict the putative subclass and extracted the top 100 class-predictor genes [6]. These 100 class-predictor genes form the basis of the current study (see Supplemental Table).
Expression mosaics representing the 100 class-defining genes were generated using the Gene Expression Dynamics Inspector (GEDI) [16, 17]. The signature graphical outputs of GEDI are expression mosaics that give microarray data a “face” that is intuitively recognizable via human pattern recognition. The algorithm for creating the mosaics is a self-organizing map. Additional technical details regarding GEDI can be found at: http://www.childrenshospital.org/research/ingber/GEDI/gedihome.htm.
Computer-based classification
The reference mosaics representing the 100 class-defining genes are published [7], and represent the average expression patterns of the individual patients within a given subclass of the derivation cohort. For each patient in the validation cohort, the respective expression data for the 100 class-defining genes were uploaded to GEDI and individual expression mosaics were generated for each patient. Validation cohort patients were then subclassified using a public image analysis platform (ImageJ, http://rsbweb.nih.gov/ij/). The reference and the individual patient mosaics from the validation group were loaded onto ImageJ. The absolute difference in RGB pixel to pixel intensity was calculated for each individual patient mosaic in the validation cohort, relative to each of the three reference mosaics. Final subclassification was based on the “least difference” between the individual patient mosaic and one of the three reference mosaics.
Clinician-based subclassification
We also conducted clinician-based subclassification as previously described [7]. Twenty-one pediatric intensivists participated as evaluators. Each evaluator was shown the 82 individual patient mosaics from the validation cohort, and asked to classify each patient as subclass “A”, “B”, or “C”, based on color and pattern similarity relative to the reference mosaics. The clinicians were not provided with any additional instructions and were blinded to clinical data. Clinician responses were catalogued and subsequently used to arrive at a final consensus subclassification based on the “majority call” (i.e. ≥8 calls for a given class).
Clinical phenotyping and data analysis
After the patients in the validation cohort were subclassified, we mined the clinical database to assess clinical differences between the 3 subclasses. Ordinal and continuous clinical variables not normally distributed were analyzed via 3 group ANOVA on Ranks. Dichotomous clinical variables were analyzed using a 2 by 3 contingency table and Chi-square test (SigmaStat Software, Systat Software, Inc., San Jose, CA).
RESULTS
Baseline Data
The characteristics of the derivation and validation cohorts are provided in Table 1. The validation cohort had a higher proportion of males, a lower proportion of patients with immune suppression, and a lower proportion of patients with negative microbiological cultures, compared to the derivation cohort. All other variables in Table 1 were not significantly different between the two cohorts.
Table 1.
Demographics and clinical characteristics of the derivation and validation cohorts.1
| Derivation Cohort | Validation Cohort | P value | |
|---|---|---|---|
| No. of patients | 98 | 82 | -- |
| Median age in years (IQR)2 | 2.2 (1.0–5.0) | 2.4 (0.8–6.5) | 0.860 |
| No. of males/females | 52/46 | 57/25 | 0.036 |
| No. of deaths (%) | 18 (18) | 12 (15) | 0.639 |
| Maximum # of organ failures (IQR) 3 | 2 (2–3) | 2 (2–3) | 0.859 |
| Median PRISM score (IQR) | 16 (11–21) | 14 (9–19) | 0.081 |
| No. with co-morbidity (%)4 | 41 (42) | 33 (40) | 0.949 |
| No. with immune suppression (%)5 | 23 (23) | 7 (8) | 0.013 |
| No. receiving hydrocortisone (%)6 | 35 (36) | 34 (42) | 0.525 |
| No. with gram pos. bacteria (%)7 | 23 (23) | 24 (29) | 0.477 |
| No. with gram neg. bacteria (%) | 20 (20) | 26 (32) | 0.119 |
| No. with negative cultures (%) | 45 (46) | 25 (30) | 0.050 |
| Median WBC count × 103/mm3 (IQR) | 13 (4–20) | 14 (7–18) | 0.446 |
| Median neutrophil count × 103/mm3 (IQR) | 7 (3–13) | 10 (4–16) | 0.110 |
| Median lymphocyte count × 103/mm3 (IQR) | 1.7 (0.8–3.1) | 2.1 (0.9–3.8) | 0.605 |
| Median monocyte count × 103/mm3 (IQR) | 0.5 (0.2–1.3) | 0.5 (0.3–1.0) | 0.878 |
Continuous variables are analyzed by ANOVA on Ranks, and proportions are analyzed by Chi-square.
Interquartile range (IQR).
Refers to the maximum number of organ failures during the initial 7 days of PICU admission.
Refers to patients having any major diagnosis in addition to septic shock (e.g. trauma, sickle cell disease, congenital heart disease, liver failure, etc.)
Refers to patients with immune deficiency secondary to an intrinsic documented defect of the immune system, or patients receiving immune-suppressive medications (e.g. calcineurin inhibitors or high dose corticosteroids).
For cardiovascular shock.
All bacterial culture data refer to samples obtained from bodily fluids that are normally sterile (i.e. blood, urine, cerebral spinal fluid, broncho-alveolar lavage, and / or peritoneal fluid).
The reference mosaics for the subclassification protocol are shown in the upper panel of Figure 1. The reference mosaics depict the average expression patterns of the same 100 class-defining genes and represent the 3 previously published derivation cohort septic shock subclasses [6, 7]. Examples of individual patient mosaics in the validation cohort, representing the same 100 class-defining genes, are shown in the lower panel of Figure 1.
Figure 1.
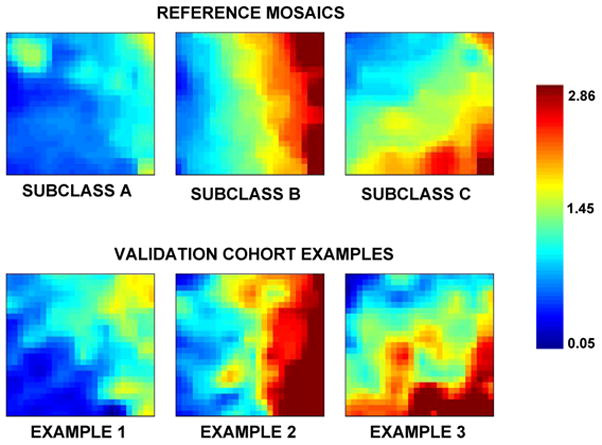
GEDI-generated reference mosaics (top panel) and examples of GEDI-generated individual patient mosaics from the validation cohort (bottom panel). The reference mosaics are derived from the previously published derivation cohort (Refs. 6 and 7) and represent the mean expression values for patients in the respective subclasses. Both the reference mosaics and the individual validation cohort examples depict the expression levels of the same 100 class-defining genes. The color bar on the right provides the relative gene expression based on the respective color intensities.
Computer-Based Subclassification
Table 2 provides the characteristics of the validation cohort subclasses based on the computer algorithm. Subclass A patients had higher illness severity than subclass B and C patients, as measured by maximum number of organ failures and PRISM scores. Subclass A patients also had higher illness severity than subclass B patients, as measured by fewer PICU free days. There was a trend toward a higher mortality odds ratio in subclass A patients, relative to subclass B and C patients. Subclass A patients were also younger, and had a lower peripheral total white blood cell count and neutrophil count, compared to subclass B patients. All other variables in Table 2 were not significantly different across the 3 septic shock subclasses in the validation cohort. These data demonstrate that computer-based image analysis can allocate patients into gene expression-based septic shock subclasses having relevant clinical differences.
Table 2.
Demographics and clinical data for the validation cohort septic shock subclasses using computer-based image analysis.1
| Subclass A | Subclass B | Subclass C | P value | |
|---|---|---|---|---|
| No. of patients | 27 | 24 | 31 | -- |
| Median age in years (IQR) 2 | 1.1 (0.3–3.4) | 6.0 (1.6–8.6) | 2.4 (1.2–5.3) | 0.006 |
| No. of males/females | 20/7 | 14/10 | 23/8 | 0.933 |
| No. of deaths (%) | 6 (22)3 | 2 (8) | 4 (13) | 0.353 |
| Maximum # of organ failures (IQR) 4 | 3 (2–4) | 2 (2–3) | 2 (2–3) | 0.001 |
| Median PRISM score (IQR) | 17 (11–27) | 10 (7–12) | 14 (8–18) | 0.003 |
| Median PICU free days (IQR) | 14 (0–20) | 21 (18–23) | 17 (7–24) | 0.048 |
| No. with co-morbidity (%)5 | 8 (30) | 12 (50) | 13 (42) | 0.324 |
| No. with immune suppression (%)6 | 2 (7) | 2 (8) | 3 (10) | 0.953 |
| No. receiving hydrocortisone (%)7 | 10 (37) | 13 (54) | 11 (35) | 0.321 |
| No. with gram pos. bacteria (%)8 | 9 (33) | 7 (29) | 8 (26) | 0.821 |
| No. with gram neg. bacteria (%) | 11 (41) | 6 (25) | 9 (29) | 0.445 |
| No. with negative cultures (%) | 5 (18) | 8 (33) | 12 (39) | 0.234 |
| Median WBC count × 103/mm3 (IQR) | 9 (4–14) | 15 (11–24) | 16 (8–18) | 0.009 |
| Median neutrophil count × 103/mm3 (IQR) | 7 (3–11) | 14 (10–22) | 10 (4–16) | 0.010 |
| Median lymphocyte count × 103/mm3 (IQR) | 1.8 (0.9–4.3) | 2.2 (0.9–3.2) | 2.1 (1.0–3.9) | 0.857 |
| Median monocyte count × 103/mm3 (IQR) | 0.5 (0.3–0.8) | 0.7 (0.3–1.0) | 0.5 (0.2–1.6) | 0.899 |
Continuous variables are analyzed as 3 group comparisons using ANOVA on Ranks and 2 degrees of freedom. Proportions are analyzed as 3 group comparisons using a 2 by 3 contingency table and Chi-square with 2 degrees of freedom.
Interquartile range (IQR).
Odds ratio for mortality vs. Subclass B: 3.1 (0.6–17.3); vs. Subclass C: 1.9 (0.5–7.7).
Refers to the maximum number of organ failures during the initial 7 days of PICU admission.
Refers to patients having any major diagnosis in addition to septic shock (e.g. trauma, sickle cell disease, congenital heart disease, liver failure, etc.)
Refers to patients with immune deficiency secondary to an intrinsic documented defect of the immune system, or patients receiving immune-suppressive medications (e.g. calcineurin inhibitors or high dose steroids).
For cardiovascular shock.
All bacterial culture data refer to samples obtained from bodily fluids that are normally sterile (i.e. blood, urine, cerebral spinal fluid, broncho-alveolar lavage, and/or peritoneal fluid).
Clinician-Based Subclassification
In this analysis the clinical evaluators conducted the subclassification as described in the Methods. For 31 of the validation cohort patients (38%), all 21 evaluators provided the same subclassification. The other potential majority calls (ranging from 8 to 20) were relatively evenly distributed (Figure 2). Four patients received < 11 majority calls (i.e. less than 50% of the evaluators agreed to the same classification). The individual expression mosaics of these 4 patients and their respective 28 day outcomes are provided in Figure 3.
Figure 2.
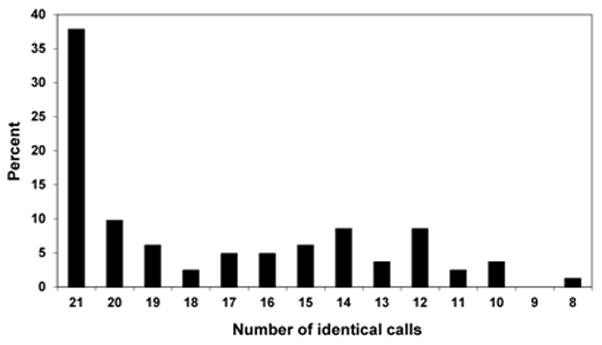
The distribution of potential majority calls (maximum = 21; minimum = 8) among the 21 clinical evaluators. These majority calls led to the final consensus classification for the 82 patients in the validation cohort.
Figure 3.
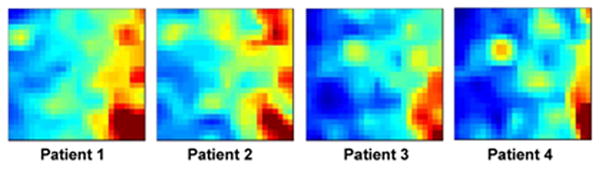
The individual expression mosaics of the four patients in the validation cohort that received < 11 majority calls by the clinical evaluators. Patient 1 (non-survivor) and Patient 2 (survivor) were allocated to subclass C by the computer algorithm. Patient 3 (non-survivor) and Patient 4 (survivor) were allocated to subclass A by the computer algorithm.
The κ coefficient to measure inter-evaluator agreement among the 21 evaluators was 0.633 (0 = no agreement; 1 = perfect agreement). Sixty-five of the validation cohort patients (79%) were allocated to the same subclass by both the computer algorithm and the consensus classification generated by the clinicians (κ coefficient = 0.688).
Table 3 provides the characteristics of the validation cohort subclasses based on the consensus classification of the 21 clinicians. Subclass A patients had higher illness severity than subclass B and C patients, as measured by maximum number of organ failures and PRISM score. Subclass A patients were also younger, and had a lower peripheral total white blood cell count and neutrophil count, compared to subclass B patients. All other variables in Table 3 were not significantly different across the 3 septic shock subclasses in the validation cohort. These data validate that clinicians can allocate patients with septic shock into gene expression-based subclasses having relevant clinical differences.
Table 3.
Demographics and clinical data for the validation cohort septic shock subclasses based on the majority call of 21 clinical evaluators.1
| Subclass A | Subclass B | Subclass C | P value | |
|---|---|---|---|---|
| No. of patients | 25 | 28 | 29 | -- |
| Median age in years (IQR)2 | 1.0 (0.2–3.1) | 5.7 (1.0–8.6) | 2.0 (1.3–5.4) | 0.005 |
| No. of males/females | 17/8 | 19/9 | 21/8 | 0.915 |
| No. of deaths (%) | 5 (20)3 | 4 (14) | 3 (10) | 0.520 |
| Maximum # of organ failures (IQR) 4 | 3 (2–4) | 2 (2–3) | 2 (2–3) | 0.032 |
| Median PRISM score (IQR) | 17 (11–28) | 11 (7–14) | 14 (9–17) | 0.014 |
| Median PICU free days (IQR) | 15 (1–20) | 21 (13–23) | 17 (9–24) | 0.191 |
| No. with co-morbidity (%)5 | 7 (28) | 16 (57) | 10 (34) | 0.071 |
| No. with immune suppression (%)6 | 1 (4) | 3 (11) | 3 (10) | 0.627 |
| No. receiving hydrocortisone (%)7 | 7 (28) | 16 (57) | 11 (36) | 0.088 |
| No. with gram pos. bacteria (%)8 | 8 (32) | 7 (25) | 9 (31) | 0.827 |
| No. with gram neg. bacteria (%) | 8 (32) | 8 (29) | 10 (34) | 0.891 |
| No. with negative cultures (%) | 6 (24) | 10 (36) | 9 (31) | 0.650 |
| Median WBC count × 103/mm3 (IQR) | 10 (4–14) | 16 (11–24) | 15 (6–18) | 0.009 |
| Median neutrophil count × 103/mm3 (IQR) | 7 (1–11) | 12 (8–21) | 10 (4–16) | 0.006 |
| Median lymphocyte count × 103/mm3 (IQR) | 1.8 (1.0–4.1) | 2.4 (0.9–3.5) | 1.6 (0.9–3.8) | 0.945 |
| Median monocyte count × 103/mm3 (IQR) | 0.5 (0.4–1.0) | 0.7 (0.3–1.0) | 0.5 (0.2–1.2) | 0.952 |
Continuous variables are analyzed as 3 group comparisons using ANOVA on Ranks and 2 degrees of freedom. Proportions are analyzed as 3 group comparisons using a 2 by 3 contingency table and Chi-square with 2 degrees of freedom.
Interquartile range (IQR).
Odds ratio for mortality vs. Subclass B: 1.5 (0.4–4.6); vs. Subclass C: 2.2 (0.5–10.2).
Refers to the maximum number of organ failures during the initial 7 days of PICU admission.
Refers to patients having any major diagnosis in addition to septic shock (e.g. trauma, sickle cell disease, congenital heart disease, liver failure, etc.)
Refers to patients with immune deficiency secondary to an intrinsic documented defect of the immune system, or patients receiving immune-suppressive medications (e.g. calcineurin inhibitors or high dose steroids).
For cardiovascular shock.
All bacterial culture data refer to samples obtained from bodily fluids that are normally sterile (i.e. blood, urine, cerebral spinal fluid, broncho-alveolar lavage, and/or peritoneal fluid).
Clinical characteristics of the combined cohorts
Combined with our previous report [6], we have allocated a total of 180 patients into expression-based subclasses. The major clinical characteristics of the respective subclasses are consistent when comparing the current validation cohort and the original derivation cohort [6]. Table 4 provides the major clinical differences between the 3 subclasses in this combined cohort. Subclass A patients have a significantly higher mortality rate and mortality odds ratio than patients in subclasses B and C. Subclass A patients also have a higher number of organ failures, a higher PRISM score, and fewer PICU free days than patients in subclasses B and C. Finally, subclass B patients are older and have a higher proportion of females, compared to subclasses A and C.
Table 4.
Major clinical characteristics of combined derivation and validation cohorts.1
| Subclass A | Subclass B | Subclass C | p value | |
|---|---|---|---|---|
| No. of patients | 55 | 69 | 56 | -- |
| Median age in years (IQR)2 | 0.7 (0.3–2.7) | 5.2 (1.9–7.3) | 2.2 (1.2–2.7) | <0.001 |
| No. of males/females | 39/16 | 33/36 | 37/19 | 0.020 |
| No. of deaths (%) | 16 (29)3 | 7 (10) | 7 (12) | 0.012 |
| Max. # of organ failures (IQR) | 3 (2–4) | 2 (2–3) | 2 (2–2) | <0.001 |
| Median PRISM score (IQR) | 19 (13–27) | 11 (10–15) | 12 (9–16) | <0.001 |
| Median PICU free days (IQR) | 11 (0–18) | 22 (17–24) | 19 (16–24) | 0.002 |
Continuous variables are analyzed as 3 group comparisons using ANOVA on Ranks and 2 degrees of freedom. Proportions are analyzed as 3 group comparisons using a 2 by 3 contingency table and Chi-square with 2 degrees of freedom.
Interquartile range (IQR).
Odds ratio for mortality vs. Subclass B: 3.6 (1.4– 9.6), p = 0.007; vs. Subclass C: 2.9 (1.1–7.7), p = 0.03.
Addressing potential confounding variables
Since we used whole blood-derived RNA it is possible that the class-defining gene expression patterns reflect differential white blood cell counts. Accordingly, we analyzed our expression data for the presence of previously published signature probe sets for neutrophils, lymphocytes, and monocytes, respectively [18]. Signature probe “presence” was defined using the following criteria: ≥100 raw expression value in a least one-half of the subjects in each subclass. Table 5 provides the results from this analysis and demonstrates that the signature probe sets were equally present across the 3 subclasses. Thus, the relative contributions of the three major leukocyte subsets, to the whole blood transcriptomic response, were relatively similar across the 3 subclasses. Accordingly, the differences in gene expression between the 3 subclasses do not appear to be artifacts of differential peripheral white blood cell counts.
Table 5.
Presence of leukocyte subset signature probes across the three gene expression-based subclasses. Data presented as number of signature probes present (see text for presence criteria).
| Neutrophil probes (n = 38) | Lymphocyte probes (n = 50) | Monocyte probes (n = 28) | |
|---|---|---|---|
| Subclass A | 33 | 43 | 20 |
| Subclass B | 33 | 43 | 21 |
| Subclass C | 33 | 44 | 20 |
The validation cohort had a significantly lower proportion of patients with negative cultures compared to the derivation cohort. Accordingly, we conducted a 2 group ANOVA (Benjamini-Hochberg False Discovery Rate = 5%) to determine if the 100 class-defining genes were differentially expressed between patients with negative cultures (n = 25) and patients with positive cultures (n = 57) in the validation cohort. This analysis revealed that none of the 100 subclass-defining genes were differentially expressed between the two groups, thus suggesting that microbiology culture status was unlikely to be a strong confounder.
The validation cohort also had a higher proportion of males compared to the derivation cohort. Accordingly, we conducted a 2 group ANOVA (Benjamini-Hochberg False Discovery Rate = 5%) to determine if the 100 class-defining genes were differentially expressed between males (n = 57) and females (n = 25) in the validation cohort. This analysis revealed that none of the 100 subclass-defining genes were differentially expressed between males and females, thus suggesting that gender was unlikely to be a strong confounder.
Since patients were recruited from multiple centers, it is possible that center-specific effects could confound the data. Table 6 provides the number of validation cohort patients in each gene expression-based subclass based on enrollment site. Thirty seven patients (45%) in the validation cohort were recruited from Site 1. Also, site 1 contributed a smaller proportion of subclass A patients, compared to subclass C patients. There were no other significant differences regarding enrollment among the other sites. Thus, we cannot rule out the possibility that the data are confounded, in part, by center-specific effects.
Table 6.
Number of patients in each gene expression-based subclass based on enrollment site.
| Subclass A | Subclass B | Subclass C | Total | |
|---|---|---|---|---|
| Site 1 | 81 | 11 | 18 | 37 |
| Site 2 | 2 | 1 | 1 | 4 |
| Site 3 | 1 | 1 | 4 | 6 |
| Site 4 | 1 | 3 | 0 | 4 |
| Site 5 | 4 | 4 | 2 | 10 |
| Site 6 | 3 | 0 | 1 | 4 |
| Site 7 | 1 | 1 | 2 | 4 |
| Site 8 | 1 | 0 | 0 | 1 |
| Site 9 | 3 | 0 | 1 | 4 |
| Site 10 | 2 | 1 | 1 | 4 |
| Site 11 | 1 | 2 | 1 | 4 |
p < 0.05 vs. subclass C (Chi-square with 2 degrees of freedom).
DISCUSSION
Tang et al. recently conducted a systematic review of microarray-based expression profiling studies in human sepsis [19]. One conclusion from this review is that the transcriptomic response is highly variable in human sepsis. Our previous data are consistent with this conclusion by demonstrating the potential existence of 3 subclasses of children with septic shock, as defined by variable gene expression. We have now prospectively validated that a 100-gene expression signature, depicted using visually intuitive mosaics, can be used to allocate patients with septic shock into subclasses having clinically relevant phenotypic differences and biologically relevant differential gene expression. While the subclassification strategy needs to be further refined, and one would expect that computer-based subclassification would be superior to that of clinician-based subclassification, we have nonetheless demonstrated that clinically relevant subclasses of children with septic shock can be identified via gene expression profiling.
The assertion that the subclasses have clinically relevant phenotypes is based on a higher level of illness severity in the subclass A patients, as measured by mortality, maximal number of organ failures, PRISM scores, and PICU free days. The profound negative impact of multiple organ failure on outcomes in critical illness is well established [20-22], and is therefore a clinically relevant measure. The reliance on PRISM scores could lead one to conclude that it would be more straightforward to calculate PRISM scores as a means of stratification. However, illness severity scores such as PRISM and APACHE are intended for population-based predictions, rather than for individual patient stratification, and do not provide biological information [23].
Patients in validation cohort subclass A had a trend toward higher mortality, which did not reach statistical significance. However, when we combined both the derivation and validation cohorts, the subclass A patients have a significantly higher mortality rate. Notably, the mortality rates of the patients in subclasses B and C are consistent with current estimates for the U.S. [24–26], whereas the mortality rate in subclass A patients is 3-fold higher.
The assertion that the subclasses have biologically relevant differences in gene expression is based on the functional significance of the 100 class-defining genes, which are enriched for genes corresponding to adaptive immunity [6, 27]. The majority of these genes corresponding to adaptive immunity are repressed in subclass A patients, relative to subclass B and C patients [6]. It is unlikely that lymphopenia accounts for this observation because the absolute lymphocyte counts were not significantly different between the 3 subclasses. Recent literature indicates that enhancement of adaptive immune function may be a rational therapeutic strategy in sepsis [28–33]. Optimization of such a strategy, however, will need to take into account the potential existence of subclass A patients, characterized by a higher illness severity and repression of adaptive immunity-related genes.
The 100 class-defining genes are also enriched for genes corresponding to glucocorticoid receptor signaling [6, 27], and these genes are also repressed in subclass A patients, relative to subclass B and C patients [6]. Glucocorticoid replacement therapy and the concept of relative adrenal insufficiency in septic shock are highly controversial topics in critical care medicine [34–36]. The potential existence of a subclass of patients with septic shock (i.e. subclass A) having a higher illness severity and repression of genes corresponding glucocorticoid receptor signaling, may have important implications for future clinical trials and may have been confounders in previous clinical trials.
Subclass A patients were significantly younger than subclass B patients in both our previous study [6] and the current study. This observation is consistent with pediatric septic shock epidemiology, which has identified young age as a risk factor for increased illness severity [24–26]. However, subclass C patients are of a similar age to subclass A patients, but have a lower illness severity in both studies. Thus, while younger age is a risk factor for illness severity, the current data indicate that the expression pattern of the 100 class-defining genes also impacts illness severity, independent of age.
Our subclassification strategy is focused on a single time point and therefore does not take into account potential temporal shifts in patient status from one subclass to another. However, the major goal of the strategy is to allocate patients into subclasses at a time point that affords earlier clinical management decisions or early stratification for clinical trials [4, 37, 38].
In conclusion, we have addressed the challenges of septic shock heterogeneity and stratification by prospectively validating the existence of septic shock subclasses based on a 100-gene expression signature. The subclasses can be identified at a clinically relevant time point and have relevant clinical differences. Finally, the expression patterns of the 100 class defining-genes may therapeutic implications.
Supplementary Material
The 100 class-defining gene probes.
Acknowledgments
Supported by grants from the National Institutes of Health: R01GM064619 and RC1HL100474
Footnotes
Dr. Thomas consulted for Discovery Laboratories. The remaining authors have not disclosed any potential conflicts of interest.
References
- 1.Wynn J, Cornell TT, Wong HR, Shanley TP, Wheeler DS. The host response to sepsis and developmental impact. Pediatrics. 125(5):1031–1041. doi: 10.1542/peds.2009-3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348(2):138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 3.Marshall JC. Sepsis: rethinking the approach to clinical research. J Leukoc Biol. 2008;83(3):471–482. doi: 10.1189/jlb.0607380. [DOI] [PubMed] [Google Scholar]
- 4.Marshall JC, Reinhart K. Biomarkers of sepsis. Crit Care Med. 2009;37(7):2290–2298. doi: 10.1097/CCM.0b013e3181a02afc. [DOI] [PubMed] [Google Scholar]
- 5.Wong HR. Pediatric septic shock treatment: new clues from genomic profiling. Pharmacogenomics. 2007;8(10):1287–1290. doi: 10.2217/14622416.8.10.1287. [DOI] [PubMed] [Google Scholar]
- 6.Wong HR, Cvijanovich N, Lin R, Allen GL, Thomas NJ, Willson DF, Freishtat RJ, Anas N, Meyer K, Checchia PA, et al. Identification of pediatric septic shock subclasses based on genome-wide expression profiling. BMC Med. 2009;7:34. doi: 10.1186/1741-7015-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong HR, Wheeler DS, Tegtmeyer K, Poynter SE, Kaplan JM, Chima RS, Stalets E, Basu RK, Doughty LA. Toward a clinically feasible gene expression-based subclassification strategy for septic shock: proof of concept. Crit Care Med. 38(10):1955–1961. doi: 10.1097/CCM.0b013e3181eb924f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldstein B, Giroir B, Randolph A. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005;6(1):2–8. doi: 10.1097/01.PCC.0000149131.72248.E6. [DOI] [PubMed] [Google Scholar]
- 9.Wong HR, Cvijanovich N, Allen GL, Lin R, Anas N, Meyer K, Freishtat RJ, Monaco M, Odoms K, Sakthivel B, et al. Genomic expression profiling across the pediatric systemic inflammatory response syndrome, sepsis, and septic shock spectrum. Crit Care Med. 2009;37(5):1558–1566. doi: 10.1097/CCM.0b013e31819fcc08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong HR, Shanley TP, Sakthivel B, Cvijanovich N, Lin R, Allen GL, Thomas NJ, Doctor A, Kalyanaraman M, Tofil NM, et al. Genome level expression profiles in pediatric septic shock indicate a role for altered zinc homeostasis in poor outcome. Physiol Genomics. 2007 doi: 10.1152/physiolgenomics.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pollack MM, Patel KM, Ruttimann UE. The Pediatric Risk of Mortality III--Acute Physiology Score (PRISM III-APS): a method of assessing physiologic instability for pediatric intensive care unit patients. J Pediatr. 1997;131(4):575–581. doi: 10.1016/s0022-3476(97)70065-9. [DOI] [PubMed] [Google Scholar]
- 12.Shanley TP, Cvijanovich N, Lin R, Allen GL, Thomas NJ, Doctor A, Kalyanaraman M, Tofil NM, Penfil S, Monaco M, et al. Genome-level longitudinal expression of signaling pathways and gene networks in pediatric septic shock. Mol Med. 2007;13(9–10):495–508. doi: 10.2119/2007-00065.Shanley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cvijanovich N, Shanley TP, Lin R, Allen GL, Thomas NJ, Checchia P, Anas N, Freishtat RJ, Monaco M, Odoms K, et al. Validating the genomic signature of pediatric septic shock. Physiol Genomics. 2008;34(1):127–134. doi: 10.1152/physiolgenomics.00025.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong HR, Freishtat RJ, Monaco M, Odoms K, Shanley TP. Leukocyte subset-derived genome-wide expression profiles in pediatric septic shock. Pediatr Crit Care Med. 2009 doi: 10.1097/PCC.0b013e3181c519b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 16.Eichler GS, Huang S, Ingber DE. Gene Expression Dynamics Inspector (GEDI): for integrative analysis of expression profiles. Bioinformatics. 2003;19(17):2321–2322. doi: 10.1093/bioinformatics/btg307. [DOI] [PubMed] [Google Scholar]
- 17.Guo Y, Eichler GS, Feng Y, Ingber DE, Huang S. Towards a holistic, yet gene-centered analysis of gene expression profiles: a case study of human lung cancers. J Biomed Biotechnol. 2006;2006(5):69141. doi: 10.1155/JBB/2006/69141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun L, Gorospe JR, Hoffman EP, Rao AK. Decreased platelet expression of myosin regulatory light chain polypeptide (MYL9) and other genes with platelet dysfunction and CBFA2/RUNX1 mutation: insights from platelet expression profiling. J Thromb Haemost. 2007;5(1):146–154. doi: 10.1111/j.1538-7836.2006.02271.x. [DOI] [PubMed] [Google Scholar]
- 19.Tang BM, Huang SJ, McLean AS. Genome-wide transcription profiling of human sepsis: a systematic review. Crit Care. 2010;14(6):R237. doi: 10.1186/cc9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baue AE, Durham R, Faist E. Systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), multiple organ failure (MOF): are we winning the battle? Shock. 1998;10(2):79–89. doi: 10.1097/00024382-199808000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Barie PS, Hydo LJ. Influence of multiple organ dysfunction syndrome on duration of critical illness and hospitalization. Arch Surg. 1996;131(12):1318–1323. doi: 10.1001/archsurg.1996.01430240072010. discussion 1324. [DOI] [PubMed] [Google Scholar]
- 22.Typpo KV, Petersen NJ, Hallman DM, Markovitz BP, Mariscalco MM. Day 1 multiple organ dysfunction syndrome is associated with poor functional outcome and mortality in the pediatric intensive care unit. Pediatr Crit Care Med. 2009;10(5):562–570. doi: 10.1097/PCC.0b013e3181a64be1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vincent JL, Opal SM, Marshall JC. Ten reasons why we should NOT use severity scores as entry criteria for clinical trials or in our treatment decisions. Crit Care Med. 38(1):283–287. doi: 10.1097/CCM.0b013e3181b785a2. [DOI] [PubMed] [Google Scholar]
- 24.Czaja AS, Zimmerman JJ, Nathens AB. Readmission and late mortality after pediatric severe sepsis. Pediatrics. 2009;123(3):849–857. doi: 10.1542/peds.2008-0856. [DOI] [PubMed] [Google Scholar]
- 25.Watson RS, Carcillo JA, Linde–Zwirble WT, Clermont G, Lidicker J, Angus DC. The epidemiology of severe sepsis in children in the United States. Am J Respir Crit Care Med. 2003;167(5):695–701. doi: 10.1164/rccm.200207-682OC. [DOI] [PubMed] [Google Scholar]
- 26.Watson RS, Carcillo JA. Scope and epidemiology of pediatric sepsis. Pediatr Crit Care Med. 2005;6(3 Suppl):S3–5. doi: 10.1097/01.PCC.0000161289.22464.C3. [DOI] [PubMed] [Google Scholar]
- 27.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437(7061):1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 28.Hotchkiss RS, Opal S. Immunotherapy for sepsis--a new approach against an ancient foe. N Engl J Med. 363(1):87–89. doi: 10.1056/NEJMcibr1004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levy Y, Lacabaratz C, Weiss L, Viard JP, Goujard C, Lelievre JD, Boue F, Molina JM, Rouzioux C, Avettand-Fenoel V, et al. Enhanced T cell recovery in HIV-1-infected adults through IL-7 treatment. J Clin Invest. 2009;119(4):997–1007. doi: 10.1172/JCI38052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg SA, Sportes C, Ahmadzadeh M, Fry TJ, Ngo LT, Schwarz SL, Stetler-Stevenson M, Morton KE, Mavroukakis SA, Morre M, et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J Immunother. 2006;29(3):313–319. doi: 10.1097/01.cji.0000210386.55951.c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sereti I, Dunham RM, Spritzler J, Aga E, Proschan MA, Medvik K, Battaglia CA, Landay AL, Pahwa S, Fischl MA, et al. IL-7 administration drives T cell-cycle entry and expansion in HIV-1 infection. Blood. 2009;113(25):6304–6314. doi: 10.1182/blood-2008-10-186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sportes C, Hakim FT, Memon SA, Zhang H, Chua KS, Brown MR, Fleisher TA, Krumlauf MC, Babb RR, Chow CK, et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J Exp Med. 2008;205(7):1701–1714. doi: 10.1084/jem.20071681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Unsinger J, McGlynn M, Kasten KR, Hoekzema AS, Watanabe E, Muenzer JT, McDonough JS, Tschoep J, Ferguson TA, McDunn JE, et al. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J Immunol. 184(7):3768–3779. doi: 10.4049/jimmunol.0903151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sprung CL, Goodman S, Weiss YG. Steroid therapy of septic shock. Crit Care Clin. 2009;25(4):825–834. x. doi: 10.1016/j.ccc.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 35.Annane D, Sebille V, Charpentier C, Bollaert PE, Francois B, Korach JM, Capellier G, Cohen Y, Azoulay E, Troche G, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. Jama. 2002;288(7):862–871. doi: 10.1001/jama.288.7.862. [DOI] [PubMed] [Google Scholar]
- 36.Sprung CL, Annane D, Keh D, Moreno R, Singer M, Freivogel K, Weiss YG, Benbenishty J, Kalenka A, Forst H, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358(2):111–124. doi: 10.1056/NEJMoa071366. [DOI] [PubMed] [Google Scholar]
- 37.Osuchowski MF, Connett J, Welch K, Granger J, Remick DG. Stratification is the key: inflammatory biomarkers accurately direct immunomodulatory therapy in experimental sepsis. Crit Care Med. 2009;37(5):1567–1573. doi: 10.1097/CCM.0b013e31819df06b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong HR, Cvijanovich N, Wheeler DS, Bigham MT, Monaco M, Odoms K, Macias WL, Williams MD. Interleukin-8 as a stratification tool for interventional trials involving pediatric septic shock. Am J Respir Crit Care Med. 2008;178(3):276–282. doi: 10.1164/rccm.200801-131OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The 100 class-defining gene probes.