

Abstract
Copy number variations (CNVs) within human 15q11.2–13.3 show reduced penetrance and variable expressivity in a range of neurologic disorders. Therefore, characterizing 15q11.2–13.3 chromatin structure is important for understanding the regulation of this locus during normal neuronal development. Deletion of the Prader–Willi imprinting center (PWS-IC) within 15q11.2–13.3 disrupts long-range imprinted gene expression resulting in Prader–Willi syndrome. Previous results establish that MeCP2 binds to the PWS-IC and is required for optimal expression of distal GABRB3 and UBE3A. To examine the hypothesis that MeCP2 facilitates 15q11.2–13.3 transcription by linking the PWS-IC with distant elements, chromosome capture conformation on chip (4C) analysis was performed in human SH-SY5Y neuroblastoma cells. SH-SY5Y neurons had 2.84-fold fewer 15q11.2–13.3 PWS-IC chromatin interactions than undifferentiated SH-SY5Y neuroblasts, revealing developmental chromatin de-condensation of the locus. Out of 68 PWS-IC interactions with15q11.2–13.3 identified by 4C analysis and 62 15q11.2–13.3 MeCP2-binding sites identified by previous ChIP-chip studies, only five sites showed overlap. Remarkably, two of these overlapping PWS-IC- and MeCP2-bound sites mapped to sites flanking CHRNA7 (cholinergic receptor nicotinic alpha 7) encoding the cholinergic receptor, nicotinic, alpha 7. PWS-IC interaction with CHRNA7 in neurons was independently confirmed by fluorescent in situ hybridization analysis. Subsequent quantitative transcriptional analyses of frontal cortex from Rett syndrome and autism patients revealed significantly reduced CHRNA7 expression compared with controls. Together, these results suggest that transcription of CHRNA7 is modulated by chromatin interactions with the PWS-IC. Thus, loss of long-range chromatin interactions within 15q11.2–13.3 may contribute to multiple human neurodevelopmental disorders.
INTRODUCTION
Imprinting centers (ICs) are loci that control imprinted gene expression in cis over several megabases (Mb) of genomic DNA. The Prader–Willi IC (PWS-IC) within 15q11.2–13.3 is genetically defined as the smallest region of overlap of paternal deletions in Prader–Willi patient DNA samples (1). Prader–Willi syndrome (PWS, OMIM 176270) is characterized by hypotonia, obesity, hypogonadism and mild mental retardation (2). The PWS-IC maps to a 4.3 kilobase (kb) region in the promoter, first exon and part of the first intron of SNRPN (1) and forms a bipartite imprinting control center with the Angelman syndrome IC (AS-IC) (3).
15q11.2–13.3 provides an ideal locus for studying the epigenetic effect of chromatin structure on neuronal gene expression for several reasons. First, the PWS-IC acts in cis to regulate paternal expression of a 2 Mb gene cluster within 15q11.2–13.3 encompassing MKRN3, MAGEL2, NDN, SNRPN, the small nucleolar RNA transcripts (snoRNAs) HBII-436, HBII-13, HBII-437, HBII-438, HBII-85, HBII-52 and UBE3A-AS (reviewed in 4). Second, 15q11.2–13.3 contains numerous genes with critical neurologic function such as CHRNA7 (cholinergic receptor nicotinic alpha 7), UBE3A and GABRB3 (5–7). Third, deletions and duplications of 15q11.2–13.3 are observed in 1–3% of autism patients (OMIM 209850), revealing the importance of this locus for normal neurologic development (8). Fourth, a neuron-specific 460 kb paternal transcription unit extends through SNRPN and the snoRNA clusters to UBE3A-AS (9). Post-natal neurons also show paternal allele-specific chromatin decondensation of the SNRPN-UBE3A-AS locus that corresponds to increased snoRNA transcription (10). Fifth, the PWS-IC has been shown in previous studies to contain the strongest MeCP2-binding site in the 15q11.2–13.3 locus (11,12). Finally, the PWS-IC has been shown to be necessary for normal post-natal development in mice (13).
Proximal 15q is enriched for blocks of segmental duplications that extend to 15q13.3 and predispose the locus for chromosomal rearrangements. 15q13.3 contains the gene CHRNA7, encoding the cholinergic receptor nicotinic alpha 7 subunit (5,14) and its partial duplication CHRFAM7A, a gene that appears to be transcribed but has unknown function (15). The protein product of CHRNA7, CHRNA7, homo-oligomerizes to form a ligand-gated ion channel, thereby allowing passage of calcium ions when bound by acetylcholine or other ligands such as nicotine (5). CHRNA7/CHRNA7 appears to be critical for normal neurodevelopment as copy number variation (CNV) of 15q13.3 is associated with autism (16,17), mental retardation (17–19) as well as schizophrenia and epilepsy (20–22). Furthermore, mice with targeted genetic deletion of Chrna7 exons 8–10 have learning and memory deficits (23–25). For these reasons, genetic alterations in 15q13.3 may contribute to multiple human neurodevelopmental disorders.
Epigenetic mechanisms have recently been shown to underlie complex neurologic processes (26). MECP2 loss of function mutations result in Rett syndrome (27) (OMIM 312750), an X-linked dominant neurodevelopmental disorder characterized by regression in late infancy similar to autism (28,29). MeCP2 is a key interpreter of epigenetic marks as a member of a family of proteins that contain a methyl CpG-binding domain (MBD) (reviewed in 30). The MBD of MeCP2 allows the protein to bind to as little as a single methylated CpG di-nucleotide in a DNA sequence (31) and recruit co-factors such as Sin3a and histone de-acetylase HDAC activity (32,33). Therefore, MeCP2 was originally hypothesized to be a transcriptional silencer of methylated gene promoters in cis (32,33). However, recent evidence suggests that MeCP2 has other important epigenetic function including a role in long-range chromatin organization (34). Studies show that MeCP2 can form a molecular bridge between chromatin structures in vitro (35–37). In addition, MeCP2 ChIP-chip studies in SH-SY5Y neurons revealed predominately intergenic MeCP2-binding patterns particularly in 15q13, a region encompassing CHRFAM7A and CHRNA7, suggesting a role in chromatin loop organization and gene expression of specific loci (11). Furthermore, loss of MeCP2 leads to dysregulation of the 15q11.2–13.3-encoded genes GABRB3 and UBE3A (27,38). Thus, MeCP2 with multiple epigenetic functions including organization of chromatin structure affects gene expression at 15q11.2–13.3 in developing neurons.
In this study, the role of the PWS-IC and MeCP2 as long-range epigenetic regulators of 15q11.2–13.3 chromatin organization was assayed in human neuronal cells by chromatin conformation capture on microarray (4C) and fluorescent in situ hybridization (FISH) analyses. Unexpectedly, a specific PWS-IC interaction with the 15q13.3 genes CHRNA7 and CHRFAM7A overlapped with MeCP2-binding sites. 4C analysis also demonstrated that 15q11.2–13.3 undergoes dynamic chromatin decondensation with neuronal maturation. Quantitative transcriptional analyses showed significantly reduced CHRNA7 transcripts in human frontal cortex samples from Rett syndrome or autism patients with reduced levels of MeCP2. Together, these results suggest that recruitment of CHRNA7 to the PWS-IC by MeCP2 during neuronal development modulates its expression. Therefore, by establishing a link between MeCP2, chromatin organization and CHRNA7 transcript levels, these results are relevant to understand the epigenetic complexity of neurodevelopmental disorders with genetic association with the 15q11.2–13.3 locus and to develop pharmacological therapies common to multiple neurodevelopmental disorders.
RESULTS
To assay 15q11.2–13.3 chromatin structure during neuronal differentiation by identifying chromatin interactions with the PWS-IC, 4C analysis (39,40) was performed. For these experiments, a model human cell culture system for neuronal maturation was used. Treatment of cycling SH-SY5Y neuroblasts with phorbol 12-myristate 13-acetate (PMA) induces cell-cycle arrest, a 2-fold increase in MeCP2 expression (41), neurite outgrowth and synapse formation with surface NeuN expression, a marker of mature neurons (42). 4C analysis identifies genomic loci that physically interact with a defined locus of interest or ‘anchor locus’ and thereby form chromatin loops (40). Specific interactions of the PWS-IC internal ‘anchor’ site with surrounding 15q11.2–13.3 loci were assayed by hybridizing ‘captured’ loci to a custom microarray containing 13 Mb of 15q11.2–13.3 and 13 Mb of non-15q11.2–13.3 control loci tiled at high density. As a positive 4C control for chromatin interaction, a known binding of the H19 locus with Igf2 (43,44) was assayed and observed in parallel experiments (Supplementary Material, Fig. S1) providing proof of principle for 4C results.
Identification of PWS-IC interactions with flanking 15q11.2–13.3 sites by 4C analysis was used to characterize overall chromatin organization. First, PWS-IC interactions with 15q11.2–13.3 were determined by bioinformatic analysis of log 2 microarray signal intensities from three replicate 4C libraries using the Tamalpais peak calling algorithm (http://genomics.ucdavis.edu/farnham) (45). PWS-IC interactions are represented as bars (Fig. 1A, rows 2 and 4) shown above the log 2 signal peaks (Fig. 1A, rows 3 and 5) that were uploaded as custom tracks to the UCSC Genome Browser (http://genome.ucsc.edu). Thus, every identified interaction of a 15q11.2–13.3 site with the PWS-IC represents a chromatin loop. Therefore, chromatin loop organization as assayed by 4C is a high-resolution representation of overall chromatin structure (40).
Figure 1.

Dynamic 15q11.2–13.3 chromatin decondensation during neuronal maturation. (A) 4C analysis of 15q11.2–13.3 chromatin structure in developing neurons shows developmentally regulated chromatin decondensation in maturing neurons. 15q11.2–13.3 loci that contact the PWS-IC (red box) are shown as a series of log 2 signal peaks from representative hybridization of 4C libraries isolated from developing SH-SY5Y neurons (row 3) and untreated SH-SY5Y neuroblasts (row 5). 4C peaks corresponding to sites of PWS-IC interaction were determined using a bioinformatic analysis of three independent replicate experiments for both PMA-treated neurons and untreated neuroblasts and are shown as a series of black bars above the corresponding peaks (rows 2 and 4). MeCP2-binding sites determined from previous studies are shown as a series of purple bars in row 1. 15q11.2–13.3 was subdivided into regions A–C based on Giemsa banding patterns in metaphase chromosomes (top row). Log 2 signal ratios are shown as red and green histograms in the UCSC Genome Browser with known genes and transcripts shown in blue font (row 6). A black line at a log 2 signal ratio of 1 is included to enable comparison of peaks in rows 3 and 5. Segmental duplications are shown as a series of colored bars below chromosome bands. (B). A high-resolution example of PWS-IC interactions flanking the HBII-85 snoRNA cluster in neuroblasts (bottom histogram) that are lost during neuronal differentiation (top histogram). Arrows show PWS-IC-binding interactions. (C). PWS-IC interactions shown as black lines above the log 2 signal peaks with the autism candidate gene GABRB3 are also altered by neuronal differentiation. PWS-IC interactions are indicated by red arrows.
The PWS-IC forms chromatin loops with heterogeneous elements of 15q11.2–13.3
4C analysis of the entire 15q11.2–13.3 locus during neuronal maturation revealed several insights into chromatin organization of this locus. Closer examination of PWS-IC interaction patterns revealed a dynamic chromatin landscape as chromatin organization was found to be heterogeneous throughout 15q11.2–13.3 both in terms of number and location of PWS-IC interactions forming chromatin loops. To facilitate this analysis, the entire 13 Mb 15q11.2–13.3 region was partitioned based on Giemsa banding pattern as an unbiased means of dividing the locus into three, approximately equivalently sized, subregions (Fig. 1A, boxes A–C, data in Table 1).
Table 1.
Quantitative analysis of 15q11.2–13.3 chromatin condensation
| Subregion | 15q11.2 | 15q12–13.1 | 15q13.2–13.3 | Total 15q11.2–13.3 | Control 6p22.3 |
| PWS-IC-binding sites in neurons | 25 | 16 | 27 | 68 | 5 |
| PWS-IC-binding sites in neuroblasts | 85 | 42 | 66 | 193 | 19 |
| Total base pairs (TBP) | 4 900 000 | 4 800 000 | 3 400 000 | 13 100 000 | 2 915 400 |
| Neuronal PWS-IC-binding frequency (sites/106 TBP, row 2/row 4) | 5.10 | 3.33 | 7.94 | 5.19 | 1.71 |
| P= probability that the number of PWS-IC interactions is at least as low by chance | 0.77288 | 0.09760 | 0.99873 | 0.94464 | 0.00629 |
| P= probability that the number of PWS-IC interactions is at least as high by chance | 0.30126 | 0.93962 | 0.00266 | 0.07900 | 0.99800 |
| Neuroblast PWS-IC-binding frequency (sites/106 TBP, row 3/row 4) | 17.35 | 8.75 | 19.41 | 14.73 | 6.52 |
| P= probability that the number of PWS-IC interactions is at least as low by chance | 0.99997 | 0.02555 | 0.99999 | 0.99999 | 0.00298 |
| P= probability that the number of PWS-IC interactions is at least as high by chance | 0.00004 | 0.98258 | 0.00001 | 0.0000007 | 0.99848 |
| Decondensation ratio (row 8/row 5) | 3.40 | 2.63 | 2.44 | 2.84 | 3.81 |
| MeCP2-binding sites in neurons | 9 | 15 | 38 | 62 | 10 |
| MeCP2-binding frequency (sites/106 TBP, row 12/row 4) | 1.84 | 3.12 | 11.18 | 4.73 | 3.43 |
| MeCP2 sites/106 TBP in 15q13.2–13.3 divided by the total number of MeCP2 sites | 6.07 | 3.58 | 1.0 | 2.36 | 3.26 |
The table summarizes chromatin condensation and MeCP2-binding density in subregions of 15q11.2–13.3 chosen based on PWS-IC interaction density in neuroblasts and a control 6p22.3 locus. 15q11.2–13.3 subregions 15q11.2, 15q12–15q13.3 plus 15q13.2–15q13.3 as shown in Figure 1 were assayed for PWS-IC interactions in neurons and neuroblasts (rows 2 and 3). Next the total number of PWS-IC sites was divided by the total number of base pairs in 15q11.2–13.3 subregions (row 4) to give the frequency of interaction for each region in rows 5 and 8. Next, a probability of obtaining the number of PWS-IC interactions at least as low or as high as observed was calculated in rows 6, 7, 9 and 10. To determine levels of chromatin decondensation with neuronal differentiation for each region, the frequency of PWS-IC sites per base in neuroblasts was divided by the frequency of PWS-IC sites in neurons as shown in row 11. Total MeCP2-binding sites (row 12) were divided by total base pairs (row 4) to calculate MeCP2 sites per TBP (row 13). MeCP2 sites/TBP for 15q13.2–15q13.3 were divided by MeCP2 sites/TBP for the other subregions as a means of comparison (row 14).
In general, initial 15q11.2–13.3 chromatin organization was heterogeneous. Surprisingly, the central 15q12–15q13.1 subregion adjacent to the PWS-IC (Fig. 1A, box B) displayed a more open chromatin conformation in neuroblasts as shown by the significant probability (P= 0.02555) that the number of PWS-IC interactions observed were less frequent than the number expected by random chance (Table 1, row 9) and than either of the more distal subregions (Fig. 1A, box A or C). In contrast, PWS-IC interactions occurred significantly more frequently than random in subregions 15q11.2 and 15q11.2–13.3 (P= 0.00004 and P= 0.00001, respectively) as shown in Table 1, row 10. As a control, PWS-IC trans-interactions with a different autosomal locus 6p22.3 occurred with significantly lower frequency than random in both neuroblasts (P= 0.00298) and neurons (P= 0.00629) as expected (Table 1, rows 6 and 9). Surprisingly, there was no obvious correlation between intensity of 15q11.2–13.3 Giemsa bands and density of PWS-IC interactions.
Not only did the initial level of chromatin condensation vary considerably between 15q11.2–13.3 subregions in neuroblasts, but the level of decondensation upon neuronal differentiation also varied by subregion. In the 15q11.2–13.3 locus, 4C analysis of PWS-IC interaction sites revealed marked chromatin decondensation 48 h after PMA treatment in SH-SY5Y neurons (Fig. 1A). This is shown by an overall 2.84-fold reduction (Table 1, row 11) in PWS-IC interactions with flanking 15q11.2–13.3 loci in neurons (Fig. 1A, rows 4 and 5) versus PWS-IC interactions observed in differentiated neuroblasts (Fig. 1A, rows 2 and 3). Interestingly, subregion 15q13.2–15q13.3, which displayed the most compact chromatin in neuroblasts (Fig. 1A, box C), not only showed the lowest level of induced chromatin decondensation at 2.44-fold (Table 1, row 11) during the neuroblast-to-neuron transition but also displayed significantly higher levels of PWS-IC binding in neurons (P= 0.00266, Table 1, row 7) than expected by random chance. Therefore, PWS-IC interaction was particularly abundant in the 15q13.2–13.3 subregion (Fig. 1A, box C), containing the CHRFAM7A and CHRNA7 loci, thereby marking this subregion for further study.
The PWS-IC forms chromatin loops containing key neurodevelopmental genes
4C analyses also revealed a number of novel PWS-IC interactions with genes necessary for normal neurologic function. For example, the HBII-85 small snoRNA cluster, the loss of which contributes to the Prader–Willi phenotype (9,46), is flanked by PWS-IC interactions. These results suggest incorporation of the snoRNA loci into a distinct 100 kb chromatin loop in undifferentiated SH-SY5Y neuroblasts and an open structure in maturing neurons (Fig. 1B), similar to what has previously been demonstrated by FISH (10).
The autism candidate gene GABRB3 (47) contains a PWS-IC interaction site in the first intron in neuroblasts that divides the gene into two loop domains (Fig. 1C, bottom histogram), but in differentiated neurons this is replaced by an interaction proximal to the transcriptional start site (Fig. 1C, top histogram). This chromatin structure suggests that GABRB3 chromatin adapts an altered configuration with neuronal maturation. Although single-representative experiments are shown in Figure 1A–C, chromatin structure patterns as assayed by 4C analysis were consistent between three replicate experiments for SH-SY5Y neuroblasts and PMA-treated SH-SY5Y neurons (Supplementary Material, Fig. S2).
MeCP2-binding sites are more frequent in the 15q13.2 and 15q13.3 subregions
To address the hypothesis that MeCP2 recruits discrete 15q11.2–13.3 sub-loci to the PWS-IC, MeCP2-binding sites mapped in a previous study (11) were compared with PWS-IC interaction sites in 15q11.2–13.3. In Figure 1A, MeCP2-binding sites as determined from ChIP-chip data are depicted as the top row of bars (Fig. 1A, row 1) in the UCSC Genome Browser as a custom track. On a 15q11.2–13.3 locus wide scale, 38 MeCP2-binding sites clustered in 15q13.2–15q13.3 (Fig. 1A, row 1, box C), where there were also discrete clusters of sites that were bound to the PWS-IC in both neurons (Fig. 1A, row 2) and neuroblasts (Fig. 1A, row 4). Therefore, MeCP2-binding sites are 6.07 times more frequent in the 15q13.2–15q13.3 subregion than in the 15q11.2 subregion and 3.58 times more frequent than in the 15q12–13.1 subregion (Table 1, row 14). These results suggest that, of all the 15q subregions, MeCP2 was most likely to affect chromatin organization of the 15q13.2–15q13.3 subregion and this was therefore the most likely region to contain genes affected by MeCP2 binding.
MeCP2-binding sites in CHRNA7 interact with the PWS-IC in maturing neurons
Previous studies had identified 62 high-confidence MeCP2-binding sites in the 13 Mb comprising the non-repetitive fraction of 15q11.2–13.3, particularly around 15q13.2 and 15q13.3, the majority of which were intergenic (11). These results and in vitro studies on chromatin templates (36,37) suggest that MeCP2 might be involved in higher order chromatin organization of 15q11.2–13.3. To test this hypothesis, bioinformatic analysis of the relationship between MeCP2-binding sites and sites of PWS-IC chromatin loop interactions in 15q11.2–13.3 was performed. Analysis of the total 15q11.2–13.3 MeCP2-binding sites and total PWS-IC interaction sites revealed that distribution of MeCP2-binding sites is random relative to PWS-IC interaction sites overall (data not shown). However, 5 out of 68 or 7.3% of discrete PWS-IC chromatin interactions were also bound by MeCP2 (Supplementary Material, Table S2) identifying them as overlapping binding sites. Furthermore, the proportion of MeCP2 sites that are also sites of interaction with the PWS-IC is much higher than expected by random chance according to Fisher's exact test (Supplementary Material, Table S2), suggesting that these specific PWS-IC and MeCP2 interaction sites have an important biologic function in neurons. As expected, one of the PWS-IC and MeCP2 overlapping binding sites corresponds to the 4C anchor sequence in the PWS-IC itself, where MeCP2 had previously been shown to bind (11,12). However, the remaining four of the PWS-IC and MeCP2 overlapping binding sites in neurons were located in 15q13.2 and 15q13.3 at CHRNA7/CHRFAM7A (Fig. 1A, box C). Two of the PWS-IC- and MeCP2-binding sites flanked the CHRNA7 gene (Fig. 2A), whereas two sites were found in an intron and upstream of the partial CHRNA7 duplication gene CHRFAM7A (Fig. 2B). Thus, both CHRNA7 and CHRFAM7A interact with the PWS-IC located >7 Mb distal via MeCP2-binding sites, consistent with a long-range chromatin loop structure in neurons.
Figure 2.

PWS-IC- and MeCP2-bound sites map to CHRNA7 and CHRFAM7A. 4C analysis reveals PWS-IC interaction sites that are also bound by MeCP2 in 15q13.3. (A) A 250 kb scale view of CHRNA7 chromatin structure reveals two sites (arrows) that are bound both by the PWS-IC (PWS-IC peaks) and MeCP2 (top bars) as determined by previous ChIP-chip analysis. 4C interactions shown as log 2 signal ratio peaks of differentiated SH-SY5Y 4C libraries identify one bivalent site 50 kb upstream of CHRNA7 near the AK097050 transcript in neurons undergoing differentiation (upper histogram) and another site ∼30 kb downstream. (B) Overlap between MeCP2-bound and PWS-IC-bound sites was also observed within an intron of CHRFAM7A (left arrow) and upstream of the translational start site and near the AK127208 transcript (right arrow) in maturing neurons. (C) MeCP2 knockdown by siRNA in SH-SY5Y cells reduces interaction of CHRNA7 with the PWS-IC. Insets show PWS-IC interactions in neurons with normal levels of MeCP2 (top histogram) as peaks compared with neurons with reduced MeCP2 levels (bottom histogram) with diminished PWS-IC interaction signals.
To further examine the connection between CHRNA7/PWS-iC interactions and MeCP2, 4C analysis was performed in neurons following knockdown of MeCP2 (Fig. 2C). For these studies, SHSY-5Y neurons were treated with siRNA targeting the 3′ UTR, and MeCP2 knockdown was confirmed by western blot (Supplementary Material, Fig. S8). Knockdown of MeCP2 protein levels in neurons correlated with disrupted PWS-IC interactions with CHRNA7 as shown by reduction in peak height compared with neurons with normal levels of MeCP2 (Fig. 2C). These results suggest that MeCP2 connects CHRNA7 to the PWS-IC through long-range chromatin interactions.
CHRNA7 interactions with the PWS-IC are confirmed by FISH
In order to confirm the interaction of the PWS-IC with CHRNA7 detected by 4C analyses, an independent assay using FISH analysis was performed. For this assay, differentially labeled genomic probes to the PWS-IC (green) and CHRNA7 genomic loci (red) (Fig. 3A) were hybridized to SH-SY5Y cells treated with the same neuronal differentiation protocol established for 4C analyses (10). Consistent with 4C results, >90% of neuroblast nuclei showed PWS-IC signals overlapping with CHRNA7 signals on both 15q11.2–13.3 alleles (Fig. 3B, bar graph), indicating close physical proximity of the two loci (Fig. 1A, rows 4 and 5). In contrast, FISH analysis of SH-SY5Y neurons revealed that 50% of nuclei had a clear separation of PWS-IC (green) and CHRNA7 (red) signals on one (40%) or both (10%) of 15q11.2–13.3 alleles consistent with chromatin decondensation on those alleles compared with neuroblasts (Fig. 3B, bar graph).
Figure 3.

PWS-IC interaction with CHRNA7 is validated by FISH. Two-color FISH analysis confirms the interaction of the PWS-IC with CHRNA7. (A) BAC clones complementary to the PWS-IC (RP11-125E1) and CHRNA7 loci (RP11-717124 as shown on the 15q11.2–13.3 map were differentially detected as green and red fluorescent signals, respectively). (B) PWS-IC and CHRNA7 signals were considered associated if there was a spatial overlap or contact between the red and green signals. One hundred nuclei from undifferentiated and maturing (SH − SY5Y + PMA) neurons were scored for signal overlap in each allele and the possible combinations (middle diagram) and were graphed as a percentage of total nuclei (left bar graph). Representative images of each PWS-IC/CHRNA7 allelic combination are shown in the right panel with scale bars.
As controls for FISH analysis of PWS-IC interactions, the 15q11.2–13.3 internal locus TJP1 and an external PML locus in 15q24.1 revealed that only in 8.7% in SH-SY5Y neuroblasts and 4.1% neuronal nuclei was there any association of the PWS-IC with TJP1 on one or both alleles, whereas only 18.8% of one or both PML alleles in neuroblasts or 23.5% of neurons showed association with the PWS-IC (Supplementary Material, Figs S3 and S5). This suggests that TJP1, which is 2.3 Mb closer, and PML, which is over 50 Mb distant from the PWS-IC than CHRNA7 rarely interact with the PWS-IC unlike CHRNA7 and that neuronal maturation did not lead to increased association. Together, these results independently confirm that the PWS-IC is in close proximity to CHRNA7 on both alleles in neuroblasts and that neuronal maturation correlates with dissociation of the PWS-IC from CHRNA7 on one and sometimes both alleles. These results also show that the PWS-IC interaction with CHRNA7 is not a general property of the distal chromosome 15 PML locus. Most importantly, these results are consistent with the 4C analyses that reveal specific interactions between the PWS-IC and CHRNA7 primarily in neuroblasts and 15q11.2–13.3 chromatin decondensation with differentiation to neurons.
CHRNA7 transcript levels in human cortex decrease with age
As CHRFAM7A is reported to have very low levels of expression in the brain (48) and its biologic function is obscure, quantitative reverse transcriptase–polymerase chain reaction (QRT–PCR) analyses focused on CHRNA7 transcript levels. Although CHRNA7 has been previously assayed in brains of adult schizophrenics (48,49), little is known about its transcript levels during early human developmental stages. For CHRNA7 analysis, RNAs were extracted post-mortem, from Brodmann area 9 obtained from a wide age range of normally developing human subject brains, 7 brains from classic Rett patients and 11 patients diagnosed with autism. QRT–PCR analysis of CHRNA7 transcript levels normalized to GAPDH levels from typically developing cortices revealed high levels of expression in subjects younger than 1 year of age and a reduction in transcript with increasing post-natal age before leveling off around 20 years of age (Fig. 4). Strikingly, CHRNA7 transcript levels were roughly 10-fold higher in the youngest (<1 year of age) than the oldest typically developing (control) brain samples (57 years of age). Therefore, a one-way analysis of co-variance (ANCOVA) was performed, which revealed a significant (P< 0.0001) effect of age on CHRNA7 expression. Interestingly, CHRNA7 transcripts appeared to be reduced in Rett and autism cortices (Fig. 4, filled symbols).
Figure 4.

CHRNA7 transcript levels decline significantly with age in the human cortex. QRT–PCR analyses of CHRNA7 expression in normally developing control (CTRL, N = 20), Rett (RTT, N = 7) and autism (AUT, N = 12) were performed on RNAs isolated from frozen, post-mortem Brodmann area 9 cortices using CHRNA7- and GAPDH-specific primers. Fold change of CHRNA7 expression relative to GAPDH was calculated using the comparative Ct method for each sample. CHRNA7 expression declines rapidly from 4.0-fold relative to GAPDH in 4-month-old cortex to ∼0.4-fold in 57-year-old cortex. Analysis of CHRNA7 expression levels was done using a one-way ANCOVA with age as a covariate showing that age has a significant relationship to CHRNA7 expression (P< 0.0001).
CHRNA7 transcript levels are significantly reduced in Rett and autism cortices
To test the hypothesis that MeCP2 modulates expression of CHRNA7 in vivo, QRT–PCR analysis was extended to RNAs isolated from frontal cortices of classic Rett syndrome patients and autism samples, where MeCP2 has been shown to be significantly less abundant (50). Since our findings in Figure 4 suggest that there is an apparent reduction of CHRNA7 in Rett and autism cortical samples, a one-way ANCOVA using age as a covariate was performed. This age covariate analysis determined that CHRNA7 transcript levels were significantly reduced (P< 0.05) compared with control samples for both Rett (RTT, P= 0.022) and autism (AUT, P= 0.012) (Fig. 5) even when cortical samples of all ages were included. Also, when cortical RNA samples were segregated into two age groups based on our findings in Figure 4, brain cortices of subjects 20 years of age and younger displayed a significant reduction in CHRNA7 transcripts compared with controls (Supplementary Material, Fig. S7). The results shown in Figure 5 and Supplementary Material, Figure S7, are consistent with results in Mecp2-deficient mice, where Chrna7 transcripts are significantly decreased (P< 0.05) at 1 week of post-natal development in total RNAs isolated from the whole brain (Supplementary Material, Figure S4). Age was found to have a significant effect on Chrna7 expression (P< 0.001) by ANCOVA (data not shown). Together, these novel results suggest that MeCP2 positively regulates CHRNA7 expression in human cortex during neuronal maturation by long-range interaction with the PWS-IC.
Figure 5.

CHRNA7 transcripts are significantly reduced in Rett and autism corticies. QRT–PCR analyses identify significant reductions in CHRNA7 expression in Rett and autism frontal cortices. RNAs were isolated from Brodmann area 9 of patients with classic Rett syndrome (RTT, N = 7) and autism patients (AUT, N = 11). RNAs were amplified and quantified using CHRNA7- and GAPDH-specific primers, and CHRNA7/GAPDH fold changes were calculated using the comparative Ct method and graphed, and normalized to the typically developing controls (CTRL, N = 20). Significant reductions (P< 0.05) in CHRNA7 transcripts are observed in both RTT and AUT samples compared with CTRL while controlling for the effect of age. Fold changes for RTT and AUT samples are normalized to CRTL samples and error bars correspond to standard error of the mean. P-values are derived from ANCOVA with age as a covariate.
DISCUSSION
This study used a novel 4C analysis in order to gain insight into the role that chromatin structure plays in the regulation of 15q11.2–13.3 genes. These results provide many insights into the organization and regulation of this locus. Primarily, these results indicate that PWS-IC interactions within 15q11.2–13.3 are dynamic during neuronal maturation, suggesting dramatic chromatin remodeling at this chromosomal subregion. These results complement studies of the 15q11.2–13.3 syntenic region 7q5 in the mouse brain that show a developmentally regulated decondensation of Snrpn-Ube3A preceding an exponential rise in SnoRNA accumulation and maturation (10). Together, these in vitro and in vivo results suggest that chromatin decondensation and remodeling regulates 15q11.2–13.3 gene transcription during neuronal maturation.
The PWS-IC is a well-characterized long-range control element of 15q11.2–13.3 (51). 4C chromatin structure results confirmed by FISH analyses showed that the PWS-IC makes specific contact with sites flanking CHRFAM7A and CHRNA7 in maturing neurons. FISH analysis of 15q11.2–13.3 conformation in developing SH-SY5Y neurons also revealed PWS-IC and CHRNA7 signal disassociation, consistent with decondensation of the intervening chromatin at this locus during neuronal maturation on one allele. Again, this result is consistent with previous in vivo results by Leung et al. (10), where the 15q11.2–13.3/7q5 paternal allele visualized by FISH analysis becomes strikingly decondensed during neuronal development . 4C chromatin structure analysis also reveals a PWS-IC interaction site upstream of GABRB3, a neuro-developmentally essential gene known to be dysregulated by loss of MeCP2 (27,38). Therefore, chromatin structure analysis directed our hypothesis that CHRNA7 makes a functional molecular connection with the PWS-IC. Although ICs can regulate imprinted gene expression over several megabases, it is surprising that the PWS-IC may epigenetically regulate CHRNA7, which is over 7 Mb away.
In addition, 4C results indicate that clusters of PWS-IC interaction sites in the 15q13.2–15q13.3 subregion overlap with regions containing segmental duplications in both neurons and neuroblasts. In particular, PWS-IC interactions cluster in two 300 kb regions of segmental duplication including portions of the CHRFAM7A and CHRNA7 genes. It has recently been shown that low copy repeat elements predispose the 15q13.3 locus to duplications and deletions that are often found in patients with neurodevelopmental disorders (reviewed in 52). Therefore, it is tempting to speculate that PWS-IC interactions form the bases of chromatin loops bearing the low copy number repeats, making them susceptible to deletion or duplication by homologous recombination.
4C analysis in SH-SY5Y neurons with and without MeCP2 knockdown suggests that MeCP2 has a role in linking the 15q13.3 region, located over 7 Mb distant, with the PWS-IC. Support for the role of MeCP2 in chromatin loop formation comes from Horike et al., who described a role for MeCP2 in chromatin loop formation and transcriptional regulation of the Dlx5/Dlx6 locus in the developing striatum (34,53). Also, in vitro chromatin assays show that MeCP2 can bridge and condense chromatin arrays (35) through multiple DNA-binding domains to form complex looped structures (37). Interestingly, a neuronal enhancer of Chrna7 expression has been recently identified in the Chrna7 intron 4 (54) near the human PWS-IC/MeCP2 interaction site observed in this study, further suggesting a potential site of action for MeCP2 binding to Chrna7. Recent results from Skene et al. (55) also indicate a function for MeCP2 in neuronal chromatin structure and repression of genome-wide endogenous retrovirus transcription. Together, the results shown here are consistent with independent studies examining the role of MeCP2 as a neuronal chromatin structural organizer at high abundance in maturing neurons.
The 15q11.2–13.3 chromatin organization studies revealed a potential and clinically relevant MeCP2 target gene CHRNA7. CHRNA7 levels were found to be significantly reduced in Rett and autism brain frontal cortices. In these studies, CHRNA7 expression was first found to be developmentally regulated with postnatal age in humans. This is shown by a 10-fold reduction in CHRNA7 from a high at 4 months of age to a leveling off of transcript levels around 20 years of age in normal cortices. Intriguingly, CHRNA7 levels in the frontal cortex were found to decline rapidly around 1–2 years of age, corresponding with the age of onset of autistic symptoms (56) and age of diagnosis of Rett symptoms (57). These initial results led to the discovery of reduced CHRNA7 levels in the human cortex from autism and Rett brains, where MeCP2 is reduced or mutated as described previously (50). For these studies, Brodmann area 9 was selected, as it is part of the dorsolateral prefrontal cortex, where previous studies have linked changes in this area to loss of function in autism (58). Analysis of this cortical region is also justified by transgenic mouse studies where genetically engineered loss of MeCP2 expression in neurons is sufficient to cause symptoms similar to Rett syndrome (59,60). These results, along with the previous finding that MECP2 levels are also significantly reduced in these samples (50), suggest that MeCP2 positively modulates expression of CHRNA7 and that MeCP2 deficiency results in CHRNA7 deficiency in childhood and adolescence. Most importantly, these results are the first in vivo evidence that MeCP2 may positively regulate CHRNA7 through long-range chromatin interaction and suggest that reduced expression may lead to abnormal brain function observed in autism and Rett syndrome.
Independent support for the role of MeCP2 as a positive modulator of Chrna7 comes from RNA expression profiling that revealed significant up regulation of Chrna7 expression in 6 week post-natal mouse hypothalamus engineered to overexpress MeCP2 (61). As CHRNA7/Chrna7 expression is highest in the earliest stage of postnatal development examined, it may also be dysregulated in embryonic stages of development, where reduced abundance may have a persistent effect on later neuronal development and function. In support of this hypothesis, CHRNA7 has reported to have high expression in nuclei that receive sensory input such as the hippocampus and cortex in the fetal brain (62). In contrast, MeCP2 has very low levels of expression in mouse embryonic brain outside of the thalamus and early post-natal cortex (63). Therefore, it is tempting to speculate that a rising concentration of MeCP2 protein in cortical neuronal nuclei during post-natal life dampens the expression of CHRNA7/Chrna7 during late infancy through adolescence. However, the results presented here indicate that loss or reduction in MeCP2 protein is concomitant with reduced CHRNA7/Chrna7 expression in developing humans and mice. Therefore, MeCP2 may act to keep CHRNA7 at an optimal level during postnatal development by both positive and negative modulation. Although the evidence of direct MeCP2-binding sites around CHRNA7 suggests that MeCP2 is a direct transcriptional modulator of CHRNA7 similar to the case of Bdnf (64), it is possible that the effect of MeCP2 deficiency on CHRNA7 is indirect. Nonetheless, these novel results suggest that reduced CHRNA7/Chrna7 expression may contribute to neurologic abnormalities observed in Rett and autism patients as well as in Mecp2-deficient mice.
Epigenetic mechanisms contribute to multiple neurologic disorders (26,65). Specifically, previous results have clearly demonstrated that loss or altered expression of the epigenetic factor MeCP2 leads to severe defects in neurologic development characteristic of Rett syndrome (57). Although the mechanisms underlying the Rett phenotype are not completely understood, the results presented here suggest that MeCP2 acting through the PWS-IC may contribute to the precise epigenetic 15q11.2–13.3 chromatin remodeling process that regulates expression of CHRNA7 and other key genes such as GABRB3 and UBE3A during neuronal maturation. Therefore, aberrant 15q11.2–13.3 chromatin remodeling caused by defects in MeCP2 mutation or deficiency could lead to neurodevelopmental gene dysregulation and thereby contribute significantly to the overlapping Rett and autism phenotypes. A recent phase 2 clinical trial of a CHRNA7 partial agonist in schizophrenic patients significantly reduced negative symptoms, and suggests a potential therapeutic strategy for Rett and autism patients (66).
MATERIALS AND METHODS
4C analysis of chromatin structure
4C analysis was performed according to Simonis et al. (40). Briefly, SH-SY5Y neurons were treated with formaldehyde to crosslink chromatin, followed by quenching of the reaction with 1 m glycine and cell lysis by resuspension in hypotonic buffer and dounce homogenization. Cell nuclei were resuspended in restriction buffer and digested overnight with BglII (NEB, Ipswich, MA, USA) at 37°C followed by ligation using T4 ligase (Promega, Madison, WI, USA). DNA was purified from nuclei using proteinase K and RNAseA digestion, followed by phenol/chloroform extraction. Purified DNA was digested using DpnII (NEB), re-ligated with T4 ligase (NEB), re-precipitated and then re-digested with Mnl1 (NEB) overnight at 37°C. 4C libraries were created by linear amplification of DNA fragments with Expand Long Template PCR System (Roche Applied Sciences, Indianapolis, IN, USA). Amplified 4C library fragments were evaluated by gel electrophoresis and column-purified using a PCR clean-up kit (Qiagen, Valencia, CA, USA).
A custom DNA microarray tiling 26.3 Mb of NCBI34/hg17 human genome sequence including 13.3 Mb of 15q11.2–13.3, 2.6 Mb of 11p15.5, 3.8 Mb of 7q21.3, 2.9 Mb of 19p13.2, 0.3 Mb of 20q11.21, 1.5 Mb of 2p25.1, 0.4 Mb of 1p36.12, 2.9 Mb of 6p22.3 and 0.2 Mb of 11p14.1 with repetitive sequences removed was designed and produced by Roche-NimbleGen, Madison, WI, USA (11).
Labeling and hybridization of DNA for 4C analysis were performed according to the Nimblegen Array User's Guide version 3.1 (Nimblegen). One microgram of each DNA sample was denatured in the presence of 5′-Cy3- or Cy5-labeled random nonamers (TriLink Biotechnologies) and incubated with 100 units of (exo-) Klenow fragment (NEB) and dNTP mix (6 mm) each in TE buffer (10 mm Tris/1 mm EDTA, pH 7.4) (Invitrogen) for 2 h at 37°C. Reactions were terminated by addition of 0.5 m EDTA (pH 8.0), precipitated with isopropanol and resuspended in water. Then, 13 μg of the Cy5-labeled 4C sample and 13 µg of the Cy3-labeled genomic input sample were mixed together, dried down and resuspended in 40 μl of Nimblegen Hybridization Buffer (Nimblegen) plus 1.5 µg of human COT1 DNA. Hybridization was carried out in a MAUI Hybridization System (BioMicro Systems, Salt Lake City, UT, USA) for 18 h at 42°C.
The arrays were washed using Nimblegen Wash Buffer System, dried by centrifugation and scanned at 5 μm resolution using the GenePix 4000B scanner (Axon Instruments, Union City, CA, USA). Fluorescence intensity raw data were obtained from scanned images of the arrays, using NIMBLESCAN 2.0 extraction software (Nimblegen). For each spot on the array, log 2 ratios of the Cy5-labeled test sample versus the Cy3-labeled reference sample were calculated. Then, the bi-weight mean of this log 2 ratio was subtracted from each point by the software. To control for labeling effects, Cy3/5 dye labels were also swapped between the 4C samples and total input DNA prior to hybridization. 4C peaks in three biologic replicate experiments were determined using Tamalpais peak-finder algorithm (45) and categorized into levels L1, L2 or L3 based on peak height, peak width and presence in two out of three replicates (11).
MeCP2 knockdown by siRNA
MeCP2 levels were knocked down in SH-SY5Y neurons, using an siRNA hairpin targeting the GAGGGGGAGTGTAAAGACA sequence in MECP2 designed and produced by iGENE Therapeutics (Tsukuba, Japan). SH-SY5Y cells were subjected to siRNA treatment and tested for MeCP2 knockdown by western blot (Supplementary Material, Fig. S8). For western blot analysis, 25 μg of SH-SY5Y protein was resolved by SDS–PAGE, transferred to PVDF nylon membranes and probed for MeCP2 using a custom carboxyl terminal antibody raised in chicken and GAPDH using a mouse monoclonal antibody (Advanced Immunochemical, Inc., Long Beach, CA, USA).
PWS-IC regional binding analysis
In order to determine whether the probability that the number of 4C peaks identified in a subregion is greater or less than expected by chance, the following binomial probability formula was used:
where N denotes the total number of peaks; k, the peak (number ranging from 0 to N); p, percentage of area subsection takes in array; 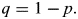
Thus, this equation calculates the chance that a number of peaks (k) would be identified in a subregion with probability (P) with the total number of peaks found (N). To determine the chance that at least the number of peaks would be found by chance, all values of P were summed from k equaling the peaks found to k equaling N. To determine the chance that no more than the number of peaks would be found by chance, all values of P were summed from k equaling 0 to k equaling the number of peaks found. Therefore, all summed P-values <0.05 were called as significant because the 4C peaks were not random with >95% certainty.
Data analysis of 4C peak relationship with MeCP2-binding sites
To determine whether MeCP2 peaks (at the L3 level) were significantly closer or further from 4C peaks (at the L3 level) than expected by chance, each of the 68 4C peaks were assigned a nearest MeCP2 peak neighbor. The 68 distances were then categorized as either near or far. To determine how often these nearest-neighbor distances would be expected by chance, 100 random peak data sets were generated over the same genomic interval, each with 62 peaks as in the MeCP2 peak data set. Nearest neighbors and distances to the 4C peaks were computed for each data set and the results were combined for use in the two-sided Fisher's exact test shown in Supplementary Table S1.
FISH analysis
DNA FISH analysis was performed as described in Leung et al. (10). Briefly, FISH was performed in SH-SY5Y neuroblasts and SH-SY5Y neurons differentiated by 48 h treatment with PMA (11,12). Bacterial artificial chromosome (BAC) DNA corresponding to the PWS-IC and 5′ region of CHRNA7 (Fig. 3) was isolated from clones obtained through BacPacResources (Oakland Children's Hospital, Oakland, CA, USA). BAC DNA was labeled by nick translation to create probes, hybridized to cells fixed in Histochoice (Amresco, Solon, OH, USA) and then dehydrated in ethanol. FISH probes were denatured with the fixed cells at 80°C for 3 min and then hybridized overnight at 37°C on cover-slipped slides. Cells were washed three times in 50% formamide/50% 2× SSC for 5 min, 2× SSC for 5 min and 2× SSC/0.1% IGEPAL for 5 min, all at 46°C prior to drying and staining with Rhodamine and Oregon Green-labeled secondary antibodies, washing three times with 1× PBS/0.5% Tween 20 and mounting in Vectashield (Vector Laboratories, Burlingame, CA, USA) containing 5 µg of DAPI.
Microscopy
FISH slides were analyzed on a Zeiss Axioplan 2 fluorescence microscope (Carl Zeiss, Inc., NY, USA) equipped with a digital camera. The microscope and peripherals were controlled by a computer-running iVision (Scanalytics, Vienna, VA, USA) software. Images were taken from the top of the specimen, and then repeated at 0.5–1 µm sections through the depth of the tissue. Each image stack was digitally de-convolved to remove out-of-focus light using HazeBuster software (Vaytek, Fairfield, IA, USA). Following haze removal, image stacks were merged and stacked to create a two-dimensional image representing all of fluorescence within the section. Images were taken using a 100× oil objective with a 1× zoom. FISH spots were counted as associated if the signals were touching or overlapping. All other red and green signal distances were counted as not associated.
CHRNA7/Chrna7 RT–PCR analysis
Seven classic Rett syndrome, 11 autism and 20 typically developing (controls), post-mortem brain cortices were obtained through the Autism Speaks-supported Autism Tissue Program (ATP) from the Harvard Brain Tissue Resource Center (HBTRC) and the University of Maryland Brain Tissue Resource Center or from the University of Miami Brain and Tissue Bank for Neurodevelopmental Disorders (Supplementary Material, Table S3). For mice, whole brains were isolated from Mecp2-deficient B6.129P2(C)-Mecp2tm1.1Bird/J mice.
RNAs were isolated using TRIzol® Reagent (Invitrogen) from Brodmann area 9 of the frontal cortices. To remove DNA contamination, the total RNA was treated with DNaseI (New England Biolabs, Ipswich, MA, USA) according to the manufacturer's instructions. Single-stranded complementary DNA (cDNA) was synthesized using Quantitect Reverse Transcription Kit (Qiagen). For each reaction, a tube without reverse transcriptase was used as a control for genomic DNA contamination. Primers were designed to cross an intron or span intron/exon boundaries to further limit the effect of possible genomic DNA contamination using Primer3 (http://frodo.wi.mit.edu/primer3/) Online PCR primer design software or Biosearch Technologies Real Time Design software (http://www.biosearchtech.com/realtimedesign). PCR amplification of cDNA was performed using 200 nm primer concentration and Express Sybr® Green ER Universal Master mix (Invitrogen). Cycling conditions were 20 s at 95°C followed by 40 cycles of 3 s at 95°C and 30 s at 60°C. PCR amplification was performed using the Mastercycler® Ep realplex (Eppendorf, Hamburg, Germany) and crossing points were analyzed using Realplex software (Eppendorf). For each reaction, a well without reverse transcriptase and a well without control cDNA were amplified to evaluate genomic DNA contamination, non-specific product formation or other contamination. All samples were normalized to GAPDH expression using the comparative CT method (Applied Biosystems, Foster City, CA, USA) to measure fold change relative to calibrator, following normalization to GAPDH. Melting curve analysis was also performed to determine homogenous product formation and to detect non-specific products.
Student's t-test was performed to verify that there were no differences in the ages of disease groups prior to running the one-way analysis of covariance (ANCOVA). Statistical analyses were performed using Mann–Whitney U and one-way ANCOVA tests. All P-values were considered significant if the associated value was <0.05.
Primers employed for QRT–PCR
Human CHRNA7—forward: GACGTGGATGAGAAGAACCAA; reverse: TGGGAAACGAACAGTCTTCA. Human GAPDH —forward: TGAACCATGAGAAGTATGACAAC; reverse: GTCCTTCCACGATACCAAAG.
Mouse Chrna7—forward: TGGGCATTGCCAGTATCT; reverse: AAAGGGAACCAGCGTACATC. Mouse Gapdh—forward: GCGGAGATGATGACCCTT; reverse GCTGAGTATGTCGTGGAGT.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
These studies were supported by the National Institutes of Health (NIH) through the Eunice Kennedy Shriver National Institute for Child Health and Human Development (NICHD) funding through grants (NIH 2R01HD041462 and 1R01HD048799).
Supplementary Material
ACKNOWLEDGEMENTS
Human brain cortical tissues were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, and the Harvard Brain Tissue Resource Center (R24MH068855).
REFERENCES
- 1.Ohta T., Gray T.A., Rogan P.K., Buiting K., Gabriel J.M., Saitoh S., Muralidhar B., Bilienska B., Krajewska-Walasek M., Driscoll D.J., et al. Imprinting-mutation mechanisms in Prader–Willi syndrome. Am. J. Hum. Genet. 1999;64:397–413. doi: 10.1086/302233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holm V.A., Cassidy S.B., Butler M.G., Hanchett J.M., Greenswag L.R., Whitman B.Y., Greenberg F. Prader–Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91:398–402. [PMC free article] [PubMed] [Google Scholar]
- 3.Buiting K., Lich C., Cottrell S., Barnicoat A., Horsthemke B. A 5-kb imprinting center deletion in a family with Angelman syndrome reduces the shortest region of deletion overlap to 880 bp. Hum. Genet. 1999;105:665–666. doi: 10.1007/s004399900196. [DOI] [PubMed] [Google Scholar]
- 4.Horsthemke B., Wagstaff J. Mechanisms of imprinting of the Prader–Willi/Angelman region. Am. J. Med. Genet. A. 2008;146A:2041–2052. doi: 10.1002/ajmg.a.32364. [DOI] [PubMed] [Google Scholar]
- 5.Peng X., Katz M., Gerzanich V., Anand R., Lindstrom J. Human alpha 7 acetylcholine receptor: cloning of the alpha 7 subunit from the SH-SY5Y cell line and determination of pharmacological properties of native receptors and functional alpha 7 homomers expressed in Xenopus oocytes. Mol. Pharmacol. 1994;45:546–554. [PubMed] [Google Scholar]
- 6.Kishino T., Lalande M., Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 7.Buxbaum J.D., Silverman J.M., Smith C.J., Greenberg D.A., Kilifarski M., Reichert J., Cook E.H., Jr., Fang Y., Song C.Y., Vitale R. Association between a GABRB3 polymorphism and autism. Mol. Psychiatry. 2002;7:311–316. doi: 10.1038/sj.mp.4001011. [DOI] [PubMed] [Google Scholar]
- 8.Schroer R.J., Phelan M.C., Michaelis R.C., Crawford E.C., Skinner S.A., Cuccaro M., Simensen R.J., Bishop J., Skinner C., Fender D., et al. Autism and maternally derived aberrations of chromosome 15q. Am. J. Med. Genet. 1998;76:327–336. doi: 10.1002/(sici)1096-8628(19980401)76:4<327::aid-ajmg8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 9.Runte M., Varon R., Horn D., Horsthemke B., Buiting K. Exclusion of the C/D box snoRNA gene cluster HBII-52 from a major role in Prader–Willi syndrome. Hum. Genet. 2005;116:228–230. doi: 10.1007/s00439-004-1219-2. [DOI] [PubMed] [Google Scholar]
- 10.Leung K.N., Vallero R.O., DuBose A.J., Resnick J.L., LaSalle J.M. Imprinting regulates mammalian snoRNA-encoding chromatin decondensation and neuronal nucleolar size. Hum. Mol. Genet. 2009;18:4227–4238. doi: 10.1093/hmg/ddp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yasui D.H., Peddada S., Bieda M.C., Vallero R.O., Hogart A., Nagarajan R.P., Thatcher K.N., Farnham P.J., Lasalle J.M. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl Acad. Sci. USA. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peddada S., Yasui D.H., LaSalle J.M. Inhibitors of differentiation (ID1, ID2, ID3 and ID4) genes are neuronal targets of MeCP2 that are elevated in Rett syndrome. Hum. Mol. Genet. 2006;15:2003–2014. doi: 10.1093/hmg/ddl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang T., Adamson T.E., Resnick J.L., Leff S., Wevrick R., Francke U., Jenkins N.A., Copeland N.G., Brannan C.I. A mouse model for Prader–Willi syndrome imprinting-centre mutations. Nat. Genet. 1998;19:25–31. doi: 10.1038/ng0598-25. [DOI] [PubMed] [Google Scholar]
- 14.Chini B., Raimond E., Elgoyhen A.B., Moralli D., Balzaretti M., Heinemann S. Molecular cloning and chromosomal localization of the human alpha 7-nicotinic receptor subunit gene (CHRNA7) Genomics. 1994;19:379–381. doi: 10.1006/geno.1994.1075. [DOI] [PubMed] [Google Scholar]
- 15.Gault J., Robinson M., Berger R., Drebing C., Logel J., Hopkins J., Moore T., Jacobs S., Meriwether J., Choi M.J., et al. Genomic organization and partial duplication of the human alpha7 neuronal nicotinic acetylcholine receptor gene (CHRNA7) Genomics. 1998;52:173–185. doi: 10.1006/geno.1998.5363. [DOI] [PubMed] [Google Scholar]
- 16.Miller D.T., Shen Y., Weiss L.A., Korn J., Anselm I., Bridgemohan C., Cox G.F., Dickinson H., Gentile J., Harris D.J., et al. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J. Med. Genet. 2009;46:242–248. doi: 10.1136/jmg.2008.059907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben-Shachar S., Lanpher B., German J.R., Qasaymeh M., Potocki L., Nagamani S.C., Franco L.M., Malphrus A., Bottenfield G.W., Spence J.E., et al. Microdeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J. Med. Genet. 2009;46:382–388. doi: 10.1136/jmg.2008.064378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shinawi M., Schaaf C.P., Bhatt S.S., Xia Z., Patel A., Cheung S.W., Lanpher B., Nagl S., Herding H.S., Nevinny-Stickel C., et al. A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat. Genet. 2009;41:1269–1271. doi: 10.1038/ng.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharp A.J., Mefford H.C., Li K., Baker C., Skinner C., Stevenson R.E., Schroer R.J., Novara F., De Gregori M., Ciccone R., et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freedman R., Coon H., Myles-Worsley M., Orr-Urtreger A., Olincy A., Davis A., Polymeropoulos M., Holik J., Hopkins J., Hoff M., et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc. Natl Acad. Sci. USA. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helbig I., Mefford H.C., Sharp A.J., Guipponi M., Fichera M., Franke A., Muhle H., de Kovel C., Baker C., von Spiczak S., et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat. Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stefansson H., Rujescu D., Cichon S., Pietilainen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orr-Urtreger A., Goldner F.M., Saeki M., Lorenzo I., Goldberg L., De Biasi M., Dani J.A., Patrick J.W., Beaudet A.L. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J. Neurosci. 1997;17:9165–9171. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levin E.D., Petro A., Rezvani A.H., Pollard N., Christopher N.C., Strauss M., Avery J., Nicholson J., Rose J.E. Nicotinic alpha7- or beta2-containing receptor knockout: effects on radial-arm maze learning and long-term nicotine consumption in mice. Behav. Brain Res. 2009;196:207–213. doi: 10.1016/j.bbr.2008.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernandes C., Hoyle E., Dempster E., Schalkwyk L.C., Collier D.A. Performance deficit of alpha7 nicotinic receptor knockout mice in a delayed matching-to-place task suggests a mild impairment of working/episodic-like memory. Genes Brain Behav. 2006;5:433–440. doi: 10.1111/j.1601-183X.2005.00176.x. [DOI] [PubMed] [Google Scholar]
- 26.Tsankova N., Renthal W., Kumar A., Nestler E.J. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 27.Samaco R.C., Hogart A., LaSalle J.M. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lasalle J.M., Yasui D.H. Evolving role of MeCP2 in Rett syndrome and autism. Epigenomics. 2009;1:119–130. doi: 10.2217/epi.09.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neul J.L., Zoghbi H.Y. Rett syndrome: a prototypical neurodevelopmental disorder. Neuroscientist. 2004;10:118–128. doi: 10.1177/1073858403260995. [DOI] [PubMed] [Google Scholar]
- 30.Fatemi M., Wade P.A. MBD family proteins: reading the epigenetic code. J. Cell. Sci. 2006;119:3033–3037. doi: 10.1242/jcs.03099. [DOI] [PubMed] [Google Scholar]
- 31.Lewis J.D., Meehan R.R., Henzel W.J., Maurer-Fogy I., Jeppesen P., Klein F., Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 32.Jones P.L., Veenstra G.J., Wade P.A., Vermaak D., Kass S.U., Landsberger N., Strouboulis J., Wolffe A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 33.Nan X., Ng H.H., Johnson C.A., Laherty C.D., Turner B.M., Eisenman R.N., Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 34.Horike S., Cai S., Miyano M., Cheng J.F., Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 35.Nikitina T., Ghosh R.P., Horowitz-Scherer R.A., Hansen J.C., Grigoryev S.A., Woodcock C.L. MeCP2-chromatin interactions include the formation of chromatosome-like structures and are altered in mutations causing Rett syndrome. J. Biol. Chem. 2007;282:28237–28245. doi: 10.1074/jbc.M704304200. [DOI] [PubMed] [Google Scholar]
- 36.Nikitina T., Shi X., Ghosh R.P., Horowitz-Scherer R.A., Hansen J.C., Woodcock C.L. Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol. Cell. Biol. 2007;27:864–877. doi: 10.1128/MCB.01593-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghosh R.P., Nikitina T., Horowitz-Scherer R.A., Gierasch L.M., Uversky V.N., Hite K., Hansen J.C., Woodcock C.L. Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry. 2010;49:4395–4410. doi: 10.1021/bi9019753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hogart A., Nagarajan R.P., Patzel K.A., Yasui D.H., Lasalle J.M. 15q11–13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum. Mol. Genet. 2007;16:691–703. doi: 10.1093/hmg/ddm014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simonis M., Klous P., Splinter E., Moshkin Y., Willemsen R., de Wit E., van Steensel B., de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat. Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 40.Simonis M., Kooren J., de Laat W. An evaluation of 3C-based methods to capture DNA interactions. Nat. Methods. 2007;4:895–901. doi: 10.1038/nmeth1114. [DOI] [PubMed] [Google Scholar]
- 41.Jung B.P., Jugloff D.G., Zhang G., Logan R., Brown S., Eubanks J.H. The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J. Neurobiol. 2003;55:86–96. doi: 10.1002/neu.10201. [DOI] [PubMed] [Google Scholar]
- 42.Pahlman S., Mamaeva S., Meyerson G., Mattsson M.E., Bjelfman C., Ortoft E., Hammerling U. Human neuroblastoma cells in culture: a model for neuronal cell differentiation and function. Acta Physiol. Scand. Suppl. 1990;592:25–37. [PubMed] [Google Scholar]
- 43.Murrell A., Heeson S., Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat. Genet. 2004;36:889–893. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- 44.Ohlsson R., Gondor A. The 4C technique: the ‘Rosetta stone’ for genome biology in 3D? Curr. Opin. Cell Biol. 2007;19:321–325. doi: 10.1016/j.ceb.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 45.Bieda M., Xu X., Singer M.A., Green R., Farnham P.J. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 2006;16:595–605. doi: 10.1101/gr.4887606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sahoo T., del Gaudio D., German J.R., Shinawi M., Peters S.U., Person R.E., Garnica A., Cheung S.W., Beaudet A.L. Prader–Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 2008;40:719–721. doi: 10.1038/ng.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abrahams B.S., Geschwind D.H. Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mexal S., Berger R., Logel J., Ross R.G., Freedman R., Leonard S. Differential regulation of alpha7 nicotinic receptor gene (CHRNA7) expression in schizophrenic smokers. J. Mol. Neurosci. 2010;40:185–195. doi: 10.1007/s12031-009-9233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guan Z.Z., Zhang X., Blennow K., Nordberg A. Decreased protein level of nicotinic receptor alpha7 subunit in the frontal cortex from schizophrenic brain. Neuroreport. 1999;10:1779–1782. doi: 10.1097/00001756-199906030-00028. [DOI] [PubMed] [Google Scholar]
- 50.Nagarajan R.P., Hogart A.R., Gwye Y., Martin M.R., LaSalle J.M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:e1–e11. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicholls R.D., Knepper J.L. Genome organization, function, and imprinting in Prader–Willi and Angelman syndromes. Annu. Rev. Genomics Hum. Genet. 2001;2:153–175. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- 52.Girirajan S., Eichler E.E. Phenotypic variability and genetic susceptibility to genomic disorders. Hum. Mol. Genet. 2010;19:176–187. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bond A.M., Vangompel M.J., Sametsky E.A., Clark M.F., Savage J.C., Disterhoft J.F., Kohtz J.D. Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat. Neurosci. 2009;12:1020–1027. doi: 10.1038/nn.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stefan M., Portis T., Longnecker R., Nicholls R.D. A nonimprinted Prader–Willi syndrome (PWS)-region gene regulates a different chromosomal domain in trans but the imprinted pws loci do not alter genome-wide mRNA levels. Genomics. 2005;85:630–640. doi: 10.1016/j.ygeno.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 55.Skene P.J., Illingworth R.S., Webb S., Kerr A.R., James K.D., Turner D.J., Andrews R., Bird A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klin A., Lang J., Cicchetti D.V., Volkmar F.R. Brief report: interrater reliability of clinical diagnosis and DSM-IV criteria for autistic disorder: results of the DSM-IV autism field trial. J. Autism Dev. Disord. 2000;30:163–167. doi: 10.1023/a:1005415823867. [DOI] [PubMed] [Google Scholar]
- 57.Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 58.Silk T.J., Rinehart N., Bradshaw J.L., Tonge B., Egan G., O'Boyle M.W., Cunnington R. Visuospatial processing and the function of prefrontal-parietal networks in autism spectrum disorders: a functional MRI study. Am. J. Psychiatry. 2006;163:1440–1443. doi: 10.1176/ajp.2006.163.8.1440. [DOI] [PubMed] [Google Scholar]
- 59.Gemelli T., Berton O., Nelson E.D., Perrotti L.I., Jaenisch R., Monteggia L.M. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol. Psychiatry. 2006;59:468–476. doi: 10.1016/j.biopsych.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 60.Adachi M., Keefer E.W., Jones F.S. A segment of the Mecp2 promoter is sufficient to drive expression in neurons. Hum. Mol. Genet. 2005;14:3709–3722. doi: 10.1093/hmg/ddi402. [DOI] [PubMed] [Google Scholar]
- 61.Chahrour M., Jung S.Y., Shaw C., Zhou X., Wong S.T., Qin J., Zoghbi H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Agulhon C., Abitbol M., Bertrand D., Malafosse A. Localization of mRNA for CHRNA7 in human fetal brain. Neuroreport. 1999;10:2223–2227. [PubMed] [Google Scholar]
- 63.Samaco R.C., Nagarajan R.P., Braunschweig D., LaSalle J.M. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum. Mol. Genet. 2004;13:629–639. doi: 10.1093/hmg/ddh063. [DOI] [PubMed] [Google Scholar]
- 64.Chang Q., Khare G., Dani V., Nelson S., Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 65.Jiang Y., Langley B., Lubin F.D., Renthal W., Wood M.A., Yasui D.H., Kumar A., Nestler E.J., Akbarian S., Beckel-Mitchener A.C. Epigenetics in the nervous system. J. Neurosci. 2008;28:11753–11759. doi: 10.1523/JNEUROSCI.3797-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tregellas J.R., Tanabe J., Rojas D.C., Shatti S., Olincy A., Johnson L., Martin L.F., Soti F., Kem W.R., Leonard S., et al. Effects of an alpha 7-nicotinic agonist on default network activity in schizophrenia. Biol. Psychiatry. 2010;69:7–11. doi: 10.1016/j.biopsych.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.