

Abstract
Oral infection of C57BL/6 mice with Toxoplasma gondii triggers severe necrosis in the ileum within 7–10 days of infection. Lesion development is mediated by Th-1 cytokines, CD4+ T cells, and sub-epithelial bacterial translocation. As such, these features share similarity to Crohn’s disease. Recently, we uncovered a role for intraepithelial lymphocytes (IEL) in mediating pathology after Toxoplasma infection. We show here that αβ and not γδ T cell IELs mediate intestinal damage. By adoptive transfer of mucosal T cells into naive Rag1−/− mice, we demonstrate that IEL do not function alone to cause inflammatory lesions, but act with CD4+ T lymphocytes from the lamina propria. Furthermore, recipient mice pretreated with broad-spectrum antibiotics to eliminate intestinal flora resisted intestinal disease after transfer of IEL and lamina propria lymphocytes. Our data provide valuable new insight into mechanisms of intestinal inflammation, findings that have important implications for understanding human inflammatory bowel disease.
INTRODUCTION
Inflammatory bowel diseases (IBD) in humans are chronic inflammatory disorders that most often emerge during the second or third decade of life. Crohn’s disease and ulcerative colitis are the most common types of idiopathic IBD 1. Crohn’s disease affects the terminal ileum in over two-thirds of patients, and is characterized by transmural inflammation and macrophage aggregation forming non-caseous granulomas. The incidence of IBD, particularly Crohn’s disease, has been rising in recent decades in developed regions of the world 2. While the etiology of IBD is unclear, a combination of genetic and environmental factors is believed to lead to dysregulated immunity, altered intestinal barrier function, and dysfunctional responses to gut flora 3.
IBD in mice can be induced genetically, chemically, and by certain infections. Included in the latter group is infection with the protozoan parasite Toxoplasma gondii, a microorganism infecting up to 30–50% of the human population 4. While normally asymptomatic, Toxoplasma emerges as an opportunistic pathogen in immunodeficient populations 5. The parasite induces a strong Th1 response 6. Oral infection of certain inbred mice, typified by the C57BL/6 strain, triggers proinflammatory pathology centered in the ileum that is characterized by necrosis of the villi and mucosal cells 7, 8. Lesion development is dependent upon CD4+ T cells, IFN-γ and TNF-α, and is therefore primarily a Th-1-mediated disease. Endogenous intestinal bacteria are involved in lesion induction, because infection is associated with a shift in gut flora to predominantly gram-negative E. coli, increased adherence, and subepithelial bacterial translocation. Moreover, gut flora-depleted, as well as Tlr4−/− and Tlr9−/− mice, are protected from parasite-induced ileitis 9–11. Overall, as an IBD model, T. gondii-induced ileitis bares many striking similarities to Crohn’s disease 12.
We recently uncovered a role for chemokine (C-C motif) receptor (CCR) 2-dependent intraepithelial lymphocytes in parasite-induced ileitis during T. gondii infection 13. By transferring wild-type IEL into Ccr2−/− mice, we converted the knockout strain from a resistant to susceptible phenotype with regard to induction of small intestinal lesions. These results were of great interest because, although some studies suggest that IEL can play a pathogenic role in intestinal inflammatory disorders 14, 15, these lymphocytes are more commonly associated with homeostasis in the gut and maintenance of barrier function 16.
Here, we show that amongst IEL, those expressing αβ T cell receptor (TCR) mediate disease, whereas γδTCR IEL are inert in this regard. We develop and validate a new model of parasite-induced ileitis, in which transfer of IEL in combination with lamina propria (LP) cells triggers rapid ileitis in immunodeficient Rag1−/− recipients without the presence of parasites. We show that intestinal microflora in recipient mice is essential for development of ileitis following adoptive transfer in this model. Further, we show that IEL recruit LP CD4+ T lymphocytes into intraepithelial regions of the mucosa, and that these cells are effectors of intestinal damage. Our data shed new light on pathogenesis of small intestinal inflammation triggered by microbial infection, and they have important implications with regard to understanding human IBD.
RESULTS
Peroral Toxoplasma infection triggers rapid conversion to a proinflammatory cytokine profile in the small intestine
T. gondii infection in C57BL/6 strain mice induces proinflammatory pathology in the small intestine ileum 8. Here, we measured cytokines in overnight cultures of ileal biopsy samples collected over a time course of infection (Fig. 1). Increasing amounts of IFN-γ (A), TNF-α (B) and IL-12/23p40 (C) were detected as infection progressed in biopsy samples with and without Peyer’s patches (PP). Examination of transcriptional responses in the ileum further confirmed these data, because transcripts for Ifn-γ (D), Tnf-α (E) and Il-12p40 (F) increased in ileal tissue over the course of infection with a similar kinetics to that seen with cytokine secretion. These data are consistent with previous studies that have shown involvement of each of these cytokines in Toxoplasma-induced immunopathology in the small intestine 7, 8.
Figure 1.

Emergence of a proinflammatory cytokine profile in the small intestine during T. gondii infection. Biopsy samples of small intestine with or without Peyer’s patches (PP) were cultured overnight and supernatants assayed for IFN-γ (A) and TNF-α (B) and IL-12/23p40 (C) by ELISA. Data, representative of 2 independent experiments, are plotted as the mean and standard error between individual mice (n = 3). Real-time RT-PCR analysis of intestinal biopsy samples revealed increased transcription genes encoding Ifn-γ (D), Tnf-α (E) and Il-12p40 (F). Examination of the T cell transcription factor T-bet revealed a steady increase over the course of infection (G) and a concomitant decrease in Rorγt expression (H). Transcription factors Foxp3 (I), Eomes (J) and Gata-3 (K) showed no consistent change or decreased expression over the course of infection. These experiments were repeated twice with similar results.
We also examined expression of T cell master transcriptional factors. The Th-1 transcription factor, T-bet increased steadily over the course of infection (Fig. 1G). Interestingly, this was paralleled by a decrease in Rorγt, a master regulator of Th-17 cells (Fig. 1H). Transcription factors associated with Treg cells (Foxp3; Fig. 1I), CD8+ cytotoxic T cells (Eomes; Fig. 1J) and Th-2 (Gata-3; Fig. 1K) remained relatively unchanged or showed no consistent pattern over the time course of infection. Overall, the data show that Toxoplasma induces a dominant Th-1 type response in the ileum.
T cell receptor (TCR)αβ+ but not TCRγδ+ IEL mediate inflammation in infected mice
Previously, we and others showed that Ccr2−/− mice are protected from T. gondii-induced ileitis 13, 17 (Fig. S1). Importantly, transfer of parasite-elicited wild-type (WT) IEL to infected Ccr2−/− mice triggered ileal damage 13. The majority of IEL express CD8, but the cells can be subdivided into TCRαβ and TCRγδ positive sub-populations (Fig. 2A). To examine whether the pathological activity of parasite-induced IEL could be attributed exclusively to either of these populations, we separated IEL from Day 4-infected WT mice into TCRαβ (Fig. 2B) and TCRγδ (Fig. 2C) populations, adoptively transferred these cells into Day 4-infected Ccr2−/− animals, then collected tissues 5 days later for histopathological evaluation. As shown in Fig. 2D and F, mice receiving TCRαβ+ IEL developed ileitis resembling that occurring in WT mice, marked by fusion, necrosis and sloughing of villus tips, and sub-epithelial inflammation. In marked contrast, mice receiving TCRγδ+ IEL failed to display severe lesion development (Fig. 2E and F), although some mild inflammatory changes were found in these animals (villus fusion being the most notable and visible in Fig. 2E). This result is consistent with others showing lack of involvement of TCRγδ+ T lymphocytes in Toxoplasma-triggered ileitis 8.
Figure 2.

TCRαβ and not TCRγδ+ IEL from WT mice induce damage in Ccr2−/− hosts. IEL were collected from WT Day 4-infected mice (A) and fractionated into TCRαβ+ (B) and TCRγδ+ (C) sub-populations by antibody labeling and magnetic bead separation. Day 4-infected Ccr2−/− mice received either TCRαβ+ IEL or TCRγδ+ IEL, and 5 days later intestines were collected and examined for pathological changes. Panels D and E show representative images of Ccr2−/− mice receiving TCRαβ+ and TCRγδ+ IEL, respectively. Scale bar in E indicates 50 μm. The graph (F) represents pathology scores for individual mice (n = 5 per group), where ** indicates p<0.05. The experiment was repeated twice with the same result.
Emergence of a CCR2+ CD4+ IEL population coincident with maximal inflammation
Our data associate Toxoplasma-induced ileitis with IEL, the majority of which are CD8 positive. Yet, studies in experimental models of colitis, as well as in Crohn’s disease, implicate a prominent role for CD4+ T lymphocytes in damage to the intestine 3, 18. To investigate this issue, we examined changes in T cell populations in the IEL compartment over the course of infection in WT mice. IEL were gated on expression of CCR2 (Fig. 3A), then expression of CD8α and CD4 was examined in CCR2 positive and CCR2 negative populations. IEL isolated from naive and day 4 post-infection mice displayed a similar expression pattern for CD8α and CD4 and other T cell markers amongst CCR2 positive and CCR2 negative cells. However, by Day 8 post-infection there was a dramatic increase in the CD4+ IEL, with the CD4+CCR2− population increasing 3-fold and the CD4+CCR2+ cells increasing 10-fold (Fig. 3B).
Figure 3.

Emergence of CCR2+ TCRαβ+CD4+ IEL correlates with maximum pathology during Toxoplasma infection. IEL were isolated and stained with anti-CCR2 mAb (A, which shows CCR2 expression on IEL from Day 4-infected mice), in combination with Ab specific for CD4, CD8α, CD8β, αβTCR and γδTCR. Panel B shows respective expression of CD8α and CD4 in CCR2− and CCR2+ populations over the course of infection. Essentially identical results were obtained in 3 independent experiments. To examine if Toxoplasma-triggered recruitment of TCRαβ+CD4+ to the IEL compartment is dependent upon CCR2, IEL were isolated from WT and Ccr2−/− mice 8 days after infection and expression of CD8α and CD4 was assessed. Representative staining profiles are shown in panel C, which also shows the average percent of CD4+ IEL in day 8-infected mice in 3 independent experiments (*, p<0.05). Panel D shows TCRαβ and TCRγδ expression on CD4+ and CD8α+ IEL, respectively, from Day 8-infected WT mice.
Emergence of CCR2+ CD4+ cells in the IEL compartment suggested that recruitment was CCR2-dependent. Accordingly, we examined recruitment of CD4+ cells in Ccr2−/− mice at day 8 post-infection. In comparison to IEL from infected WT mice, there was a substantial defect in CD4 cell recruitment in the absence of CCR2 (Fig. 3C). Over multiple experiments comparing WT and Ccr2−/− mice there was approximately 50% decreased recruitment of CD4 cells associated with lack of CCR2 (Fig. 3C). Virtually all CD4+ IEL expressed TCRαβ (Fig. 3D). In contrast, CD8α+ IEL expressed either αβTCR or γδTCR (Fig. 3D).
Both IEL and LP compartments are required for maximal intestinal inflammation
An important consideration for the adoptive transfer experiments in Ccr2−/− mice was the potential contribution of host LP cells to the development of intestinal lesions. Another consideration was the potential contribution of endogenous IEL in Ccr2−/− recipients. A final complicating factor was that Ccr2−/− recipients were themselves infected with Toxoplasma. To avoid these complications, we tested the effect of transferring IEL and LP from day 4-infected WT mice into uninfected Rag1−/− mice that lack lymphocyte populations. We chose day 4 as the time point to isolate cells reasoning that at this time infection would be well established, but intestinal lesions would still be absent. As shown in Fig. 2 and 13, IEL from day 4-infected mice were composed of CD8 T cells expressing TCRαβ and TCRγδ. The LP compartment from day 4-infected mice was a heterogeneous mix of B cells, CD4+ and CD8+ T cells, dendritic cells, monocytes/macrophages and a small number of neutrophils (Fig. S2A). In accord with other studies where T. gondii induced a collapse in the regulatory T cell population in the intestinal mucosa 19, we found no detectable Foxp3-positive cells in the IEL or LP compartment at this time point (data not shown). Likewise, our preparations contained very low numbers of NK and NKT cells (data not shown).
IEL and LP compartments were isolated from day 4-infected WT mice, injected alone or in combination into Rag1−/− recipients, and small intestine collected 5 days later for histological evaluation. Noninfected Rag1−/− mice displayed little or no evidence of intestinal lesions (Fig 4A and B). Rag1−/− recipients of the LP compartment also displayed minimal evidence of intestinal damage. Interestingly (given our previous transfer experiments in Ccr2−/− mice), adoptive transfer of the IEL compartment alone induced only minor inflammation in Rag1−/− recipients, characterized mostly by epithelial tip sloughing. However, transfer of IEL in combination with LP triggered ileal lesions in Rag1−/− recipients (Fig. 4A and B), and pathology closely resembled that seen in infected WT mice (e. g., Fig. S1).
Figure 4.

IEL and LP from infected mice synergize to induce intestinal lesions and bacterial translocation after transfer into uninfected Rag1−/− recipients. IEL and LP were isolated from day 4 infected WT mice and transferred alone or in combination into Rag1−/− mice. Intestines were harvested 5 days after transfer, fixed, stained and examined for inflammatory changes. Groups of noninfected Rag1−/− mice received no WT cells, LP alone, IEL alone or equal numbers of the two cell types (Panel A). Scale bar denotes 50 μm. Pathology scores from individual mice in each group are shown in B (*, p<0.05). The experiment was repeated twice with the same result. Noninfected Rag-1−/− animals received IEL, LP or IEL + LP from Day 4-infected WT mice. Small intestines were collected 5 days after transfer and tissue sections were subjected to fluorescence in situ hybridization (FISH) analysis employing a Cy3-labelled 16S ribosomal RNA pan-bacterial probe (red) to detect the presence and localization of intestinal bacteria (Panel C). The staining also includes a 6FAM-labeled nonsense probe so that non-specific binding is visible as yellow fluorescence. Cell nuclei were counterstained with DAPI (blue). The scale bar in represents 50 μm. This experiment was performed three times with the same result.
Increased adherence and translocation of luminal bacteria into the mucosal tissue are features of small intestinal lesions in Toxoplasma-infected mice 9, 10. Accordingly, we used fluorescence in situ hybridization employing a probe specific for bacterial 16S ribosomal RNA to assess translocation of gut flora following adoptive transfer in Rag1−/− animals. We found no evidence of bacterial translocation after transfer of IEL or LP into Rag1−/− recipients (Fig. 4C). In marked contrast, co-transfer of IEL and LP cells resulted in increased bacterial translocation into lamina propria regions of the Rag1−/− small intestine (Fig. 4C).
We considered the possibility that lesions in Rag1−/− recipients were caused by co-transfer of parasites rather than IEL and LP cells per se. Accordingly, infection levels were assessed in the transfer populations following cell permeabilization and antibody staining for intracellular T. gondii. Neither IEL (Fig. S2B) nor LP (Fig. S2C) contained detectable parasites. In contrast, the same Toxoplasma-reactive antibody was highly effective at detecting infected cells in the peritoneal cavity of wild type mice following i. p. injection of Toxoplasma (Fig. S2D). Furthermore, tissues prepared from Rag1−/− recipients of WT IEL and LP five days after adoptive transfer showed no evidence of infection by immunohistochemical staining for Toxoplasma, despite induction of lesions in the small intestine (Fig. S2E and Fig. 4A). This contrasted with anti-Toxoplasma staining of small intestine of day 8-infected WT mice, where parasites were easily detected (Fig. S2F). We also inoculated Day 4-infected LP and IEL samples onto human fibroblast monolayers and found no evidence of plaque formation even after 2 weeks of culture (data not shown). Importantly, Rag1−/− mice did not develop intestinal lesions following direct T. gondii infection (Fig. S3B, compare to WT animals infected with the same parasite preparation in Fig. S3A) despite presence of parasites in the mucosa (Fig. S3C). We conclude that intestinal lesions in Rag1−/− recipients of IEL and LP cells were not generated as a result of co-transfer of parasites, but instead as result of synergistic interactions between these two cell populations.
Rag1−/− IEL/LP recipients possess ileal cytokine profiles closely matching infected WT mice
To examine the cytokine profile in Rag1−/− animals receiving IEL + LP, we cultured ileal biopsies and measured cytokine levels in the resulting supernatants. Ileal samples from Day 8-infected WT mice released substantial amounts of IFN-γ, IL-12/23p40 and TNF-α (Fig. 5A–C). Ileal samples from naïve Rag1−/− mice released low levels of each respective cytokine. In marked contrast, ilea from Rag1−/− IEL/LP recipients secreted IFN-γ, IL-12/23p40 and TNF-γ at levels similar to that measured in WT samples (Fig. 5A–C). We also examined transcriptional profiles and found that Il-12p40, Tnf-α and Ifn-γ genes were each highly upregulated after IEL/LP transfer relative to ileal samples from non-transferred Rag1−/− mice (Fig. 5D). Analysis of the transcription factors in IEL/LP Rag-1−/− recipients revealed a pattern broadly similar to that seen in infected WT mice (Fig. 5E and Fig. 1).
Figure 5.

Reconstitution of Rag1−/− mice with IEL and LP from infected WT mice induces a proinflammatory cytokine profile in recipient animals. Ileal explants from Day 8-infected WT (excluding Peyer’s patches), naive Rag1−/−, and Rag1−/− IEL/LP recipients were cultured overnight and levels of TNF-α (A), IL-12/23p40 (B) and IFN-γ (C) were determined by ELISA. For Rag1−/− adoptively transferred recipients, IEL and LP were prepared from Day 4-infected WT mice, transferred into noninfected Rag1−/− mice, and ileal biopsy samples were isolated 5 days post-transfer. RNA was isolated from ileal tissues of adoptively transferred mice and examined by real-time RT-PCR for Il-12p40, Tnf-α and Ifn-γ (D), as well as T-bet, Rorγt, Gata-3, Foxp3, and Eomes (E). The data are plotted as change relative to samples from normal Rag1−/− mice. These experiments were repeated twice with similar results. Ileal samples were also analyzed for proinflammatory gene transcripts in a real-time RT-PCR gene array (Panel F). Transcripts displaying an average of 5-fold or greater over uninfected Rag1−/− ileal samples are represented as a clustering image. AT, Rag1−/− recipients of LP/IEL; WT, day 8-infected WT ilea. Each column represents an individual mouse, with the last two columns representing mean values of the AT and WT groups relative to noninfected Rag1−/− ileal samples.
We next employed a real-time RT-PCR gene array targeted to inflammatory cytokines, chemokines and their receptors to analyze transcriptional responses in the ilea of Rag1−/−/IEL+LP mice in comparison to infected WT mice and naive Rag1−/− control animals. The results are summarized as a heat map of the genes with greater than or equal to 5-fold change over non-infected Rag1−/− values (Fig. 5F). Applying this threshold, 20 gene transcripts were upregulated in both infected WT ileum and ileum from adoptively transferred (AT) Rag1−/− recipients. Twelve transcripts were upregulated 5-fold or more only in AT recipients, whereas 10 transcripts were upregulated to similar levels only in infected WT ileum (Fig. 5F). Amongst the common transcripts, Ifn-γ was highly upregulated. In both WT and AT recipients we also detected increased CCL2 and CCL8, major ligands for CCR2. The latter chemokine receptor is implicated in development of ileal inflammation during Toxoplasma infection 13, 17. Transcripts encoding several other chemokines and receptors involved in Th-1 inflammatory responses (CCL7, CCR3, CCR5, CCR9, CXCL9) were also elevated in both infected and Rag1−/− IEL/LP recipients.
Fig. S4 shows linear scatter plots of the complete data sets. Relative to levels in control Rag1−/− mice, transcript levels in adoptively transferred (Fig. S4A) and infected WT mice (Fig. S4B) remained within or were raised above the 5-fold threshold, with no evidence for down-regulation of transcripts. When transcript levels of AT vs. infected WT were compared, the majority of transcripts fell within the 5-fold threshold, emphasizing the similarity of the ileal chemokine/cytokine environments in these two groups (Fig. S4C). In sum, the chemokine/cytokine profile in Rag1−/− IEL/LP animals is remarkably similar to that of infected WT mice.
Lesion development in Rag1−/− recipients requires IEL but not LP from Toxoplasma-infected mice
We next determined whether LP and IEL populations from noninfected WT mice were capable of inducing pathology following transfer into Rag1−/− recipients. Adoptive transfer of IEL and LP from noninfected mice did not cause damage to the small intestine (Fig. 6A). When IEL from noninfected mice were co-transferred with LP from infected animals, Rag1−/− recipients developed a low, but significant (p < 0.05) level of intestinal damage (Fig. 6B and D). However, by far the most severe lesions were triggered by combined transfer of IEL from infected mice with LP from noninfected animals (Fig. 6C and D, p < 0.01). Pathology scores in the latter group were similar to scores obtained from transfers that were performed when both LP and IEL compartments were obtained from infected mice (Fig. 4B).
Figure 6.
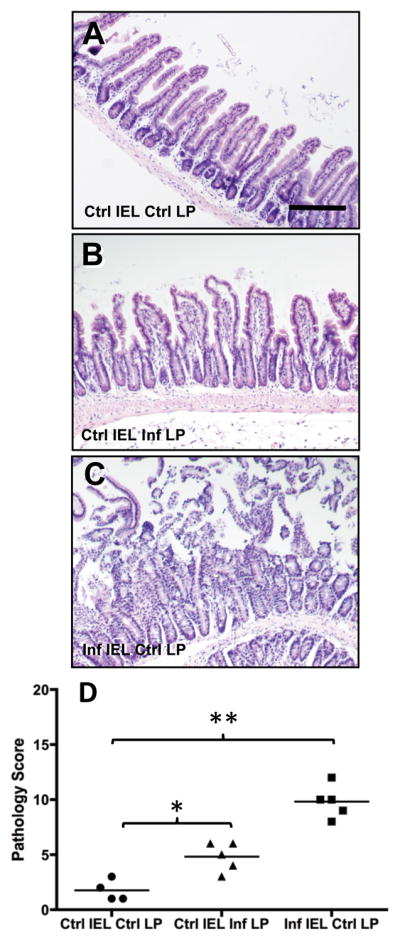
IEL from infected mice induce disease in Rag1−/− mice when co-transferred with LP from noninfected animals. Noninfected Rag1−/− recipients received combinations of IEL and LP from infected (Inf) and noninfected (Ctrl) mice, then 5 days later tissues were collected for histopathological evaluation in the ileum. A, Rag1−/− mice receiving IEL and LP from noninfected mice; B, Rag1−/− recipients of IEL from noninfected mice and LP from infected animals; C, Rag1−/− recipients of IEL from infected mice in combination with LP from noninfected animals. Scores for individual mice are shown in D, where * indicates p < 0.05 and ** indicates p < 0.01. Scale bar in A represents 50 μm.
CD4+ T cells emerging in the IEL compartment derive from the LP and synergize with IEL to cause lesions
We employed CD45 congenic animals to examine the dynamic behavior of LP cells after co-transfer with IEL into Rag1−/− mice. Groups of WT (CD45.2) and CD45.1 congenic mice were infected with 100 ME49 cysts and the IEL and LP compartments were isolated 4 days later. Naive Rag1−/− recipients were injected with IEL from WT (CD45.2) mice along with LP from CD45.1 congenic animals. Five days after the adoptive transfer the IEL were harvested from the Rag1−/− mice and stained with antibodies to determine the presence of CD4+ T cells and the LP congenic marker, CD45.1 as outlined in the schematic shown in Fig. 7A. Transfer of LP alone resulted in little or no detectable CD4+ cells in the IEL compartment (Fig. 7B). However when IEL and LP were co-transferred there was a 15-fold increase in CD4+ cells in the IEL compartment (with the remainder being epithelial cells; data not shown). Gating on CD4+ cells revealed that the majority were CD45.1 positive and therefore derived from the LP compartment (Fig. 7C). A smaller population of CD45.2 positive cells derived from transferred IEL was also present (Fig. 7C). We conclude that IEL drive recruitment of CD4+ cells in the LP, most likely through production of chemokines.
Figure 7.

Parasite-elicited CD4+ LP lymphocytes are recruited into the IEL compartment in dependence upon CCR2. A, Uninfected Rag1−/− mice (CD45.2 background) were adoptively transferred with Day 4-infected LP cells (PepBoy/J; CD45.1) with or without Day 4-infected IEL (C57BL/6; CD45.2) to trace the origin of cells emerging in the IEL compartment. B, Five days post-transfer, the IEL compartment was isolated and CD4 expression was examined in Rag1−/− recipients of LP alone (LP) and LP in combination with IEL (LP+IEL). C, Expression of CD45.1 (LP donor origin) and CD45.2 (IEL donor origin) by CD4 cells from mice receiving LP+IEL. D, Percent of CD4+ T cells in the IEL compartment in Rag1−/− recipients of WT IEL in combination with LP from WT or Ccr2−/− mice. Donor cells were derived from Day 4-infected animals. *, p< 0.05.
Because the CD4+ T cells emerging in the IEL compartment express CCR2 (Fig. 3B), we asked whether presence of this receptor on LP CD4+ T lymphocytes was necessary to enable recruitment. CD45.2 positive cells could be recovered from the IEL compartment following transfer of WT CD45.2 LP along with WT CD45.1 IEL into Rag1−/− recipients. We examined the phenotype of these cells and found that after co-transfer with WT LP cells, a population of CD45.2 positive CD4+ T cells appeared in the IEL compartment, and this population was significantly lower following co-transfer with Ccr2−/− LP cells (Fig. 7D). We conclude that CCR2 expression on LP CD4+ T cells is required for maximal recruitment to the IEL compartment.
Previous data showed that CD4+ T cells mediate intestinal pathology during Toxoplasma infection. Therefore, we asked whether CD4+ T cells accounted for the pathogenic activity of the LP compartment when co-transferred with IEL. We isolated LP cells from day 4-infected mice which in this experiment were composed of approximately 23% CD4+ and 15% CD8α+ T cells (Fig. 8A). Using immunomagnetic beads, this population was separated into CD4 positive and CD4 negative fractions (Fig. 8A). These populations were then each co-transferred along with day 4-infected IEL and tissues were collected 5 days later. Although overall ileal damage was relatively mild, we found that pathology tracked with the CD4+ T cell fraction (Fig. 8B) and not with the CD4 negative population of LP cells. We also observed an expansion in intestinal flora in the CD4+ recipients, most of which was tightly associated with the mucosal surface (Fig 8C). This was in contrast to the CD4− recipient mice. Here we saw far less bacteria and there were minimal indications of bacterial tissue adherence (Fig 8C). In sum, these data show CCR2-dependent αβTCR+CD8+ IEL mediate recruitment of LP-derived αβTCR+CD4+ T cells into the epithelium resulting in damage to intestinal tissue.
Figure 8.

Ileal lesion development and bacterial overgrowth are associated with CD4+ LP lymphocytes. Day 4-infected LP cells were fractionated using immunomagnetic beads into CD4 positive and CD4 negative populations (A). Each was transferred with Day 4-infected IEL into Rag1−/− recipients and pathology assessed 5 days later. Representative ileal sections from a recipient of CD4+ LP/IEL or a recipient of CD4−LP/IEL and pathology scores of individual mice are shown in B (**, p<0.05). FISH analysis using a 16S ribosomal RNA pan-bacterial probe (red) was performed on ileal tissue from CD4+LP/IEL and CD4−LP/IEL Rag1−/− recipients (C). A 6FAM-labeled nonsense probe was included to control for non-specific staining, and cell nuclei were stained with DAPI (blue). Scale bars indicate 50 μm. These experiments were repeated twice with similar results.
IEL and LP cells do not cause lesions in recipient Rag1−/− mice that have been treated with broad-spectrum antibiotics
Intestinal bacterial flora is recognized to play a role in exacerbation of inflammation. Therefore, we asked whether the endogenous microbiota in recipient mice promoted the pathogenic effector activity of IEL/LP transfer populations. Naïve Rag1−/− mice were treated with a cocktail of broad-spectrum antibiotics prior to and after adoptive transfer of IEL/LP. The effectiveness of the antibiotic treatment regimen was confirmed by LB agar cultures of fecal pellets isolated at the termination of the experiment (Fig S5).
Ileal damage was minimal in antibiotic treated mice that were recipients of IEL/LP cells (Fig. 9A and C) when compared to lesions in bacteria-replete mice receiving IEL/LP cells (Fig 9B and C). Importantly, the antibiotic treatment itself did not induce and pathological changes in the intestines (Fig. 9C). As a confirmation of disease induction in the intestine, biopsy cultures from antibiotic-treated mice receiving IEL/LP showed dramatically lower levels of IFN-γ (Fig. 9D) and TNF-α (Fig. 9E) compared with nontreated animals receiving IEL/LP cells. We conclude that intestinal flora in recipient mice plays a role in promoting the tissue-damaging effects of adoptively transferring IEL and LP cells.
Figure 9.

Elimination of intestinal flora in Rag1−/− mice prevents intestinal inflammation following IEL+LP transfer. Recipient mice were pre-treated with a broad-spectrum antibiotic cocktail for 3 weeks prior to cell transfer. Day 4 parasite-elicited IEL/LP populations were subsequently adoptively transferred into recipients and intestinal inflammation and cytokine production was evaluated 5 days later. A, Rag1−/− recipients treated with broad-spectrum antibiotics and adoptively transferred with LP/IEL. B, In parallel, nontreated Rag1−/− animals received the same populations of IEL/LP. Because depletion of flora results in mucous overproduction and interference with H&E staining, tissue sections in A and B were stained with Alcian blue/PAS. C, Pathology scores for commensal depleted, commensal depleted with IEL/LP cells, and non-commensal depleted with IEL/LP cells. Each symbol represents a single mouse. IFN-γ (D) and TNF-α (E) production in intestinal biopsy samples from commensal depleted or non-depleted Rag1−/− recipients of IEL/LP populations. In these experiments, * indicates p< 0.05. These experiments were repeated twice with similar results.
DISCUSSION
Toxoplasma triggers proinflammatory lesions in the intestine of C57BL/6 mice. This destructive pathology has many features of Crohn’s disease, insofar as both encompass ileal involvement, CD4+ T lymphocyte pathogenic activity, inflammatory cytokine overproduction, and dysbiosis of gut microflora 12. Moreover, both Crohn’s inflammation and Toxoplasma-triggered ileitis can be ameliorated by blocking TNF-α 7, 20. Recently, we uncovered a role for CCR2- and IFN-γ-dependent CD8α+ IEL in gut lesion development during oral T. gondii infection 13. Here, we establish that αβ and not γδ TCR+ IEL mediate intestinal damage. We also show that transfer of IEL together with LP cells from infected mice into naïve Rag1−/− mice triggers proinflammatory intestinal pathology in the absence of parasites. We show that IEL recruit LP-derived αβTCR+CD4+ T cells into intraepithelial regions in partial dependence on CCR2, and that these cells are mediators of intestinal damage. Transfer of IEL alone in this model induced mild inflammation, suggesting that in addition to recruitment of LP cells, they may also directly contribute to damage in the intestine. We also found that endogenous intestinal flora in recipient mice was required for the pathogenic effects of cell transfer. These results, which are summarized in Fig. S6, are the first to demonstrate that microbial induced interactions between IEL and LP lymphocytes promote proinflammatory cytokine responses and inflammatory lesions in the small intestine.
Important issues that await resolution are the MHC requirements and antigen specificity of the IEL and LPL subsets. The requirement for gut flora in recipient Rag1−/− mice suggests that one or both subsets could be reactive to intestinal bacteria. With regard to the IEL compartment, some CD8+ IEL subsets display unusual MHC requirements 16, and it will therefore be important in the future to examine responses of MHC class I and β2-microglobulin knockout mice.
Our results show pathological activity of IEL induced by Toxoplasma. Nevertheless, under homeostatic conditions the IEL compartment is generally associated with immunoregulation and immune quiescence in the intestinal mucosa 16, 21. For example, transfer of CD8ααTCRαβ IEL protects immunodeficient mice from subsequent transfer of pathogenic CD4+CD45RBhigh splenocytes 22. In parallel, it has been shown that γδTCR+ IEL have a protective function in the dextran sodium sulfate mouse colitis model23. Yet, IEL isolated from Crohn’s disease patients have been shown to display abnormally enhanced cytotoxic activity and overproduction of IFN-γ compared to IEL from normal donors 24, 25. The latter observations, together with our data, suggest that dysfunctional IEL form part of the constellation that constitutes IBD.
Related to our studies is elegant work by others who examined early mucosal responses during low dose Toxoplasma infection, a situation that avoids immunopathology, enabling host survival, protective immunity, and persistent infection. Here, IEL protect against ileitis and subsequent parasite challenge after adoptive transfer 26. In the LP compartment, CD4+ T cells synergize with intestinal epithelial cells resulting in protective proinflammatory responses that control infection 27. At least part of the beneficial activity of IEL in this model stems from TGF-β production that down-regulates LP CD4+ T lymphocyte responses and controls ileitis 28, 29. We hypothesize that high dose T. gondii infection triggers a switch in IEL function from mediators of protection and immunoregulation to effectors of pathology.
In our model, transfer of IEL and LP cells from Toxoplasma-infected mice triggered fulminant pathology in noninfected Rag1−/− recipients. It seems most unlikely that parasites were the direct cause of disease in these mice as we could not detect parasites in either the cells injected into the naïve Rag1−/− or in intestinal tissue isolated 5 days after transfer. Furthermore, Rag1−/− mice are resistant to lesion development when directly infected with T. gondii (Fig. S3 and ref. 30). In wild-type mice, development of intestinal lesions during Toxoplasma infection is associated with a shift from gram-positive to predominantly gram-negative bacteria in the small intestine, and fulminant pathology is accompanied by increased adherence and bacterial translocation into the sub-epithelium. Host responses to gut bacteria are implicated in intestinal inflammation because both Tlr4−/− and Tlr9−/− mice are resistant to lesion development 10, 11, 31.
We show here that Ccr2−/− LP cells do not re-locate to the IEL compartment after transfer into Rag1−/− mice. Yet, previously we found that IEL transfer into T. gondii infected Ccr2−/− recipients (where the LP compartment would be CCR2 negative) were able to induce intestinal damage. We speculate that in the latter case other chemokines and cytokines induced as a result of Toxoplasma infection in the recipients compensate for loss of CCR2 responsiveness.
What is the triggering event that drives Toxoplasma-mediated intestinal lesion development? Possibly, the parasite induces early barrier damage as a result of invasion and egress from epithelial cells, enabling translocation of bacteria and in turn triggering pathogenic responses. Arguing against this scenario are data suggesting that Toxoplasma crosses the intestinal barrier using a paracellular pathway that involves transmigration without membrane disruption 32. It is also possible that the parasite directly activates mucosal lymphocytes for pathogenic activity, although this does not fully account for the requirement for bacterial flora in lesion development.
Another possibility is that disease development is a consequence of immunosuppression by Toxoplasma. The parasite is known to downregulate cytokine and chemokine responses during intracellular macrophage and dendritic cell infection 33–36. This may have relevance to induction of IBD, because recent data reveal impaired cytokine secretion by macrophages from Crohn’s patients 37. Older studies have shown impaired neutrophil recruitment in individuals with Crohn’s disease 38, 39. Thus, a model for Crohn’s pathogenesis is that defective sentinel responses to occasional bacterial ingress in the gut results in failure to recruit neutrophils, leading to loss of control of infection and chronic secondary inflammation 37. We are currently examining the functional status of intestinal dendritic cells and macrophages at early time points of T. gondii infection.
The results of this study highlight the value of Toxoplasma infection in C57BL/6 mice as a model for induction of Crohn’s like pathology in the ileum. Our results reveal for the first time that LP T cells and IEL synergize to cause inflammatory ileitis. By exploiting Toxoplasma as a trigger, we can distinguish cell and cytokine requirements required for inflammation, and in so doing we can expect to gain further mechanistic insight into immunological factors that are important in human IBD.
METHODS
Mice
Eight to 12 week old female C57BL/6 and Swiss Webster mice were purchased from Taconic Farms (Germantown, NY). B6.SJL-Ptprca Pep3b/BoyJ (CD45.1 congenic; backcrossed for 22 generations), B6.129S7-Rag1tm1Mom/J (Rag1−/−; backcrossed for 10 generations) and B6.129S4-Ccr2tm1lfc/J (Ccr2−/−; backcrossed for 9 generations) mice were purchased from The Jackson Laboratory. Animals were housed under specific pathogen-free conditions at the Cornell University College of Veterinary Medicine animal facility, which is accredited by the International Association for Assessment and Accreditation of Laboratory Animal Care.
Antibiotic Treatment
Mice were treated with sterile water supplemented with ampicillin (1 g/L, Sandoz Inc.), vancomycin (500 mg/L, Hospira Inc.), enrofloxacin (200 mg/L, Bayer) and imipenem (250 mg/L, Merck) ad libitum for 3–4 weeks. Antibiotics were changed weekly and cages changed every 3 days.
Parasites and infections
Swiss-Webster mice were used to generate cysts of the low virulence type II Toxoplasma strain, ME49. Mice were infected with 100 cysts by oral gavage.
LP and IEL isolation
Small intestines were removed, cleaned of mesentery and fat, and flushed with sterile PBS. Isolation of IEL was performed as previously described 13. Absence of contaminating LP cells was tested by antibody staining for B lymphocytes, which are absent from the IEL compartment. LP leukocytes were isolated as described 40. After removal of the mucosal layer, small intestine was cut into 5 mm fragments. Cells were subsequently liberated from tissues by digestion (37°C, 20 min) in RPMI containing 0.2 mg/ml Liberase CI (Roche), 15 mg/ml DNAse (Sigma-Aldrich). Leukocytes were separated from contaminating debris by discontinuous Percoll gradient centrifugation.
Lymphocyte isolation
For CD4+ T cell separation, single cell suspensions of LP leukocytes were incubated with anti-CD4 magnetic beads according to manufacturer’s instructions (Miltenyi Biotech). The cells were washed and separated by positive selection on an AutoMACs separator. Similarly, αβ IEL were separated from γδ IEL using anti-γδ magnetic beads (Miltenyi Biotech).
Gut biopsy culture
Intestines were removed from mice and flushed with sterile PBS containing 300 U/ml penicillin and 300 μg/ml streptomycin. Intestines were opened longitudinally and a 6 mm dermal biopsy punch (Miltex) was used to collect tissue pieces. Intestinal sections were incubated overnight in DMEM supplemented with 10% bovine growth serum, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 0.05 mM β-mercaptoethanol, 300U/ml penicillin, 300 μg/ml streptomycin, and 30 mM HEPES (37°C, 4% CO2). Cell-free supernatants were collected and cytokine levels measured as previously described 41.
Flow cytometry
Single cell suspensions were stained in buffer (PBS, 1% BSA, 0.01% NaN3) containing 10% mouse serum. Anti-CCR2 (clone MC-21) was used as previously described 13. Antibodies employed were phycoerythrin (PE)- and allophycocyanin (APC)-conjugated anti-CD4 mAb, FITC- and APC-conjugated anti-CD8α mAb, PE-conjugated anti-CD8β mAb, PerCP-Cy5.5-conjugated anti-TCRβ, FITC- and APC-conjugated anti-TCRγδ (all from eBioscience). Anti-SAG-1 (p30) directly conjugated to FITC (Argene) was used to stain intracellular parasites following permeabilization with 0.075% saponin. Cells were analyzed on a BD FACSCalibur flow cytometer.
Adoptive transfer
Mice received 5 × 106 IEL, 5 × 106 LP or a 1:1 mixture of both (totaling 1 × 107 cells) by intravenous retro-orbital injection. Mice receiving IEL and CD4+ or CD4− LP received 5 × 106 IEL and 1 × 106 LP. Intestines were subsequently removed and fixed in 10% neutral buffered formalin. Groups of paraffin-embedded tissue sections were stained with Hematoxylin and Eosin or Alcian blue/PAS and examined for pathological changes, and sequential sections were subjected to fluorescence in situ hybridization.
RNA isolation, microarray analysis and semi-quantitative PCR
RNA was prepared from 1 cm ileum segments utilizing an RNA purification kit (Omega Bio-Tek). RNA was converted into cDNA following the manufacturer’s recommendations (SABiosciences), and samples were run on the mouse “Inflammatory Cytokines & Receptors PCR Array” (SABiosciences) using an ABI 7500 Fast machine. Data were normalized to housekeeping genes present of the array and compared to uninfected Rag-1 knockout mice. Semi-quantitative PCR was performed utilizing the ABI-Prism 7500 fast cycle real-time PCR system (Applied Biosystems). Gene expression levels were normalized to Gapdh expression and differences between groups were analyzed by the ΔΔ CT method. Transcriptional responses were expressed relative to noninfected mice.
Fluorescence in situ hybridization (FISH)
FISH was performed as previously described 13, using probes (EUB338, or non-EUB338) labeled on the 5′ end with either FITC or Cy3 (Integrated DNA Technologies). Images were collected with a BX51 microscope (Olympus) equipped with a DP70 camera using DP Controller Software (version 1.1.1.65; Olympus) and DP Manager software (version 1.1.1.71; Olympus).
Pathology scoring
Damage to the intestine was scored as described 13. Briefly, 5 criteria (villus fusion/blunting, lamina propria inflammation, epithelial tip sloughing, villus tip necrosis, and transmural inflammation) were each assigned a score of 0 (not apparent) to 4 (severe). Each mouse was assigned a cumulative score out of a maximum of 20. Scoring was carried out in double-blinded fashion.
Statistical analyses
Pathology scores were analyzed using an ANOVA with a Bonferroni post-test correction to calculate statistical differences between groups. Where appropriate, student’s t test was used to analyze statistical differences between two groups. Values of p <0.05 were considered significant.
Supplementary Material
Acknowledgments
This work was funded by NIH grants to EYD (AI083526) and KJM (DK077728), and a CCFA Senior Fellowship Award (KWS). SBC was supported by the Cornell University Graduate Program in Biological and Biomedical Sciences. The technical assistance of F. Davis and M. Hossain is gratefully acknowledged.
Footnotes
Conflicts of interest statement: No conflicts
Supplementary Material is linked to the online version of the paper at http://www.nature.com/mi
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 3.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dubey JP. The history and life-cycle of Toxoplasma gondii, in Toxoplasma gondii. In: Weiss LM, Kim K, editors. The model apicomplexan: Perspective and methods. Academic Press; San Diego: 2007. pp. 1–17. [Google Scholar]
- 5.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 6.Denkers EY, Gazzinelli RT. Regulation and function of T cell-mediated immunity during Toxoplasma gondii infection. Clin Microbiol Rev. 1998;11:569–588. doi: 10.1128/cmr.11.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liesenfeld O, et al. TNF-a, nitric oxide and IFN-g are all critical for development of necrosis in the small intestine and early mortality in genetically susceptible mice infected perorally with Toxoplasma gondii. Parasite Immunol. 1999;21:365–376. doi: 10.1046/j.1365-3024.1999.00237.x. [DOI] [PubMed] [Google Scholar]
- 8.Liesenfeld O, Kosek J, Remington JS, Suzuki Y. Association of CD4+ T cell-dependent, IFN-g-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J Exp Med. 1996;184:597–607. doi: 10.1084/jem.184.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heimesaat MM, et al. Gram-Negative Bacteria Aggravate Murine Small Intestinal Th1-Type Immunopathology following Oral Infection with Toxoplasma gondii. J Immunol. 2006;177:8785–8795. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- 10.Heimesaat MM, et al. Exacerbation of Murine Ileitis By Toll-Like Receptor 4 Meditated Sensing of Lipopolysaccharide From Commensal Escherichia coli. Gut. 2007;56:941–948. doi: 10.1136/gut.2006.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minns LA, et al. TLR9 Is Required for the Gut-Associated Lymphoid Tissue Response following Oral Infection of Toxoplasma gondii. J Immunol. 2006;176:7589–7597. doi: 10.4049/jimmunol.176.12.7589. [DOI] [PubMed] [Google Scholar]
- 12.Liesenfeld O. Oral infection of C57BL/6 mice with Toxoplasma gondii: a new model of inflammatory bowel disease? J Infect Dis. 2002;185:S96–101. doi: 10.1086/338006. [DOI] [PubMed] [Google Scholar]
- 13.Egan CE, et al. CCR2-dependent intraepithelial lymphocytes mediate inflammatory gut pathology during Toxoplasma gondii infection. Mucosal Immunol. 2009;2:527–535. doi: 10.1038/mi.2009.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El-Asady R, et al. TGF-{beta}-dependent CD103 expression by CD8(+) T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J Exp Med. 2005;201:1647–1657. doi: 10.1084/jem.20041044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hue S, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004;21:367–377. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 16.Kunisawa J, Takahashi I, Kiyono H. Intraepithelial lymphocytes: their shared and divergent immunological behaviors in the small and large intestine. Immunol Rev. 2007;215:136–153. doi: 10.1111/j.1600-065X.2006.00475.x. [DOI] [PubMed] [Google Scholar]
- 17.Benevides L, et al. CCR2 receptor is essential to activate microbicidal mechanisms to control Toxoplasma gondii infection in the central nervous system. Am J Pathol. 2008;173:741–751. doi: 10.2353/ajpath.2008.080129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uhlig HH, Powrie F. The role of mucosal T lymphocytes in regulating intestinal inflammation. Springer Semin Immunopathol. 2005;27:167–180. doi: 10.1007/s00281-005-0206-6. [DOI] [PubMed] [Google Scholar]
- 19.Oldenhove G, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrante M, et al. Efficacy of infliximab in refractory pouchitis and Crohn’s disease-related complications of the pouch: a Belgian case series. Inflamm Bowel Dis. 2010;16:243–249. doi: 10.1002/ibd.21037. [DOI] [PubMed] [Google Scholar]
- 21.Ishikawa H, et al. Curriculum vitae of intestinal intraepithelial T cells: their developmental and behavioral characteristics. Immunol Rev. 2007;215:154–165. doi: 10.1111/j.1600-065X.2006.00473.x. [DOI] [PubMed] [Google Scholar]
- 22.Das G, et al. An important regulatory role for CD4+CD8 alpha alpha T cells in the intestinal epithelial layer in the prevention of inflammatory bowel disease. Proc Natl Acad Sci U S A. 2003;100:5324–5329. doi: 10.1073/pnas.0831037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Chou K, Fuchs E, Havran WL, Boismenu R. Protection of the intestinal mucosa by intraepithelial gamma delta T cells. Proc Natl Acad Sci U S A. 2002;99:14338–14343. doi: 10.1073/pnas.212290499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nussler NC, et al. Enhanced cytolytic activity of intestinal intraepithelial lymphocytes in patients with Crohn’s disease. Langenbecks Arch Surg. 2000;385:218–224. doi: 10.1007/s004230050268. [DOI] [PubMed] [Google Scholar]
- 25.Watanabe M, et al. Preferential activation of CD4+V beta 5.2/5.3+ intestinal intraepithelial lymphocytes in the inflamed lesions of Crohn’s disease. Clin Immunol Immunopathol. 1996;78:130–139. doi: 10.1006/clin.1996.0022. [DOI] [PubMed] [Google Scholar]
- 26.Lepage AC, Buzoni-Gatel D, Bout DT, Kasper LH. Gut-derived intraepithelial lymphocytes induce long term immunity against Toxoplasma gondii. J Immunol. 1998;161:4902–4908. [PubMed] [Google Scholar]
- 27.Mennechet FJD, et al. Lamina propria CD4+ T lymphocytes synergize with murine intestinal epithelial cells to enhance proinflammatory response against an intracellular pathogen. J Immunol. 2002;168:2988–2996. doi: 10.4049/jimmunol.168.6.2988. [DOI] [PubMed] [Google Scholar]
- 28.Buzoni-Gatel D, et al. Murine ileitis after intracellular parasite infection is controlled by TGF-b-producing intraepithelial lymphocytes. Gastroenterol. 2001;120:914–924. doi: 10.1053/gast.2001.22432a. [DOI] [PubMed] [Google Scholar]
- 29.Mennechet FJ, et al. Intestinal intraepithelial lymphocytes prevent pathogen-driven inflammation and regulate the Smad/T-bet pathway of lamina propria CD4+ T cells. Eur J Immunol. 2004;34:1059–1067. doi: 10.1002/eji.200324416. [DOI] [PubMed] [Google Scholar]
- 30.Rachinel N, et al. The induction of acute ileitis by a single microbial antigen of Toxoplasma gondii. J Immunol. 2004;173:2725–2735. doi: 10.4049/jimmunol.173.4.2725. [DOI] [PubMed] [Google Scholar]
- 31.Benson A, Pifer R, Behrendt CL, Hooper LV, Yarovinsky F. Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe. 2009;6:187–196. doi: 10.1016/j.chom.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barragan A, Sibley LD. Transepithelial migration of Toxoplasma gondii is linked to parasite migration and virulence. J Exp Med. 2002;195:1625–1633. doi: 10.1084/jem.20020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bierly AL, Shufesky WJ, Sukhumavasi W, Morelli A, Denkers EY. Dendritic cells expressing plasmacytoid marker PDCA-1 are Trojan horses during Toxoplasma gondii infection. J Immunol. 2008;181:8445–8491. doi: 10.4049/jimmunol.181.12.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee CW, Bennouna S, Denkers EY. Screening for Toxoplasma gondii regulated transcriptional responses in LPS-activated macrophages. Infect Immun. 2006;74:1916–1923. doi: 10.1128/IAI.74.3.1916-1923.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luder CGK, Algner M, Lang C, Bleicher N, Gross U. Reduced expression of the inducible nitric oxide synthase after infection with Toxoplasma gondii facilitates parasite replication in activated murine macrophages. Internat J Parasitol. 2003;33:833–844. doi: 10.1016/s0020-7519(03)00092-4. [DOI] [PubMed] [Google Scholar]
- 36.McKee AS, Dzierszinski F, Boes M, Roos DS, Pearce EJ. Functional inactivation of immature dendritic cells by the intracellular parasite Toxoplasma gondii. J immunol. 2004;173:2632–2640. doi: 10.4049/jimmunol.173.4.2632. [DOI] [PubMed] [Google Scholar]
- 37.Smith AM, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med. 2009;206:1883–1897. doi: 10.1084/jem.20091233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marks DJ, et al. Defective acute inflammation in Crohn’s disease: a clinical investigation. Lancet. 2006;367:668–678. doi: 10.1016/S0140-6736(06)68265-2. [DOI] [PubMed] [Google Scholar]
- 39.Segal AW, Loewi G. Neutrophil dysfunction in Crohn’s disease. Lancet. 1976;2:219–221. doi: 10.1016/s0140-6736(76)91024-2. [DOI] [PubMed] [Google Scholar]
- 40.Lefrancois L, Lycke N. Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer’s patch, and lamina propria cells. Curr Protoc Immunol. 2001;Chapter 3(Unit 3):19. doi: 10.1002/0471142735.im0319s17. [DOI] [PubMed] [Google Scholar]
- 41.Sukhumavasi W, et al. TLR adaptor MyD88 is essential for pathogen control during oral toxoplasma gondii infection but not adaptive immunity induced by a vaccine strain of the parasite. J Immunol. 2008;181:3464–3473. doi: 10.4049/jimmunol.181.5.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.