

Abstract
BACKGROUND
Uterine leiomyomas (fibroids) are the most common pelvic tumors in women. We assessed the potential therapeutic utility of Ro 41-0960, a synthetic catechol-O-methyl transferase inhibitor (COMTI), in the Eker rat.
METHODS
We randomized uterine fibroid-bearing Eker rats for treatment with Ro 41-0960 (150 mg/kg/12 h) versus vehicle for 2 and 4 weeks. The fibroids were measured by caliper and subjected to histological evaluation. Urinary levels of 2-hydroxy estrogen (E2), 16-hydroxy E2 and DPD (osteoporosis marker) and serum liver enzymes were evaluated. Expressions of Cyclin D1, proliferating cell nuclear antigen (PCNA), Poly [ADP-ribose] polymerase1 (PARP1), tumor suppressor gene (P53) and transforming growth factor (TGFβ3) were assessed in fibroids using immunohistochemical analysis or RT–PCR. Apoptosis was confirmed using terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL).
RESULTS
Ro 41-0960-treated rats exhibited fibroid volumes of 86 ± 7% and 105 ± 12% of initial burden, at 2 and 4 weeks post-treatment, respectively, significantly lower than control group (240 ± 15% and 300 ± 18%; P< 0.01). Ro 41-0960 increased the urinary 2-hydroxy E2/16-hydroxy E2 ratio, level of p53 mRNA and TUNEL positivity (P< 0.05) and decreased PARP1, PCNA and cyclin D1 proteins and TGFβ3 mRNA (P< 0.05). Ro 41-0960 did not change normal tissue histology, liver functions or urinary DPD level.
CONCLUSIONS
Ro 41-0960 (COMTI) arrested growth/shrunk uterine fibroids in Eker rats. This result may be related to modulation of estrogen-dependent genes involved in apoptosis, proliferation and extracellular matrix deposition via accumulation of 2-hydroxy estrogen. The efficacy and safety of Ro 41-0960 in rats suggest its candidacy for treatment of uterine fibroids.
Keywords: uterine leiomyomas, catechol-O-methyl transferase inhibitor, Eker rats, estradiol, catechol estrogen
Introduction
Uterine leiomyomas, commonly known as fibroids, are the most common pelvic tumors in women, symptomatically affecting at least 25% of women of reproductive age (Healy et al., 1986; Lumsden and Wallace, 1998). Moreover, the prevalence of uterine leiomyomas may be >77% overall (i.e. presence of symptomatic and asymptomatic fibroids in women of reproductive age; Cramer and Patel, 1990). Uterine leiomyomas are the most common indication for hysterectomy in the USA (Wilcox et al., 1994), and they commonly cause severe irregular prolonged menstrual bleeding, infertility and abortion (Hart et al., 2001; Surrey et al., 2001).
The current treatment options for leiomyomas are far from satisfactory, and the gold standard treatment remains surgical removal through either hysterectomy or myomectomy.
Currently, there is no optimal medical treatment option which has been approved by the Federal Drug Agency for uterine leiomyomas. Non-surgical treatment of leiomyomas has been primarily through the use of GnRH agonists that suppress circulating levels of estradiol (E2) and progesterone (Al-Hendy and Salama, 2006a). Unfortunately current treatment options have serious drawbacks, including the severe—adverse side effects associated with GnRH agonist (GnRHa), such as osteoporosis and menopausal symptoms (Andreyko et al., 1987; Al-Hendy and Salama, 2006a), and loss of procreative potential as well as the cost and post-operative complications of surgical approaches (VeKaut, 1993; Payne and Haney, 2003; Al-Hendy and Salama, 2006a).
The role of ovarian hormones in the pathogenesis of uterine leiomyoma is well established (Otubu et al., 1982; Maruo et al., 2004). E2 and its metabolites play a critical role in initiating and promoting leiomyoma growth (Dixon et al., 2002; Yue et al., 2003). Furthermore, there is compelling evidence that endogenous levels of catechol estrogen (CE) metabolites are significantly different between the uterine leiomyoma and adjacent normal myometrium (Reddy et al., 1981). Consequently, estrogen-metabolizing enzymes may be exploited as a potential therapeutic target for the non-surgical treatment of uterine fibroids.
Catechol-O-methyl transferase (COMT) metabolizes many endobiotics, including CE (Creveling, 2003). The COMT enzyme catalyzes the conversion of hydroxylated estrogen metabolites (2-hydroxy E2 and 4-hydroxy E2) into their methylated counterparts. We and others have demonstrated that 2-hydroxy E2, a substrate for COMT, exhibits an anti-estrogenic effect in multiple biological systems including human uterine cells (Vandewalle, 1989; Bradlow, 1996; Wentz et al., 2006). We have also demonstrated that the decrease or inhibition of COMT activity is associated with decreased clearance of 2-hydrox E2, leading to a hypoestrogenic milieu and may result in the inhibition of human leiomyoma cell growth in vitro (Salama et al., 2006). In addition, 2-methoxy E2, a product of COMT action, may have a mitogenic effect in several cell systems (Banerjee et al., 2003; Liu and Zhu, 2004). Additionally, our previous laboratory studies demonstrated that COMT expression is higher in human leiomyoma tissue compared with adjacent normal myometrium (Salama et al., 2006). Therefore, the inhibition of COMT activity may indirectly modulate the biological effects of E2 and produce an imbalance between E2 and its metabolites, which may favor leiomyoma shrinkage.
Female Eker rats, which carry a germline mutation in the tuberous sclerosis-2 tumor suppressor gene, develop uterine leiomyomas with high frequency by 12–16 months of age (Everitt et al., 1995). Eker rats exhibit uterine fibroid lesions that are benign and share many of the same phenotypic, histological and pathological characteristics as the cognate human disease (Everitt et al., 1995; Howe et al., 1995). Therefore, we and others have used the Eker rat experimentally as a model system for human uterine leiomyoma to study the role of hormones in fibroid development and to test the utility of new therapeutic modalities (Everitt et al., 1995; Howe et al., 1995; Hassan et al., 2009, 2010).
The present study was designed to assess the potential therapeutic benefits of Ro 41-0960, a synthetic highly specific COMT inhibitor (COMTI), on uterine leiomyoma lesions in the Eker rat as a preclinical model for developing medical non-invasive treatment of uterine fibroids in women.
Materials and Methods
Drug
The specific, selective COMT inhibitor Ro 41-0960 was purchased from Sigma-Aldrich (St. Louis, MO, USA). Ro 41-0960 was suspended in saline using a few drops of Tween 20 just before injection.
Animals and treatment protocol
Female Eker rats from our own breeding colony, which was started with breeders provided by Dr Cheryl L. Walker's closed colony (M.D. Anderson Cancer Center, Smithville, TX, USA) were used. Uterine tumors were initially confirmed and measured by calipers using survival surgery and also at the time of euthanization. Twenty-four, 14–16-month-old rats bearing uterine fibroids were then randomized to receive s.c. injection with either COMTI (Ro 41-0960) at 150 mg/kg twice per day for 28 days (treated group) (Wentz et al., 2007) or vehicle (negative control group). Animals were observed daily for any post-treatment complications. Total 24-h urine samples were collected daily during the treatment course using special metabolic cages. Sample rats (6/group) were selected randomly and euthanized at the following time points: 14 days post-treatment and 28 days post-treatment (end of the experiment). All animals were euthanized by CO2 asphyxiation. Blood was collected using cardiac puncture, and tissue samples were harvested from the tumor and the following organs: uterus, vagina, cervix, ovary, liver, kidney, lungs, brain and long bones. The care and handling of the rats were in accord with guidelines from the National Institutes of Health and the Association for the Accreditation of Laboratory Animal Care-accredited facilities, and all of the protocols involving the use of these animals were approved by the local Institutional Animal Care and Use Committees.
Histopathological evaluation of tumor tissue
Each tumor tissue was divided into two parts: one part was formalin-fixed and paraffin-embedded and subjected to hematoxylin and eosin (H&E) histological analysis using standard protocols (Bancroft and Stevens, 1996) and was also used for immunohistochemical analysis of proteins that control proliferation and apoptosis; the other part was immediately preserved in liquid nitrogen for later studies. Cell proliferation was assessed in the paraffin-embedded tissue preparations by detection of proliferating cell nuclear antigen (PCNA) and cyclin D1 protein using rat-specific monoclonal antibodies (Santa Cruz Biotech, Santa Cruz, CA, USA) at 1:6000 and 1:3000 dilutions, respectively. Apoptosis was evaluated with the ApopTag Kit (Intergen, Purchase, NY, USA) following the manufacturer's instructions. The death substrate Poly (ADP-ribose) (PAR) polymerase (PARP1) was also evaluated using a rat-specific anti-PARP1 antibody at 1:2000 dilution.
The percentage of apoptotic cells and the protein levels were established by counting the number of ApopTag-labeled nuclei or the number of positively stained cells (brown in color) in three 400× microscopic fields for each tissue, and these numbers were compared with the total number of cells. At least 2000 cells were counted per specimen, and the numbers shown represent the average of three counts.
Determination of urinary levels of 2-hydroxy and 16-hydroxy estrogen metabolites using enzyme immunosorbent assay
The level of estrogen metabolites 2- and 16-hydroxy E2 were estimated using competitive solid-phase enzyme immunoassays (EIA) ESTRAMET 2/16 KIT (Immuna Care Corporation, Sentry Parkway, PA, USA) according to the supplier's protocol. The values of the estrogen metabolites were normalized against the urinary creatinine content measured in our local clinical chemistry laboratory.
RT–PCR analysis
Total RNA was isolated from the tumor tissues preserved in liquid nitrogen using the RNAqueous Kit (Ambion Inc, Austin, TX, USA). The first-strand cDNA was synthesized from 2 µg total RNA using the TaqMan Reverse Transcription Kit (Applied Biosystems, Foster City, USA). PCR amplification of transforming growth factor (TGFβ3) and tumor suppressor gene P53 were performed using a rat-specific TaqMan Gene Expression Assay (Applied Biosystem, Foster City, USA) according to the supplier's protocol. Glycerol-3-phosphate dehydrogenase (G3PDH) was used as an endogenous control. The potential effects of COMTI on mRNA levels were calculated as described earlier (Pfaffl, 2001; Salama et al., 2007; Othman et al., 2007) and were normalized against the G3PDH mRNA level, and changes resulting from COMTI treatment are presented as fold-change compared with control animals.
Evaluation of the safety of COMTI treatment in the Eker rat model
Samples from a wide variety of tissues, including the uterus (myometrium and endometrium), ovary, cervix, vagina, liver and long bones (estrogen-responsive tissues) as well as other organs, such as spleen, lung, kidney and brain, were collected and sent for histopathological assessment by a pathologist (blinded to the assignment of treatment) for signs of tissue damage, inflammation, necrosis or other pathological changes using H&E staining (Bancroft and Stevens, 1996). Blood samples were centrifuged for 10 min at 2000g to obtain the serum. Serum samples were carefully placed into clean dry Wassermann tubes for the determination of liver function tests [aspartate aminotransferase (AST), alanine aminotransferase (ALT) and total bilirubin] using standard techniques (Reitman and Frankel, 1957; Sbrana et al., 2006).
Deoxypyridinoline (DPD) cross-links, a marker of bone resorption, was determined by a sensitive competitive EIA in a microtiter stripwell format utilizing a monoclonal anti-DPD-antibody coated on a strip to capture the DPD using the Metra DPD Kit (Quidal corporation, San Diego, Cal, USA) according to the supplier's instructions and the method described previously (Acil et al., 1996; Seibel et al., 1998). DPD was expressed in nanomole/millimole creatinine.
Statistics
The data are presented as mean ± SE. Results were analyzed using two-tailed unpaired t-tests (Sigma Stat version 3; SPSS Inc., Chicago, IL, USA); P< 0.05 was considered statistically significant.
Results
Ro 41-0960 (COMTI) shrinks Eker rat uterine leiomyoma
As shown in Fig. 1, the Eker rats treated with s.c. injection of Ro 41-0960 (COMTI) at a dose of 150 mg/kg twice/day exhibited significant shrinkage or slowing of growth of the total tumor volume compared with the vehicle-treated group (P< 0.01). Animals treated with Ro 41-0960 (COMTI) exhibited a uterine fibroid volume of 86 ± 7% and 105 ± 12% of pretreatment volume at 2 and 4 weeks post-treatment, respectively. Conversely, tumor size in the control animals continued to increase and reached 240 ± 15% and 300 ± 18% of pretreatment size at the 2- and 4-week time points, respectively.
Figure 1.

Treatment with Ro 41-0960, a synthetic COMTI, in the Eker rat shrinks total leiomyoma volume compared with vehicle treatment. Tumor volume was calculated as a percentage of the corresponding day zero (pretreatment) volume and presented as mean ± SE of six animals at each time point. a: indicates a significant difference between the control and treated animals (P < 0.01).
Ro 41-0960 (COMTI) changes uterine leiomyoma architecture in the Eker rat model
H&E sections of fibroid lesions prepared from control Eker rat groups and stained with H&E showed bundles of smooth muscle cells of various sizes running in different directions. Those muscle cells that were cut longitudinally showed cylindrical nuclei, eosinophilic cytoplasm and ill-defined cell outlines. The nuclei were uniform, and the mitotic figures were absent or sparse. The cells that were transversely cut had nuclei that appeared to be circular with clear surrounding cytoplasm. The muscle fibers were intermixed with fibrous connective tissue (Fig. 2). Conversely, the H&E sections of Ro 41-0960-treated rats showed bundles of edematous smooth muscle cells. The nuclei were smaller and rounded to oval, in shape. No mitotic activity was seen and the cellularity was remarkably decreased (Fig. 2).
Figure 2.

Representative micrographs of fibroid lesions (hematoxylin-eosin stain) from vehicle control and Ro 41-0960 (COMTI)-treated Eker rats at 2 and 4 weeks. Original magnifications 40×. Nuclei in control rats are well defined with clear nuclear membranes (arrow). Tissue generally demonstrates high cellularity. After 2 and 4 weeks of treatment with Ro 41-0960 (COMTI), tissues demonstrate less cellularity and mostly consist of ECM. The few cells remaining are distorted in shape with irregular cellular and nuclear membranes (arrow heads). The control micrograph representing the 4-week time point.
Ro 41-0960 (COMTI) treatment induces apoptosis in Eker rat uterine leiomyoma
As shown in Fig. 3A, Ro 41-0960 (COMTI) induced a significant decrease in death substrate PARP1 protein at 2 and 4 weeks compared with control animals at the same time points. At the 2- and 4-week time points, the percentage of PARP1-positive cells were 42 ± 2.5% and 18 ± 1% compared with 73 ± 2% and 69 ± 2.75% in control tissues, respectively.
Figure 3.

Ro 41-0960 (COMTI) treatment of Eker rats modulates P53 and PARP1 apoptotic pathways. Data represent uterine leiomyoma tissues collected at 2 and 4 weeks after Ro 41-0960 treatment compared with those of the vehicle-treated group: (A) Ro 41-0960 decreases the death substrate poly (ADP-ribose) (PAR) polymerase (PARP1) protein as assessed by immunohistochemistry; original magnification: 20×; (B) Ro 41-0960 increases the percentage of TUNEL-positive cells; original magnification: 20×; and (C) Ro 41-0960 increases P53 mRNA levels. a: indicates a significant difference between the vehicle versus treated rats at P < 0.05, using a two-tailed Student's t-test. Control data are from the 4-week time point.
We further evaluated apoptosis induction using the terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) test. The Ro 41-0960 (COMTI)-treated fibroid lesions exhibited a strong positive TUNEL reaction: 25 ± 2% and 40 ± 3% at 2 and 4 weeks, respectively. This positivity was significantly higher (P < 0.05) compared with that of the vehicle-treated group at the 2- and 4-week time points (2.5 ± 0.7% and 2.8 ± 1.5%, respectively; Fig. 3B).
As shown in Fig. 3C, Ro 41-0960 (COMTI) significantly increased P53 mRNA levels by 1.22 ± 0.09 and 1.9 ± 0.09-fold over the vehicle control (P< 0.05) at 2 and 4 weeks after treatment, respectively.
Ro 41-0960 (COMTI) inhibits the expression of proliferation- and extracellular matrix-related genes
Immunohistochemical analysis using rat-specific antibodies against PCNA and cyclin D1 showed that treatment of Eker rat with Ro 41-0960 (COMTI) resulted in a significant (P < 0.05) decrease in the expression of proliferation-related genes PCNA and cyclin D1 in sections of uterine leiomyoma tissue collected at 2 and 4 weeks post-treatment, compared with the control. The percentages of PCNA-positive cells in the tumor sections prepared from Ro 41-0960-treated animals euthanized after 2 and 4 weeks post-treatment were 47 ± 2.4% and 38 ± 2% compared with 80 ± 4% and 72 ± 3.5% in the control animals, respectively (P< 0.05; Fig. 4A).
Figure 4.

Ro 41-0960 (COMTI) treatment decreases the expression of proliferation- and ECM formation-related genes. Immunohistochemical analysis of proliferation-related (A) PCNA and (B) Cyclin-D1 in the Eker rat uterine leiomyoma tissue sections collected from animals treated with vehicle only (control) or Ro 41-0960 at 2 and 4 weeks post-treatment. Original magnification: 20×. (C) Levels of TGFβ3 mRNA. a: indicates a significant difference between the Ro 41-0960-treated and vehicle-treated rats, P < 0.05. Control data represent the 4-week time point.
Interestingly, the sections from Ro 41-0960 (COMTI) -treated animals showed 35 ± 2% and 18 ± 0.95% cyclin D1-positive cells compared with 55 ± 4% and 60 ± 3% in the vehicle controls at 2 and 4 weeks post-treatment, respectively (Fig. 4B).
Additionally, Ro 41-0960 (COMTI) significantly (P< 0.05) decreased the mRNA levels for TGFβ3 to 0.8 ± 0.03 and 0.5 ± 0.04-fold of that in the control animals at 2 and 4 weeks post-treatment, respectively (Fig. 4C).
Treatment with Ro 41-0960 (COMTI) modulates levels of urinary estrogen metabolites
As shown in Fig. 5A, Eker rats injected with Ro 41-0960 (COMTI) at 150 mg/kg showed a significant (P < 0.05) increase in the urinary levels of 2-hydroxy E2 metabolites compared with the vehicle-treated control animals. On the other hand, the urinary level of 16-hydroxy E2, which is not part of the COMT pathway, was not affected by such treatment (data not shown), which in turn resulted in an increase in the ratio of 2-hydroxy E to 16-hydroxy E versus control (Fig. 5B).
Figure 5.

Ro 41-0960 (COMTI) increases the urinary level of 2-hydroxy estrogen metabolite (A) and increases the ratio of 2-hydroxy estrogen to 16-hydroxy estrogen metabolites (B). Data are calculated as mean and SE for six animals at each time point and are represented as (A) a percentage of the data at day zero; and (B) a ratio of absolute values. A indicates a significant difference between the Ro 41-0960-treated rats and the vehicle-treated rats (P < 0.05).
Safety of Ro 41-0960 (COMTI) treatment in the Eker rat model
Close monitoring of the animals on a daily basis during the treatment course did not reveal any abnormal observations. All of the animals tolerated the treatment and survived the experiment with no apparent signs of toxicity.
Interestingly, careful histological evaluation using H&E staining of the tissue section obtained from various Eker rat organs at different time points (2 and 4 weeks) exhibited no signs of tissue damage or necrosis (some examples are shown in Supplementary data, Fig. S1). Moreover, Ro 41-0960 did not change the urinary levels of the bone decalcification marker DPD during the treatment period compared with the control (Fig. 6).
Figure 6.
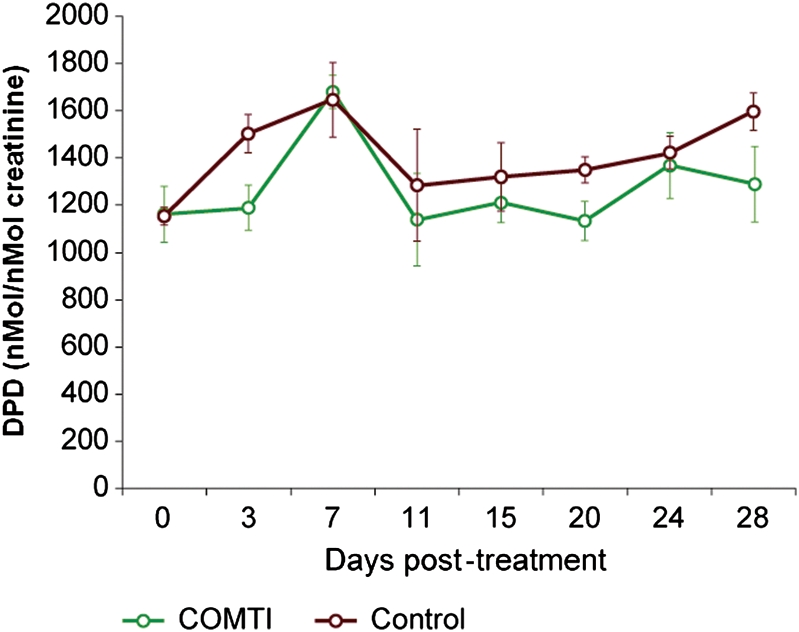
Effect of Ro 41-0960 (COMTI) injection at 150 mg/kg bid on Eker rat urinary levels of the bone decalcification marker Deoxy Pyridinoline (DPD). DPD was evaluated using a competitive enzyme immunoassay and normalized against creatinine. Data are mean ± SE of six animals for each time point. There is no significant difference between the Ro 41-0960-treated rats and the vehicle-treated rats.
Most importantly, Ro 41-0960 treatment had no significant effect on the liver function tests (AST, ALT and total bilirubin) in Eker rats at 2 and 4 weeks post-treatment compared with vehicle-treated controls (Fig. 7A and B).
Figure 7.

Effect of Ro 41-0960 (COMTI) on liver function tests in Eker rats. Eker rats were treated with Ro 41-0960 at a dose of 150 mg/kg bid or vehicle. Ro 41-0960 produce no significant changes in liver function tests: (A) AST, ALT and (B) total bilirubin. Liver function was evaluated 2 and 4 weeks after Ro 41-0960 injection and compared with vehicle-treated control animals using Student's two-tailed t-test.
Discussion
Estrogen plays a pivotal role in leiomyoma pathogenesis and growth (Murphy and Ghahary, 1990; Rein et al., 1995; Chegini et al., 2003; Yue et al., 2003; Al-Hendy et al., 2004; Maruo et al., 2004). However, results published earlier, which have recently been confirmed in our laboratory, suggest that in addition to the parent molecule the oxidative metabolites of estrogen, including the CEs 2-hydroxy-E2, 4-hydroxy E2 and 16-hydroxy E2, may also affect estrogen-induced human uterine leiomyoma growth (Reddy et al., 1981; Al-Hendy and Salama, 2006b). Recent reports from our lab demonstrated that the development of uterine leiomyoma is associated with high COMT expression, and that specific COMT genotypes are associated with higher enzymatic activity (Al-Hendy and Salama, 2006b; Salama et al., 2006). Therefore, interrupting estrogen metabolism or the estrogen receptor (ER) signaling pathway may be a viable strategy for many therapeutic modalities. One of these approaches exploits COMTI to interfere with estrogen metabolism in many estrogen-dependent neoplasms, such as breast cancer (Lavigne et al., 2001).
In the present report, we aimed to develop an alternative localized non-surgical conservative treatment approach for uterine leiomyoma utilizing this novel COMTI approach. To that end, we evaluated the therapeutic modality of COMTI in the Eker rat, the only authentic immunocompetent animal model that spontaneously develops uterine leimyoma lesions and shares many of the same characteristics as human leiomyoma (Everitt et al., 1995). Our goal was to explore the treatment efficiency and safety of COMTI in the Eker rat model as a first preclinical step in preparation for a possible future clinical trial.
Our studies revealed that Ro 41-0960 (COMTI) treatment was able to significantly shrink pre-existing Eker rat uterine leiomyoma lesions compared with a vehicle-treated group (Fig. 1). The shrinkage/growth arrest reached a maximum level after 2 weeks of treatment and appeared to be sustained for the full treatment period (28 days). Furthermore, Ro 41-0960 (COMTI) modulated the expression of several estrogen-regulated genes that control various cellular functions, such as apoptosis (P53, PARP1 and TUNEL assay; Fig. 3), proliferation (PCNA and cyclin-D1; Figs. 4A and B) and extracellular matrix (ECM) formation (TGF-β3; Fig. 4C; Imai et al., 1998; Dixon et al., 2002; Gao et al., 2002; Sozen and Arici, 2002; Chegini et al., 2003). In addition, Ro 41-0960 (COMTI) treatment changed the ratio of 2-hydroxy E2 to 16-hydroxy E2 in favor of a systemic hypoestrogenic status (high 2-hydroxy E2/16-hydroxy E2), which provides a potential mechanism of action for this COMTI-mediated therapeutic approach and possibly explains the rapid and impressive tumor shrinkage in the uterine fibroid lesions (Fig. 5B). Notably, this treatment approach appeared to be safe and well tolerated in rats; for example, it did not induce any macroscopic or microscopic tissue damage in the organs tested. COMTI treatment also did not cause any significant change in the liver function tests or in urinary levels of the bone decalcification marker DPD, suggesting no major effects of COMTI on liver and bone in the 28-day study period (Figs. 6 and 7).
Accumulating evidence indicates that uterine leiomyoma growth is, in part, regulated by the balance between the genes controlling proliferation and apoptosis (Dixon et al., 2002; Loy et al., 2005). PCNA and cyclin D1 have been reported as markers of cellular proliferation (Kurki et al., 1986; Baldin et al., 1993; Gao et al., 2002) and are expressed at a higher level in leiomyoma compared with adjacent normal myometrium (Dixon et al., 2002). As shown in Fig. 4A and B, in the COMTI-treated rats, tumor lesions exhibited significantly decreased PCNA and cyclin-D1 expression, which might reflect a cell cycle arrest (Kurki et al., 1986; Baldin et al., 1993; Dixon et al., 2002) mediated by active p53 overexpression (Fig. 3C).
The tumor suppressor P53 has been categorized as both a caretaker and a gatekeeper for the cell cycle (Lane, 1992). Additionally, P53 controls multiple downstream targets that regulate variable cellular pathways, such as cell cycle, apoptosis, DNA repair and replication (Bargonetti and Manfredi, 2002; Haupt et al., 2003). Interestingly, estrogen stimulates leiomyoma growth in part by decreasing p53 protein levels in the nucleus and suppressing normal p53 function. Conversely, GnRH agonist treatment, one of the treatment modalities currently available for uterine leiomyoma, increases leiomyoma p53 tumor suppressor content (Gao et al., 2002). P53-mediated apoptosis has been documented by two separate pathways (Haupt et al., 2003): the extrinsic death-receptor pathway and the intrinsic mitochondrial pathway, which shift the balance in the Bcl-2 family toward the proapoptotic members, such as Bax. Both pathways lead to activation of caspases as the common final pathway, which degrades cellular components (Haupt et al., 2003). In accordance with this mechanism, our results indicated that the COMTI-treated animals had a significantly lower death substrate PARP1 protein levels at 2 and 4 weeks compared with the control animals, suggesting an activation of the caspase cascade (Shanmugathasan and Jothy, 2000; Chiarugi, 2002; Soldani and Scovassi, 2002). Apoptosis was also confirmed in our study by the TUNEL assay, and all of the tissue sections prepared from Ro 41-0960 (COMTI)-treated animals exhibited a significant increase in the percentage of apoptotic cells compared with those from the control group at both time points (Fig. 3B). On the other hand, Ro 41-0960 (COMTI) treatment lowered the protein level of proliferation-related genes, namely PCNA, an endogenous marker of proliferation (Kurki et al., 1986; Dixon et al., 2002) and cyclin D1, a cell cycle regulating gene (Baldin et al., 1993), which may indicate an overall growth arrest in uterine leiomyoma after treatment with Ro 41-0960.
The TGF-β family includes pleiotrophic cytokines that play key roles in tissue morphogenesis and growth (|Ingman and Robertson, 2002). TGF-β increases the expression of ECM proteins (Ignotz and Massague, 1986; Ignotz et al., 1987). Leiomyomas contain abundant quantities of ECM and contain increased levels of estrogen-induced TGF-β3 relative to adjacent normal myometrium (Sozen and Arici, 2002). In the present study, we demonstrated that the interference of estrogen metabolism by Ro 41-0960 (COMTI) resulted in a marked decrease in the level of TGF-β3 mRNA (Fig. 4C), which possibly led to a decrease in leiomyoma ECM formation and ultimately contributed to a decrease in leiomyoma volume.
Microsomes from human uterine leiomyoma tumor tissue have been shown to metabolize E2 to catechol metabolites, including 2-hydroxy E2 and 4-hydroxy E2, and non-catechol metabolites, such as 16-hydroxy E2 (Vandewalle, 1989; Bradlow, 1996; Creveling, 2003; Wentz et al., 2006). Ro 41-0960 (COMT) converts the CEs 2- and 4-hydroxyE to 2- and 4-methoxyestrogen, respectively (Creveling, 2003). The CE 2-hydroxy E2 can act as an estrogen antagonist (Vandewalle and Lefebvre, 1989; Bradlow, 1996; Devanesan et al., 2001; Al-Hendy and Salama, 2006b), while the methylated counterparts (2- or 4-methoxy E2) can act as estrogen agonists in multiple biologic assays (Banerjee et al., 2003; Lippert et al., 2003; Liu and Zhu, 2004; Sutherland et al., 2005). Both rat and human COMT have a higher catalytic activity toward 2-hydroxy E2 compared with 4-hydroxy E2 (Zhu and Liehr, 1993; Goodman et al., 2001). Thus we elected to focus on the more abundant 2- and 16-derivatives as possible ER modulators (Vandewalle and Lefebvre, 1989; Martucci and Fishman, 1993; Zhu and Liehr, 1993; Bradlow, 1996; Cavalieri et al., 2000; Devanesan et al., 2001; Goodman et al., 2001; Lord et al., 2002; Banerjee et al., 2003, Lippert et al., 2003, Liu and Zhu, 2004; Sutherland et al., 2005)
Eker rats injected with 150 mg/kg Ro 41-0960 (COMT) showed a remarkable increase in urinary levels of 2-hydroxy estrogen metabolites compared with the vehicle-treated animals. On the other hand, the urinary level of 16-hydroxy E2, a potent estrogenic metabolite associated with increased cancer risk (Martucci and Fishman, 1993; Muti et al., 2000), was not affected by such treatment. This, in turn, resulted in an increase in the 2/16-hydroxy E2 metabolite ratio, which leads to a low estrogenic environment and may eventually lead to the shrinkage of uterine leiomyoma lesions.
Several prior reports have shown that the ratio of 2-hydroxy E2 to 16-hydroxy E2 can be used as a biomarker that correlates with carcinogenicity as well as tumor follow-up, and the ratio was described as more representative of the intrinsic tissue estrogen milieu than each metabolite separately (Fishman et al., 1995; Kabat et al., 1997; Meilahn et al., 1998; Muti et al., 2000; Lord et al., 2002). This was described in several types of cancer in various anatomical sites, such as breast cancer, mammary tumor, head and neck cancer as well as endometrial cancer (Persson, 1985; Taioli et al., 1996; Wong et al., 1997; Auborn et al., 1998; Ursin et al., 1999; Yoo et al., 2001; Lord et al., 2002).
From a clinical point of view, COMTI therapy for uterine leiomyoma must be safe and well tolerated and have no or minimal toxic side effects on other normal organs, especially the liver, a common target for systemically injected drugs (Thirunavukkarasu and Sakthisekaran, 2001). Interestingly, careful histologic and pathologic examination of the various Eker rat organs after treatment with Ro 41-0960 (COMTI), especially the liver and estrogen-dependent organs such as the uterus, revealed no evidence of adverse effects or organ damage. Furthermore, the liver function tests were unaffected after 4 weeks of daily Ro 41-0960 (COMTI) treatment (Fig. 7). Because of the deleterious effects of GnRHa on bone mineral density, which is secondary to hypoestrogenemia, we wanted to evaluate the effects of Ro 41-0960 (COMTI) on bone health. As shown in Fig. 6, the urinary level of DPD, a sensitive osteolytic bone marker that cross-links collagen molecules in bones (Colwell et al., 1993; Eastell et al., 1997), showed no significant change in the Ro 41-0960 (COMTI)-treated rats compared with the vehicle-treated control animals, which supported the favorable safety profile of this Ro 41-0960 (COMTI) treatment regimen on bone health (Fig. 6).
The role of COMT in mammary tissue neoplasia is controversial however, with some studies showing a higher COMT activity associated with breast cancer risk (Thompson et al., 1998; Mitrunen et al., 2001), others describing a possible protective effect (Davis et al., 1997; Lavigne et al., 2001; Salama et al., 2007) and many reporting no association between COMT and increased cancer risk (Millikan et al., 1998; Hamajima et al., 2001; Kocabas et al., 2002; Wedren et al., 2003; Ahsan et al., 2004; Wen et al., 2005). Thus we are currently planning a detailed toxicity evaluation following a long period of treatment with COMTI, including the effect on the mammary gland.
It is noteworthy to mention that most of the currently available medical treatment options for uterine fibroids, such as selective progesterone receptor modulators and oral GnRH antagonists, are being developed for chronic therapy of this condition (Kettle et al., 1998; Hara et al., 2003; Armer and Smelt, 2004; Sankaran and Manyonda, 2008; Engman et al., 2009). It is mechanistically inconceivable that a medication would totally eliminate all fibroid lesions in a short period of time, after which the drug could be stopped. A developing theme is gradually being accepted in the reproductive medicine community that uterine fibroids could, and should, be considered as a chronic disease, just as hypertension or diabetes mellitus is, in which a medication will be used chronically. As COMT expression is higher in leiomyoma tissues than adjacent normal myometrium (Reddy et al., 1981; Salama et al., 2006), a low dose of COMTI is expected to affect human fibroids while sparing normal myometrium and other normal organs (as supported by data in the current study), which might provide a safe therapeutic window of favorable anti-fibroid effects. In addition COMTIs are relatively inexpensive compared with GnRHa, and are available as an oral formula, which would encourage patient compliance. All of these advantages would suggest COMTI as a potential candidate for further evaluation in future clinical trials in women with symptomatic uterine fibroids.
In conclusion, Ro 41-0960, a highly specific and selective synthetic COMT inhibitor, produced a dramatic reduction in uterine leiomyoma volumes in Eker rats over 4 weeks of treatment with 150 mg/kg/12 h. The Ro 41-0960-mediated tumor shrinkage may be in part related to the modulation of certain estrogen-dependent genes regulating leiomyoma apoptosis (P53 and PARP1), proliferation (PCNA and cyclin D1) and ECM formation (TGFβ3). These mechanisms are conceivably affected by a decrease in the estrogenic milieu, by increasing the anti-estrogenic metabolite 2-hydroxy E2. Daily administration of Ro 41-0960 was well tolerated by all of the animals and had a favorable safety profile for target and non-target tissues.
This study has provided some essential preclinical data to support the development of an alternative safe non-surgical, non-invasive treatment option for uterine leiomyoma.
Supplementary data
Supplementary data are available at http://humrep.oxfordjournals.org/.
Authors' roles
M.H.H. made Substantial contributions to the conception and design, acquisition of data, analysis and interpretation of data. Participated in drafting the article and revising it critically for important intellectual content. He made substantial contribution in the physical work. He was involved in the final approval of the version to be published. H.F. made substantial contributions to acquisition of data, analysis and interpretation of data. Was involved with revising the article critically for important intellectual content, examining the histopathological finding and the final approval of the version to be published. S.B. made substantial contributions to acquisition of data, analysis and interpretation of data. Was involved in revising the article critically for important intellectual content and the final approval of the version to be published. A.A.-H. made Substantial contributions to conception and design, acquisition of data, analysis and interpretation of data. Was involved in drafting the article and revising it critically for important intellectual content and supplying the laboratory space and facilities. Made substantial contribution in physical work and financial support. Participated in the final approval of the version to be published.
Funding
NIH/NICHD R01 HD046228 to A.A.-H.
Supplementary Material
Acknowledgements
We are grateful to Dr Cheryl L. Walker (M.D. Anderson Cancer Center, Smithville, Texas) for providing us with Eker rat breeder stock.
References
- Acil Y, Brinckman J, Nothbohm H, Muller K, Batge B. Changes with age in the urinary excretion of hydroxylysylpyridinoline (HP) and lysylpyridinoline (LP) Scand J Clin Lab Invest. 1996;56:275–283. doi: 10.3109/00365519609088617. doi:10.3109/00365519609088617. [DOI] [PubMed] [Google Scholar]
- Ahsan H, Chen Y, Whittemore AS, Kibriya MG, Gurvich I, Senie RT, Santella RM. A family-based genetic association study of variants in estrogen-metabolism genes COMT and CYP1B1 and breast cancer risk. Breast Cancer Res Treat. 2004;85:121–131. doi: 10.1023/B:BREA.0000025401.60794.68. doi:10.1023/B:BREA.0000025401.60794.68. [DOI] [PubMed] [Google Scholar]
- Al-Hendy A, Salama S. Gene therapy and uterine leiomyoma: a review. Hum Reprod Update. 2006a;16:385–400. doi: 10.1093/humupd/dml015. doi:10.1093/humupd/dml015. [DOI] [PubMed] [Google Scholar]
- Al-Hendy A, Salama SA. Ethnic distribution of estrogen receptor-alpha polymorphism is associated with a higher prevalence of uterine leiomyomas in black Americans. Fertil Steril. 2006b;86:686–693. doi: 10.1016/j.fertnstert.2006.01.052. doi:10.1016/j.fertnstert.2006.01.052. [DOI] [PubMed] [Google Scholar]
- Al-Hendy A, Lee EJ, Wang HQ, Copland JA. Gene therapy of uterine leiomyomas: adenovirus-mediated expression of dominant negative estrogen receptor inhibits tumor growth in nude mice. Am J Obstet Gynecol. 2004;191:1621–1631. doi: 10.1016/j.ajog.2004.04.022. doi:10.1016/j.ajog.2004.04.022. [DOI] [PubMed] [Google Scholar]
- Andreyko JL, Marshall LA, Dumesic DA, Jaffe RB. Therapeutic uses of gonadotropin releasing hormone analogs. Obstet Gynecol Surv. 1987;42:1–21. [PubMed] [Google Scholar]
- Armer RE, Smelt KH. Non-peptidic GnRH receptor antagonists. Curr Med Chem. 2004;11:3017–3028. doi: 10.2174/0929867043363983. [DOI] [PubMed] [Google Scholar]
- Auborn K, Abramson A, Bradlow HL, Sepkovic D, Mullooly V. Estrogen metabolism and laryngeal papillomatosis: a pilot study on dietary prevention. Anticancer Res. 1998;18:4569–4573. [PubMed] [Google Scholar]
- Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812–821. doi: 10.1101/gad.7.5.812. doi:10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- Bancroft JD, Stevens A. Theory and Practice of Histological Techniques. 4th edn. New York, USA: Churchill Livingstone; 1996. [Google Scholar]
- Banerjee SN, Sengupta K, Banerjee S, Saxena NK, Banerjee SK. 2-Methoxyestradiol exhibits a biphasic effect on VEGF-A in tumor cells and upregulation is mediated through ER-alpha: a possible signaling pathway associated with the impact of 2-ME2 on proliferative cells. Neoplasia. 2003;5:417–426. doi: 10.1016/s1476-5586(03)80044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargonetti J, Manfredi JJ. Multiple roles of the tumor suppressor p53. Curr Opin Oncol. 2002;14:86–91. doi: 10.1097/00001622-200201000-00015. doi:10.1097/00001622-200201000-00015. [DOI] [PubMed] [Google Scholar]
- Bradlow LH. 2-Hydroxyesterone: the ‘good’ estrogen. J Endocrinol. 1996;150:S259–S265. [PubMed] [Google Scholar]
- Cavalieri E, Frenkel K, Liehr JG, Rogan E, Roy D. Estrogens as endogenous genotoxic agents – DNA adducts and mutations. J Natl Cancer Inst Monogr. 2000;27:75–93. doi: 10.1093/oxfordjournals.jncimonographs.a024247. [DOI] [PubMed] [Google Scholar]
- Chegini N, Verala J, Luo X, Xu J, Williams RS. Gene expression profile of leiomyoma and myometrium and the effect of gonadotropin releasing hormone analogue therapy. J Soc Gynecol Invest. 2003;10:161–171. doi: 10.1016/s1071-5576(03)00004-2. doi:10.1016/S1071-5576(03)00004-2. [DOI] [PubMed] [Google Scholar]
- Chiarugi A. Poly(ADP-ribose) polymerase: killer or conspirator? The suicide hypothesist revisited. Trends Pharmacol Sci. 2002;23:122–129. doi: 10.1016/S0165-6147(00)01902-7. doi:10.1016/S0165-6147(00)01902-7. [DOI] [PubMed] [Google Scholar]
- Colwell A, Russell RG, Eastell R. Factors affecting the assay of urinary 3-hydroxy pyridinium crosslinks of collagen as markers of bone resorption. Eur J Clin Invest. 1993;23:341–349. doi: 10.1111/j.1365-2362.1993.tb02034.x. doi:10.1111/j.1365-2362.1993.tb02034.x. [DOI] [PubMed] [Google Scholar]
- Cramer SF, Patel A. The frequency of uterine leiomyomas. Am J Clin Pathol. 1990;94:435–438. doi: 10.1093/ajcp/94.4.435. [DOI] [PubMed] [Google Scholar]
- Creveling CR. The role of catechol-O-methyltransferase in the inactivation of catecholestrogen. Cell Mol Neurobiol. 2003;23:289–291. doi: 10.1023/A:1023680302975. doi:10.1023/A:1023680302975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DL, Telang NT, Osborne MP, Bradlow HL. Medical hypothesis: bifunctional genetic-hormonal pathways to breast cancer. Environ Health Perspect. 1997;105:571–576. doi: 10.1289/ehp.97105s3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanesan P, Todorovic R, Zhao J, Gross MI, Rogan EG, Cavalieri EL. Catechol estrogen conjugates and DNA adducts in the kidney of male Syrian golden hamsters treated with 4-hydroxyestradiol: potential biomarkers for estrogeninitiated cancer. Carcinogenesis. 2001;22:489–497. doi: 10.1093/carcin/22.3.489. doi:10.1093/carcin/22.3.489. [DOI] [PubMed] [Google Scholar]
- Dixon D, Flake GP, Moore AB, He H, Haseman JK, Risinger JI, Lancaster JM, Berchuck A, Barrett JC, Robboy SJ. Cell proliferation and apoptosis in human uterine leiomyomas and myometria. Virchows Arch. 2002;441:53–62. doi: 10.1007/s00428-001-0568-7. doi:10.1007/s00428-001-0568-7. [DOI] [PubMed] [Google Scholar]
- Eastell R, Colwell A, Hampton L, Reeve J. Biochemical markers of bone resorption compared with estimates of bone resorption from radiotracer kinetic studies in osteoporosis. J Bone Miner Res. 1997;12:59–65. doi: 10.1359/jbmr.1997.12.1.59. doi:10.1359/jbmr.1997.12.1.59. [DOI] [PubMed] [Google Scholar]
- Engman M, Granberg S, Williams ARW, Meng CX, Lalitkumar PGL, Gemzell-Danielsson K. Mifepristone for treatment of uterineleiomyoma. A prospective randomized placebo controlled trial. Hum Reprod. 2009;24:1870–1879. doi: 10.1093/humrep/dep100. doi:10.1093/humrep/dep100. [DOI] [PubMed] [Google Scholar]
- Everitt JI, Wolf DC, Howe SR, Goldsworthy TL, Walker CL. Rodent model of reproductive tract leiomyomata. Clinical and pathological features. Am J Pathol. 1995;146:1556–1567. [PMC free article] [PubMed] [Google Scholar]
- Fishman J, Osborne MP, Telang NT. The role of estrogen in mammary carcinogenesis. Ann N Y Acad Sci. 1995;768:91–100. doi: 10.1111/j.1749-6632.1995.tb12113.x. doi:10.1111/j.1749-6632.1995.tb12113.x. [DOI] [PubMed] [Google Scholar]
- Gao Z, Matsuo H, Satoshi N, Kurachi O, Maruo T. p53 tumor suppressor protein content in human uterine leiomyomas and its down-regulation by 17B-estradiol. J Clin Endocrinol Metab. 2002;87:3915–3920. doi: 10.1210/jcem.87.8.8711. doi:10.1210/jc.87.8.3915. [DOI] [PubMed] [Google Scholar]
- Goodman JE, Lavigne JA, Wu K, Helzlsouer KJ, Strickland PT, Selhub J, Yager JD. COMT genotype, micronutrients in the folate metabolic pathway and breast cancer risk. Carcinogenesis. 2001;22:1661–1665. doi: 10.1093/carcin/22.10.1661. doi:10.1093/carcin/22.10.1661. [DOI] [PubMed] [Google Scholar]
- Hamajima N, Matsuo K, Tajima K, Mizutani M, Iwata H, Iwase T, Miura S, Oya H, Obata Y. Limited association between catechol-O-methyltransferase (COMT) polymorphism and breast cancer risk in Japan. Int J Clin Oncol. 2001;6:13–18. doi: 10.1007/pl00012073. doi:10.1007/PL00012073. [DOI] [PubMed] [Google Scholar]
- Hara T, Araki H, Kusaka M, Harada M, Cho N, Suzuki N, Furuya S, Fujino M. Suppression of a pituitary-ovarian axis by chronic oral administration of a novel nonpeptide gonadotropin-releasing hormone antagonist, TAK-013, in cynomolgus monkeys. J Clin Endocrinol Metab. 2003;88:1697–1704. doi: 10.1210/jc.2002-021065. doi:10.1210/jc.2002-021065. [DOI] [PubMed] [Google Scholar]
- Hart R, Khalaf Y, Yeong CT, Seed P, Taylor A, Braude P. A prospective controlled study of the effect of intramural uterine fibroids on the outcome of assisted conception. Hum Reprod. 2001;16:2411–2417. doi: 10.1093/humrep/16.11.2411. [DOI] [PubMed] [Google Scholar]
- Hassan MH, Zhang D, Salama S, Hamada F, Arafa H, Fouad H, Walker C, Al-Hendy A. Towards fibroid gene therapy: adenovirus-mediated delivery of herpes simplex virus 1 thymidine kinase gene/ganciclovir shrinks uterine leiomyoma in the Eker rat model. Gynecol Obstet Invest. 2009;68:19–32. doi: 10.1159/000209675. doi:10.1159/000209675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan MH, Salama SA, Zhang D, Arafa HM, Hamada FM, Fouad H, Walker CC, Al-Hendy A. Gene therapy targeting leiomyoma: adenovirus-mediated delivery of dominant-negative estrogen receptor gene shrinks uterine tumors in Eker rat model. Fertil Steril. 2010;93:239–250. doi: 10.1016/j.fertnstert.2008.09.086. doi:10.1016/j.fertnstert.2008.09.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis—the p53 network. J Cell Sci. 2003;116:4077–4085. doi: 10.1242/jcs.00739. doi:10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- Healy DL, Lawson SR, Abbott M, Baird DT, Fraser HM. Toward removing uterine fibroids without surgery: subcutaneous infusion of a luteinizing hormone-releasing hormone agonist commencing in the luteal phase. J Clin Endocrinol Metab. 1986;63:619–625. doi: 10.1210/jcem-63-3-619. doi:10.1210/jcem-63-3-619. [DOI] [PubMed] [Google Scholar]
- Howe SR, Gottardis MM, Everitt JI, Goldsworthy TL, Wolf DC, Walker C. Rodent model of reproductive tract leiomyomata. Establishment and characterization of tumor-derived cell lines. Am J Pathol. 1995;146:1568–1579. [PMC free article] [PubMed] [Google Scholar]
- Ignotz RA, Massague J. Transforming growth-factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular-matrix. J Biol Chem. 1986;261:4337–4529. [PubMed] [Google Scholar]
- Ignotz RA, Endo T, Massague J. Regulation of fibronectin and type-I collagen messenger-RNA levels by transforming growth-factor-beta. J Biol Chem. 1987;262:6443–6446. [PubMed] [Google Scholar]
- Imai A, Takagi A, Horibe S, Takagi H, Tamaya T. Evidence for tight coupling of gonadotropin-releasing hormone receptor to stimulated fas ligand expression in reproductive tract tumors: possible mechanism for hormonal control of apoptotic cell death. J Clin Endocrinol Metab. 1998;83:427–431. doi: 10.1210/jcem.83.2.4530. doi:10.1210/jc.83.2.427. [DOI] [PubMed] [Google Scholar]
- Ingman WV, Robertson SA. Defining the actions of transforming growth factor beta in reproduction. Bioessays. 2002;24:904–914. doi: 10.1002/bies.10155. doi:10.1002/bies.10155. [DOI] [PubMed] [Google Scholar]
- Kabat GC, Chang CJ, Sparano JA, Sepkovie DW, Hu XP, Khalili A, Rosenblatt R, Braddlow HL. Urinary estrogen metabolites and breast cancer: a case–control study. Cancer Epidemiol Biomarkers Prev. 1997;6:505–509. [PubMed] [Google Scholar]
- Kettle LM, Murphy AA, Morales AJ, Yen SC. Clinical efficacy of the antiprogesterone RU486 in the treatment of endometriosis and uterine fibroids. Hum Reprod. 1998;9:116–120. doi: 10.1093/humrep/9.suppl_1.116. [DOI] [PubMed] [Google Scholar]
- Kocabas NA, Sardas S, Cholerton S, Daly AK, Karakaya AE. Cytochrome P450 CYP1B1 and catechol O-methyltransferase (COMT) genetic polymorphisms and breast cancer susceptibility in a Turkish population. Arch Toxicol. 2002;76:643–649. doi: 10.1007/s00204-002-0387-x. doi:10.1007/s00204-002-0387-x. [DOI] [PubMed] [Google Scholar]
- Kurki P, Vanderlaan M, Dolbeare F, Gray J, Tan EM. Expression of proliferating cell nuclear antigen (PCNA)/cyclin during the cell cycle. Exp Cell Res. 1986;166:209–219. doi: 10.1016/0014-4827(86)90520-3. doi:10.1016/0014-4827(86)90520-3. [DOI] [PubMed] [Google Scholar]
- Lane DP. Cancer—p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. doi:10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- Lavigne JA, Goodman JE, Fonong T, Odwin S, He P, Roberts DE, Yager JD. The effects of catechol-O-methyltransferase inhibition on estrogen metabolite and oxidative DNA damage levels in estradiol-treated MCF-7 cells. Cancer Res. 2001;61:7488–7494. [PubMed] [Google Scholar]
- Lippert C, Seeger H, Mueck AO. The effect of endogenous estradiol metabolites on the proliferation of human breast cancer cells. Life Sci. 2003;72:877–883. doi: 10.1016/s0024-3205(02)02305-6. doi:10.1016/S0024-3205(02)02305-6. [DOI] [PubMed] [Google Scholar]
- Liu ZJ, Zhu BT. Concentration-dependent mitogenic and antiproliferative actions of 2-methoxyestradiol in estrogen receptorpositive human breast cancer cells. J Steroid Biochem Mol Biol. 2004;88:265–275. doi: 10.1016/j.jsbmb.2003.12.003. doi:10.1016/j.jsbmb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Lord RS, Bongiovanni B, Bralley JA. Estrogen metabolism and the diet-cancer connection: rationale for assessing the ratio of urinary hydroxylated estrogen metabolites. Altern Med Rev. 2002;7:112–129. [PubMed] [Google Scholar]
- Loy CJ, Evelyn S, Lim FK, Liu MH, Yong EL. Growth dynamics of human leiomyoma cells and inhibitory effects of the peroxisome proliferator- activated receptor-gamma ligand, pioglitazone. Mol Hum Reprod. 2005;11:561–566. doi: 10.1093/molehr/gah199. doi:10.1093/molehr/gah199. [DOI] [PubMed] [Google Scholar]
- Lumsden MA, Wallace EM. Clinical presentation of uterine fibroids. Baillieres Clin Obstet Gynaecol. 1998;12:177–195. doi: 10.1016/s0950-3552(98)80060-6. doi:10.1016/S0950-3552(98)80060-6. [DOI] [PubMed] [Google Scholar]
- Martucci CP, Fishman J. P450 enzymes of estrogen metabolism. Pharmacol Ther. 1993;57:237–257. doi: 10.1016/0163-7258(93)90057-k. doi:10.1016/0163-7258(93)90057-K. [DOI] [PubMed] [Google Scholar]
- Maruo T, Ohara N, Wang J, Matsuo H. Sex steroidal regulation of uterine leiomyoma growth and apoptosis. Hum Reprod Update. 2004;10:207–220. doi: 10.1093/humupd/dmh019. doi:10.1093/humupd/dmh019. [DOI] [PubMed] [Google Scholar]
- Meilahn EN, De Stavola B, Allen DS, Fentiman I, Bradlow HL, Sepkovic DW, Kuller LH. Do urinary oestrogen metabolites predict breast cancer? Guernsey III cohort follow-up. Br J Cancer. 1998;78:1250–1255. doi: 10.1038/bjc.1998.663. doi:10.1038/bjc.1998.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millikan RC, Pittman GS, Tse CK, Duell E, Newman B, Savitz D, Moorman PG, Boissy RJ, Bell DA. Catechol-O-methyltransferase and breast cancer risk. Carcinogenesis. 1998;19:1943–1947. doi: 10.1093/carcin/19.11.1943. doi:10.1093/carcin/19.11.1943. [DOI] [PubMed] [Google Scholar]
- Mitrunen K, Jourenkova N, Kataja V, Eskelinen M, Kosma VM, Benhamou S, Kang D, Vainio H, Uusitupa M, Hirvonen A. Polymorphic catechol-O-methyltransferase gene and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2001;10:635–640. [PubMed] [Google Scholar]
- Murphy LJ, Ghahary A. Uterine insulin-like growth factor-1: regulation of expression and its role in estrogen-induced uterine proliferation. Endocr Rev. 1990;11:443–453. doi: 10.1210/edrv-11-3-443. doi:10.1210/edrv-11-3-443. [DOI] [PubMed] [Google Scholar]
- Muti P, Bradlow HL, Micheli A, Krogh V, Freudenheim JL, Schünemann HJ, Stanulla M, Yang J, Sepkovic DW, Trevisan M, et al. Estrogen metabolism and risk of breast cancer: a prospective study of the 2:16alpha-hydroxyestrone ratio in premenopausal and postmenopausal women. Epidemiol. 2000;11:635–640. doi: 10.1097/00001648-200011000-00004. doi:10.1097/00001648-200011000-00004. [DOI] [PubMed] [Google Scholar]
- Othman ER, Salama SA, Ismail N, Al-Hendy A. Gene therapy of endometriosis: adenovirus mediated expression of dominant negative estrogen receptor induces apoptosis in human endometriotic cells. Fertil Steril. 2007;88:462–471. doi: 10.1016/j.fertnstert.2006.11.046. doi:10.1016/j.fertnstert.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Otubu JA, Buttram VC, Besch NF, Besch PK. Unconjugated steroids in leiomyomas and tumor-bearing myometrium. Am J Obstet Gynecol. 1982;143:130–133. doi: 10.1016/0002-9378(82)90640-8. [DOI] [PubMed] [Google Scholar]
- Persson I. The risk of endometrial and breast cancer after estrogen treatment. A review of epidemiological studies. Acta Obstet Gynecol Scand Suppl. 1985;130:59–66. doi: 10.3109/00016348509157149. doi:10.3109/00016348509157149. [DOI] [PubMed] [Google Scholar]
- Payne JF, Haney AF. Serious complications of uterine artery embolization for conservative treatment of fibroids. Fertil Steril. 2003;79:128–131. doi: 10.1016/s0015-0282(02)04398-4. doi:10.1016/S0015-0282(02)04398-4. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantifications in real-time RT-PCR. Nucleic Acids Res. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy VV, Hanjani P, Rajan R. Synthesis of catechol estrogens by human uterus and leiomyoma. Steroids. 1981;37:195–203. doi: 10.1016/s0039-128x(81)80017-7. doi:10.1016/S0039-128X(81)80017-7. [DOI] [PubMed] [Google Scholar]
- Rein MS, Barbieri RL, Friedman AJ. Progesterone: a critical role in the pathogenesis of uterine myomas. Am J Obstet Gynecol. 1995;172:14–18. doi: 10.1016/0002-9378(95)90077-2. doi:10.1016/0002-9378(95)90077-2. [DOI] [PubMed] [Google Scholar]
- Reitman S, Frankel SA. Colormetric method for the determination of serum glutamic oxalocetic and glutamic pyruvic transaminases. A J Clin Path. 1957;28:56–63. doi: 10.1093/ajcp/28.1.56. [DOI] [PubMed] [Google Scholar]
- Salama SA, Ho S, Wang H, Tenhunen J, Tilgmann C, Al-Hendy A. Hormonal regulation of catechol-O-methyl transferase activity in women with uterine leiomyomas. Fertil Steril. 2006;86:259–262. doi: 10.1016/j.fertnstert.2005.12.049. doi:10.1016/j.fertnstert.2005.12.049. [DOI] [PubMed] [Google Scholar]
- Salama SA, Jamaluddin M, Kumar R, Hassan MH, Al-Hendy A. Progesterone regulates catechol-O-methyl transferase gene expression in breast cancer cells: distinct effect of progesterone receptor isoforms. J Steroid Biochem Mol Biol. 2007;107:253–261. doi: 10.1016/j.jsbmb.2007.03.049. doi:10.1016/j.jsbmb.2007.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran S, Manyonda IT. Medical management of fibroids. Best Pract Res Clin Obstet Gynaecol. 2008;22:655–676. doi: 10.1016/j.bpobgyn.2008.03.001. doi:10.1016/j.bpobgyn.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Sbrana E, Mateo RI, Xiao SY, Popov VL, Newman PC, Tesh RB. Clinical laboratory, virologic, and pathologic changes in hamsters experimentally infected with Pirital virus (Arenaviridae): a rodent model of Lassa fever. Am J Trop Med Hyg. 2006;74:1096–1102. [PubMed] [Google Scholar]
- Seibel MJ, Woitge HW, Farahmand I, Oberwittler H, Ziegler R. Automated and manual assays for urinary crosslinks of collagen: Which assay to use? Exp Clin Endocrinol Diabetes. 1998;106:143–148. doi: 10.1055/s-0029-1211967. doi:10.1055/s-0029-1211967. [DOI] [PubMed] [Google Scholar]
- Shanmugathasan M, Jothy S. Apoptosis, anoikis and their relevance to the pathobiology of colon cancer. Pathol Int. 2000;50:273–279. doi: 10.1046/j.1440-1827.2000.01047.x. doi:10.1046/j.1440-1827.2000.01047.x. [DOI] [PubMed] [Google Scholar]
- Soldani C, Scovassi AI. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis. 2002;7:321–328. doi: 10.1023/a:1016119328968. doi:10.1023/A:1016119328968. [DOI] [PubMed] [Google Scholar]
- Sozen I, Arici A. Interactions of cytokines, growth factors, and the extracellular matrix in the cellular biology of uterine leiomyomata. Fertil Steril. 2002;78:1–12. doi: 10.1016/s0015-0282(02)03154-0. doi:10.1016/S0015-0282(02)03154-0. [DOI] [PubMed] [Google Scholar]
- Surrey ES, Lietz AK, Schoolcraft WB. Impact of intramural leiomyomata in patients with a normal endometrial cavity on in vitro fertilization–embryo transfer cycle outcome. Fertil Steril. 2001;75:405–410. doi: 10.1016/s0015-0282(00)01714-3. doi:10.1016/S0015-0282(00)01714-3. [DOI] [PubMed] [Google Scholar]
- Sutherland TE, Schuliga M, Harris T, Eckhardt BL, Anderson RL, Quan L, Stewart AG. 2-methoxyestradiol is an estrogen receptor agonist that supports tumor growth in murine xenograft models of breast cancer. Clin Cancer Res. 2005;11:1722–1732. doi: 10.1158/1078-0432.CCR-04-1789. doi:10.1158/1078-0432.CCR-04-1789. [DOI] [PubMed] [Google Scholar]
- Taioli E, Garte SJ, Trachman J, Garbers S, Sepkovic DW, Osborne MP, Mehi S, Bradlow HL. Ethnic differences in estrogen metabolism in healthy women. J Natl Cancer Inst. 1996;88:617. doi: 10.1093/jnci/88.9.617. doi:10.1093/jnci/88.9.617. [DOI] [PubMed] [Google Scholar]
- Thirunavukkarasu C, Sakthisekaran D. Effect of selenium on N-nitrosodiethylamine-induced multistage hepatocarcinogenesis with reference to lipid peroxidation and enzymic antioxidants. Cell Biochem Funct. 2001;19:27–35. doi: 10.1002/cbf.895. doi:10.1002/cbf.895. [DOI] [PubMed] [Google Scholar]
- Thompson PA, Shields PG, Freudenheim JL, Stone A, Vena JE, Marshall JR, Graham S, Laughlin R, Nemoto T, Kadlubar FF, Ambrosone CB. Genetic polymorphisms in catechol-O-methyltransferase, menopausal status, and breast cancer risk. Cancer Res. 1998;58:2107–2110. doi:10.1093/jnci/91.12.1067. [PubMed] [Google Scholar]
- Ursin G, London S, Stanczyk FZ, Gentzschein E, Paganini-Hill A, Ross RK, Pike MC. Urinary 2-hydroxyestrone/16alphahydroxyestrone ratio and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 1999;91:1067–1072. doi: 10.1093/jnci/91.12.1067. doi:10.1093/jnci/91.12.1067. [DOI] [PubMed] [Google Scholar]
- Vandewalle B. Opposite effects of estrogen and catecholestrogen on hormone-sensitive breast cancer cell growth and differentiation. Mol Cell Endocrinol. 1989;61:239–246. doi: 10.1016/0303-7207(89)90135-4. doi:10.1016/0303-7207(89)90135-4. [DOI] [PubMed] [Google Scholar]
- Vandewalle B, Lefebvre J. Opposite effects of estrogen and catecholestrogen on hormone-sensitive breast cancer cell growth and differentiation. Mol Cell Endocrinol. 1989;61:239–246. doi: 10.1016/0303-7207(89)90135-4. doi:10.1016/0303-7207(89)90135-4. [DOI] [PubMed] [Google Scholar]
- VeKaut BS. Changing trends in treatment of leiomyomata uteri [abstract] Curr Opin Obstet Gynecol. 1993;5:301. [PubMed] [Google Scholar]
- Wedren S, Rudqvist TR, Granath F, Weiderpass E, Ingelman-Sundberg M, Persson I, Magnusson C. Catechol-O-methyltransferase gene polymorphism and post-menopausal breast cancer risk. Carcinogenesis. 2003;24:681–687. doi: 10.1093/carcin/bgg022. doi:10.1093/carcin/bgg022. [DOI] [PubMed] [Google Scholar]
- Wen W, Cai Q, Shu XO, Cheng JR, Parl F, Pierce L, Gao YT, Zheng W. Cytochrome P450 1B1 and catechol-O-methyltransferase genetic polymorphisms and breast cancer risk in Chinese women: results from the shanghai breast cancer study and a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2005;14:329–335. doi: 10.1158/1055-9965.EPI-04-0392. doi:10.1158/1055-9965.EPI-04-0392. [DOI] [PubMed] [Google Scholar]
- Wentz MJ, Jamaluddin M, Garfield RE, Al Hendy A. Regulation of catechol-O-methyltransferase expression in human myometrial cells. Obstet and Gyn. 2006;108:1439–1447. doi: 10.1097/01.AOG.0000243775.73788.11. doi:10.1097/01.AOG.0000243775.73788.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentz MJ, Shi S, Shi L, Salama SA, Harirah HM, Fouad H, Garfield RE, Al-Hendy A. Treatment with an inhibitor of catechol-O-methyltransferase activity reduces preterm birth and impedes cervical resistance to stretch in pregnant rats. Reprod. 2007;134:831–839. doi: 10.1530/REP-07-0245. doi:10.1530/REP-07-0245. [DOI] [PubMed] [Google Scholar]
- Wilcox LS, Koonin LM, Pokras R, Strauss LT, Xia Z, Peterson HB. Hysterectomy in the United States, 1988–1990. Obstet Gynecol. 1994;83:549–555. doi: 10.1097/00006250-199404000-00011. doi:10.1097/00006250-199404000-00011. [DOI] [PubMed] [Google Scholar]
- Wong GY, Bradlow L, Sepkovic D, Mehl S, Mailman J, Osborne MP. Dose-ranging study of indole-3-carbinol for breast cancer prevention. J Cell Biochem Suppl. 1997;29:111–116. doi: 10.1002/(sici)1097-4644(1997)28/29+<111::aid-jcb12>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Yoo HJ, Sepkovic DW, Bradlow HL, Yu GP, Sirilian HV, Schantz SP. Estrogen metabolism as a risk factor for head and neck cancer. Otolaryngol Head Neck Surg. 2001;124:241–247. doi: 10.1067/mhn.2001.113507. doi:10.1067/mhn.2001.113507. [DOI] [PubMed] [Google Scholar]
- Yue W, Santen RJ, Wang JP, Li Y, Verderame MF, Bocchinfuso WP, Korach KS, Devanesan P, Todorovic R, Rogan EG, et al. Genotoxic metabolites of estradiol in breast: potential mechanisms of estradiol induced carcinogenesis. J Steroid Biochem Mol Biol. 2003;86:477–486. doi: 10.1016/s0960-0760(03)00377-7. doi:10.1016/S0960-0760(03)00377-7. [DOI] [PubMed] [Google Scholar]
- Zhu BT, Liehr JG. Inhibition of the catechol-O-methyltransferase-catalyzed O-methylation of 2- and 4-hydroxyestradiol by catecholamine: implications for the mechanism of estrogen-induced carcinogenesis. Arch Biochem Biophys. 1993;304:248–256. doi: 10.1006/abbi.1993.1346. doi:10.1006/abbi.1993.1346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.