

Abstract
Oncolytic virotherapy has shown substantial promises as an alternative therapeutic modality for solid tumors in both preclinical studies and clinical trials. The main therapeutic activity of virotherapy derives from the direct lytic effect associated with virus replication and the induction of host immune responses to the infected tumor cells. Here we show that some human and murine tumor cell lines are highly resistant to the lytic effect of a type II herpes simplex virus-derived oncolytic virus, FusOn-H2, which was constructed by deleting the N-terminal region of the ICP10 gene. However, these tumor cells still respond exceptionally well to FusOn-H2 virotherapy in vivo. Histological examination of the treated tumors revealed that, in contrast to tumors supporting FusOn-H2 replication, implants of these highly resistant lines showed massive infiltration of neutrophils after virotherapy. Further analysis showed that there is a correlation between an intrinsically strong interferon response activity and the recruitment of neutrophils in these tumors. These results suggest that an innate immune response mainly represented by neutrophils may be part of the virotherapy-mediated antitumor mechanism in these tumors.
Keywords: oncolytic virus, cancer virotherapy, herpes simplex virus, neutrophil infiltration, innate immunity
Introduction
A persistent criticism of many emerging cancer treatments is that their beneficial effects extend only to a subset of patients. This limitation tends to be more common with biotherapeutic interventions such as immunotherapy and gene therapy. For example, studies by Morgan et al showed that only 2 of 15 patients receiving infusions of their own modified T-cells responded with clearly objective regressions of metastatic melanoma 1. With a few notable exceptions, such as the strong link between a mutated epidermal growth factor (EGFR) gene and clinical responses to its tyrosine kinase inhibitor Iressa 2, 3, the mechanisms accounting for the heterogeneous responses of tumors to biotherapy remain poorly understood. New insight into these mechanisms could greatly accelerate progress in the development of effective biotherapeutic agents for use in cancer patients.
Virotherapy is a strategy in which a virus that preferentially replicates in tumor cells is applied either locally or systemically to lyse such cells 4. Unlike typical forms of gene-based cancer therapy, oncolytic viruses are thought to kill tumor cells directly through selective replication/cytolysis and consequent spread to surrounding tumor tissues. These properties represent a major advantage over the inherent inefficiency of gene delivery and the resultant limited tumor cell killing of conventional gene therapies. Several viruses, including adenovirus 5, herpes simplex virus 6, retrovirus 7, vaccinia virus 8, measles virus 9 and vesicular stomatitis virus 10 have been modified for oncolytic purposes. These viruses can be derived either from naturally occurring viruses that preferentially target tumor cells 10 or from genetically engineered viruses that target cancer cells by a defined molecular mechanism 6, 11-13. Despite only a relatively short history of research and development, several oncolytic viruses are being tested in clinical trials against tumors of different tissue origins; in general, they have shown excellent safety profiles and some have produced indications of efficacy 14. However, as with many other biotherapeutic approaches, the clinical utility of virotherapy is restricted by the generally small group of patients with favorable responses. In one recent clinical trial, 26 patients were treated with an oncolytic virus derived from a type I herpes simplex virus (HSV-1), but only three had favorable responses 15. This and similar outcomes underscore the need to understand why some tumors (but not others) respond well to treatment with oncolytic viruses.
We recently developed an oncolytic virus based on a type II herpes simplex virus (HSV-2) by deleting the N-terminal region of the ICP10 gene from the viral genome 16. Designated FusOn-H2, it has multiple antitumor mechanisms and has shown potent oncolytic activity against tumor cells of different tissue origins 16-18. Yet, several tumor cell lines that we have screened in vitro show almost highly resistant to the replication of FusOn-H2 and thus its oncolytic effect. This suggests that patients whose tumor cells are nonpermissive to FusOn-H2 replication in vitro would be unresponsive to FusOn-H2 virotherapy. Here we show that an intrinsically strong interferon response activity underlies the resistance of murine and human tumors to FusOn-H2 lytic activity, but does not preclude a favorable therapeutic response. To the contrary, treatment of implanted tumors with FusOn-H2 virotherapy led to their massive infiltration and destruction by neutrophils, suggesting a need to re-examine current strategies of virotherapy for cancer patients.
Materials and Methods
Cell lines and viruses
The EC9706 cell line was kindly provided by Dr. Mingrong Wang (Chinese Academy of Medical Sciences). Panc02-H7 and MiaPaCa-2 cells were gifts from Dr. Min Li (Baylor College of Medicine). The remainder of the cell lines were purchased from ATCC. EC9706 designates a human esophageal carcinoma line 19, LLC a murine Lewis lung carcinoma line, and Panco2-H7 a murine pancreatic cancer line 20. All of the cells were cultured in DMEM containing 10% fetal bovine serum (FBS).
FusOn-H2 was derived from the wild-type HSV-2 strain 186 (wt186). The details of its construction are described elsewhere 16. Viral stocks were prepared by infecting Vero cells with 0.01 plaque-forming units (pfu) per cell. Viruses were harvested 2 days later and purified as described 21. The purified viruses were titrated, divided into aliquots and stored at -80°C until use.
Plasmid construction and assays of endogenous interferon response activity
pJ-ISRE-P contained a murine ISG56 promoter with five copies of interferon-stimulated response element (ISRE; AGTTTCACTTTCCAGTCTCAGTTTCAGTTTCT ) that were synthesized by DNA 2.0 (Menlo Park, CA). The sequence of the ISG56 promoter with ISREs was derived from the Gene bank (#S77714S1). pJ-ISRE-SEAP was constructed by inserting the gene encoding the secreted form of alkaline phosphatase (SEAP) and SV40-polyA signal into the HindIII and HpaI site of pJ-ISRE-P.
To measure ISRE activity, we seeded tumor cells in 24-well plates in duplicate and transfected them with 1 μg of pJ-ISRE-SEAP or control plasmid (pcDNA-GFP). In some experiments, 1000 units of IFN-α, β or γ were added to the medium immediately after the plasmid transfection. Supernatants were collected daily from day 1 to day 13 according to experimental design. SEAP in the supernatants was quantified with the Great EscAPe SEAP Chemiluminescence Detection Kit from Clontech Laboratories, Inc. (Mountain View, CA).
Characterization of viral growth
Cells were seeded in triplicate into 24-well plates at 50% density. On the next day, they were infected with the test viruses at 0.1 pfu/cell for 1 h and washed once with PBS to remove unabsorbed and uninternalized viruses before fresh medium was added. At 48 h postinfection, the cells were harvested, and viruses were released by repeated freezing and thawing and sonication. Virus titers were determined on Vero cells by a plaque assay.
Real-time PCR assays
RNA was extracted from tumor cells with Trizol Reagent from Invitrogen (Carlsbad, CA). Human IFN-β and ISG56 transcripts were detected by two-step real-time PCR (RT-PCR), using the TaqMan® Gene Expression Assay kit. ISG56 primer sequences were: ISG56 forward: GGGAGTTATCCATTGATGACGATGA; ISG56 reverse: GGTGTCTAGGAATTCAATCTGATCCAA; FAM-labeled ISG56 probe: ATGCCTGATTTAGAAAACA. RT-PCR was performed according to the TaqMan® Gene Expression Assay protocol with the ABI Prism® 7700 Sequence Detection System (Foster City, CA). The reaction was run with the following cycling conditions. After being held at 50°C for 2 min and at 95°C for 10 min, the samples were run for 40 cycles including 95°C for 15 sec and 60°C for 1 min. RT-PCR data were analyzed with the ABI Prism® 7700 Sequence Detection System.
Experimental Animals
All animal experiments and procedures were approved by Animal Care and Use Committees of Baylor College of Medicine and University of Houston. Female Hsd athymic (nu/nu) mice (obtained from Harlan, Indianapolis, Indiana) were kept under specific pathogen-free conditions and used in experiments when they attained the age of 5 to 6 weeks. EC9706 cells were harvested from subconfluent cultures by a brief exposure to 0.25% trypsin and 0.05% EDTA. After trypsinization was stopped with medium containing 10% FBS, the cells were washed once in serum-free medium and resuspended in PBS. On day 0, 5×106 EC9706 cells were inoculated into the right flank of nude mice. Two weeks after tumor cell implantation, when the tumors reached approximately 3-5 mm in diameter, mice received a single intratumor injection of either 3×106 or 6×104 pfu of FusOn-H2 in a volume of 100 μl, or the same volume of PBS. The tumors were measured weekly and their volumes determined by the formula: tumor volume [mm3] = (length [mm]) × (width [mm])2 × 0.52. For histological examination, mice were euthanized by CO2 exposure at days 1, 2, 3 and 5 after receiving intratumoral injection of FusOn-H2. Tumor tissues were explanted and sectioned for H&E staining. For immunohistochemical staining for neutrophils, tumor sections were initially incubated with a rat anti-mouse neutrophil monoclonal antibody (NIMP-R14) purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The second antibody is goat-anti-rat IgG (Invitrogen, Carlsbad, CA) and the third antibody is HRP-labeled donkey-anti-goat IgG (Vector Laboratories, Burlingame, CA). The slides were then stained with Vectastain ABC kit (Vector Laboratories, Burlingame, CA).
For isolation of neutrophils from tumors, excised tumor tissues were minced in 1 ml of Hanks’ balanced saline solution (HBSS) containing 0.05% collagenase and 0.002% DNase I and filtered through a 70 μm cell strainer (BD Falcon, Bedford, MA). After centrifugation (400× g, 10 min, 4°C), the pellets were resuspended in HBSS with 15 mM EDTA and 1% bovine serum albumin (BSA). The cell suspensions were then loaded onto a three-layer Percoll gradient of 78%, 69%, and 52%, respectively (Amersham Pharmacia Biotech, Uppsala, Sweden), and centrifuged at 1,500× g for 30 min at room temperature without braking. Neutrophils were retrieved from the 69%/78% interface and transferred into BSA 1%-coated tubes. The collected neutrophils were washed twice with HBSS–EDTA–BSA buffer and resuspended in 1640 medium. To isolate neutrophils from the peritoneal cavity, we injected mice intraperitoneally with EC9706 cells infected with FusOn-H2 or mock-infected. Twenty-four h later, the peritoneal cavity was washed with 1640 medium, and the cells collected by centrifugation (400× g, 10 min, 4°C), resuspended in HBSS with 15 mM EDTA and 1% bovine serum albumin (BSA) and loaded onto a Percoll gradient for separation and collection.
For measurement of neutrophil cytotoxicity against tumor cells, EC9706 cells (at 20×106 cells/ml) were initially labeled with 10 μM CFSE (Molecular Probes Europe, Leiden, the Netherlands) for 10 min at 37°C. The labeling was stopped by addition of an equal volume of fetal calf serum. After 2 washes with PBS, the labeled tumor cells were resuspended in 1640 medium. Then, 3×105 tumor cells were mixed with the purified neutrophils at different ratios in a total 200 μl volume per well in 96-well plates and incubated at 37°C for 24 h. The cells were harvested and further stained with propidium iodide (PI, 1 μg/ml) before they were analyzed with BD FACSAria flow cytometer (BD Biosciences, San Jose, California) to quantify dead (double staining for PI and CFSE) and viable (single staining for CFSE) EC9706 cells. The percentage of surviving cells was calculated by dividing the number of input viable CFSE cells by the number of CFSE-stained viable cells 24 h after incubation with neutrophils.
For in vitro neutrophil migration assay, neutrophils were freshly purified from healthy C57BL6 mouse blood as described by Boxio et al 22. The viability of neutrophil population was confirmed as being >95% by trypan blue exclusion staining. The purified neutrophils were used immediately in the matrigel invasion assay with the BD BioCoat Matrigel Invasion Chambers (BD Biosciences, cat. no. 354480) as described elsewhere 23. Briefly, tumor cells (with or without FusOn-H2 infection) were seeded on the lower wells of the chambers at a density of 5 × 105. The chambers were agitated for 30 min in a shaker to allow adsorption. After that, neutrophils at a density of 25 × 104 were seeded into the top wells of the inserts in serum free medium. After 22 h of incubation at 37°C in a 5% CO2 atmosphere, the non-migrating cells were removed from the upper surface of the inserts with a cotton swab. The cells clinging on the lower side of the insert were fixed with ice-cold methanol and stained with Hoechst 33258 (300nM) (Sigma-Aldrich, cat. no. 861405). Six images covering the membrane were randomly taken under microscope, and the mean number of migrating cells was determined. Neutrophils were identified by staining and recognizing the nucleus's characteristic multilobulated shape with Türk's solution (Cat # 109277, Merck, Whitehouse Station, NJ).
Statistical analysis
Quantitative results are reported as means and standard errors. Statistical analyses were performed by the two-way t test. P values of less than 0.05 were considered statistically significant.
Results
Permissiveness of tumor cells to FusOn-H2 replication
Since the construction of FusOn-H2, we have characterized this oncolytic virus in more than two dozen tumor cell lines derived from different tissues of both humans and mice. FusOn-H2 efficiently lysed most of the tumor cells that we screened and effectively shrank tumors established from these cells when injected either locally or systemically 16-18. However, approximately 20% of the tumor cell lines were highly resistant to FusOn-H2 replication. In contrast to the fully permissive tumor cells, in which the input virus replicated as much as 100-fold within 48 h after infection (Fig. 1), the yield of FusOn-H2 in each of five tumor cell lines representing esophageal carcinoma, cervical cancer, lung carcinoma, melanoma and pancreatic cancer, barely increased over the same time period (Fig. 1). In most cases, the oncolytic virus can infect the tumor cells, as indicated by the expression of green fluorescent protein (GFP) gene was inserted into the viral genome during its construction 16. The blockage of virus growth in these tumor cells mainly occurred during virus replication. Because the therapeutic effect of an oncolytic virus is believed to depend mainly on its ability to replicate and spread, these results indicate that FusOn-H2 would be largely ineffective against tumors established from these cell lines.
Figure 1. FusOn-H2 replication in tumor cells of different tissue origins.
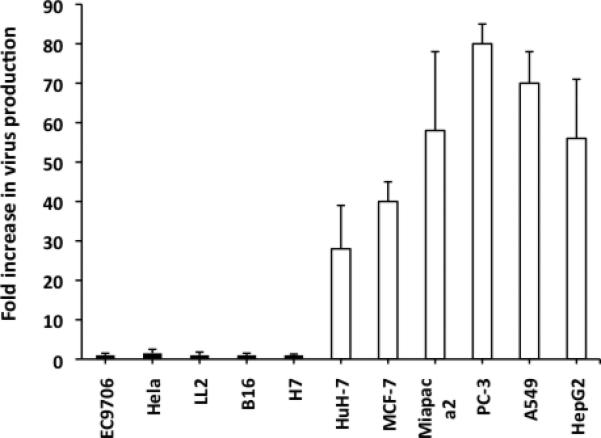
Cells were seeded in 24-well plates in duplicate and infected with FusOn-H2 at 0.1 pfu per cell for 1 h. Cells were washed and harvested with or without 24 h incubation. The fold increase in viral replication was calculated by dividing the virus titer at 24 h after infection by the values of titer for the same cells harvested immediately after washing without incubation. The data are reported as means of triplicate experiments.
FusOn-H2 is highly effective against implanted tumors established from some of the cancer cell lines resistant to viral replication
As tumor cell resistance to viral replication generally predicts a poor response to virotherapy, we initially excluded tumors established from such resistant cells from in vivo evaluation of the antitumor effects of FusOn-H2. Recently, however, we elected to include several highly resistant tumor cell lines in our in vivo experiments, primarily as negative controls. Surprisingly, a single injection of FusOn-H2 at 3×106 plaque-forming units (pfu) produced a dramatic antitumor effect, nearly eradicating tumors established from implants of EC9706 cells, which are resistant to FusOn-H2 replication (Fig. 2). This effect was essentially duplicated when the virus dose was reduced 50-fold, to as low as 6 × 104 pfu. Other than tumor disappearance, the animals showed no sign of toxicity during the virotherapy. Together, these observations suggest that FusOn-H2 destroyed the highly resistant tumor cells in vivo through mechanisms other than direct oncolysis.
Figure 2. Therapeutic effect of FusOn-H2 against established EC9706 tumors.
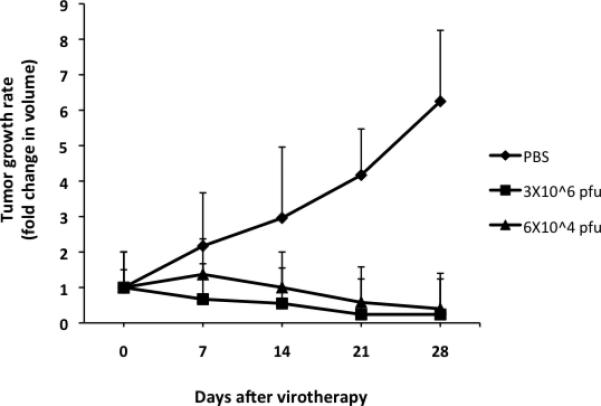
Tumors were initially established by implanting 5×106 EC9706 cells in the right flank of nude mice. Once tumors reached the approximate size of 5 mm in diameter, they were injected with FusOn-H2 at a dose of 3×106 or 6×104 pfu. Tumors were measured weekly post-treatment, and the tumor growth rate was determined by dividing the tumor volume before treatment by the tumor volume after treatment.
FusOn-H2 induces massive infiltration of neutrophils into resistant tumors
To account for the unexpected antitumor effects of FusOn-H2 virotherapy, we initially established tumors from EC9706 or 4T1 cells (a murine mammary tumor line that is significantly more permissive than EC9706 to FusOn-H2 replication but otherwise is similar to EC9706 in that they both form tumors aggressively once implanted into mice). After their injection with FusOn-H2, the tumors were harvested at days 1, 2, 3 and 5 for histological examination. The results revealed a massive infiltration of neutrophils in EC9706 tumors treated with FusOn-H2 (Fig. 3, blue arrows). The inner areas of the tumors were almost entirely filled with neutrophils; the few remaining tumor cells did not appear healthy (white arrow in Fig. 3a). Tumor cells near the periphery seemed viable and formed a ring surrounding the inflamed interior (Fig. 3d). Infiltrating neutrophils were much less common in EC9706 tumors treated with PBS (Fig. 3b) and were virtually undetectable in 4T1 tumors, which showed obvious oncolytic effects due to robust FusOn-H2 replication (Fig. 3c). In a subsequent experiment, we inoculated EC9706 tumor cells into both flanks and treated tumors on the right flank with FusOn-H2. The treated tumors shrank, but tumor growth on the opposite flank was not affected (data not shown). Histological examination of the untreated tumors did not reveal any increases in neutrophil infiltration (Fig. 3e), indicating that the massive infiltration of neutrophils was a regional effect that was directly associated with virus infection.
Figure 3. Massive infiltration of resistant tumors by neutrophils after treatment with FusOn-H2.
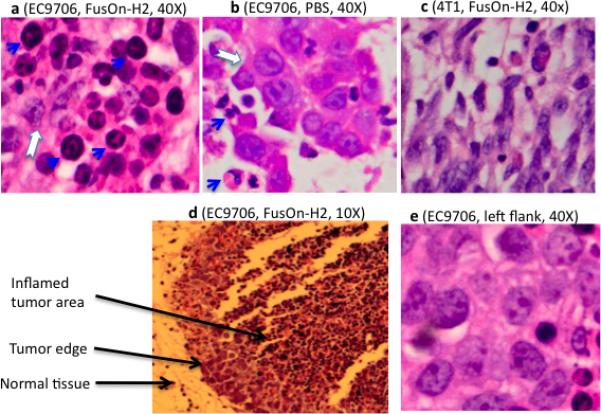
Highly resistant EC9706 (a, b, d and e) or permissive 4T1 (c) tumor cells were implanted on the right flank a, b, c, d) or on both flanks (e) of female nu/nu mice. Once tumors reached the approximate size of 5 mm in diameter, FusOn-H2 or PBS was injected into the tumors on the right flank, as indicated. Tumors were explanted on days 1, 2, 3 and 5 and sectioned for H&E staining. The sections shown here represent day 2 after virus or PBS administration. Blue arrows indicate infiltration; the white arrow marks degenerating tumor cells.
To further characterize the infiltrating neutrophils, we conducted another in vivo experiment. Established EC9706 tumors were injected with either 3×106 pfu of FusOn-H2 or the same amount of virus that had been inactivated by UV radiation, or PBS. Tumors were explanted 2 days later and divided into halves. One half was used for preparation of frozen sections for visualization of virus infection by examining GFP expression under a fluorescent microscope. As FusOn-H2 contains the GFP gene, the virus infectivity could be conveniently determined by this way. The other half of tumors was used for preparation of paraffin sections for immunohistochemical staining of neutrophils. The results were shown in Fig. 4. The micrographs, taken at a low magnitude (10X) from sections immunohistochemically stained for neutrophils, showed that there was a widespread neutrophil infiltration in tumors treated with FusOn-H2 (Fig. 4a). However, the extent of neutrophil infiltration was drastically reduced in tumors treated with the inactivated virus (Fig. 4b,f), indicating that virus infectivity was probably necessary for the induction of neutrophil infiltration. Neutrophils were not readily visible in untreated tumors, suggesting that these tumors were not intrinsically associated with neutrophil infiltration. Similar neutrophil infiltration was also detected in tumors established from another highly resistant tumor cells, B16 murine melanoma, after FusOn-H2-virotherapy (supplement data).
Fig. 4. Further characterization of the infiltrating neutrophils.
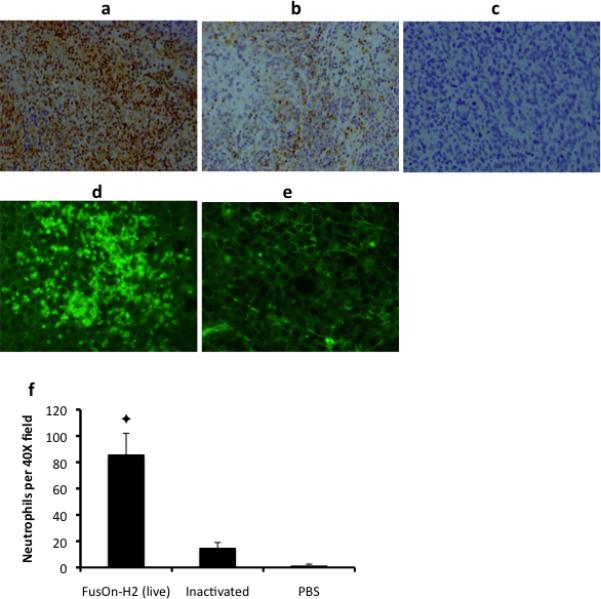
EC9706 tumors were established on the right flank of nude mice and injected with 3×106 pfu of FusOn-H2 (a, d) or the same of FusOn-H2 that had been inactivated by UV radiation (b, e), or PBS (c). Tumors were explanted two days later and divided into halves; one half for preparation of frozen sections for examining GFP expression under a fluorescent microscope (d, e) and the other half for immunohistochemical staining of neutrophils (a-c). The infiltrating neutrophils from a-c were quantitated by counting 10 microscopic fields (40X) and the average numbers are plotted in f. ✦p<0.01 vs. inactivated FusOn-H2.
Next, to examine the effect of the infiltrating neutrophils on EC9706 cells more directly, we performed an additional in vivo experiment similar to that illustrated in Fig. 3. After harvesting infiltrating neutrophils from established tumors at 2 days post-treatment, we immediately mixed them with EC9706 tumor cells at different ratios and measured cytolysis 24 h later (FusOn-H2 was undetectable in the purified neutrophils). The neutrophils retrieved from the FusOn-H2-treated EC9706 tumors had a significantly higher killing activity against EC9706 tumor cells than did those isolated from untreated tumors (Fig. 5a). These results demonstrate a critical difference in cytolytic capacity between neutrophils in FusOn-H2-treated versus untreated EC9706 tumors.
Figure 5. In vitro assay for killing and migration activity of neutrophils.
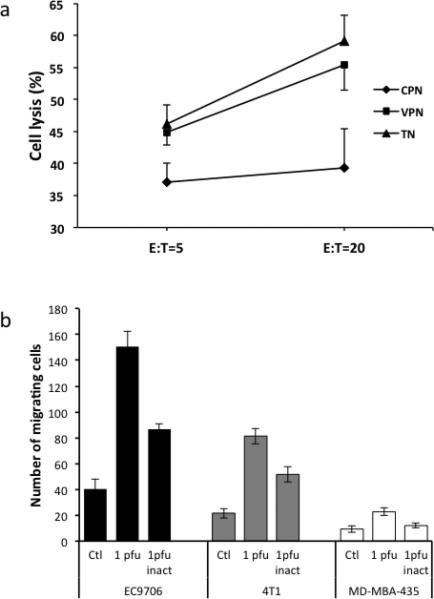
a. Killing activity of neutrophils isolated from tumor tissues or from peritoneal cavity. Neutrophils were isolated from either established EC9706 tumors that had been treated with FusOn-H2 (TN) or from peritoneal cavity that had been injected with EC9706 cells infected with FusOn-H2 (VPN) or mock infected (CPN). The purified neutrophils were then mixed with EC9706 cells at the ratio of either 5 or 20, and cytolysis was determined 24 h later. b. In vitro neutrophil migration assay using the BD BioCoat Matrigel Invasion Chambers. Tumor cells (with or without infection with FusOn-H2) were seeded on the lower wells and neutrophils were seeded into the top wells. After 22h of incubation, the non-migrating cells were removed and the cells clinging on the lower side of the insert were fixed and quantified.
We also measured the effect of FusOn-H2-infected tumor cells on the migration ability of neutrophils in an in vitro experiment. Freshly isolated neutrophils and tumor cells of different preparations (mock-infected, infected with 1 pfu/cell of FusOn-H2 or UV-inactivated FusOn-H2) were seeded in matrigel invasion chambers for cell migration assay as described 23. The results show that significantly more neutrophils were migrating toward the well seeded with FusOn-H2-infected EC9706 cells than to the wells seeded with two permissive cell lines, 4T1 and MD-MBA-435 (Fig. 5b). As compared with the mock-infected cells, cells infected with UV-inactivated FusOn-H2 can increase neutrophil migration. However, to achieve the maximal chemoattractant effect, full infectivity of the virus is required.
Strong endogenous interferon response activity in tumor cells with resistance to FusOn-H2 replication
The observation that only FusOn-H2-resistant tumors showed massive neutrophil infiltration after virotherapy indicates an intrinsic biological difference between the resistant and nonresistant tumors. To pursue this notion, we first evaluated the interferon response status of the tumor cells, as this response functions as a critical innate antiviral mechanism and could explain the failure of FusOn-H2 to replicate well in some tumor lines but not others. For this purpose, we constructed a test plasmid, pJ-ISRE-SEAP, in which SEAP gene is driven by a minimal promoter linked to 3 tandem repeats of ISRE, derived from the ISG56 promoter region. When transfected into tumor cells, this construct enabled us to measure SEAP levels in the culture medium and hence to monitor the cells’ interferon response activity.
Figure 6a shows the results of transfecting pJ-ISRE-SEAP into the five lines of tumor cells that showed resistance to FusOn-H2 as well as a panel of tumor cells that are permissive to the virus. Supernatants were collected and the secreted SEAP quantified at different time points after transfection. All five highly resistant cell lines had much higher levels of SEAP secretion than did the permissive lines (Fig. 6a). Among the five resistant lines, EC9706 (human esophageal carcinoma) and B16 (murine melanoma) showed an extremely high level of ISRE activity. We also monitored SEAP secretion by EC9706 cells for an extended time, demonstrating that it peaked on day 5 after transfection. Thereafter, it declined slightly but remained at a relatively high level for up to 2 weeks, the longest time span that we monitored (Fig. 6b).
Figure 6. Endogenous interferon response activity.
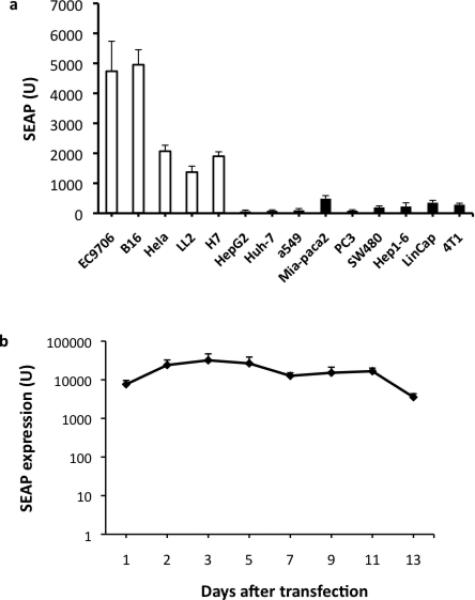
a. Both resistant and nonresistant tumor cells were transfected with 1μg of pJ-ISRE-SEAP, which contains the SEAP gene driven by a minimal promoter linked to 4 copies of ISRE. Twenty-four h after transfection, supernatants were collected and the SEAP concentration was quantified. b. The duration of SEAP expression from pJ-ISRE-SEAP was further characterized in a separate experiment. Again EC9706 cells were transfected with 1μg of pJ-ISRE-SEAP. Supernatants were collected at the indicated times for quantification of SEAP. The near two-fold difference in SEAP level between 6a and 6b is probably due to variations in either cell density or transfection efficiency between these two experiments.
Because SEAP is an exogenous marker gene that was introduced into tumor cells by transfection, we thought it important to measure the expression of endogenous genes that are normally regulated via ISRE. For this purpose, we chose to measure ISG56 expression, again by RT-PCR. We also asked if FusOn-H2 infection would further regulate ISRE activity. Thus, total RNA was extracted from the highly resistant EC9706 cell line and from two permissive lines (MCF-7 and HuH-7) with or without FusOn-H2 infection. When quantified by RT-PCR, ISG56 transcripts were significantly more abundant in EC9706 cells than in the other two lines (Fig. 7a). FusOn-H2 infection further increased the level of ISG56 transcripts in EC9706 cells, in contrast to results in the MCF-7 and HuH-7 lines, where FusOn-H2 infection led to reductions in the levels of these transcripts. These results further confirm the high-level intrinsic interferon response activity in these highly resistant tumor lines.
Figure 7. Endogenous ISG56 and interferon expression in tumor cells with or without FusOn-H2 infection and the effect of externally added IFNs on ISRE activity.
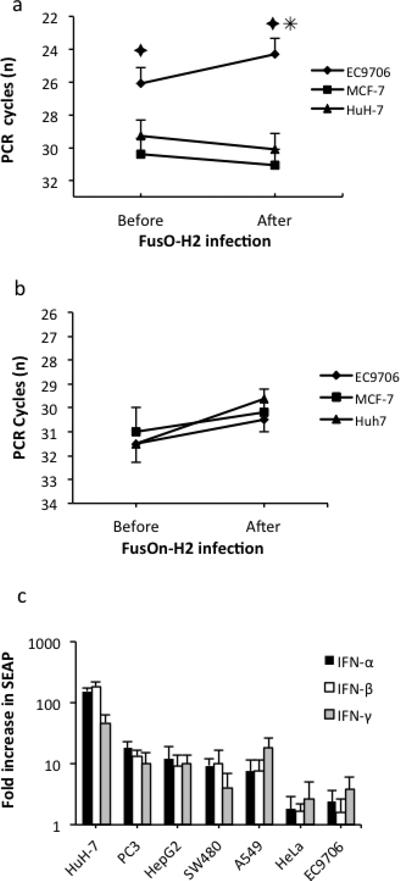
a and b. Endogenous ISG56 and interferon expression in tumor cells with or without FusOn-H2 infection. Tumor cells were seeded into six-well plates in duplicate and incubated overnight at 37°C. One set of cells was harvested and the others were infected with 1 pfu/cell of FusOn-H2 for 24 h before harvesting. The total RNA was prepared for RT-PCR quantification of ISG56 transcripts (a) or IFN-β transcripts (b) as described in Materials and Methods. ✦p<0.01 vs. MCF-7 or HuH-7, ✳p<0.05 vs. before infection. c. Effect of externally added IFNs on ISRE activity. Tumor cells were initially transfected with pJ-ISRE-SEAP. Then, different types of IFNs were added to the medium followed by 24 h of incubation before the collection of supernatants for SEAP assay. The fold of increase in SEAP activity was calculated by dividing the amount of SEAP released into the medium before addition of IFNs by that measured 24 h after IFN incubation. The Huh-7, PC3, HepG2, SW480 and A549 tumor cells are all permissive to FusOn-H2 replication.
We next asked if the strong endogenous interferon response activity in the highly resistant tumor cells is interferon-dependent, by comparing IFN-α and IFN-β secretions in resistant versus permissive tumor cells with or without FusOn-H2 infection. Using an ELISA assay, we were unable to detect IFN-α in most tumor cells, whether or not they were infected with FusOn-H2 (data not shown). Although IFN-β transcripts could be detected with RT-PCR, which is thought to be more sensitive than ELISA, their amounts did not differ significantly between the highly resistant (EC9706) and permissive (MCF-7 and HuH-7) cells (Fig. 7b). FusOn-H2 infection increased the amount of IFN-β transcripts but only marginally. We then determined if addition of interferons to the culture medium would affect SEAP expression from pJ-ISRE-SEAP. After transfecting a panel of tumor cells with this construct and adding different types of IFNs to the medium, we collected the supernatants 24 h later for quantification of SEAP. Addition of both type I and type II IFNs led to varied but consistently significant increases of SEAP expression in permissive tumor cells (Fig. 7c). In the Huh-7 line, for example, SEAP expression increased more than 100-fold after addition of type I IFNs. By contrast, the presence of exogenous IFNs had little effect on SEAP expression in highly resistant tumor cells (EC9706 and HeLa lines). Together, these results suggest that the high levels of endogenous ISRE activity in these highly resistant cell lines is IFN-independent and that other, co-existing mechanisms likely contributed to this intrinsic activity.
Discussion
The majority of studies evaluating the therapeutic effects of virotherapy are conducted with implanted tumor cells selected in vitro for their permissiveness to a particular oncolytic virus, as exemplified by our previous work with the HSV-2-derived oncolytic virus FusOn-H2 16-18, 24. In our experience, approximately 20% all tumor lines show resistant to FusOn-H2 in vitro and therefore would not be expected to have appreciable responses to virotherapy in vivo. Here we report the surprising finding that some of the tumor cells unable to support FusOn-H2 replication can nonetheless respond well to therapy with the virus. Indeed, even an intermediate dose of FusOn-H2 virtually eradicated tumor implants that had shown resistance to this virus in vitro; a similar result was obtained when the virus dose was further reduced by as much as 50-fold. Our data therefore indicate that the outcome of FusOn-H2 virotherapy against these highly resistant tumors depends on mechanisms other than direct oncolysis.
The most straightforward explanation for these favorable responses to virotherapy despite intrinsic resistance to FusOn-H2 lies in the massive infiltration of neutrophils that could be seen throughout the entire tumor tissue. Indeed, subsequent studies of neutrophils extracted from the treated tumors revealed that they could lyse tumor cells ex vivo. Thus, FusOn-H2 virotherapy for certain highly resistant tumor cells appears to induce an innate antitumor immune response dominated by neutrophil infiltration. This echoes recent studies suggesting that under certain conditions, neutrophils can function as a key antitumor mediator 25, 26. For example, it has been reported that TGF-β blockade increases neutrophil-attracting chemokines, resulting in an influx of CD11b+/Ly6G+ tumor-associated neutrophils that are cytotoxic to tumor cells 27. Studies by Breitbach et al suggest that infiltrating neutrophils contributed to a reduced blood flow to tumor tissues during virotherapy with vesicular stomatitis virus and vaccinia virus – derived oncolytic viruses 28. Another studies by Grote et al show that an oncolytic measles virus engineered to express GM-CSF could enhance the antitumor effect by recruiting neutrophils to the tumor site 29. Our studies suggest that there are two types of tumor cells that respond to FusOn-H2 virotherapy by different ways. The majority of tumor cells are permissive to virus replication and these tumor cells are mainly destroyed by direct cytolysis of the virus. However, there are approximately 20% of tumor cells that are highly resistant to virus replication. Some of these resistant tumor cells are still effectively destroyed by the virotherapy, partly through induction of innate antitumor immunity including massive neutrophil infiltration.
An innate immune response dominated by neutrophils would offer some distinct and unique advantages over adaptive immunity. First, infiltration of tumors is uniformly massive, as demonstrated in Fig. 3. By contrast, during adaptive immune responses, T effector cells are usually found at low frequencies in tumor tissues, which may have limited their antitumor efficacy. Indeed, for adaptive antitumor immunity to be successful, substantial expansion of the initially generated tumor-specific T cells is crucial 30. In most cases, however, T effector cell proliferation has proved extremely inefficient within the tumor microenvironment, probably accounting, at least in part, for the disappointing overall results from an array of clinical trials of T cell-based immunotherapy. Another major advantage of neutrophils over T cells in tumor destruction is that the former has the ability to liquefy the entire tumor tissues, which include tumor cells and tumor stromas such as collagen fibrils, stromal cells, lymphatics and capillaries 31. By contrast, T cells can only lyse tumor cells and their effects are frequently limited or actively inhibited by the remaining tumor stroma. Thus, given the relative ease with which large numbers of tumor-killing neutrophils were recruited to tumor sites in this study, we suggest that FusOn-H2 virotherapy may represent a unique strategy for enhancing the impact of immunotherapy against certain subgroups of tumors.
Analyses of the tumor cells highly resistant to FusOn-H2 replication revealed that they have strong intrinsic interferon response activity, yet the release of interferon from the tumor cells did not appear to be increased, even after FusOn-H2 infection. This is probably because many tumor cells have a defective interferon pathway 32; however, external addition of interferons led to greater ISRE activity in permissive tumor cells but not in highly resistant cells, indicating that the high levels of endogenous ISRE activity we describe are almost entirely independent of type I interferons themselves. Although the mechanism underlying this enhanced activity is unclear, it was recently reported that overexpression of an intracellular cytoplasmic protein, termed MITA (or STING), can activate interferon regulatory factor 3 (IRF3) 33-35, which then forms a complex with CREB-binding protein 36. This complex subsequently translocates to the nucleus to activate interferon-induced genes through stimulation of ISRE 37. Thus, we suggest that one possibility for the elevated endogenous ISRE activity in these highly resistant tumor cells is probably due to aberrant overexpression of this protein. Another possibility may be due to more efficient sensitization by toll-like receptors (TLRs), such as TLR-9, to FusOn-H2 in these tumor cells. These possibilities are under investigation.
How the intrinsically high ISRE activity in these tumor cells contribute to the induction of massive neutrophil infiltration during FusOn-H2 virotherapy is not clear. We suggest that neutrophil recruitment results from the combinatorial effect of the endogenously inflamed tumor cells due to high ISRE activity and the subsequent infection of the cells by FusOn-H2, as neither effect alone was able to elicit this response. It is also possible that cytokines/chemokines other than interferons may be needed to trigger this event. One attractive candidate is interleukin 8 (IL-8), a proinflammatory cytokine released at sites of tissue damage by various cell types. An important function of IL-8 is to recruit neutrophils into sites of inflammation and to activate their biological activity by enhancing superoxide anion production 38. It has been reported that HSV infection of epithelial cells induces IL-8 gene expression when there is an abundance of inflammatory cytokines in the local tissue 39. Another cytokine candidate is RANTES, which is upregulated via ISRE and has been reported to preferentially recruit neutrophils 40. Indeed, if RANTES and IL-8 were overexpressed in the same cell, their protein products might act synergistically to recruit neutrophils to tumor sites. Our attempt to detect release of RANTES from FusOn-H2-infected tumor cells in an in vitro experiment turned out to be negative (data not shown). Thus, these cytokines might have been released from nontumor cells in the in vivo tumor environment after animals receiving FusOn-H2 virotherapy. Additionally, HSV-2 encodes a secreted form of glycoprotein G (gG), which has recently been described to have proinflammatory properties and can function as a chemoattractant for both monocytes and neutrophils in a dose-dependent fashion 41. As FusOn-H2 is derived from HSV-2, the secreted gG from the virus may have contributed partly to the induction of neutrophil infiltration in these resistant tumors.
Identification of tumor cells, such as the EC9706 line, that respond favorably to FusOn-H2 virotherapy despite their apparent semi-permissiveness suggests that a screening procedure could be established to identify this subset of tumors, enabling the selection of a low FusOn-H2 dose and/or for a maximal therapeutic effect. The finding of a strong endogenous interferon response activity in these tumor cells may have other clinical implications. For example, it may be possible to devise a “molecular probe” to specifically identify circulating tumor cells with a high endogenous interferon response, thus accelerating the diagnosis of cancer and providing an estimate of prognosis.
Supplementary Material
S1. Neutrophil infiltration in B16 tumors after virotherapy. 2×105 B16 cells were implanted subcutaneously to the right flank of C57BL/6 mice. Once tumors reached the approximate size of 5 mm in diameter, they were injected with 3×106 pfu of either FusOn-H2 or Baco-1 (an HSV-1-based oncolytic virus) or PBS. Tumors were explanted two days later and tumor sections were prepared for H&E staining. The infiltrating neutrophils were quantitated by counting 10 fields (40X) under a microscope and the average numbers are plotted. ✦p<0.01 vs. Baco-1.
Acknowledgements
This work was supported in part by NIH grants 2R01CA106671 and R01CA132792, and William and Ella Owens Medical Research Foundation. We thank Dr. Zihua Zeng for assistance in histological examination of tumor sections and for discussion.
Footnotes
Conflict of interest declaration
The authors declare that they have no conflict of interest.
References
- 1.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Parato KA, Senger D, Forsyth PA, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5:965–976. doi: 10.1038/nrc1750. [DOI] [PubMed] [Google Scholar]
- 5.Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells [see comments]. Science. 1996;274:373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 6.Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854–856. doi: 10.1126/science.1851332. [DOI] [PubMed] [Google Scholar]
- 7.Logg CR, Tai CK, Logg A, Anderson WF, Kasahara N. A uniquely stable replication-competent retrovirus vector achieves efficient gene delivery in vitro and in solid tumors. Hum Gene Ther. 2001;12:921–932. doi: 10.1089/104303401750195881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:8751–8757. [PubMed] [Google Scholar]
- 9.Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 2002;62:4656–4662. [PubMed] [Google Scholar]
- 10.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 11.van der Poel HG, Molenaar B, van Beusechem VW, Haisma HJ, Rodriguez R, Curiel DT, et al. Epidermal growth factor receptor targeting of replication competent adenovirus enhances cytotoxicity in bladder cancer. J Urol. 2002;168:266–272. doi: 10.1097/00005392-200207000-00089. [DOI] [PubMed] [Google Scholar]
- 12.Glasgow JN, Bauerschmitz GJ, Curiel DT, Hemminki A. Transductional and transcriptional targeting of adenovirus for clinical applications. Curr Gene Ther. 2004;4:1–14. doi: 10.2174/1566523044577997. [DOI] [PubMed] [Google Scholar]
- 13.McCormick F. Cancer-specific viruses and the development of ONYX-015. Cancer Biol Ther. 2003;2:S157–160. [PubMed] [Google Scholar]
- 14.Bell JC. Oncolytic viruses: what's next? Curr Cancer Drug Targets. 2007;7:127–131. doi: 10.2174/156800907780058844. [DOI] [PubMed] [Google Scholar]
- 15.Hu JC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12:6737–6747. doi: 10.1158/1078-0432.CCR-06-0759. [DOI] [PubMed] [Google Scholar]
- 16.Fu X, Tao L, Cai R, Prigge J, Zhang X. A Mutant Type 2 Herpes Simplex Virus Deleted for the Protein Kinase Domain of the ICP10 Gene Is a Potent Oncolytic Virus. Mol Ther. 2006;13:882–890. doi: 10.1016/j.ymthe.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 17.Fu X, Tao L, Li M, Fisher WE, Zhang X. Effective treatment of pancreatic cancer xenografts with a conditionally replicating virus derived from type 2 herpes simplex virus. Clin Cancer Res. 2006;12:3152–3157. doi: 10.1158/1078-0432.CCR-06-0045. [DOI] [PubMed] [Google Scholar]
- 18.Fu X, Tao L, Zhang X. An oncolytic virus derived from type 2 herpes simplex virus has potent therapeutic effect against metastatic ovarian cancer. Cancer Gene Ther. 2007;14:480–487. doi: 10.1038/sj.cgt.7701033. [DOI] [PubMed] [Google Scholar]
- 19.Li W, Ding F, Zhang L, Liu Z, Wu Y, Luo A, et al. Overexpression of stefin A in human esophageal squamous cell carcinoma cells inhibits tumor cell growth, angiogenesis, invasion, and metastasis. Clin Cancer Res. 2005;11:8753–8762. doi: 10.1158/1078-0432.CCR-05-0597. [DOI] [PubMed] [Google Scholar]
- 20.Wang B, Shi Q, Abbruzzese J, Xiong Q, Le X, Xie K. A Novel, Clinically Relevant Animal Model of Metastatic Pancreatic Adenocarcinoma Biology and Therapy. Int J Gastrointest Cancer. 2001;29:37–46. [PubMed] [Google Scholar]
- 21.Nakamori M, Fu X, Meng F, Jin A, Tao L, Bast RCJ, et al. Effective Therapy of metastatic ovarian cancer with an oncolytic herpes simplex virus incorporating two membrane-fusion mechanisms. Clinical Cancer Res. 2003;9:2727–2733. [PubMed] [Google Scholar]
- 22.Boxio R, Bossenmeyer-Pourie C, Steinckwich N, Dournon C, Nusse O. Mouse bone marrow contains large numbers of functionally competent neutrophils. J Leukoc Biol. 2004;75:604–611. doi: 10.1189/jlb.0703340. [DOI] [PubMed] [Google Scholar]
- 23.Borley AC, Hiscox S, Gee J, Smith C, Shaw V, Barrett-Lee P, et al. Anti-oestrogens but not oestrogen deprivation promote cellular invasion in intercellular adhesion-deficient breast cancer cells. Breast Cancer Res. 2008;10:R103. doi: 10.1186/bcr2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu X, Nakamori M, Tao L, Amato R, Zhang X. Antitumor effects of two newly constructed oncolytic herpes simplex viruses against renal cell carcinoma. Int J Oncol. 2007;30:1561–1567. [PubMed] [Google Scholar]
- 25.Challacombe JM, Suhrbier A, Parsons PG, Jones B, Hampson P, Kavanagh D, et al. Neutrophils are a key component of the antitumor efficacy of topical chemotherapy with ingenol-3-angelate. J Immunol. 2006;177:8123–8132. doi: 10.4049/jimmunol.177.11.8123. [DOI] [PubMed] [Google Scholar]
- 26.Chen YL, Chen SH, Wang JY, Yang BC. Fas ligand on tumor cells mediates inactivation of neutrophils. J Immunol. 2003;171:1183–1191. doi: 10.4049/jimmunol.171.3.1183. [DOI] [PubMed] [Google Scholar]
- 27.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breitbach CJ, Paterson JM, Lemay CG, Falls TJ, McGuire A, Parato KA, et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol Ther. 2007;15:1686–1693. doi: 10.1038/sj.mt.6300215. [DOI] [PubMed] [Google Scholar]
- 29.Grote D, Cattaneo R, Fielding AK. Neutrophils contribute to the measles virus-induced antitumor effect: enhancement by granulocyte macrophage colony-stimulating factor expression. Cancer Res. 2003;63:6463–6468. [PubMed] [Google Scholar]
- 30.van Heijst JW, Gerlach C, Swart E, Sie D, Nunes-Alves C, Kerkhoven RM, et al. Recruitment of antigen-specific CD8+ T cells in response to infection is markedly efficient. Science. 2009;325:1265–1269. doi: 10.1126/science.1175455. [DOI] [PubMed] [Google Scholar]
- 31.Jain RK. Transport of molecules, particles, and cells in solid tumors. Annu Rev Biomed Eng. 1999;1:241–263. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 32.Wong LH, Krauer KG, Hatzinisiriou I, Estcourt MJ, Hersey P, Tam ND, et al. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3gamma. J Biol Chem. 1997;272:28779–28785. doi: 10.1074/jbc.272.45.28779. [DOI] [PubMed] [Google Scholar]
- 33.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Lin R, Genin P, Mamane Y, Hiscott J. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol Cell Biol. 2000;20:6342–6353. doi: 10.1128/mcb.20.17.6342-6353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wozniak A, Betts WH, Murphy GA, Rokicinski M. Interleukin-8 primes human neutrophils for enhanced superoxide anion production. Immunology. 1993;79:608–615. [PMC free article] [PubMed] [Google Scholar]
- 39.Oakes JE, Monteiro CA, Cubitt CL, Lausch RN. Induction of interleukin-8 gene expression is associated with herpes simplex virus infection of human corneal keratocytes but not human corneal epithelial cells. J Virol. 1993;67:4777–4784. doi: 10.1128/jvi.67.8.4777-4784.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan ZZ, Parkyn L, Ray A, Ray P. Inducible lung-specific expression of RANTES: preferential recruitment of neutrophils. Am J Physiol Lung Cell Mol Physiol. 2000;279:L658–666. doi: 10.1152/ajplung.2000.279.4.L658. [DOI] [PubMed] [Google Scholar]
- 41.Bellner L, Thoren F, Nygren E, Liljeqvist JA, Karlsson A, Eriksson K. A proinflammatory peptide from herpes simplex virus type 2 glycoprotein G affects neutrophil, monocyte, and NK cell functions. J Immunol. 2005;174:2235–2241. doi: 10.4049/jimmunol.174.4.2235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1. Neutrophil infiltration in B16 tumors after virotherapy. 2×105 B16 cells were implanted subcutaneously to the right flank of C57BL/6 mice. Once tumors reached the approximate size of 5 mm in diameter, they were injected with 3×106 pfu of either FusOn-H2 or Baco-1 (an HSV-1-based oncolytic virus) or PBS. Tumors were explanted two days later and tumor sections were prepared for H&E staining. The infiltrating neutrophils were quantitated by counting 10 fields (40X) under a microscope and the average numbers are plotted. ✦p<0.01 vs. Baco-1.