

Abstract
Calcium- and integrin-binding protein 1 (CIB1) has been shown to be involved in cell spreading and migration. The signaling events regulated by CIB1 during cell migration are poorly understood. Here we found that accumulation of CIB1 at the tip of the filopodia requires an intact cytoskeleton. Depletion of CIB1 using shRNA affects formation of FAK- and phosphotyrosine-rich focal adhesions without affecting stress fiber formation. Overexpression of CIB1 results in cell migration on fibronectin and Erk1/2 MAP kinase activation. CIB1-induced cell migration is dependent upon Erk1/2 activation, since it is inhibited by the MEK-specific inhibitor PD98059. Furthermore, CIB1-induced cell migration, as well as Erk1/2 activation, is dependent on PKC, Src family kinases as well as PI-3 kinase as it is inhibited by bisindolylmaleimide 1, PP2, and wortmannin respectively in a dose-dependent manner. Co-expression of dominant-negative Cdc42 completely abolished CIB1-induced cell migration. Additionally, co-expression of constitutively active, but not dominant negative PAK1, a CIB1 binding protein, inhibited CIB1-induced cell migration. These results suggest that CIB1 positively regulates cell migration and is necessary for the recruitment of FAK to the focal adhesions. Furthermore, CIB1-induced cell migration is dependent on MAP kinase signaling and its function is attenuated by PAK1.
Keywords: CIB1, Cell migration, PAK-1, Cdc42, FAK
INTRODUCTION
The dynamic interaction of a cell with extracellular matrix (ECM) proteins plays an important role in wound healing, defense against infection, tumor metastasis, and cell migration, proliferation, and differentiation [Hynes, 2002]. Cells interact with the ECM through proteins called integrins. Integrins are heterodimers composed of α and β subunits, each of which contains a large extracellular domain, a transmembrane domain, and a short cytoplasmic domain. As a result of ligand binding, integrin clustering occurs and is critical for the activation of intracellular signaling [Miyamoto et al., 1995], which includes signal transduction events such as the activation of protein kinases, an increase of intracellular calcium levels, and cytoskeleton reorganization [Aplin et al., 1998; Juliano et al., 2004]. The integrin-mediated signals lead to the formation of focal adhesions where integrins associate with the cytoskeleton through their cytoplasmic domain [Pavalko and Otey, 1994]. These events are crucial for cell adhesion to ECM and the process of cell migration. One of the major signaling pathways activated downstream of integrins during cell migration is the MAP kinase signaling cascade [Miyamoto et al., 1996; Sastry et al., 1999; Zhu and Assoian, 1995]. Although some of the players in the signaling cascade induced by integrins leading to MAP kinase activation have been identified, significant gaps still exist.
CIB1 is a 22 kDa calcium-binding protein, which specifically binds to the cytoplasmic domain of integrin αIIb in platelets [Naik et al., 1997]. CIB1 (also known as calmyrin or kinase interacting protein Kip) contains two functional calcium-binding EF-hand domains [Blamey et al., 2005]. CIB1 is anchored to the platelet membrane through myristoylation. Upon platelet activation, CIB1 initially concentrates at the filopodia through its association with the cytoskeleton [Naik and Naik, 2003b; Shock et al., 1999]. CIB1 mRNA and protein are widely distributed in a variety of tissues, suggesting that it has cellular functions independent of integrin αIIbβ3, which is specific to platelets [Naik and Naik, 2003a; Shock et al., 1999]. In addition to integrin αIIbβ3, CIB1 has been shown to interact with a variety of proteins with diverse functions, such as kinases, phosphatases, ion channels, and cytoskeletal proteins [Heineke et al., 2010; Kauselmann et al., 1999; Tsuboi et al., 2006]. Consistent with its ability to bind and regulate these proteins, it has been shown to be involved in a variety of processes, such as spermatogenesis, thrombosis, cardiac hypertrophy, and angiogenesis [Heineke et al., 2010; Naik et al., 2009; Tsuboi, 2002; Yuan et al., 2006; Zayed et al., 2007]. The role of CIB1 in regulating some of these processes could be attributed to its ability to regulate cell spreading and cell motility [Leisner et al., 2005; Naik and Naik, 2003a; b]. In this report, we show that in adherent cells, the filopodial localization of CIB1 is actin cytoskeleton-dependent. CIB1 supports cell migration on fibronectin (Fn) by augmenting FAK- and phosphotyrosine-rich focal adhesion formation. We also show that CIB1-induced cell migration requires signaling through PI-3 kinase, PKC, and SFKs, eventually leading to the activation of the MAP kinase-signaling cascade. Furthermore, we show that PAK1, one of the binding partners of CIB1, inhibits CIB1-induced cell migration.
MATERIAL AND METHODS
REAGENTS
Fibronectin and vitronectin was purchased from BD Bioscience (San Jose, CA). Wortmannin, cytochalasin D, bisindolylmaleimide 1, BAPTA-AM, gelatin, and genistein were purchased from Sigma (St. Louis, MO); herbimycin A, PP2, and LY29002 were from Calbiochem (Darnstadt, Germany). Vinculin monoclonal antibody was a generous gift from Dr. Keith Burridge (University of North Carolina at Chapel Hill, NC). Phosphospecific Erk1/2 antibody, anti-Erk1/2, MAP Kinase inhibitor PD98059, and anti-VE-Cadherin were purchased from Cell Signaling (Danvers, MA). Polyclonal antibodies against c-Src, FAK, PAK1, and HSC-70 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Human fibrinogen (Fg) was purchased from Enzyme Research (South Bend, IN). Generation of anti-CIB1 monoclonal antibody was described previously [Naik et al., 1997]. All other reagents used were of analytical grade from Sigma.
CELL CULTURE AND TRANSFECTION
Chinese hamster ovary (CHO) and human breast cancer (T47D) cells were obtained from American Tissue Culture Collection (ATCC; Manassas, VA). Cells were maintained in DMEM or RPMI supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin or 10 μg/ml insulin. Human umbilical cord vein endothelial cells (HUVECs) were obtained from Lonza (Walkersville, MD) and maintained in the medium provided by the manufacturer. Ectopic expression of CIB1 and the empty vector (mock) transfected in CHO cells using lipofectamine 2000 reagent (Invitrogen; Carlsbad, CA) or T47D cells by Qiagen (Qiagen Science, MD) following the manufacturer’s protocol as described previously [Naik et al., 2001]. Geneticin (G418) was added to the culture medium 24 h after transfection at a final concentration of 600 μg/ml. The resistant colonies were isolated to obtain single cell clones or a pool of stably transfected cells and maintained in culture medium containing 300 μg/ml of G418. In a separate set of experiments, CHO cells expressing integrin αIIbβ3 (B7 cells) [Naik and Naik, 2003a] were transiently transfected with DN-Cdc42 (Cdc42N17) or its effector PAK1 or DN-PAK1 (PAK1K299R) alone or co-transfected with recombinant CIB1. Downregulation of CIB1 using short hairpin RNA (shRNA) was performed as previously reported [Naik and Naik, 2011]. The amounts of expression of various proteins were determined by immunoblotting. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2. All cDNA constructs used were verified by sequencing.
HUMAN PLATELET PREPARATION
Approximately 50 ml of whole blood was drawn by venipuncture from healthy aspirin-free volunteers older than 18 years, under informed consent. Approval was obtained from the University of Delaware Institutional Review Board for these studies according to the Declaration of Helsinki. Blood was collected in 6.5 ml of acidified citrate dextrose (pH 4.5) as an anticoagulant. Platelet-rich plasma (PRP) was obtained and platelets were washed as described previously [Naik and Naik, 2003b]. Washed platelet suspension was adjusted to 2 × 108 platelets/ml as required.
IMMUNOFLUORESCENCE
Immunofluorescence studies were conducted according to the procedure described previously [Naik and Naik, 2003a]. Briefly, serum-starved cells were treated with various inhibitors and their relevant controls as indicated for 20 min and than allowed to spread on Fn (10 μg/ml) pre-coated Lab-Tek II 8-chambered coverglass slides (Nunc, Naperville, IL). In a separate set of experiments washed platelets (6 μl) 0.6 to 1.0 × 106 /ml were allowed to adhere on Fg (20 μg/ml) pre-coated slides at various time points. The cells were then fixed in freshly prepared 4% paraformaldehyde (PF) in phosphate buffer saline (PBS) for 10 min, rinsed, and permeabilized in 0.2% Triton X-100 for 5 min. Cells were then blocked with 3% bovine serum albumin (BSA) in PBS (blocking solution) for 1 h, and incubated with anti-CIB1 (1:100) or anti-FAK (1:100) or 4G10 (1:100) or anti-Vinculin (1:100) at 4°C overnight. After incubation, cells were washed three times with blocking solution and incubated with rhodamine-conjugated donkey anti-mouse IgG secondary antibody (1:300) or FITC-conjugated donkey anti-rabbit antibody (1:300) for 1 h at room temperature (RT). For staining of F-actin, FITC-conjugated phalloidin (1:500) was included during incubation time along with the secondary antibody. Cells were washed three times with blocking solution, followed by a final wash of PBS. Slowfade was added to minimize fading of the fluorescence intensity. Confocal microscopy was performed using a Zeiss LSM510 laser-scanning microscope (Ithaca, NY). Images were processed using Adobe Software.
MIGRATION ASSAYS
Scratch assay
Downregulation of CIB1-induced cell migration was monitored by 2 different assays, both of which have been widely used for assessing cell migration. In a qualitative wound-healing assay [Naik et al., 2003], HUVECs transfected with shRNA alone or CIB1shRNA were serum-starved after 48 h of transfection for 8 h, detached by Versene and plated (500,000cells/ml) on 24-well plate Fn pre-coated to form a monolayer. A confluent monolayer was scratched by use of a sterile yellow pipette tip and rinsed with serum-free medium. Photographs were taken at time zero by a phase-contrast microscope (Nikon). The cells were allowed to migrate in the scratch area under serum-free conditions for 8 h at 37°C. The cells were stained with a Diff-Quick (Dade Diagnostics, Newark, DE) staining kit, following the manufacturer’s instructions. The extent of migration in the scratch area was documented.
Transwell assay
A haptotactic transwell motility assay was performed as described previously [Naik and Naik, 2003a]. Briefly, membranes of 8 μm pore size inserts (costar) in triplicates were coated underneath with various extracellular matrices (ECM) overnight at 4°C, and simultaneously the mock or CIB1-overexpressing cells were serum-starved (DMEM containing 5 mg/ml BSA) overnight. After detachment by trypsinization, enzyme activity was neutralized using 1 mg/ml soybean trypsin inhibitor. In a separate set of experiments, HUVECs transfected with CIB1shRNA or shRNA were serum starved for 8 h. Washed cells were diluted at 500,000 cells/ml and kept at 37°C for 30 min before plating. A 100 μl of cell suspension was added to the upper compartment of the pre-coated insert and serum-free medium was added in the lower compartment of the insert. Migration through the pores was allowed to proceed for 5 h at 37°C. In the case of inhibitor treatment, prior to the addition of cells into the inserts, cells in suspension were incubated with or without the appropriate inhibitor for 20 min, and then allowed to migrate in the presence of inhibitor for 5 h under serum-free condition. The unmigrated cells were removed from the top compartment of the insert with a cotton tip applicator; whereas migrated cells to the underside of the insert were fixed in freshly prepared 4% PF/PBS for 10 min and stained with the Diff-Quick solution. Inserts were dried and the stained cells were then counted from ten different views per insert in triplicates using Phase-contrast microscope. Experiments were repeated independently more than three times. Data collected were analyzed and plotted using Sigma plot software.
IMMUNOPRECIPITATION AND WESTERN BLOTTING
Immunoprecipitation studies were performed as described previously [Naik and Naik, 2003b]. Briefly, washed (2 × 108) platelets were allowed to spread on immobilized Fg pre-coated 100mm bacterial tissue culture dishes and BSA was used as a control. Platelet lysates were precleared with isotype-specific antibody and immunoprecipitated with appropriate primary antibodies as indicated. The immunocomplex was processed by Western blotting. Mock or CIB1-overexpressing cell lysates (50 μg/ml) were subjected to 10–12% SDS-PAGE and transferred onto a PVDF membrane (Bio-Rad). The membranes were blocked with 3% BSA and incubated with primary antibodies as indicated, washed and further incubated with appropriate secondary antibodies conjugated with horseradish peroxidase. The bands were visualized using Lumiglo reagent.
STATISTICAL ANALYSIS
All experiments presented are results from at least three independent experiments. Statistical analysis was performed using student’s t-test; Data are mean ± s.e.m; P< 0.05 were considered as statistically different.
RESULTS
AN INTACT CYTOSKELETON IS REQUIRED FOR THE LOCALIZATION OF CIB1 AT THE TIP OF THE FILOPODIA
When analyzed, the subcellular localization of CIB1 in CHO cells adhered to Fn; we found that it colocalizes with F-actin and concentrates at the tip of the filopodia (Fig. 1Ai). Since CIB1 is a calcium-binding protein, we asked if the presence of intracellular calcium is necessary for its localization to the filopodia. Intracellular calcium was chelated by treating the CHO cells with BAPTA-AM. We found that upon chelation of intracellular calcium, the cells appeared spherical in shape without any visible F-actin or filopodia formation and CIB1 remained uniformly associated with the membrane (Fig. 1Aii). Similar results were obtained when calcium was chelated by adding BAPTA-AM after they were attached and spread on Fn. Since calcium is important for several cellular events, including filopodia formation, it could not be concluded that the observed mislocalization is solely due to the lack of calcium binding to CIB1. To investigate if actin cytoskeletal organization is required for CIB1 to concentrate at the filopodia, we pretreated CHO cells with cytochalasin D (cyto D), the known actin-depolymerizing agent, and then allowed them to adhere to Fn. The depolymerization of actin filaments with cyto D had a significant effect on subcellular localization of CIB1. Instead of being associated with the membrane, CIB1 was now clustered at one place (Fig. 1Aiii). However, treatment of cells with cyto D after they have been allowed to spread on Fn did not cause such clustering of CIB1, suggesting that the actin cytoskeleton is necessary for the proper localization of CIB1 to the membrane and filopodia (Fig. 1Aiv).
Figure 1.

Intact cytoskeleton is necessary for CIB1 localization at the tip of the filopodia. Confocal images of serum-starved CHO cells spread on Fn matrix. A (i): Untreated cells and localization of CIB1 (red) at the tip of the filopodia is shown by arrows. (ii): Treated with (10 μM) BAPTA-AM. (iii): Treated with (1 μM) cytochalasin D (pre-cyto D) in suspension and than allowed on spread on Fn for 1 h at 37°C. (iv): Cells were allowed to attach to Fn for 1 h and than treated with cytochalasin D (post-cyto D) and kept for additional 15 min, fixed and stained with anti-CIB1 (red) and FITC-phalloidin (green). Localization of CIB1 (red) in ‘A(ii & iii)’ is shown by arrowheads. Scale bar, A (i–iii); 10 μm and (iv); 20 μm). Ectopic expression of CIB1 enhances cell migration on Fn. B: Western blot analysis indicates protein expression level in CIB1-overexpressing cells as compared to the mock cells (upper panel); equal protein loading is shown by blotting with HSC-70 (lower panel). C: Quantitation of transwell migration assay of CIB1-overexpressing or mock cells plated on various ECM coated inserts in triplicates. D: Quantitation of transwell migration assay of CIB1-overexpressing or mock T47D cells. Ten random views of each insert in triplicates were counted. Data is expressed as average number of cells per view. Each experiment was repeated independently three times.
OVEREXPRESSION OF CIB1 AUGMENTS CELL MIGRATION ON FN
In order to study the role of CIB1 in the process of cell migration, we stably overexpressed recombinant CIB1 in CHO cells (Fig. 1B). Cells transfected with an empty vector (Mock) were used as controls. Mock or CIB1-overexpressing cells adhered well to Fn, and CIB1 overexpression resulted in a significant enhancement of cell migration on Fn, but not on gelatin, collagen or vitronectin (Fig. 1C), as shown previously by our laboratory [Naik and Naik, 2003a]. In order to establish that the observed effect of CIB1 is general and not specific to the CHO cell specific, we overexpressed CIB1 in T47D, a breast carcinoma cell line. We found an enhanced migration, similar to CHO cells, suggesting that the effect of CIB1 is not cell-type specific (Fig. 1D).
DEPLETION OF CIB1 AFFECTS CELL MIGRATION
Since overexpression of CIB1 induces cell migration, we reasoned that depletion of endogenous CIB1 should attenuate cell migration. We used vector-based shRNA specific to CIB1 to deplete endogenous CIB1 from HUVE cells. We were able to deplete over 90% of CIB1 without affecting other endogenous proteins such as HSC-70, PAK1, and VE-cadherin (Fig. 2A). We next performed a scratch assay, a widely used method, in addition to the trans-well assay to assess cell migration to determine the ability of CIB1 depleted cells to migrate on Fn. As expected, cells transfected with vector alone as control migrated into the wounded area within 8 h. On the other hand, CIB1shRNA transfected cells failed to migrate into the wounded area during that time (Fig. 2B). To obtain a more quantitative measure of this effect, we performed a transwell migration assay. As expected, depletion of CIB1 attenuated cell migration by 50% (Fig. 2C), further supporting the role for CIB1 in regulating cell migration.
Figure 2.

Knockdown of endogenous CIB1 by shRNA inhibited cell migration. A: Lysates from HUVECs transfected with shRNA or CIB1shRNA were Western blotted with various antibodies to check the protein expression level as indicated. B: A scratch assay using cells as in A, were monitored from time zero for 8 h under serum-free conditions. C: Quantitation of transwell migration assay of shRNA or CIB1shRNA HUVECs. Data is expressed as average number of cells per view. Each experiment was repeated three times.
CIB1-INDUCED CELL MIGRATION ON FIBRONECTIN IS PKC AND ERK DEPENDENT
Since the Erk1/2 activation pathway is crucial for cell migration, we determined if CIB1 overexpression augments this pathway. Indeed, CIB1 overexpression markedly enhanced Erk1/2 phosphorylation (Fig. 3A). This phosphorylation of Erk1/2 was dose-dependently inhibited when cells were pretreated with bisindolylmaleimide 1, a general PKC inhibitor as well as an MEK inhibitor PD98059. When the cell morphology was observed, CIB1 overexpressing cells spread well with numerous stress fibers and membrane ruffles (Fig. 3B). As expected, CIB1 colocalized with F-actin predominantly at the membrane ruffles. Interestingly, pretreatment with bisindolylmaleimide 1 completely inhibited lamellipodia formation. CIB1, however, remained localized to the tips of the filopodia (Fig. 3B). When tested for the role of PKC in CIB1-induced cell migration, inhibition of PKC dose-dependently inhibited CIB1-induced cell migration (Fig. 3C). Furthermore, inhibition of Erk1/2 MAP kinase pathway by MEK inhibitor PD98059 also inhibited CIB1-induced cell migration (Fig. 3D). These results suggest that PKC and Erk1/2 are downstream of CIB1.
Figure 3.

CIB1-induced cell migration inhibited by PKC and MEK inhibitors. A: Western blot analysis of cell lysates from mock or CIB1- overexpressing cells treated with DMSO or with 10 μM inhibitors. Blots were incubated with phospho-specific anti-Erk1/2 (p-Erk1/2) and reprobed for total Erk1/2 to ensure equal loading of proteins in the lanes. Shown is a representative blot from three separate experiments. B: Confocal images of serum-starved cells treated with DMSO or 10 μM PKC inhibitor for 20 min. CIB1 localization at the tip of the filopodia and intact actin-fibers (upper panel), treatment with the bisindolylmaleimide I, a protein kinase C inhibitor (BIS) (lower panel; scale bar 10 μm). C&D: Quantitative analysis of transwell migration assay of mock or CIB1-overexpressing cells untreated or treated with BIS or PD98059, MEK inhibitor and allowed to migrate on Fn for 5 h. Triplicate inserts were used for each treatment. Ten random views of each insert were counted. Data is expressed as percent control. Number of mock cells migrating across the membrane was considered 100 percent. Each experiment was repeated independently three times. *P<0.05 vs untreated CIB1 overexpressed (control).
CIB1-INDUCED CELL MIGRATION IS PI-3 KINASE DEPENDENT
It has been shown that PI-3 kinase plays a significant role in cell migration [Hall, 1998]. In order to elucidate the role of this signaling pathway in CIB1-induced cell migration on Fn, we treated the cells with wortmannin, a PI-3 kinase inhibitor. Pretreatment of cells with wortmannin substantially inhibited CIB1-induced Erk1/2 activation (Fig. 4A). Furthermore, we found that, similar to PKC inhibition, PI-3 kinase inhibition affected cell spreading, but not CIB1 localization to the filopodia (Fig. 4B). Interestingly, CIB1-induced cell migration was dose-dependently inhibited without much effect on basal cell migration (Fig. 4C). Similar results were obtained when another PI-3K specific inhibitor, LY29002, was used (data not shown). These results suggest that PI-3K is downstream of CIB1.
Figure 4.

CIB1-induced cell migration is inhibited by PI-3 kinase inhibitor.
A: Western blot analysis of cell lysates from mock or CIB1-overexpressing cells treated with DMSO or with 10 nM wortmannin (Wort). Immunoblots were incubated with phospho-specific anti-Erk1/2 (p-Erk1/2) and the blots were reprobed for total Erk1/2. Shown is a representative blot from three separate experiments. B: Confocal images of DMSO or 10 nM wortmannin treated cells spread on Fn (scale bar 10 μm). C: Quantitative analysis of transwell migration assay of mock or CIB1-overexpressing cells untreated or treated with various concentrations of wortmannin as indicated were allowed to migrate on Fn for 5 h. Triplicate inserts were used for each treatment. Ten random views of each insert were counted. Data is expressed as percent control. Number of mock cells migrating across the membrane was considered 100 percent. Each experiment was repeated independently three times. *P<0.05 vs untreated CIB1 overexpressed.
CIB1-INDUCED CELL MIGRATION IS SRC FAMILY KINASE DEPENDENT
It is well accepted that a number of non-receptor tyrosine kinases such as SFKs are activated during cell migration. In order to determine if any tyrosine kinases are downstream of CIB1, we pretreated mock and CIB1-overexpressing CHO cells with genistein, a general tyrosine kinase inhibitor. We found that genistein did not inhibit CIB1-induced Erk1/2 activation (Fig. 5A). Treatment of cells with genistein, although substantially reducing cellular levels of phospho-tyrosine, it did not affect cell spreading and CIB1 localization at the filopodia and membrane ruffles (Fig. 5B). Furthermore, genistein treatment had no effect on CIB1-induced cell migration even at a concentration of 500 μg/ml (Fig. 5C). These results preliminarily suggest that CIB1-induced cell migration is independent of tyrosine kinases.
Figure 5.

Src family kinase inhibitors, but not genistein, affects CIB1-induced cell migration. A: Western blot analysis of lysates from mock or CIB1-overexpressing cells treated with DMSO or with 10 μM genistein or 1 μM PP2. Immunoblots were incubated with phospho-specific anti-Erk1/2 (p-Erk1/2) and the blots were reprobed for total Erk1/2. Shown is a representative blot from three separate experiments. B: Confocal images of DMSO or 10 μM genistein treated cells. Cells were stained for phospho-tyrosine (PY20) and CIB1. C: Quantitative analysis of transwell migration assay of mock or CIB1-overexpressing cells untreated or treated with various concentrations of genistein as indicated. D: Confocal images of DMSO or 10 μM herbimycin treated cells. Cells were stained for F-actin and CIB1. E&F: Quantitative analysis of transwell migration assay of mock or CIB1-overexpressing cells untreated or treated with various concentrations of inhibitors as indicated. Three separate inserts for each treatment were used. Ten random views of each insert were counted. Data is expressed as percent control. Number of mock cells migrating across the membrane was considered 100 percent. Each experiment was repeated three times. Scale bar 10 μm.
We next examined the role of SFKs on CIB1-induced cell migration. We found that herbimycin A, a pharmacological inhibitor of SFKs, inhibited cell spreading and CIB1 colocalization with F-actin at the membrane ruffles and filopodia (Fig. 5D). Furthermore, herbimycin A dose-dependently inhibited CIB1-induced cell migration on Fn. Interestingly, herbimycin A only had an effect on CIB1-induced cell migration and not on basal cell migration (Fig. 5E). To further confirm the role of SFKs in CIB1-induced cell migration, we used PP2, a selective and potent inhibitor of SFKs. Pretreatment of cells with PP2 significantly inhibited CIB1-induced Erk1/2 activation (Fig. 5A). Interestingly, PP2 at lower concentrations specifically inhibited CIB1-induced migration and at higher concentrations inhibited both CIB1-induced as well as basal cell migration (Fig. 5F) confirming the role of SFKs in CIB1-induced cell migration.
Src association with CIB1-FAK complex and its activity is enhanced during cell spreading
To further assess the role of c-Src, a member of SFK, we used platelets as a model system. Platelets, when allowed to attach to immobilized Fg through integrin αIIbβ3, initially form filopodia and then spread by developing lamellipodia [Naik and Naik, 2003b]. We determined the distribution of CIB1 and c-Src during various stages of platelet spreading on immobilized Fg. CIB1 and c-Src were found to colocalize around the membrane of the discoid platelets (Fig. 6A). However, as the platelets attached to Fg, both CIB1 and c-Src were accumulated and co-localized at the tip of the filopodia (Fig. 6A). As spreading continued, lamellipodia were extended and CIB1 redistributed from the tip of the filopodia to the edge of the fully spread platelet where CIB1 remained co-localized with c-Src at the membrane periphery (Fig. 6A). A population of c-Src, however, remained concentrated at the center of the spread platelet that was not associated with CIB1. This observation demonstrated that distribution of CIB1 and a subset of c-Src is localized along with the membrane of discoid platelets; upon attachment to Fg, these proteins colocalize to a defined area as platelets progress through different stages of spreading.
Figure 6.
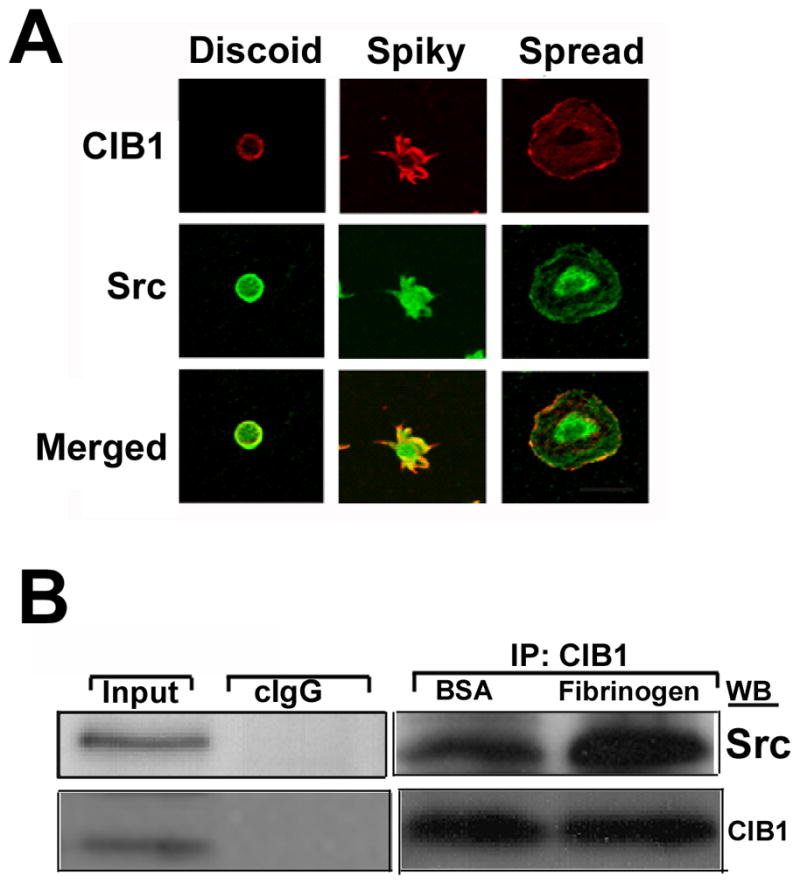
CIB1 and Src associate when platelets spread on immobilized Fg. A: Confocal images of washed platelets at various stages of spreading on immobilized Fg. Shown are the representative images of three different progressive stages of platelet spreading on immobilized Fg. Scale bar 20μm. B: Western blot analysis of platelet lysates exposed to BSA or attached on immobilized Fg. CIB1 was immunoprecipitated and the immunocomplex was subjected to Western blotting. The above membrane was probed with anti-Src (upper panel), and again reprobed with anti-CIB1 to ensure equal loading (lower panel).
Since the c-Src is a possible kinase that is involved in platelet spreading on immobilized Fg during integrin outside–in signaling, we next sought to determine if Src was present in the CIB1 immunoprecipitates. We found that the CIB1 immunoprecipitate from both BSA and Fg attached platelets contained Src kinase, but in platelets attached to Fg had ~two-fold more Src associated with CIB1 as compared to platelets on BSA (Fig. 6B).
Taken together, these results suggest that CIB1-induced cell migration on Fn is dependent upon SFKs, PKC, PI-3 kinase and Erk1/2-mediated signaling pathways.
DEPLETION OF CIB1 ATTENUATE FAK-RICH FOCAL ADHESION FORMATION
We used vector-based shRNA to deplete endogenous CIB1 expression in CHO cells. CIB1-depleted CHO cells adhere and spread normally (Fig. 7A). However, these cells do not form FAK-containing focal adhesions as indicated by the complete absence of FAK at the end of the actin stress fibers (Fig. 7A). We next tested whether depletion of CIB1 affects focal adhesion formation or if FAK fails to associate with the focal adhesions in the absence of CIB1. Mock-transfected cells readily formed focal adhesions, as visualized by phosphotyrosine staining, and FAK was present at these focal adhesions (Fig. 7B). Interestingly, in the case of CIB1-depleted HUVECs, while they did form focal adhesions, their numbers were substantially reduced as indicated by decreased vinculin containing focal adhesion (Fig. 7C). The quantification of this data suggests a significant decrease in focal adhesion formation in the cells depleted of CIB1 (Fig. 7D). Furthermore, depletion of CIB1 renders focal adhesion to be completely devoid of FAK (Fig. 7A&B). These results suggest that CIB1 is needed for the efficient formation and recruitment of FAK to the focal adhesions. The reduced amount of focal adhesion seen in CIB1-depleted cells could be due to leftover CIB1 since the depletion was not complete.
Figure 7.

Knockdown of endogenous CIB1 by shRNA impaired FAK and vinculin localization, A: Confocal images of CHO cells transiently transfected with pSuper (upper panel) or CIB1 shRNA (lower panel) and stained for F-actin and FAK. B: Same cells as in ‘A’ stained for phosphotyrosine and FAK. C: Confocal images of HUVECs transfected with shRNA (upper panel) or CIB1shRNA (lower panel) stained for F-actin and vinculin. D: Quantification of data from ‘C’. Scale Bar; 20 μm.
PAK1 INHIBITS CIB1-INDUCED CELL MIGRATION ON FN
Since CIB1 specifically localizes to the tip of the filopodia, we asked if filopodia formation is necessary for CIB1-induced cell migration. To address this, we expressed Cdc42N17, a dominant negative form of Cdc42, in CHO cells stably expressing integrin αIIbβ3. The expression was assessed by Western blot analysis. Inhibition of filopodia formation by the expression of Cdc42N17 abolished CIB1-induced cell migration on Fg without affecting basal cell migration, suggesting that filopodia formation and probably CIB1 localization at the filopodia are necessary for cell migration (Fig. 8A). PAK1 is a known downstream effector of Cdc42 and is shown to interact with CIB1 and inhibit cell migration [Leisner et al., 2005; Manser et al., 1994]. To assess the involvement of PAK1, we co-expressed PAK1 with CIB1. We found that expression of PAK1 did not affect cell migration in mock cells, but it completely abolished CIB1-induced cell migration (Fig. 8B). We next asked if PAK1 activity is necessary for this inhibition. To test this, we co-expressed a DN- PAK1 with CIB1. Interestingly, expression of dominant negative PAK1 significantly inhibited basal cell migration, but it had no inhibitory effect on CIB1-induced cell migration (Fig. 8B). In fact, the cell migration was augmented (from ~1.5 to ~3.5 fold) when dominant negative PAK1 was co-expressed with CIB1. These results suggest that CIB1-induced cell migration requires Cdc42 and is negatively regulated by PAK1.
Figure 8.

DN-Cdc42 as well as PAK1, but not DN-PAK cotransfected with recombinant CIB1 attenuated CIB1-induced cell migration. A&B: Quantitative analysis of transwell migration assay of mock or CIB1-overexpressing cells in the presence or absence of (A) DN-Cdc42, or (B) constitutively active (PAK1) or DN-PAK1. Transfected cells were allowed to migrate on Fn for 5 h. Triplicate inserts were used for each treatment. Ten random views of each insert were counted. Data is expressed as percent control. Number of mock cells migrating across the membrane was considered 100 percent. Each experiment was repeated independently three times. *P<0.05 vs untreated CIB1 overexpressed.
DISCUSSION
CIB1 is a widely expressed calcium-binding protein that interacts with a variety of signaling proteins, including the cytoplasmic domain of megakaryocytes and platelet specific integrin αIIb subunit [Naik and Naik, 2003a; Naik et al., 1997]. Previously, we have shown that upon platelet activation by agonists, CIB1 associates with the actin cytoskeleton and is involved in platelet spreading [Naik and Naik, 2003b; Shock et al., 1999]. Here we show the involvement of CIB1 in the signaling pathway that leads to motility of cells independent of its interaction with integrin αIIbβ3. We also show that CIB1 is needed for proper formation of FAK- and phosphotyrosine-rich focal adhesions. The observed induction of cell migration by CIB1 is independent of its localization to the filopodia, but it is dependent on the formation of lamellipodia. We have shown that CIB1 and FAK associate to form a functional complex as determined by co-immunoprecipitation experiments [Naik and Naik, 2003a]. Furthermore, CIB1 has been shown to colocalize at the focal adhesions and that overexpression of CIB1 augments, whereas depletion attenuates, focal adhesion formation. How CIB1 activates FAK is not well understood and is currently being investigated. A recent report suggests that SNARE-mediated membrane trafficking is required for integrin dependent focal adhesion turnover through a FAK/Src/PI-3K-dependent pathway [Skalski et al., 2010]. It is therefore possible that CIB1 may regulate cell migration through regulation of membrane trafficking.
Our observation that CIB1 did not induce cell migration on collagen or vitronectin could probably be due to the lack of expression of specific integrins that bind to these ECM proteins. The signaling pathway induced by CIB1 appears to be canonical, that includes SFKs, PI-3 kinase and PKC. These molecules have been previously implicated downstream of integrin activation leading to integrin dependent MAP kinase activation and cell migration [Ng et al., 1999; Playford and Schaller, 2004; Zhu and Assoian, 1995]. More specifically, activation of FAK has been shown to regulate MAP kinases downstream of integrin by physically associating with Src, PI-3K, and Grb2 [Zhao and Guan, 2010]. The observation that genistein failed to inhibit CIB1-induced cell migration despite its ability to reduce overall tyrosine phosphorylation is intriguing. Although genistein has been originally reported to be a general tyrosine kinase inhibitor, it appears that it is more specific to growth factor receptor tyrosine kinases [Akiyama et al., 1987; Kovacs et al., 2009]. For instance, it has been shown that protein tyrosine kinase inhibitor herbimycin A, but not genistein, specifically inhibits signaling through T-cell antigen receptor [Graber et al., 1992]. FAK is activated downstream of both growth factor receptors as well as integrins. However, in our experiments, we performed cell migration independent of growth factors; it is therefore possible that CIB1-dependent FAK activation and hence cell migration is not affected by genistein.
Small GTPase family members have been shown to play a pivotal role in cell migration. Of these, RhoA induces stress fiber formation, Rac induces lamellipodia formation, and Cdc42 induces filopodia formation. Since CIB1 specifically concentrates at the tip of filopodia, it is logical to expect a role for CIB1 in Cdc42 signaling. The observation that the DN-Cdc42 completely abolished CIB1-induced migration suggests that CIB1-induced cell migration is Cdc42 dependent. Furthermore, CIB1 has been shown to interact with PAK1, a downstream effector of Cdc42, and enhance its activity in fibroblasts and transformed epithelial cells [Leisner et al., 2005]. It was further shown that CIB1-induced PAK1 activation is inhibitory to cell migration due to an LIM kinase-dependent increase in cofilin phosphorylation [Leisner et al., 2005]. However, the same group later showed that genetic ablation of CIB1 reduced endothelial cell migration on Fn and adhesion dependent PAK1 and Erk1/2 activation [Zayed et al., 2007]. Since PAK1 is also known to promote cell migration, it appears that PAK1 is able to regulate cell migration both positively and negatively [Ching et al., 2007; Huynh et al., 2010]. It appears that the effect of PAK1 on cell migration is cell-type specific and is regulated by small GTPase-dependent and independent pathways [Zayed et al., 2007]. PI-3 kinase has been shown to interact with PAK1 and regulate cytoskeletal reorganization in a small GTPase-independent manner [Papakonstanti and Stournaras, 2002]. Our observation that DN-PAK1 inhibits endogenous cell migration, but enhances CIB1-induced cell migration supports the possibility that CIB1-induced cell migration is positively regulated by canonical Erk1/2 pathway in a Cdc42-dependent manner, which is negatively regulated by PAK1. Although beyond the scope of this work, further studies will be required to conclusively determine the role of PAK1 in CIB1-induced cell migration by knockdown of PAK1 using shRNA or PAK1 knockout mouse cells.
The results presented here show that CIB1 is a regulator of cell migration. During adhesion-dependent signaling, CIB1 concentrates at the tip of the filopodia, which requires dynamic cytoskeletal reorganization. CIB1 is required for the formation of FAK- and phosphotyrosine-rich focal adhesions. CIB1 augments the canonical signaling pathway induced by integrin binding to ECM leading to Erk1/2 activation, probably through its ability to activate FAK. Thus, inhibition of SFKs, PI-3K, PKC, and MEK affects CIB1-induced lamellipodia formation as well as cell migration. CIB1-induced cell migration is Cdc42 dependent, but is negatively regulated by its effector PAK1. How PAK1 affects CIB1-induced cell migration is the topic of future investigation.
Acknowledgments
Dr. Patricia J. Keely (University of Wisconsin, Madison, WI) for giving the constructs for DN-PAK1, PAK1, Cdc42 and DN-Cdc42. The authors thank Kushal U. Naik for critically editing the manuscript.
Contract grant sponsor: National Institutes of Health;
Contract grant number: (UPN) from HL57630
References
- Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem. 1987;262:5592–5. [PubMed] [Google Scholar]
- Aplin AE, Howe A, Alahari SK, Juliano RL. Signal transduction and signal modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol Rev. 1998;50:197–263. [PubMed] [Google Scholar]
- Blamey CJ, Ceccarelli C, Naik UP, Bahnson BJ. The crystal structure of calcium- and integrin-binding protein 1: insights into redox regulated functions. Protein Sci. 2005;14:1214–21. doi: 10.1110/ps.041270805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching YP, Leong VY, Lee MF, Xu HT, Jin DY, Ng IO. P21-activated protein kinase is overexpressed in hepatocellular carcinoma and enhances cancer metastasis involving c-Jun NH2-terminal kinase activation and paxillin phosphorylation. Cancer Res. 2007;67:3601–8. doi: 10.1158/0008-5472.CAN-06-3994. [DOI] [PubMed] [Google Scholar]
- Graber M, June CH, Samelson LE, Weiss A. The protein tyrosine kinase inhibitor herbimycin A, but not genistein, specifically inhibits signal transduction by the T cell antigen receptor. Int Immunol. 1992;4:1201–10. doi: 10.1093/intimm/4.11.1201. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–14. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Heineke J, Auger-Messier M, Correll RN, Xu J, Benard MJ, Yuan W, Drexler H, Parise LV, Molkentin JD. CIB1 is a regulator of pathological cardiac hypertrophy. Nat Med. 2010;16:872–9. doi: 10.1038/nm.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh N, Liu KH, Baldwin GS, He H. P21-activated kinase 1 stimulates colon cancer cell growth and migration/invasion via ERK- and AKT-dependent pathways. Biochim Biophys Acta. 2010;1803:1106–13. doi: 10.1016/j.bbamcr.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Juliano RL, Reddig P, Alahari S, Edin M, Howe A, Aplin A. Integrin regulation of cell signalling and motility. Biochem Soc Trans. 2004;32:443–6. doi: 10.1042/BST0320443. [DOI] [PubMed] [Google Scholar]
- Kauselmann G, Weiler M, Wulff P, Jessberger S, Konietzko U, Scafidi J, Staubli U, Bereiter-Hahn J, Strebhardt K, Kuhl D. The polo-like protein kinases Fnk and Snk associate with a Ca(2+)- and integrin-binding protein and are regulated dynamically with synaptic plasticity. Embo J. 1999;18:5528–39. doi: 10.1093/emboj/18.20.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs D, Cardinali G, Aspite N, Picardo M. Bovine colostrum promotes growth and migration of the human keratinocyte HaCaT cell line. Growth Factors. 2009;27:448–55. doi: 10.3109/08977190903211077. [DOI] [PubMed] [Google Scholar]
- Leisner TM, Liu M, Jaffer ZM, Chernoff J, Parise LV. Essential role of CIB1 in regulating PAK1 activation and cell migration. J Cell Biol. 2005;170:465–76. doi: 10.1083/jcb.200502090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–6. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Akiyama SK, Yamada KM. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science. 1995;267:883–5. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Teramoto H, Gutkind JS, Yamada KM. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: roles of integrin aggregation and occupancy of receptors. J Cell Biol. 1996;135:1633–42. doi: 10.1083/jcb.135.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik MU, Naik UP. Calcium-and integrin-binding protein regulates focal adhesion kinase activity during platelet spreading on immobilized fibrinogen. Blood. 2003a;102:3629–36. doi: 10.1182/blood-2003-05-1703. [DOI] [PubMed] [Google Scholar]
- Naik MU, Naik UP. Calcium- and integrin-binding protein 1 regulates microtubule organization and centrosome segregation through polo like kinase 3 during cell cycle progression. Int J Biochem Cell Biol. 2011;43:120–9. doi: 10.1016/j.biocel.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik MU, Nigam A, Manrai P, Millili P, Czymmek K, Sullivan M, Naik UP. CIB1 deficiency results in impaired thrombosis: the potential role of CIB1 in outside-in signaling through integrin alpha IIb beta 3. J Thromb Haemost. 2009;7:1906–14. doi: 10.1111/j.1538-7836.2009.03581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik MU, Vuppalanchi D, Naik UP. Essential role of junctional adhesion molecule-1 in basic fibroblast growth factor-induced endothelial cell migration. Arterioscler Thromb Vasc Biol. 2003;23:2165–71. doi: 10.1161/01.ATV.0000093982.84451.87. [DOI] [PubMed] [Google Scholar]
- Naik UP, Naik MU. Association of CIB with GPIIb/IIIa during outside-in signaling is required for platelet spreading on fibrinogen. Blood. 2003b;102:1355–62. doi: 10.1182/blood-2003-02-0591. [DOI] [PubMed] [Google Scholar]
- Naik UP, Naik MU, Eckfeld K, Martin-DeLeon P, Spychala J. Characterization and chromosomal localization of JAM-1, a platelet receptor for a stimulatory monoclonal antibody. J Cell Sci. 2001;114:539–47. doi: 10.1242/jcs.114.3.539. [DOI] [PubMed] [Google Scholar]
- Naik UP, Patel PM, Parise LV. Identification of a novel calcium-binding protein that interacts with the integrin alphaIIb cytoplasmic domain. J Biol Chem. 1997;272:4651–4. doi: 10.1074/jbc.272.8.4651. [DOI] [PubMed] [Google Scholar]
- Ng T, Shima D, Squire A, Bastiaens PI, Gschmeissner S, Humphries MJ, Parker PJ. PKCalpha regulates beta1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J. 1999;18:3909–23. doi: 10.1093/emboj/18.14.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papakonstanti EA, Stournaras C. Association of PI-3 kinase with PAK1 leads to actin phosphorylation and cytoskeletal reorganization. Mol Biol Cell. 2002;13:2946–62. doi: 10.1091/mbc.02-01-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavalko FM, Otey CA. Role of adhesion molecule cytoplasmic domains in mediating interactions with the cytoskeleton. Proc Soc Exp Biol Med. 1994;205:282–93. doi: 10.3181/00379727-205-43709. [DOI] [PubMed] [Google Scholar]
- Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23:7928–46. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- Sastry SK, Lakonishok M, Wu S, Truong TQ, Huttenlocher A, Turner CE, Horwitz AF. Quantitative changes in integrin and focal adhesion signaling regulate myoblast cell cycle withdrawal. J Cell Biol. 1999;144:1295–309. doi: 10.1083/jcb.144.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shock DD, Naik UP, Brittain JE, Alahari SK, Sondek J, Parise LV. Calcium-dependent properties of CIB binding to the integrin alphaIIb cytoplasmic domain and translocation to the platelet cytoskeleton. Biochem J. 1999;342(Pt 3):729–35. [PMC free article] [PubMed] [Google Scholar]
- Skalski M, Sharma N, Williams K, Kruspe A, Coppolino MG. SNARE-mediated membrane traffic is required for focal adhesion kinase signaling and Src-regulated focal adhesion turnover. Biochim Biophys Acta. 2010;1813:148–58. doi: 10.1016/j.bbamcr.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Tsuboi S. Calcium integrin-binding protein activates platelet integrin alpha IIbbeta 3. J Biol Chem. 2002;277:1919–23. doi: 10.1074/jbc.M110643200. [DOI] [PubMed] [Google Scholar]
- Tsuboi S, Nonoyama S, Ochs HD. Wiskott-Aldrich syndrome protein is involved in alphaIIb beta3-mediated cell adhesion. EMBO Rep. 2006;7:506–11. doi: 10.1038/sj.embor.7400665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Leisner TM, McFadden AW, Clark S, Hiller S, Maeda N, O’Brien DA, Parise LV. CIB1 is essential for mouse spermatogenesis. Mol Cell Biol. 2006;26:8507–14. doi: 10.1128/MCB.01488-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayed MA, Yuan W, Leisner TM, Chalothorn D, McFadden AW, Schaller MD, Hartnett ME, Faber JE, Parise LV. CIB1 regulates endothelial cells and ischemia-induced pathological and adaptive angiogenesis. Circ Res. 2007;101:1185–93. doi: 10.1161/CIRCRESAHA.107.157586. [DOI] [PubMed] [Google Scholar]
- Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2010 Nov 29; doi: 10.1016/j.addr.2010.11.001. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Assoian RK. Integrin-dependent activation of MAP kinase: a link to shape-dependent cell proliferation. Mol Biol Cell. 1995;6:273–82. doi: 10.1091/mbc.6.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]