

Abstract
Haploinsufficiency of TBX1, encoding a T-box transcription factor, is largely responsible for the physical malformations in velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome (22q11DS) patients. Cardiovascular malformations in these patients are highly variable, raising the question as to whether DNA variations in the TBX1 locus on the remaining allele of 22q11.2, could be responsible. To test this, a large sample size is needed. The TBX1 gene was sequenced in 360 consecutive 22q11DS patients. Rare and common variations were identified. We did not detect enrichment in rare SNP number in those with or without a congenital heart defect. One exception was that there was increased number of very rare SNPs between those with normal heart anatomy compared to those with right-sided aortic arch or persistent truncus arteriosus, suggesting potentially protective roles in the SNPs for these phenotype enrichment groups. Nine common SNPs (MAF >0.05) were chosen and used to genotype the entire cohort of 1,022 22q11DS subjects. We did not find a correlation between common SNPs or haplotypes and cardiovascular phenotype. This work demonstrates that common DNA variations in TBX1 do not explain variable cardiovascular expression in 22q11DS patients, implicating existence of modifiers in other genes on 22q11.2 or elsewhere in the genome.
Keywords: 22q11.2 deletion syndrome, TBX1 sequencing, cardiovascular defects, genomic disorder
Introduction
Velo-cardio-facial syndrome/DiGeorge syndrome, also known as 22q11.2 deletion syndrome (22q11DS; MIM#s 192430, 188400), is a congenital malformation disorder characterized by craniofacial anomalies, immune deficiency, neonatal hypocalcemia and cardiac outflow tract (conotruncal) defects. It occurs in approximately 1/4,000 live births (Burn and Goodship, 1996). Most affected individuals have a hemizygous 3 million base pair (Mb) de novo deletion on chromosome 22q11.2 caused by de novo meiotic non-allelic homologous recombination events. These events take place between flanking segmental duplications termed LCR22 (low copy repeats on 22q11.2) (Edelmann, et al., 1999). To help positionally clone the causative gene(s) on 22q11.2, early goals were set on identifying rare patients with smaller, nested deletions or translocations with subsets of phenotypes (Budarf, et al., 1995; Fibison, et al., 1990; Lindsay, et al., 1995; Lu, et al., 2001; Scambler, et al., 1991). The proximal 1.5 Mb region, flanked by LCR22s, was identified as the critical region of the syndrome (Lindsay, et al., 1995; Morrow, et al., 1995). Because the 1.5 Mb deletion size was large, containing many genes, it was not possible to narrow the region to a smaller interval. Mouse models containing nested deletions (Kimber, et al., 1999; Lindsay, et al., 1999) were generated to find candidate genes. Bacterial artificial chromosome (BAC) transgenic mice were created to confer genetic rescue and narrow the number of candidate genes to a few (Lindsay, et al., 2001; Merscher, et al., 2001). A single gene, Tbx1 was identified as the strongest candidate for physical malformations (Lindsay, et al., 2001; Merscher, et al., 2001) and inactivation by gene targeting approaches demonstrated that it is largely responsible (Jerome and Papaioannou, 2001). While Tbx1 heterozygous mice showed mild anomalies with reduced penetrance, homozygous null mutants died at birth with a cleft palate, absent thymus and parathyroid glands as well as a common cardiac outflow tract, termed a persistent truncus arteriosus.
Support this gene’s importance in humans came from rare non-deleted patients with a velo-cardio-facial or DiGeorge syndrome phenotype that had mutations in TBX1 (MIM# 602054), some of which alter function (Conti, et al., 2003; Gong, et al., 2001; Griffin, et al., 2010; Stoller and Epstein, 2005; Torres-Juan, et al., 2007; Yagi, et al., 2003; Zweier, et al., 2007). This indicates that haploinsufficiency of TBX1 in most, or mutation of TBX1 in a small minority of patients with related phenotypes, is responsible for the main physical malformations occurring in patients with the syndrome.
One of the most characteristic features of 22q11DS is that it occurs with variable phenotypic severity. With respect to structural malformations, some patients are mildly affected, for example, with a characteristic facies and hypernasal speech, while others are severely affected with absent thymus, cleft palate, severe hypocalcemia and/or tetralogy of Fallot. Environmental exposures, stochastic factors or genetic modifiers are thought to be responsible for variable expressivity. One hypothesis is that alteration of DNA sequence in a gene on the remaining allele of 22q11.2, possibly TBX1 is the cause of this phenotypic variability.
We have amassed a cohort of 1,022 22q11DS patients, 65% of which have a congenital heart disease (intracardiac defects and/or aortic arch defects; CHD) while 35% have a normal heart and aortic arch anatomy. The majority of those with a CHD had more than one cardiovascular anomaly. The goal of the project was to determine if there are phenotype-phenotype correlations or phenotype-genotype correlations. Unfortunately, our cohort has too few individuals per group to consider each anomaly on its own. As a compromise, we created phenotype enrichment groups.
In this study we performed genotype-phenotype correlations with TBX1. We found no correlation between common SNPs or haplotypes and the CHD phenotype suggesting that there is not a predominant common variant of TBX1 serving as a genetic modifier of cardiovascular anomalies. This work suggests that variations elsewhere on 22q11.2 or in the genome serve as cardiovascular modifiers. Alternatively, DNA variations in TBX1 might explain variable expressivity for other malformations seen in association with the syndrome.
Material and Methods
Human subjects and phenotype data
Blood or saliva samples were obtained from 22q11DS subjects, with their informed consent (Internal Review Board, 1999-201; 07-005352_CR2 CHOP IRB). A commercial fluorescence in situ hybridization test for the 22q11.2 deletion, MLPA testing (Multiplex ligation-dependent probe amplification, SALSA MLPA kit P250 DiGeorge; MRC Holland, The Netherlands) or microsatellite marker analysis was part of the work up of individuals with the syndrome for mutation analysis. TBX1 locus sequence analysis was performed on DNAs available for 372 subjects obtained consecutively that had existing cardiac phenotype data. Consistency of features is based upon comparison discussions with clinicians as to clinical definitions (Bassett, et al., 2011). Half the DNA samples were obtained from Montefiore Medical Center/Upstate Medical University (RJS) and half were obtained from the Children’s Hospital of Philadelphia. Some of the DNAs failed to provide any sequence data. A total of 360 DNA samples were sequenced completely. An International 22q11.2 Consortium was formed and DNA samples were obtained from additional subjects (de-identified) resulting in a total of 1,022 DNA samples from unrelated probands with the 22q11DS. A total of 106 DNAs were obtained from Canada, 91 DNAs from Belgium, 38 DNAs from Italy, 13 DNAs from the Netherlands, 30 DNAs from France, 10 DNAs from Atlanta, 9 DNAs from California, 18 DNAs from Montefiore, 16 DNAs from a VCFSEF conference (Utah), 276 DNAs from CHOP, 398 DNAs from Upstate Medical University and 11 from elsewhere. The cohort used for analysis was self reported as being of Caucasian ethnicity. We obtained phenotype information on intracardiac and aortic arch anatomy from echocardiogram and cardiology summary reports. The entire set of DNAs were plated and used for genotype analysis.
TBX1 resequencing
There are three isoforms of human TBX1 protein, isoform A (NM_080646), B (NM_005992) and C (NM_080647), where there are alternative terminal exons. A total of 12 exons that encode the 3 transcripts of TBX1 were sequenced in 360 consecutive 22q11.2 patients (Supp. Figure S1). All exon-intron boundaries extending >100 bp into each intron were included. The first exon containing the 5′ UTR (untranslated region) and an additional 2,500 bp of sequence that included the putative promoter region were also sequenced. The 3′ UTR was also included in the analysis (Supp. Figure S1). Transcription start site prediction programs suggest there are three alternative start sites in the TBX1 gene (UCSC Genome Browser; First exon prediction track). The CpG islands corresponding to the three putative transcription start sites in the TBX1 locus were sequenced. Short blocks of a few hundred base pairs of DNA retained among vertebrates such as identified by the 28-Way or 17-Way Most Conserved track (UCSC genome browser) with a LOD score >50, as chosen arbitrarily, were also sequenced because they may contain regulatory elements. All the evolutionary conserved non-coding regions within 5 kb of the gene body containing exons, from 18,122,096-18,152,117 (hg18; http://www.genome.ucsc.edu/) were sequenced (Supp. Figure S1). The human orthologous region to a previously defined enhancer for pharyngeal apparatus expression of mouse Tbx1 (18,115,184-18,115,529;(Garg, et al., 2001) was sequenced as well. None of the other blocks of evolutionary conserved sequences upstream or downstream of the TBX1 gene were sequenced because their functional roles are not known.
All primers are listed in Supp. Table S1. The PCR products were sequenced using the Big Dye Terminator Sequencing Kit (Applied Biosystems, Carlsbad, CA) on an automated DNA capillary sequencer (model 3730; Applied Biosystems, Carlsbad, CA). Resequencing services were provided by the J. Craig Venter Institute under U.S. Federal Government contract number N01-HV-48196 from the National Heart, Lung, and Blood Institute. Six duplicated samples were used as quality control (QC). Variants that passed the QC were used for final statistical analysis.
Genotype analysis
Common (minor allele frequency, MAF>0.05) and rare SNPs (MAF<0.05) in TBX1 were identified from DNA sequence analysis. The SNPs were assigned to linkage disequilibrium (LD) blocks by creating an LD matrix in Haploview software (Barrett, et al., 2005) using TBX1 resequencing data from the 360 patients. Based upon the LD structure of the locus, 9 of the 17 common SNPs identified were then used for genotyping the entire cohort of 1,022 22q11DS subjects. We used the Sequenom iPLEX system for genotype analysis. Reagents and protocols for multiplex PCR, single base primer extension (SBE) and generation of mass spectra, were followed as per the manufacturer’s instructions (for complete details see iPLEX Application Note, Sequenom Inc., San Diego, CA, PMC14630). A multiplexed assay containing 31 SNPs (9 from TBX1; the rest from chromosomes X and Y to check for correct gender) was designed using MassARRAY Assay Design 3.1 (Sequenom Inc., San Diego, CA). Briefly, initial multiplexed PCR was performed in 5 μl reactions on 384-well plates containing 10 ng of genomic DNA. Reactions contained 0.5 U FastStart Taq DNA Polymerase (Roche, Nutley, NJ), 100 nM primers, 1× PCR buffer, 2 mM MgCl2, and 500 μM dNTPs. Following enzyme activation at 94°C for 4 min, the DNA was amplified with 45 cycles of 94°C × 20 sec, 56°C × 30 sec, 72°C × 1 min, followed by a 3 minute extension at 72°C. Unincorporated dNTPs were removed using shrimp alkaline phosphatase (0.3 U, Sequenom Inc, San Diego, CA). Single-base extension was carried out by addition of SBE primers at concentrations from 0.625 μM (low MW primers) to 1.25 μM (high MW primers) using iPLEX enzyme and buffers (Sequenom Inc., San Diego, CA) in 9 μl reactions. Reactions were desalted and SBE products measured using the MassARRAY Compact system, and mass spectra were analyzed using TYPER 3.4 software (Sequenom, San Diego), in order to generate genotype calls and allele frequencies. Primer sequences are available on request.
Statistical analysis
Phenotype-phenotype correlations
The intracardiac as well as aortic arch phenotypes were extracted from echocardiogram and cardiology summary reports and were entered into an Oracle-based clinical database. The individual intracardiac and aortic arch defects that were scored include persistent truncus arteriosus (PTA), atrial septal defects (ASD), tetralogy of Fallot (TOF), ventricular septal defects (VSD), interrupted aortic arch type B (IAAB) and right sided aortic arch (RAA) and Pulmonary atresia or stenosis (PS). Each individual conotruncal heart defect (CTD) or atrial septal defect, was coded to a dichotomous variable for phenotype (0=absent, 1=present). VSD without other intracardiac defects was termed VSD2. Contingency tables were created for the relationship between having one individual defect and having another defect. Chi-square tests on each contingency table were performed for pair-wise correlation between individual cardiovascular defects. Cramer’s phi was used as the correlation coefficient to measure the effect size (Cramér, 1946).
Common SNPs and cardiovascular defects
Nine common SNPs, MAF >0.05, were genotyped in 1,022 DNA samples from the 22q11DS cohort as described above. Chi-square (χ2) test or Fisher’s exact test was used to test associations between the “genotype” (allele) and individual heart defects. Power calculations for CHD associations were performed using the CaTS Genetic Power Calculator (http://www.sph.umich.edu/csg/abecasis/CaTS/index.html) (Skol, et al., 2006).
Haplotype analysis
Since the TBX1 gene is located within the 22q11.2 deletion region, and all proband DNAs have one copy of the gene, their individual haplotypes were directly observed. We applied an X-chromosome type of model in Haploview (Barrett, et al., 2005) to evaluate the haplotype block structure of the TBX1 gene using 17 common SNPs (MAF >0.05) from TBX1 resequencing data for analysis as a guide. Specifically, we used the same procedure used to analyze the non-pseudoautosomal regions of the X chromosome in males.
Very rare variant enrichment analysis
Variants with minor allele frequency (MAF) less than <0.01 were treated as a very rare variations. We selected 45 very rare variants that passed quality control for the analysis in 360 DNA samples. Different variants were collapsed and tested for overall frequency differences between patients with and without cardiovascular defects (Li and Leal, 2008). The very rare variant allele counts were collapsed by assigning each individual a carrier/non-carrier status. This was based on the presence or absence of very rare variant minor alleles. A simple contingency table was constructed to test for differences in rare variant minor allele frequency between cases and controls using the Chi-squared test (or Fisher’s exact test where necessary).
Results
Phenotype enrichment groups
Clinical data from 1,022 DNA samples from patients with the 22q11.2 deletion were used for this study. In our cohort, 65% (664 patients) had a cardiovascular defect while 35% had normal intracardiac and aortic arch anatomy. Most of the subjects had more than one cardiovascular abnormality. To obtain enough patients in each phenotype category for the genetic study, we created phenotype enrichment groups for each cardiovascular malformation. We did not distinguish between individual subtypes of malformations. Phenotype enrichment groups focused on one defect at a time, while not taking other malformations into consideration. We present the numbers of subjects in each group in Figure 1 and the correlation analysis in Table 1. Due to the co-occurrence of cardiovascular phenotypes, it was not possible to make specific phenotype-phenotype correlations with one exception (Table 1). There was a significant correlation between the ASD and VSD enrichment groups, suggesting that septal defects have a similar genetic basis in association with the syndrome.
Figure 1.
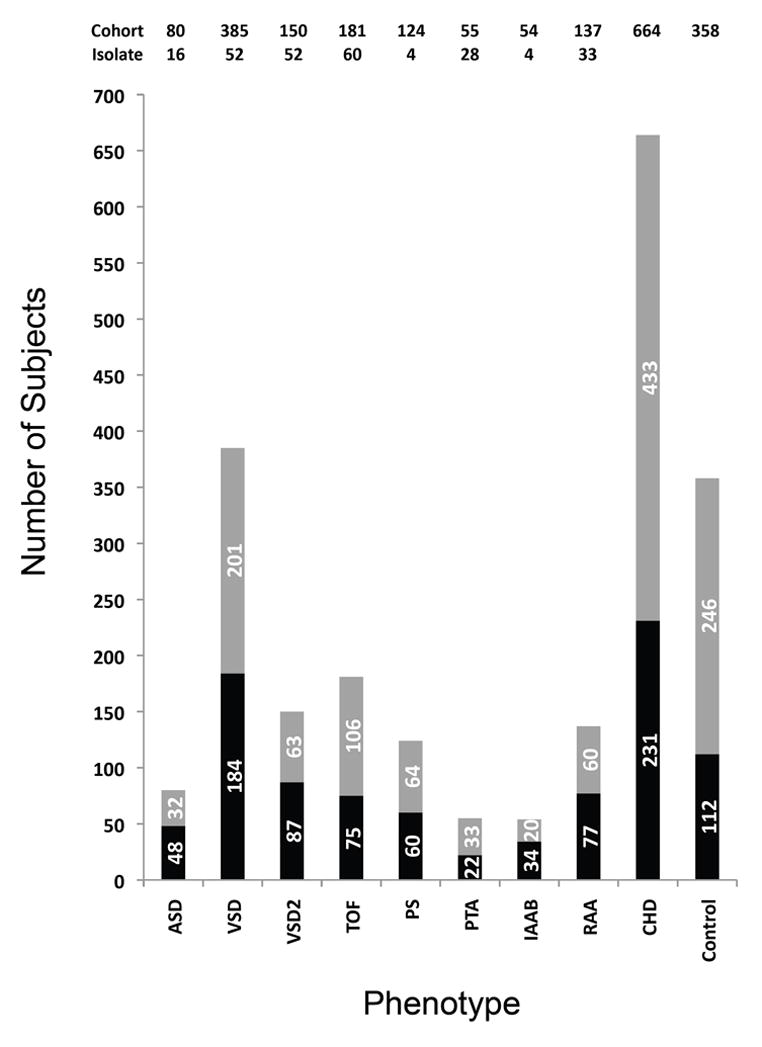
Cardiovascular phenotype enrichment groups. The stacked bar chart represents the total number of samples used for TBX1 resequencing (Cohort). The number in each group is also indicated above the bar chart (Cohort). Those with a significant intracardiac or aortic arch malformation are referred to as having a congenital heart defect (CHD). Total samples of isolated individual malformations were shown on the top of the figure (Isolate). For example, the total number of patients with atrial septal defects (ASDs) is 80. Among the 80, 16 had an ASD but no other defects (not shown in the table). The rest had ASD as well as other cardiovascular defects. TBX1 sequencing was performed on 48 ASD patients, while genotyping for common SNP variants was performed in all of them. The number 32 derives from subtracting 48 from 80. There were 385, 22q11DS subjects with VSDs (ventricular septal defects; 52 samples with VSD only). VSD occurs as part of tetralogy of Fallot (TOF) and persistent truncus arteriosus (PTA). Among those in the VSD enrichment group, 150 had a VSD that was not associated with TOF or PTA (52 samples had VSD but no other intracardiac defect). A separate designation was made for those with a VSD but not in association with TOF or PTA (VSD2). There were 150 with VSD as the only intracardiac defect. A total of 181 patients had TOF; 75 were subjected to TBX1 resequencing and all were genotyped. Sixty 22q11DS patients had TOF but no aortic arch defect or ASD (not shown in the table). Pulmonic stenosis (PS) occurs in association with TOF and it can co-occur with a VSD. In some of the echocardiogram summaries, PS and VSD were indicated but TOF wasn’t. In those situations, we did not assume the patient had a TOF. A total of 124 comprised the PS enrichment group (pulmonic atresia or stenosis). Sixty were subjected to TBX1 resequencing and all were genotyped. Among those with PS, only 4 had this malformation and no other intracardiac or aortic arch anomaly. Only 28 of 55 had a PTA but no other anomaly. Fifty-four patients had an interrupted aortic arch type B (IAAB); 34 were sequenced and all were genotyped. Only 4 total had an IAAB but no other intracardiac or aortic arch defect. A total of 137 had a right-sided aortic arch (RAA) and 77 were subjected to TBX1 resequencing, while all were genotyped. Thirty-three patients had RAA as the only cardiovascular defect. In total, 664 patients had an intracardiac and/or aortic arch malformation (CHD; cases) and were genotyped while 231 were subjected to TBX1 resequencing. A total of 358 patients had no detectable CHD (controls), and 112 were sequenced, while all were genotyped. Defects such as small patent foramen ovale or abnormal branching of the subclavian arteries were not always detected or noted and were not included in the analysis.
Table 1.
Phenotype-Phenotype correlation analysis
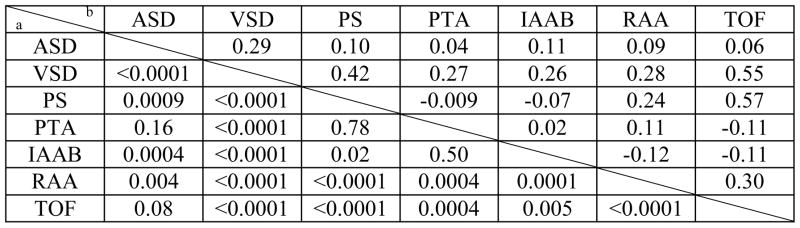
|
The left lower panel shows the Chi-square p value
the right shows Cramer’s phi correlation coefficient.
TBX1 gene resequencing
We obtained unambiguous DNA sequence from 360 22q11DS subjects. A total of 84 DNA variations including SNPs (single nucleotide polymorphisms) and small indels (insertion-deletion polymorphisms) were detected in the sequence data from all the subjects. Of these 84, only 71 DNA variations provided consistent read calls and thus passed our criteria for quality control. Seventeen of these were common variations with a MAF >0.05 (0.07–0.44, Supp. Table S2), while 54 were rare variations with a MAF <0.05. A total of 45 of the 54 rare variants were considered, comprised very rare variations with a MAF <0.01.
Among the 71 SNPs, three were missense, non-synonymous SNPs (rs41298838; rs72646967, and rs4819522, Figure 2 and Supp. Table S1). The SNP rs41298838 resulted in a Glycine to Serine amino acid substitution at position 310. This variation occurred in one patient who had no cardiovascular abnormalities. The frequency of the G allele is 0.07 in the Asian population but this SNP is monomorphic for the A allele in Caucasian and African populations as noted from dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/; submitted by Dr. Nickerson based on one population with different ethnicity). This patient is self reported as Asian. This variation was predicted to be “possibly damaging” in its effect on the protein, and had a score of 0.815 by the PolyPhen software (http://genetics.bwh.harvard.edu/pph2/) (Adzhubei, et al., 2010). The SNP rs72646967, is a common variation that occurred in 63 patients. It resulted in an Asparagine to Histidine amino acid substitution at position 397. The SNP rs4819522, which resulted in a Threonine to Methionine variation at position of 350 of TBX1 isoform A, is another common SNP. Both were predicted to be “benign” in its effects on the protein with scores of 0.000 and 0.028, respectively as determined by PolyPhen software. Protein sequence conservation across species analysis was performed using the SIFT software (http://blocks.fhcrc.org/sift/SIFT_seq_submit2.html) developed by Ng and Henikoff (Ng and Henikoff, 2001; Ng and Henikoff, 2002). The first two variations, above, were predicted to be “tolerated” with score 0.16 and 1 respectively, and the third variation was predicted to be “damaging” with score 0.02, but with lower confidence. Overall, there was not a significant increase or decrease in occurrence or frequency of missense SNPs in those with and without congenital heart defects.
Figure 2.

DNA variations in TBX1 exons. Three TBX1 isoforms (TBX1 isoform B, top, 18,124,226-18,151,112; TBX1 isoform A, bottom, 18,124,226-18,147,068; TBX1 isoform C, middle, 18,124,226-18,134,855 on human genome hg18) were shown on the “RefSeq Genes” track in the UCSC Genome Browser. Custom track “Variations in TBX1 exons” was added to visualize the DNA variations found in the exons from DNA sequence analysis of 360 22q11DS patients. Each bar represents a variation, color coded as red, green and blue to represent non-synonymous, synonymous and UTR variations, respectively.
Six synonymous SNPs (rs72646967, rs41298814, rs2301558, rs41298840, rs13054377 and rs72646968) were also found (Figure 2). The SNPs rs41298814, rs2301558, rs41298840 and rs72646967 appeared commonly in our population (MAF >0.05), while rs72646968 occurred in one patient having PS. Six SNPs were identified in the 3′-UTRs of each of the three isoforms (untranslated region). Four SNPs occurred in the 3′ UTR of TBX1 isoform C, two occurred in the 3′ UTR of isoform A, while none occurred in the 3′ UTR of isoform B (Figure 2). There was no enhancement of the synonymous SNPs in patients with congenital heart defects compared to those without in our cohort of 360.
We compared the frequency and regions where the SNPs occurred versus the same content in the emerging 1,000 Genomes Project data (Figure 3). Allele frequencies were similar, despite the fact that DNAs from different ethnicities were sequenced in the 1,000 Genomes Project. Figure 3). This suggests that there are no significant differences between allele frequencies or map locations of the SNPs in our cohort (Figure 3). Only one novel variant was identified in our study that was not present in the 1,000 Genomes data or dbSNP build 131. This was a −/TCGC indel (insertion/deletion) variant, referred to as (Novel 23) was identified. The variant occurred in the same DNA sample that had the minor allele of the non-synonymous SNP, rs41298838.
Figure 3.
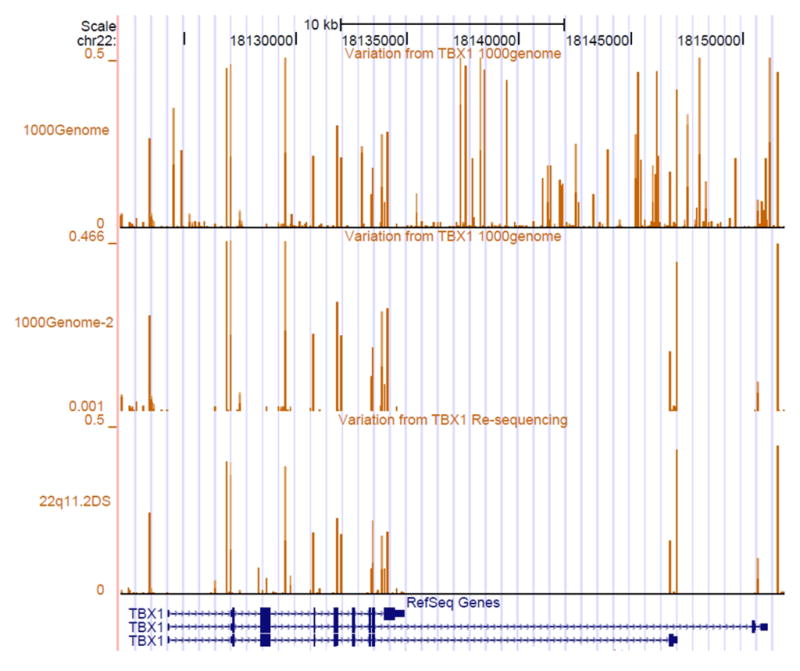
Comparison of frequency SNPs to public databases. We performed a comparison of data on the MAF (Minor allele frequency) of SNPs between the 22q11DS study and 1,000 Genome Project (http://www.1000genomes.org/;) (Durbin, et al., 2010). Three custom tracks are shown in addition to the RefSeq track in a snapshot of the UCSC Genome Browser (hg18). The “1,000Genome” track shows the MAFs of all DNA variations identified in the TBX1 locus. The data were extracted from pilot data (release 2010_07) from the 1,000 Genome Project. The next two tracks, entitled “1,000 Genome-2″ and “ 22q11DS” track show the MAFs of DNA variants indentified in the same regions of TBX1 that were sequenced in our cohort. The height of the lines representing each variant depicts the MAF, which turn out to be similar in both datasets. Detailed information on DNA variations identified by TBX1 resequencing is in Supp. Table S2.
Very rare variation enrichment analysis
One important possibility is that cases with cardiovascular defects or controls, without, might show an enrichment of rare TBX1 DNA variations (Supp. Table S2). These variants were combined and used for rare variation enrichment statistical analysis in 360 samples (Figure 1). We did not find any enrichment of the number of very rare DNA variations between 22q11DS cases with presence of congenital heart disease compared with 22q11DS controls, with normal anatomy (p=0.44).
Phenotype enrichment group analysis was performed to identify subtle differences. Controls showed enrichment of rare variants compared to two phenotype enrichment groups. Groups with RAA and PTA had fewer rare variants than the group with normal cardiovascular anatomy (Figure 4A). There were no rare variants in those with a RAA but no other malformation (n=11). Based upon this, is it possible that some very rare DNA variations might serve to protect against congenital heart disease in 22q11DS patients.
Figure 4.

(A) Enrichment of very rare DNA variations. “Control”, “ CHDs” (Congenital heart defects=CTDs+ASD), “RAA” and “PTA” custom tracks were added to the UCSC Genome Browser as shown. The custom tracks indicate the type of rare variations (MAF<0.01) identified by TBX1 resequencing analysis in each phenotype enrichment group (total number of subjects and definition of each phenotype group is in Figure 1). Each line or bar represents a different rare SNP. The orange vertical lines represent variations from the putative promoter region, black represents intronic variants and blue are variants from the 3′-UTR. The red bar represents a nonsynonymous variant and a green bar denotes a synonymous variation. Detailed information of rare variations used for DNA enrichment analysis is shown in Supp. Table S2. (B) Multiple sequence alignment of 45 very rare TBX1 variants (MAF<0.01). Multiple alignment data from 44 vertebrate species generated using multiz and other tools in the UCSC/Penn State Bioinformatics comparative genomics alignment pipeline were extracted using Galaxy (http://main.g2.bx.psu.edu/), a web-based genome analysis tool (Blankenberg, et al., 2010; Goecks, et al., 2010). Both Galaxy and then Jalview (Waterhouse, et al., 2009) were used to edit alignments. The species in the database with significant missing sequence were discarded and only results from 24 mammals are shown. The first two rows indicate the major or minor allele identified in the 22q11DS patient cohort. Each row below represents one of the 24 mammalian species. In general, each column represents a rare DNA variation except for the 8th variation that has a three base and 24th that has a four base indel. The most intense blue color corresponds to the most conserved SNPs. A total of 30 major alleles from the 22q11DS cohort show evolutionary conservation. Five minor alleles show conservation as indicated. The minor allele (TCGC), the only novel variation (novel 23) found in the sample, was conserved among most mammalian species. Detailed information on the 45 rare variations is shown in Supp. Table S2.
One way to detect the functional importance of rare variations is to determine whether the major allele shows evolutionary conservation. Figure 4B shows an evolutionary pile-up of the major/minor allele from 45 very rare variants identified in the TBX1 locus. Thirty-five of the 45 SNPs (78%) SNPs showed evolutionary conservation among mammals, suggesting potential functional roles. Thirty of 35 major alleles and 5 of 35 minor alleles were conserved among different mammalian species. The minor alleles of five SNPs as well as the minor allele (TCGC), the only novel variation (novel 23) found, were conserved among primate species and some mammalian species.
Common SNPs and congenital heart defects
The next goal was to genotype the larger cohort of 22q11DS patients to test for association between common SNPs and phenotype. The common TBX1 SNPs fell into three linkage disequilibrium (LD) blocks (Figure 5). The estimate of LD structure was consistent with data from the general population of Caucasians (CEU HapMap III; data not shown).
Figure 5.
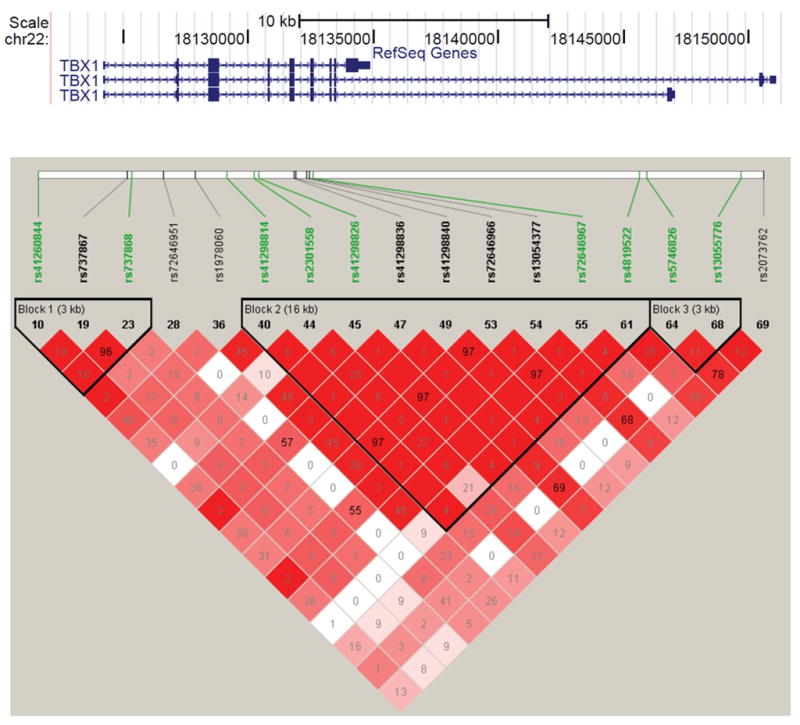
Linkage disequilibrium (LD) plot of 17 common SNPs from the TBX1 resequencing data. An LD matrix was generated from DNA sequence data, using the X-chromosome model in Haploview 4.2. Seventeen common SNPs with a MAF >0.05 were used to generate the plot. The SNPs highlighted in green are those selected to genotype in the whole cohort of 1,022 patients. Standard (D′/LOD) models were used to create the LD color scheme. The value in each pairwise box is the r2 value. We found that there were three LD blocks in the TBX1 locus.
Based upon the LD structure, nine common SNPs were selected to represent the three LD blocks. They were used to genotype the remaining subjects, so that we had data on all 1,022 samples. We found that there was no significant difference between the allele frequencies of these SNPs in cases compared to controls with the 22q11.2 deletion (p>0.05) (Table 2). Analysis of haplotypes gave similar non-significant association results (data not shown). In order to control the bias of population stratification due to different ethnicities, the self reported Asian (5 subjects) and African American (10 subjects) and Moroccan (1 subject) were removed, however none of the common SNPs or haplotypes in these patients was associated with CHDs (data not shown). The analysis was also performed between each phenotype enrichment group versus the control group (no intracardiac and no aortic arch defects). None of the common SNPs showed association among these groups (Supp. Table S3)
Table 2.
Association analysis for representative common SNPs in TBX1
| Marker | Location (hg18) | A1/A2a | Minor Allele Freq. | Chi-Squared P | CHDs | Controls | ||
|---|---|---|---|---|---|---|---|---|
| A1 | A2 | A1 | A2 | |||||
| rs41260844 | 18123423 | T/C | 0.28 | 0.91 | 162 | 425 | 88 | 227 |
| rs737868 | 18127081 | G/C | 0.38 | 0.81 | 209 | 337 | 107 | 179 |
| rs41298814 | 18130772 | C/T | 0.21 | 0.50 | 117 | 465 | 68 | 241 |
| rs2301558 | 18131828 | T/C | 0.24 | 0.84 | 140 | 455 | 76 | 239 |
| rs41298826 | 18132006 | C/T | 0.20 | 0.41 | 108 | 465 | 65 | 242 |
| rs72646967 | 18134090 | C/A | 0.18 | 0.89 | 92 | 425 | 48 | 228 |
| rs4819522 | 18146781 | T/C | 0.16 | 0.50 | 95 | 490 | 45 | 265 |
| rs5746826 | 18147050 | T/G | 0.45 | 0.59 | 254 | 313 | 142 | 162 |
| rs13055776 | 18150706 | T/C | 0.10 | 0.08 | 50 | 540 | 39 | 284 |
A1 is the minor allele and A2 is the major allele.
Rare variants and haplotypes generated by common SNPs
The TBX1 gene is hemizygously deleted in 22q11DS individuals, making it possible to directly determine haplotypes from the genotype data. Existence of the DNA sequence and genotyping data make it possible to further investigate the relationship between haplotype and rare variants directly (Cohen, et al., 2004; Frazer, et al., 2009; Kryukov, et al., 2007; Nejentsev, et al., 2009; Pritchard, 2001; Pritchard and Cox, 2002).
We investigated the relationship between common and rare haplotypes generated by 17 common SNPs and 54 rare SNPs using TBX1 resequencing data. Taking this approach, 49 haplotypes, containing common and rare SNPs were generated from the 360 samples (Figure 6). As seen in the Figure, 21 of 49 haplotypes captured 54 rare variants. The first haplotype (Hap1: CAGGGTCTCAAAACGCA) is a common haplotype (frequency 0.272). It captured nine rare variants with allele frequency of 0.3–0.6%. The only rare amino acid change (rs41298838; Figure 2) was captured by this common haplotype. The second haplotype (Hap2: CTCGGTCTCAAAACGCA, frequency 0.189) was less common than Hap1, but it captured five rare variants including two variants with a minor allele frequency of 4.5%–4.8%. However, the common haplotypes had different frequency of capture of rare alleles than of rare haplotypes. Rare haplotypes including Hap11 (CTCGACCCCGGACCTCA, frequency 0.006), Hap15 (CTCGATTTCAAAACTCG, frequency 0.003), Hap16 (CTCGGTTTCAAAACTTG, frequency 0.003), Hap19 (CACGGTTTCAAAATTCG, frequency 0.003) and Hap20 (CAGGATTTCAAAACTCG, frequency 0.003) captured 13 rare variants with the same frequency. The data suggest that rare SNP variations could be captured by common haplotypes.
Figure 6.
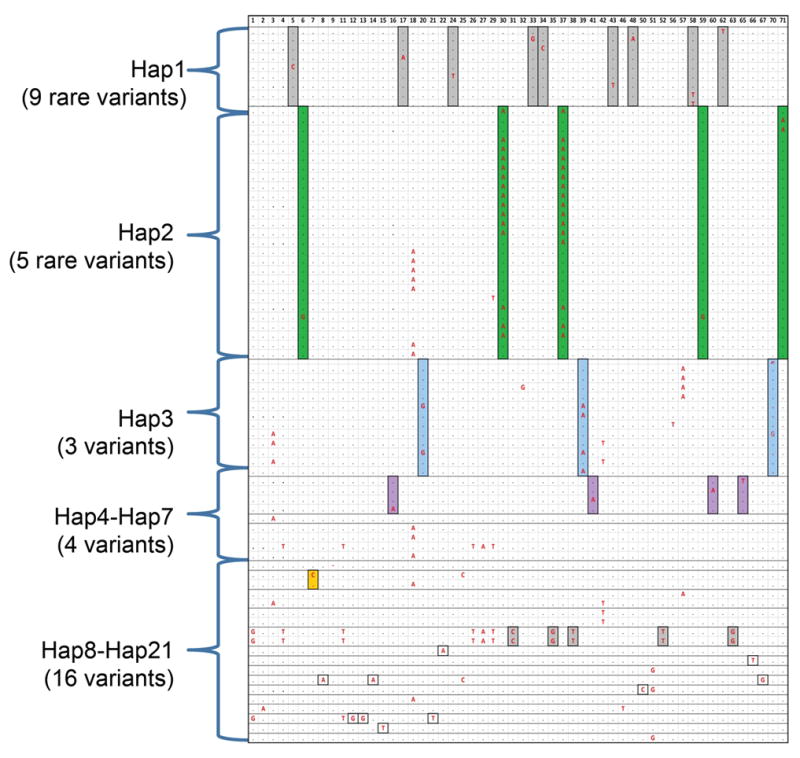
Haplotypes of common SNPs (MAF>0.05) and 54 rare variants (MAF<0.05). A total of 54 rare variants (MAF<0.05) identified by TBX1 resequencing were shown in each column (the order of variants is according to Supp. Table S2). Each row represents a patient that carries one or more of the 54 rare variants (the major allele is a black dot; the minor allele is indicted in bold red font). Each haplotype group is indicated as Hap1–21 (Hap22–49 which didn’t occur in any patient with rare variations, is not shown.). They were determined by the allele pattern of the 17 common SNPs. Some patients shared common haplotypes. Twenty-one of 49 haplotypes captured all rare variations (MAF<0.05) in a total of 77 subjects. Hap1 (CAGGGTCTCAAAACGCA), Hap2 (CTCGGTCTCAAAACGCA) and Hap3 (TTCGACCCCGGACCTCG) were common haplotypes (frequency>0.05) generated by 17 common SNPs from TBX1 resequencing data. Hap4 to Hap7 were less common haplotypes (0.01<frequency<0.05) and hap8 to hap21 were very rare haplotypes (frequency<0.01). For each variant, the boxes highlighted in different colors represent the minor allele of rare variants observed in the subjects with the given haplotypes indicated (Hap1–21).
However, the rare variants did not occur with equal frequency within common haplotypes. This suggests that it is possible to capture most but not all the rare SNPs within haplotype information. Sixteen of 54 rare variations could be captured by rare haplotypes generated by surrounding common variants, and 15 of 16 rare variants could be observed with equal frequencies with haplotypes. However, the ratio of the number of rare variants with equal frequencies with haplotypes to rare variants captured by rare haplotypes could be exaggerated because 14 of 16 rare variants only occurred in one patient.
Discussion
In this report, we present an analysis of TBX1 DNA sequence variations in 1,022 22q11DS patients to determine whether they modify cardiovascular defects. We found that neither common DNA variations themselves nor common haplotypes in TBX1 influence the presence or severity of significant cardiovascular phenotypes. There was no enrichment of very rare variations (MAF<0.01) in 22q11DS patients with cardiovascular defects (cases) versus patients with normal cardiovascular anatomy (controls). Collectively, these results suggest that common and rare variations elsewhere on 22q11.2 or in the genome influence expression of cardiovascular phenotypes. We did observe an enrichment of very rare variations in controls without cardiovascular malformations versus 22q11DS patients with RAA and PTA phenotypes that may be protective by altering expression or function of TBX1. This is supported by the fact that 67% of the SNPs show evolutionary conservation among mammals suggesting that they may be functionally relevant. Larger studies will be needed to understand the importance of rare SNP enrichment. In addition, functional analysis will be required to understand how each variation could influence phenotypic expression.
Common DNA variations
There have been previous studies performed with similar goals to find variations in TBX1 influencing cardiovascular phenotypes. In one study, a total of 39 22q11DS patients were examined with 23 possessing a congenital heart defect while 16 had no defect (Voelckel, et al., 2004). Single stranded conformational analysis (SSCP) was performed on TBX1 exons 2–9, followed by DNA sequencing of PCR products showing any possible variation. They found polymorphisms occurring in healthy individuals, but did not find any missense variations, and suggested that mutations in TBX1 do not alter cardiac phenotypes in 22q11DS patients (Voelckel, et al., 2004). In a second study, the entire 43,776 bp TBX1 locus (hg18: 18,109,240-18,153,016) was sequenced in 29 22q11DS patients and 95 unrelated healthy controls (Heike, et al., 2010). They identified rare and common SNPs and haplotypes, but there were too few subjects to determine if any influenced expressivity. In the third study, an analysis was done of 174 22q11DS patients, where 123 had a cardiovascular defect and 51 had a normal heart (Rauch, et al., 2004). They also compared allele frequencies to that of 96 normal dizygous healthy individuals (Rauch et al., 2004) and did not detect a significant difference in the frequency of common recurrent SNPs (Rauch, et al., 2004). Although previous studies suggest that common TBX1 variants are not modifiers of cardiovascular phenotypes, the studies were relatively small in size and thus, had relative low power to detect an association. Our study generated from over a thousand patients had more than 80% power to detect an association between CHD and TBX1 SNPs with a genotype relative risk (GRR) of 1.2 in an additive genetic model.
Rare DNA variations
One of the greatest challenges in human genetics is to make genotype-phenotype correlations to find genetic modifiers. In order to evaluate rare DNA variations with significant power to draw conclusions, it was necessary to take a different approach. We determined whether the total number of rare DNA variations in TBX1 was altered in frequency between 22q11DS cases with a cardiovascular phenotype and 22q11DS controls without. We found that the total number of rare variations was not enhanced in affected cases versus controls with the 22q11.2 deletion, overall. Normal intracardiac and aortic arch anatomy occurred in less than 1/3 of the 22q11DS cohort. It is possible that rare variants might be enriched in controls versus cases of specific sub-phenotypes. We found rare variations in TBX1 occur more frequently in controls than those with two sub-phenotypes, RAA and PTA, both involving the aortic arch. None of the rare variations were deemed pathogenic mutations. We did not find an association between common or rare haplotypes and RAA or PTA.
Existence of rare pathogenic mutations on the remaining allele of TBX1 might lead to neonatal lethality and would be missed in this type of cohort. Mutations in TBX1, albeit rare, have been uncovered in a small subset of individuals with conotruncal heart defects (Conti, et al., 2003; Gong, et al., 2001; Griffin, et al., 2010; Paylor, et al., 2006; Rauch, et al., 2010; Torres-Juan, et al., 2007; Yagi, et al., 2003; Zweier, et al., 2007). Based upon these studies in which mutations on one allele only occurred, it is likely that severe mutations on the single remaining allele of TBX1 in 22q11DS patients would be lethal. Supporting this idea, mice that are homozygous for Tbx1 null mutations die at birth with a PTA (Jerome and Papaioannou, 2001; Lindsay, et al., 2001; Merscher, et al., 2001). Thus, we do not expect to identify premature stop codons or highly pathogenic missense changes. We did detect two missense changes, one was a Glycine to Serine change in a single patient and the other was an Asparagine to Histidine change that occurred commonly, showing no association to cardiovascular phenotype.
To determine the potential functional consequences of rare variations as a whole, we determined whether rare variations comprised specific haplotypes, thereby serving to proxy rare variations among the entire cohort of 1,022 samples.
To determine the possible functional consequence of rare variants enrichment, we ascertained whether they alter nucleotides that were highly conserved among vertebrate species. Approximately 78% of the rare SNPs showed strong evolutionary conservation suggesting that they could influence expression levels of TBX1. In our case, it is possible that rare variations serve to enhance TBX1 expression levels. TBX1 function in mice and humans is sensitive to both decreased and increased gene copy number.
TBX1 function in mice and humans is sensitive to both decreased and increased gene copy number. Increase or decrease of Tbx1 dosage in mice results in related cardiovascular malformations as occurring in patients (Liao, et al., 2004; Zhang and Baldini, 2008). Humans with the 22q11.2 duplication have been described and are increasing in diagnosis due to application of microarray comparative genome hybridization technologies. The duplication is the reciprocal product of an inter-chromosomal non-allelic homologous recombination event in meiosis (Edelmann, et al., 1999). A developmental delay disorder with associated congenital malformations including related heart defects occurs as a result of the duplication, but with reduced penetrance (Ensenauer, et al., 2003; Ou, et al., 2008; Yobb, et al., 2005). It is possible that three copies of TBX1 instead of two copies cause similar types of physical malformations. This is relevant to the results obtained in which 22q11DS patients with RAA or PTA have fewer rare TBX1 DNA variations than those with no cardiovascular defects and suggests that these variants might alter TBX1 levels or function and serve as protective roles. A few years ago, human TBX1 missense mutations were found to cause gain of function resulting in 22q11DS related phenotypes (Zweier, et al., 2007). In the study by Zweier et al., they followed up the mutations in non-deleted patients by testing transcriptional activation of the mutants in cell culture and found enhanced activity (Zweier, et al., 2007). Overall, these results suggest that the TBX1 gene is sensitive to altered dosage and variations that effect protein expression or function could alter phenotypes.
Other genes on 22q11.2 could influence expressivity
Studies in mice suggest that modifiers may exist because genetic background strongly influences phenotype. There may be many different causative modifiers in humans, some of which might be DNA variations in genes on the remaining allele of 22q11.2 as the other copy is no longer present. A recent release of the newest human genome assembly (hg19) has been annotated in the UCSC and Ensembl genome browsers. Currently, there are over 60 known and predicted genes in the 3 Mb region (Hg19, chr22:18,809,439-21,436,728; 2,627,290bp; http://genome.ucsc.edu) and 39 in the nested 1.5 Mb critical region (chr22:18,809,439-20,328,689; 1,519,251 bp). It is possible that DNA variations in other genes on the remaining allele influence expressivity of cardiovascular defects to a greater extent than TBX1. Among the genes in the 22q11.2 interval, CRKL (MIM# 602007), encoding a cytoplasmic adaptor to receptor tyrosine kinases, has been shown in mouse models to interact genetically with TBX1 (Guris, et al., 2006). Tbx1+/− mouse embryos had VSDs and mild aortic arch branching defects such as abnormal origin of the right subclavian artery at reduced penetrance (Jerome and Papaioannou, 2001; Lindsay, et al., 2001; Merscher, et al., 2001), whereas Tbx1−/− mouse embryos had a PTA with obligate VSD (Jerome and Papaioannou, 2001; Lindsay, et al., 2001; Merscher, et al., 2001). Inactivation of both copies of Crkl in mice resulted in similar defects as in humans with the deletion or related mouse models that act in the Tbx1 genetic pathway (Guris, et al., 2006). Crkl−/− null mutants had a VSD, aortic arch anomalies (IAAB, abnormal origin of the right subclavian artery) and double outlet right ventricle. Tbx1+/−; Crkl+/− mice had the full spectrum of cardiovascular defects as seen in 22q11DS patients. One may suggest based upon mouse genetic data that DNA variations in CRKL might alter expressivity of cardiovascular malformations.
Another important gene on the remaining 22q11.2 allele to investigate is the Ubiquitin fusion degradation 1 homolog to yeast Ufd1 gene (UFD1L; MIM# 601754). It is a component of a complex that facilitates degradation of polyubiquitin-tagged proteins (Meyer, et al., 2000; Ye, et al., 2001). A patient was identified with a hemizygous mutation in UFD1L that had similar cardiovascular malformations as in 22q11DS patients (Yamagishi, et al., 1999), although its role as a candidate gene for non-deleted patients with a 22q11DS phenotype is not clear (Wadey, et al., 1999). In addition to UFD1L, another gene, histone cell cycle regulation defective homolog A (HIRA; MIM# 600237 [S. cerevisiae]); TUPLE1; TUP1-like enhancer of split protein 1), has been studied for its role in etiology of the main clinical features of 22q11DS (Halford, et al., 1993; Magnaghi, et al., 1998). Inactivation of one allele of Hira in mouse models did not result in a phenotype while inactivation of both copies resulted in gastrulation defects (Roberts, et al., 2002). Similar to UFD1L, HIRA has had limited follow up studies in comparison to that of TBX1 and CRKL, which when inactivated in the mouse cause cardiac outflow tract defects. Overall, it would be useful to perform DNA sequencing of exons of all the genes on the remaining allele of chromosome 22q11.2 to identify common or rare variants that alter the phenotypic spectrum.
Genes elsewhere in the genome that can alter expressivity
Previous mouse genetic studies have suggested that it is not genes on the other allele of 22q11.2 that can influence expressivity but variants elsewhere in the genome. In a study of a mouse model containing a deletion of the region of synteny to 22q11.2, termed Df1/+, it was found that varying the background of the remaining allele had no alteration in phenotype (Taddei, et al., 2001). Genetic factors due to varying genetic background elsewhere, were hypothesized to influence expressivity (Taddei, et al., 2001).
To identify genetic modifiers, several candidate gene approaches were undertaken by using mouse models and intercrossing mutant mice with Tbx1 mouse mutants. One important gene downstream of Tbx1 is Fgf8 (Fibroblast growth factor ligand 8). Foxa2, a forkhead transcription factor gene, might act upstream of Tbx1, which might regulate Fgf8 protein levels (Hu, et al., 2004). Hypomorphic Fgf8 mutant mouse embryos had similar malformations as homozygous Tbx1 null mutant embryos suggesting a possible shared genetic pathway (Park, et al., 2006) (Ilagan, et al., 2006). Epistasis studies by intercrossing Tbx1+/− mice with Fgf8+/− mice showed a much more severe phenotype than either alone (Vitelli, et al., 2002). Of interest, there is also a genetic interaction in the mouse between Crkl and Fgf8 (Park, et al., 2006); as well as one between all three genes, Tbx1, Crkl and Fgf8 (Guris, et al., 2006), indicating the importance of the three genes for development of the outflow tract. In addition to these, a genetic interaction was identified between several additional genes including Pitx2, Gbx2, Chd7 and Hes1, among others (Calmont, et al., 2009; Nowotschin, et al., 2006; Randall, et al., 2009) (van Bueren, et al., 2010). Mutations in PITX2 causes Reiger syndrome (MIM# 180500), however cardiac malformations are not a hallmark feature of the disorder in humans (Semina, et al., 1996). Among the others, Chd7 is particularly interesting because mutation or deletion of CHD7 in humans causes CHARGE syndrome (MIM# 214800) with associated cardiac outflow tract defects such as VSD and TOF [(Vissers, et al., 2004) CHARGE, coloboma, heart defects, atresia choanae, retarded growth, genital abnormalities and ear defects)]. A genetic interaction between the two genes indicates that the two diseases, 22q11DS and CHARGE syndrome share common pathways (Randall, et al., 2009)
Unbiased gene profiling approaches have been performed to identify new genes or pathways downstream of Tbx1 (Ivins, et al., 2005; Liao, et al., 2008). From this, there are many genes to pursue in human DNAs by genotyping for common SNPs or sequencing to uncover rare variations. Nonetheless, truly unbiased genome wide approaches must be undertaken to identify common or rare variations influencing the expressivity of cardiovascular defects. One approach would be to perform genome wide association studies (GWAS) to identify loci harboring common SNP variations. This would involve genotyping many hundreds if not thousands of 22q11DS patients with high-density microarrays containing up to a million or more SNPs. The goal would be to perform genotype-phenotype correlations in a similar manner as for TBX1.
Another outcome from performing a GWAS is the ability to also identify common and rare genomic deletions or duplications referred to as copy number variations (CNVs). Such CNVs might uncover second hit loci that interact with genes on the 22q11.2 region. Finally, whole exome sequencing analysis has to potential to uncover rare DNA variations that act as second hit mutations influencing expressivity. For exome sequence analysis, mutations will be found but the challenge will be to determine which are responsible for influencing expressivity.
Environmental exposures, stochastic factors as well genetic modifiers are likely responsible for variable expressivity in 22q11DS. It is clear that both environmental (intra-uterine) and stochastic factors play a significant role in influencing expressivity. Exposure to high or low levels of retinoic acid (activated form of vitamin A) or alcohol during pregnancy results in similar malformations as those occurring in 22q11DS patients (Burd, et al., 2007; Ruckman, 1990). Further, evidence for non-DNA based etiologies derives from studies of monozygotic twins with the disorder. In a few studies of monozygotic twins with the syndrome it was found that most have discordant phenotypes (Lu, et al., 2001; Vincent, et al., 1999; Yamagishi, et al., 1998). One problem with the studies is that not many monozygotic twin pairs have been analyzed. Nonetheless, this indicates that environmental and stochastic factors influence expressivity.
In summary, we found that neither common SNPs nor haplotypes in TBX1 could explain the basis of phenotypic expression variability of congenital heart disease in 1,022 patients with 22q11DS. In our cohort, 65% of 22q11DS patients with the 22q11.2 deletion have a congenital heart defect while 35% do not. The finding that rare variations were enriched in those with no cardiovascular defect as compared to those with RAA and PTA, and that a subset of those DNA variants were highly conserved among vertebrate species suggests possible functional roles. More work needs to be done examining common and rare DNA variations of additional genes on 22q11.2 and elsewhere to determine their role in altering expressivity of 22q11DS. It is also possible that although TBX1 variations do not influence cardiovascular phenotypes, they may be important for craniofacial or other malformations associated with the syndrome.
Supplementary Material
Acknowledgments
This work could not be done without the families affected with 22q11DS as well as numerous clinicians that participated in the study. The genetic assays and DNA isolation was performed in the Genomics and Molecular Cytogenetics Cores at Einstein, respectively. We thank Dr. Silvia Racedo, Dr. Ping Kong, Maria Delio, Sean Herman, Dennis Monks, David Sweet, Noah Kolatch, Raquel Castellanos, Stephania Macchiarulo and Laina Freyer, for technical assistance. We gratefully acknowledge funding from NIH grant HL084410 that provided support.
Footnotes
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, et al. Practical Guidelines for Managing Patients with 22q11.2 Deletion Syndrome. The Journal of pediatrics. 2011 doi: 10.1016/j.jpeds.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. In: Ausubel Frederick M, et al., editors. Current protocols in molecular biology. Unit 19. Chapter 19. 2010. pp. 101–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budarf ML, Konkle BA, Ludlow LB, Michaud D, Li M, Yamashiro DJ, McDonald-McGinn D, Zackai EH, Driscoll DA. Identification of a patient with Bernard-Soulier syndrome and a deletion in the DiGeorge/velo-cardio-facial chromosomal region in 22q11.2. Human molecular genetics. 1995;4(4):763–6. doi: 10.1093/hmg/4.4.763. [DOI] [PubMed] [Google Scholar]
- Burd L, Deal E, Rios R, Adickes E, Wynne J, Klug MG. Congenital heart defects and fetal alcohol spectrum disorders. Congenital heart disease. 2007;2(4):250–5. doi: 10.1111/j.1747-0803.2007.00105.x. [DOI] [PubMed] [Google Scholar]
- Burn J, Goodship J. Developmental genetics of the heart. Current opinion in genetics & development. 1996;6(3):322–5. doi: 10.1016/s0959-437x(96)80009-8. [DOI] [PubMed] [Google Scholar]
- Calmont A, Ivins S, Van Bueren KL, Papangeli I, Kyriakopoulou V, Andrews WD, Martin JF, Moon AM, Illingworth EA, Basson MA, et al. Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development. 2009;136(18):3173–83. doi: 10.1242/dev.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305(5685):869–72. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- Conti E, Grifone N, Sarkozy A, Tandoi C, Marino B, Digilio MC, Mingarelli R, Pizzuti A, Dallapiccola B. DiGeorge subtypes of nonsyndromic conotruncal defects: evidence against a major role of TBX1 gene. European journal of human genetics: EJHG. 2003;11(4):349–51. doi: 10.1038/sj.ejhg.5200956. [DOI] [PubMed] [Google Scholar]
- Cramér H. Mathematical Methods of Statistics. Princeton: Princeton University Press; 1946. [Google Scholar]
- Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann L, Pandita RK, Morrow BE. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. American journal of human genetics. 1999;64(4):1076–86. doi: 10.1086/302343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensenauer RE, Adeyinka A, Flynn HC, Michels VV, Lindor NM, Dawson DB, Thorland EC, Lorentz CP, Goldstein JL, McDonald MT, et al. Microduplication 22q11.2, an emerging syndrome: clinical, cytogenetic, and molecular analysis of thirteen patients. American journal of human genetics. 2003;73(5):1027–40. doi: 10.1086/378818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fibison WJ, Budarf M, McDermid H, Greenberg F, Emanuel BS. Molecular studies of DiGeorge syndrome. American journal of human genetics. 1990;46(5):888–95. [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nature reviews. Genetics. 2009;10(4):241–51. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- Garg V, Yamagishi C, Hu TH, Kathiriya IS, Yamagishi H, Srivastava D. Tbx1, a DiGeorge syndrome candidate gene, is regulated by Sonic hedgehog during pharyngeal arch development. Developmental Biology. 2001;235(1):62–73. doi: 10.1006/dbio.2001.0283. [DOI] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome biology. 2010;11(8):R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W, Gottlieb S, Collins J, Blescia A, Dietz H, Goldmuntz E, McDonald-McGinn DM, Zackai EH, Emanuel BS, Driscoll DA, et al. Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. Journal of medical genetics. 2001;38(12):E45. doi: 10.1136/jmg.38.12.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin HR, Topf A, Glen E, Zweier C, Stuart AG, Parsons J, Peart I, Deanfield J, O’Sullivan J, Rauch A, et al. Systematic survey of variants in TBX1 in non-syndromic tetralogy of Fallot identifies a novel 57 base pair deletion that reduces transcriptional activity but finds no evidence for association with common variants. Heart. 2010;96(20):1651–5. doi: 10.1136/hrt.2010.200121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guris DL, Duester G, Papaioannou VE, Imamoto A. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Developmental cell. 2006;10(1):81–92. doi: 10.1016/j.devcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Halford S, Wadey R, Roberts C, Daw SC, Whiting JA, O’Donnell H, Dunham I, Bentley D, Lindsay E, Baldini A, et al. Isolation of a putative transcriptional regulator from the region of 22q11 deleted in DiGeorge syndrome, Shprintzen syndrome and familial congenital heart disease. Human molecular genetics. 1993;2(12):2099–107. doi: 10.1093/hmg/2.12.2099. [DOI] [PubMed] [Google Scholar]
- Heike CL, Starr JR, Rieder MJ, Cunningham ML, Edwards KL, Stanaway IB, Crawford DC. Single Nucleotide Polymorphism Discovery in TBX1 in Individuals with and without 22q11.2 Deletion Syndrome. Birth Defects Research Part a-Clinical and Molecular Teratology. 2010;88(1):54–63. doi: 10.1002/bdra.20604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T, Yamagishi H, Maeda J, McAnally J, Yamagishi C, Srivastava D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development. 2004;131(21):5491–502. doi: 10.1242/dev.01399. [DOI] [PubMed] [Google Scholar]
- Ilagan R, Abu-Issa R, Brown D, Yang YP, Jiao K, Schwartz RJ, Klingensmith J, Meyers EN. Fgf8 is required for anterior heart field development. Development. 2006;133(12):2435–45. doi: 10.1242/dev.02408. [DOI] [PubMed] [Google Scholar]
- Ivins S, Lammerts van Beuren K, Roberts C, James C, Lindsay E, Baldini A, Ataliotis P, Scambler PJ. Microarray analysis detects differentially expressed genes in the pharyngeal region of mice lacking Tbx1. Developmental biology. 2005;285(2):554–69. doi: 10.1016/j.ydbio.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nature genetics. 2001;27(3):286–91. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Kimber WL, Hsieh P, Hirotsune S, Yuva-Paylor L, Sutherland HF, Chen A, Ruiz-Lozano P, Hoogstraten-Miller SL, Chien KR, Paylor R, et al. Deletion of 150 kb in the minimal DiGeorge/velocardiofacial syndrome critical region in mouse. Human molecular genetics. 1999;8(12):2229–37. doi: 10.1093/hmg/8.12.2229. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: Implications for complex disease and association studies. American Journal of Human Genetics. 2007;80(4):727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. American journal of human genetics. 2008;83(3):311–21. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Aggarwal VS, Nowotschin S, Bondarev A, Lipner S, Morrow BE. Identification of downstream genetic pathways of Tbx1 in the second heart field. Developmental biology. 2008;316(2):524–37. doi: 10.1016/j.ydbio.2008.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Kochilas L, Nowotschin S, Arnold JS, Aggarwal VS, Epstein JA, Brown MC, Adams J, Morrow BE. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Human molecular genetics. 2004;13(15):1577–85. doi: 10.1093/hmg/ddh176. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Botta A, Jurecic V, Carattini-Rivera S, Cheah YC, Rosenblatt HM, Bradley A, Baldini A. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature. 1999;401(6751):379–83. doi: 10.1038/43900. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Shaffer LG, Carrozzo R, Greenberg F, Baldini A. De novo tandem duplication of chromosome segment 22q11–q12: clinical, cytogenetic, and molecular characterization. American journal of medical genetics. 1995;56(3):296–9. doi: 10.1002/ajmg.1320560316. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410(6824):97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- Lu JH, Chung MY, Hwang B, Chien HP. Monozygotic twins with chromosome 22q11 microdeletion and discordant phenotypes in cardiovascular patterning. Pediatric cardiology. 2001;22(3):260–3. doi: 10.1007/s002460010219. [DOI] [PubMed] [Google Scholar]
- Magnaghi P, Roberts C, Lorain S, Lipinski M, Scambler PJ. HIRA, a mammalian homologue of Saccharomyces cerevisiae transcriptional co-repressors, interacts with Pax3. Nature genetics. 1998;20(1):74–7. doi: 10.1038/1739. [DOI] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al. TBX1 is responsible for cardiovascular defects in Velo-Cardio-Facial/DiGeorge syndrome. Cell. 2001;104(4):619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. The EMBO journal. 2000;19(10):2181–92. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow B, Goldberg R, Carlson C, Das Gupta R, Sirotkin H, Collins J, Dunham I, O’Donnell H, Scambler P, Shprintzen R, et al. Molecular definition of the 22q11 deletions in velo-cardio-facial syndrome. American journal of human genetics. 1995;56(6):1391–403. [PMC free article] [PubMed] [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324(5925):387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome research. 2001;11(5):863–74. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome research. 2002;12(3):436–46. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S, Liao J, Gage PJ, Epstein JA, Campione M, Morrow BE. Tbx1 affects asymmetric cardiac morphogenesis by regulating Pitx2 in the secondary heart field. Development. 2006;133(8):1565–73. doi: 10.1242/dev.02309. [DOI] [PubMed] [Google Scholar]
- Ou Z, Berg JS, Yonath H, Enciso VB, Miller DT, Picker J, Lenzi T, Keegan CE, Sutton VR, Belmont J, et al. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genetics in medicine: official journal of the American College of Medical Genetics. 2008;10(4):267–77. doi: 10.1097/GIM.0b013e31816b64c2. [DOI] [PubMed] [Google Scholar]
- Park EJ, Ogden LA, Talbot A, Evans S, Cai CL, Black BL, Frank DU, Moon AM. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development. 2006;133(12):2419–33. doi: 10.1242/dev.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, Sparks C, Choi CH, Oghalai J, Curran S, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: Implications for 22q11 deletion syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(20):7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? American journal of human genetics. 2001;69(1):124–37. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Cox NJ. The allelic architecture of human disease genes: common disease - common variant... or not? Human Molecular Genetics. 2002;11(20):2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- Randall V, McCue K, Roberts C, Kyriakopoulou V, Beddow S, Barrett AN, Vitelli F, Prescott K, Shaw-Smith C, Devriendt K, et al. Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. The Journal of clinical investigation. 2009;119(11):3301–10. doi: 10.1172/JCI37561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Devriendt K, Koch A, Rauch R, Gewillig M, Kraus C, Weyand M, Singer H, Reis A, Hofbeck M. Assessment of association between variants and haplotypes of the remaining TBX1 gene and manifestations of congenital heart defects in 22q11.2 deletion patients. Journal of Medical Genetics. 2004;41(4) doi: 10.1136/jmg.2003.010975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch R, Hofbeck M, Zweier C, Koch A, Zink S, Trautmann U, Hoyer J, Kaulitz R, Singer H, Rauch A. Comprehensive genotype-phenotype analysis in 230 patients with tetralogy of Fallot. Journal of medical genetics. 2010;47(5):321–31. doi: 10.1136/jmg.2009.070391. [DOI] [PubMed] [Google Scholar]
- Roberts C, Sutherland HF, Farmer H, Kimber W, Halford S, Carey A, Brickman JM, Wynshaw-Boris A, Scambler PJ. Targeted mutagenesis of the Hira gene results in gastrulation defects and patterning abnormalities of mesoendodermal derivatives prior to early embryonic lethality. Molecular and cellular biology. 2002;22(7):2318–28. doi: 10.1128/MCB.22.7.2318-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckman RN. Cardiovascular defects associated with alcohol, retinoic acid, and other agents. Annals of the New York Academy of Sciences. 1990;588:281–8. doi: 10.1111/j.1749-6632.1990.tb13217.x. [DOI] [PubMed] [Google Scholar]
- Scambler PJ, Carey AH, Wyse RK, Roach S, Dumanski JP, Nordenskjold M, Williamson R. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics. 1991;10(1):201–6. doi: 10.1016/0888-7543(91)90501-5. [DOI] [PubMed] [Google Scholar]
- Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nature genetics. 1996;14(4):392–9. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies (vol 38, pg 209, 2006) Nature genetics. 2006;38(3):390–390. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- Stoller JZ, Epstein JA. Identification of a novel nuclear localization signal in Tbx1 that is deleted in DiGeorge syndrome patients harboring the 1223delC mutation. Human molecular genetics. 2005;14(7):885–92. doi: 10.1093/hmg/ddi081. [DOI] [PubMed] [Google Scholar]
- Taddei I, Morishima M, Huynh T, Lindsay EA. Genetic factors are major determinants of phenotypic variability in a mouse model of the DiGeorge/del22q11 syndromes. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(20):11428–31. doi: 10.1073/pnas.201127298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Juan L, Rosell J, Morla M, Vidal-Pou C, Garcia-Algas F, de la Fuente MA, Juan M, Tubau A, Bachiller D, Bernues M, et al. Mutations in TBX1 genocopy the 22q11.2 deletion and duplication syndromes: a new susceptibility factor for mental retardation. European journal of human genetics: EJHG. 2007;15(6):658–63. doi: 10.1038/sj.ejhg.5201819. [DOI] [PubMed] [Google Scholar]
- van Bueren KL, Papangeli I, Rochais F, Pearce K, Roberts C, Calmont A, Szumska D, Kelly RG, Bhattacharya S, Scambler PJ. Hes1 expression is reduced in Tbx1 null cells and is required for the development of structures affected in 22q11 deletion syndrome. Developmental Biology. 2010;340(2):369–380. doi: 10.1016/j.ydbio.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent MC, Heitz F, Tricoire J, Bourrouillou G, Kuhlein E, Rolland M, Calvas P. 22q11 deletion in DGS/VCFS monozygotic twins with discordant phenotypes. Genetic counseling. 1999;10(1):43–9. [PubMed] [Google Scholar]
- Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nature genetics. 2004;36(9):955–7. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development. 2002;129(19):4605–4611. doi: 10.1242/dev.129.19.4605. [DOI] [PubMed] [Google Scholar]
- Voelckel MA, Girardot L, Giusiano B, Levy N, Philip N. Allelic variations at the haploid TBX1 locus do not influence the cardiac phenotype in cases of 22q11 microdeletion. Annales de genetique. 2004;47(3):235–40. doi: 10.1016/j.anngen.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Wadey R, McKie J, Papapetrou C, Sutherland H, Lohman F, Osinga J, Frohn I, Hofstra R, Meijers C, Amati F, et al. Mutations of UFD1L are not responsible for the majority of cases of DiGeorge Syndrome/velocardiofacial syndrome without deletions within chromosome 22q11. American journal of human genetics. 1999;65(1):247–9. doi: 10.1086/302468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–91. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362(9393):1366–73. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- Yamagishi H, Garg V, Matsuoka R, Thomas T, Srivastava D. A molecular pathway revealing a genetic basis for human cardiac and craniofacial defects. Science. 1999;283(5405):1158–61. doi: 10.1126/science.283.5405.1158. [DOI] [PubMed] [Google Scholar]
- Yamagishi H, Ishii C, Maeda J, Kojima Y, Matsuoka R, Kimura M, Takao A, Momma K, Matsuo N. Phenotypic discordance in monozygotic twins with 22q11.2 deletion. American journal of medical genetics. 1998;78(4):319–21. [PubMed] [Google Scholar]
- Ye YH, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414(6864):652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- Yobb TM, Somerville MJ, Willatt L, Firth HV, Harrison K, MacKenzie J, Gallo N, Morrow BE, Shaffer LG, Babcock M, et al. Microduplication and triplication of 22q11.2: a highly variable syndrome. American journal of human genetics. 2005;76(5):865–76. doi: 10.1086/429841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Baldini A. In vivo response to high-resolution variation of Tbx1 mRNA dosage. Human molecular genetics. 2008;17(1):150–7. doi: 10.1093/hmg/ddm291. [DOI] [PubMed] [Google Scholar]
- Zweier C, Sticht H, Aydin-Yaylagul I, Campbell CE, Rauch A. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. American journal of human genetics. 2007;80(3):510–7. doi: 10.1086/511993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.