

Abstract
A channelopathy mutation in segment IIS6 of CaV1.4 (I745T) has been shown to cause severe visual impairment by shifting the activation and inactivation curves to more hyperpolarized voltages and slowing activation and inactivation kinetics. A similar gating phenotype is caused by the corresponding mutation, I781T, in CaV1.2 (midpoint of activation curve (V0.5) shifted to −37.7 ± 1.2 mV). We show here that wild-type gating can partially be restored by a helix stabilizing rescue mutation N785A. V0.5 of I781T/N785A (V0.5 = −21.5 ± 0.6 mV) was shifted back towards wild-type (V0.5 = −9.9 ± 1.1 mV). Homology models developed in our group (see accompanying article for details) were used to perform Molecular Dynamics-simulations (MD-simulations) on wild-type and mutant channels. Systematic changes in segment IIIS6 (M1187-F1194) and in helix IIS6 (N785-L786) were studied. The simulated structural changes in S6 segments of I781T/N785A were less pronounced than in I781T. A delicate balance between helix flexibility and stability enabling the formation of hydrophobic seals at the inner channel mouth appears to be important for wild-type CaV1.2 gating. Our study illustrates that effects of mutations in the lower part of IIS6 may not be localized to the residue or even segment being mutated, but may affect conformations of interacting segments.
Keywords: calcium channel, mutagenesis, channelopathy, MD simulation, CaV1.2, channel activation
Introduction
Mutations inducing channelopathies cause changes in electrical excitability and calcium entry, thereby increasing susceptibility to migraine, ataxia, periodic paralysis, cardiac arrhythmias, hypoglycemia and incomplete congenital stationary night blindness (reviewed in refs. 1-4). Point mutations in CaV1 and CaV2 have been shown for example to accelerate or decelerate inactivation kinetics, to shift the threshold of channel activation, to decelerate activation and to change recovery from inactivation.5-8
We have recently analyzed the consequences of a point mutation (I745T) in segment IIS6 of the CaV1.4 α1-subunit that causes a retinal disorder by shifting the voltage-dependence of CaV1.4 channel activation and inactivation by about −30 mV with slowed activation, deactivation and inactivation kinetics.5,8 Affected males and females of a large New Zealand family suffer from severe visual impairment.5
Latter studies revealed that substitutions of I781, the CaV1.2 residue corresponding to I745 in CaV1.4, by residues of different hydrophobicity, size and polarity shifted activation and steady state inactivation in the hyperpolarizing direction. The changes in channel gating indicated that amino acid substitutions for I781 and adjacent residues destabilize the closed conformation and/or stabilize the open conformation of CaV1.2.9
In the present study we analyzed possible consequences of I781T and other amino acid substitutions in segment IIS6, using Molecular Dynamics simulations, which suggest systematic changes in segment IIS6 and distortions in the neighboring segment IIIS6. The “allosterically” affected region in segment IIIS6 (starting at residue M1187-F1194) has previously been shown to play a crucial role in Ca2+ channel inactivation.10. A rescue mutation (I781T/N785A) substantially reduced the gating changes caused by I781T in both segments and, remarkably, also diminished the helix distortions in both S6 segments in MD simulations.
Results
Interactions of I781 with the environment
Based on our homology model of the open conformation (see accompanying article), I781 is located at the lower third of the IIS6 segment and faces helix IIS5. The L4-5 helix of domain II is located in close proximity. I781 is surrounded by hydrophobic amino acids, and interacts with highly conserved amino acids L684 and L687 of helix IIS5. The backbone nitrogen atom of I781 forms a hydrogen bond to the backbone oxygen of F778 of the same helix, and the backbone oxygen atom of I781 bonds to the backbone nitrogen atom of residue N785, which is located one helical turn below. Additionally, the side chain of N785 forms hydrogen bonds with two conserved serines from helix IIS5 (Fig. 1).
Figure 1.
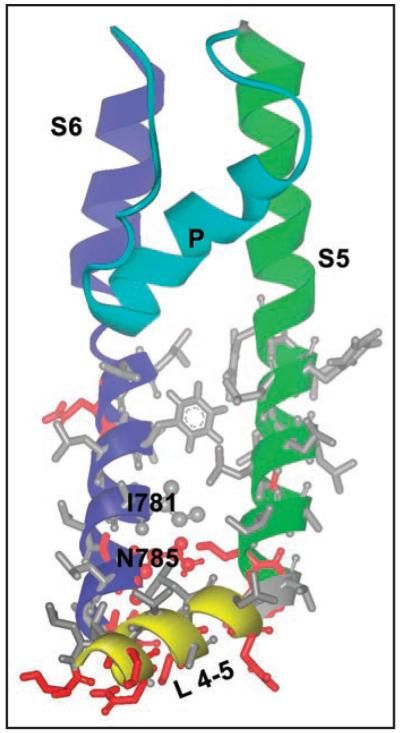
Structural details and location of I781 hotspot in the open Cav1.2 channel. Hydrophobic amino acids are shown as gray sticks, polar or charged amino acids are coloured red. I781 and N785 are in scaled ball and stick representation. From this figure it can be clearly seen that I781 is located in a hydrophobic area, which is followed by polar amino acids one turn below.
MD simulations of I781T and I781P suggest helix distortions in segments IIS6 and IIIS6
Replacement of I781 by various amino acids severely influences channel gating. Polar or charged amino acid replacements induce the strongest changes in channel gating (with the exception of proline), while hydrophobic residues such as alanine or leucine have less severe consequences on channel activation and inactivation.9
The static structure models only partially disclosed the molecular mechanism of the gating distortions. Therefore, the dynamics of calcium channel mutants were analyzed, using molecular dynamics simulations. The stability of the mutant channels, which can be measured by calculating the RMSD (root mean square displacement), was only slightly increased compared to wild-type channels (~0.5 Å, data not shown).
Detailed analysis of the trajectories revealed distortions in the pore-forming helices IIS6 and IIIS6 for the threonine and proline mutant compared to wild-type, where no such distortions could be detected. Surprisingly, effects associated with replacement of the medium size hydrophobic isoleucine with a small polar threonine, extend beyond domain II. We consistently observed an unwinding of helix IIIS6 at the helix crossing interface. To analyze this phenomenon in more detail, backbone angles of the corresponding region in helix IIS6 and IIIS6 were studied (Fig. 2A, B and E). Amino acids in the middle of helix IIIS6 (M1187 to F1191) lost their helical conformation within the first 300 ps (Fig. 3C and D). During the rest of the 10 ns simulation runs distortions in this region remained, while the rest of the structure stayed intact. Interestingly, the overall helical structure of IIS6, where mutations have been introduced, remained intact during the whole simulation. Detailed analysis of the backbone angles of IIS6 segment revealed unchanged behaviour of the backbone angles (φ/ψ) for residue 781, irrespective of the amino acid at this position (Fig. 2A). However, one helical turn below, the backbone angles for mutant channels change. The backbone angles of residue 785 vary only slightly (data not shown), but considerable changes are observed in the next residue, the highly conserved L786 (Fig. 2B). Interestingly, in the wild-type channel this amino acid is near (within 3-5 Å) the distorted amino acids in helix IIIS6 (Fig. 3B). Simultaneously, the interacting amino acid from IIIS6, I1190 looses its hydrophobic contact with L786 from IIS6 (Fig. 3D). As a consequence large fluctuations of the adjacent backbone dihedral angle occurred (see Fig. 2E).
Figure 2.

Backbone angles of wild-type and mutant channels. (A) Backbone angle (ψ) of position 781 (domain II). Interestingly, there are no differences observable between wild-type (black line) and threonine mutant (gray, dotted line). This is also true for other mutant channels (data not shown). (B) Backbone angle (ψ) for amino acid L786 (domain II), which is located downwards of I781. Interestingly, this amino acid is relatively stable in wild-type but fluctuates drastically (up to 150°) in the I781T mutant channel. (C) Behaviour of L786 in the rescuing double mutant I781T/N785A channel. The backbone of L786 is more flexible in the I781T/N785A double mutant than wild-type but less flexible than in the I781T mutant and stabilizes gradually (B, above). (D) Backbone angle (ψ) of the non rescuing double mutant I781T/N785G channel. The backbone angle is more flexible than wild-type and adopts a conformation, usually seen in beta sheets. (E) Backbone angle of I1190 in IIIS6. This amino acid is involved in hydrophobic interactions to the neighboring IIS6 residue L786 in the open conformation of the wild-type channel (see also Fig. 4B). Because of the higher flexibility in this area introduced by single point mutations in position 781, interactions to IIS6 are lost (Fig. 4D) and the backbone conformation of I1190 fluctuates. (F) In the double mutant the interactions between L786 (IIS6) and I1190 (IIIS6) remain intact and the backbone of helix III remains stable. (G) Backbone angle of I1190 in the non rescuing double mutant I781T/N785G channel. In agreement with the higher flexibility at position L786 (domain II), I1190 looses hydrophobic contacts to L786 and shows considerable fluctuations.
Figure 3.

Structural consequences of mutations on pore helix stability revealed by MD simulations. (A) Representative snapshot of the wild-type channel pore at the end of six ns simulation in ribbon presentation. No distortions in IIIS6 segment appear during our repeated simulations. (B) Details of amino acids involved in structural rearrangements upon mutation in position 781 to threonine. Polar amino acids are coloured red, while hydrophobic residues are gray. Position 781 is facing the opposite side and cannot directly interact with helix IIIS6. N785 is closer to the neighboring S6 segment and is connected to L786, which interacts with the highly conserved I1190 from helix IIIS6. The distance between L786 and I1190 is less than 4 Å. (C) Ribbon presentation of pore forming S6 helices for the threonine mutant channel. Note that helix IIIS6 is distorted in the middle, while the rest of the helix remains helical throughout the simulation. Snapshot after 300 ps of unconstrained MD. (D) Details of amino acid interactions in the pore forming helices of segment II of the wild-type channel. Hydrophobic interactions between L786 (IIS6) and I1190 (IIIS6) are lost and I1190 is facing towards the opposite side, as a consequence of the I781T mutation.
Our simulations suggest changes in hydrophobic-hydrophobic interactions between neighboring S6 segments as a consequence of distortions in segment IIIS6. In the wild-type channel hydrophobic interactions between helix IIIS6 and IVS6 are formed between residues V1192, I1196 and F1199 from IIIS6 and F1495, L1496, N1499 and L1500 from helix IVS6 (Fig. 5A). In the threonine mutant hydrophobic interactions between these two helices are formed by I1190, F1191, F1194, V1195 and F1199 from segment IIIS6 and residues M1491, L1492, F1495 and L1496 from segment IVS6 (Fig. 5B).
Figure 5.

Changes in hydrophobic-hydrophobic helix interactions. (A) Ribbon presentation of pore forming S6 segments of the wild-type channel after 10 ns (average structure over last 500 ps). Interacting residues between segments IIIS6 and IVS6 are shown in stick and surface representation. (B) Ribbon presentation of I781T mutant channel after 10 ns simulation (average structure over last 500 ps). Interacting residues between helix IIIS6 and IVS6 are shown in stick and surface representation. As a consequence of the increased flexibility between residues M1187-F1194, and the thereby induced loss of helical conformation, residues following F1194, which remain in α-helical conformation, are rotated. This leads to different helix-helix contacts, as described in the Results section.
Rescue mutation N785A
If a threonine or proline (data not shown) in position I781 induces structural changes in IIS6 one helical turn below, as suggested by MD simulations (see changes in backbone angles of residue L786 in Fig. 2B), we reasoned that introducing a helix stabilizing alanine might rescue the gating of wild-type channels. Since we did not want to abolish hydrophobic interactions between L786 (IIS6) and I1190 (IIIS6), we choose to mutate the neighboring residue 785 (I781 + 4, to force α-helical configuration).The double mutant (I781T/N785A) was created and tsA-201 cells were co-transfected with mutant α1 and β1a and α2-δ1 subunits (see Methods for details). Remarkably, the activation curve of the corresponding double mutant (V0.5 = −21.5 ± 0.6 mV, n = 6) was shifted back towards the position of wild-type CaV1.2 (V0.5 = −9.9 ± 1.1 mV) compared to I781T (V0.5 = −37.7 ± 1.2 mV, Fig. 6A). The kinetics of activation and deactivation of the double mutant were faster than in I781T, at negative voltages, slightly slower than in wild-type (Fig. 6C).9 Thus, our functional analysis of double mutant I781T/N785A suggests a partial restoration of wild-type kinetics. Substitution of N785 by glycine, which introduces greater flexibility in the α-helix, did not rescue wild-type gating. Instead, the position of the activation curve remained shifted (I781T/N785G V0.5 = 41.2 ± 0.7 mV), and activation and deactivation kinetics at hyperpolarized voltages remained slow, as observed for mutants I781T/P (Fig. 6D and E).
Figure 6.

Functional analysis of CaV1.2 mutants in positions I781 and N785. Averaged voltage dependences of activation (A) and steady state inactivation (B) of wild-type, I781T,9 double mutants I781T/N785A, I781T/N785G and single mutant N785G. N785A did not conduct current (see Table 1 and Fig. 6). (C) Illustration of activation and deactivation time courses of IBa through I781T/N785A. Activation and deactivation kinetics of I781T/N785A were slower than in wild-type but significantly faster than in I781T (compare with 26). Time courses were fitted by mono-exponential function (solid lines) with time constants of 4.6 ms (−30 mV), 3.1 ms (−20 mV) and 2.5 ms (−10 mV) for activation (C, left) and with time constants of 1.5 ms (−70 mV), 2.6 ms (−60 mV) and 4.1 ms (−50 mV) for deactivation (C, right). Mean time constants of channel activation (D) and deactivation (E) are plotted versus the test potential for wild-type, I781T (dotted lines, data from 26), N785G (filled squares), I781T/N785G (filled circles) and I781T/N785A (open circles) mutant CaV1.2 channels.
In order to elucidate the rescue mechanism we analyzed the gating properties of the corresponding single point mutants. N785G slightly shifted the voltage dependence of channel activation in the depolarizing direction (Fig. 6A), while N785A was not functional (Table 1).
Table 1.
Activation and inactivation gating characteristics
| Mutation | Half-activation, mV |
Slope, mV | Half-inactivation mV |
r300, % |
|---|---|---|---|---|
| Wild-type* | −9.9 ± 1.1 | 6.3 ± 0.7 | −38.7 ± 1.0 | 65.4 ± 4 |
| I781T* | −37.7 ± 1.2 | 7.2 ± 1.0 | −57.8 ± 0.7 | 47 ± 3 |
| N785A | No currents | |||
| N785G | −1.2 ± 0.5 | 5.6 ± 0.6 | −34.0 ± 0.7 | 66 ± 7 |
| N785L | −12.3 ± 1.1 | 6.7 ± 0.7 | −42.2 ± 0.8 | 58 ± 8 |
| I781T/ N785A |
−21.5 ± 0.6 | 5.8 ± 0.7 | −44.1 ± 1.0 | 48 ± 8 |
| I781T/ N785G |
−41.2 ± 0.7 | 4.2 ± 0.6 | −65.2 ± 0.7 | 40 ± 7 |
| I781T/ N785L |
−34.4 ± 0.9 | 7.6 ± 0.9 | −53.8 ± 1.2 | 45 ± 7 |
Data from ref. 9.
To elucidate the possible structural implications, we performed MD simulations. The double mutants I781T/N785A and I781/N785G were built using the program WHAT IF.14 The mutant channels were energy minimized using 10000 steps of steepest descent with the minimization tool in Gromacs,15 as described above. As shown in Figure 5A, the double mutant I781T/N785A prevents unwinding of residues M1187 to F1191 in segment IIIS6. Detailed analysis of backbone angles, in agreement with the above described threonine mutant, revealed unchanged behaviour at position 781 (data not shown). The C-terminal backbone angle at position L786 is still more flexible than wild-type, but does not lead to conformational changes in IIIS6 (Figs. 2C and 4). Consequently, the interacting amino acid I1190 does not show any different behaviour compared to wild-type (Fig. 2F). In agreement with experiments, the double mutant I781T/N785G did not rescue wild-type gating, and MD simulations reveal similar behaviour of I781T/N785G and I781T single mutant channel, with analogous distortions of segments IIS6 and IIIS6, as described above (Figs. 2D and G and 4B).
Figure 4.

Pore helices of I781T/N785A and I781T/N785G double mutants. (A) Ribbon presentation of the double mutant I781T/N785A. Only pore forming S6 segment is shown. T781 and A785 are coloured red and gray, according to their hydrophobicity/polarity. During five independent 6ns simulations, helix IIIS6 always remained intact, confirming the rescuing effect of this double mutant. (B) Ribbon presentation of the double mutant I781T/N785G. A 500 ps average structure at the end of 10 ns simulation of S6 segments are shown. In agreement with experiment, this mutant channel shows similar behaviour as I781T single mutant channel, with comparable distortions in segment IIIS6, which have been observed in repeated simulations (five runs).
These data may suggest that higher helix stability induced by the additional mutation N785A can shift the activation and inactivation curves back towards the positions of wild-type CaV1.2 (Fig. 6A and B), whereas higher flexibility facilitated by the additional N785G prevents efficient channel closure. Interestingly, mutating the hydrophilic N785 to the hydrophobic leucine did not significantly rescue wild-type gating (V0.5(I781T/N785L) = −34.4 ± 0.9 mV, p > 0.05, not significantly different from I781T, Table 1). The activation curve of the single mutant N785L (V0.5 = −12.3 ± 1.1 mV, Table 1, not significantly different from wild-type, p > 0.05) was also not shifted compared to wild-type, suggesting that hydrophobic interactions in position 785 play no major role.
Evidence for membrane incorporation of none conducting mutant channel N785A
Mutant N785A did not conduct Ba2+ currents. We have therefore examined the subcellular distribution of the green fluorescent protein-tagged (GFP-tagged) mutant N785A by confocal microscopy in order to analyze plasma membrane targeting. N781A GFP-tagged CaV1.2 α1-subunits were co-expressed together with β1a and α2-δ1 subunits in tsA-201 cells. The plasma membrane was visualized by staining with FM4-64 (Fig. 7, see also ref. 9).
Figure 7.
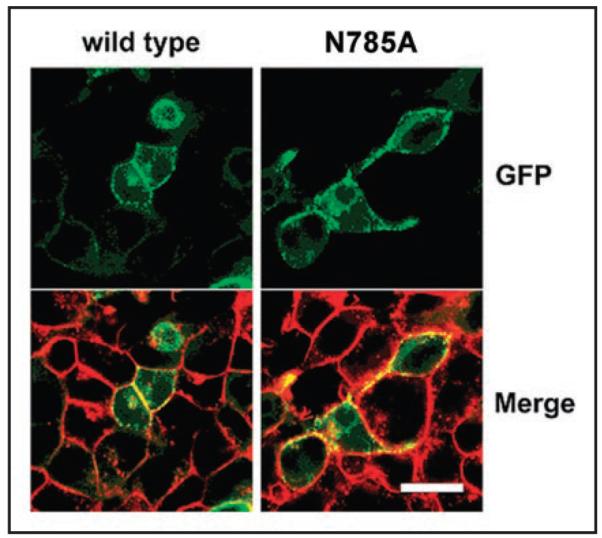
Evidence for membrane localization of mutant N785A. Transiently transfected ts-A201 cells expressing wild-type and two representative non-functional CaV1.2 channel N785A are shown. The wild-type and the mutant GFP-tagged (green) CaV1.2 α1-subunit were co-expressed with β1a and α2-δ1 subunits and the cells stained with FM4-64 (red), a plasma membrane marker. In the merged images, GFP-tagged α1-subunits located in the plasma membrane are seen as punctuated yellow clusters. Scale bar, 20 μm.
As previously shown for wild-type CaV1.2 α1-subunits, we observed significant localization of N785A at the plasma membrane, suggesting that the lack of current observed for these mutants can not be attributed to a failure of the mutant CaV1.2 α1-subunits to reach the plasma membrane.
Discussion
Mutational effects on closed and open channel conformations
Studies on KV have shown that the closed state in voltage gated ion channels may be stabilized by hydrophobic interactions between amino acids in the lower third of the “bundle crossing” region of S6 segments.17,18 In such a scenario fast closure of CaV1.2 is likely to require intact S6 helical conformations enabling efficient hydrophobic interactions in the lower third of the channel pore. Helix deformations caused by I781T/P in IIS6, with possible allosteric effects on IIIS6, lower the probability of a tight fit closed conformation during repolarization, which could explain the deceleration of tail currents (Fig. 6). Furthermore, hydrophobic interactions in the open channel conformation of mutant channels I781T, I781P and I781T/N785G are changed, as a consequence of the structural rearrangements, caused by the increased flexibility of residues M1187-F1194 in segment IIIS6. Residues following F1194 remain in α-helical conformation, but, as a consequence of the preceding structural disturbances, the side-chains are rotated equivalent to a single gap. As a result helix-helix interactions between segments IIIS6 and IVS6 are changed, as can be seen in Figure 5A and B. Our simulations suggest that hydrophobic interactions between these two segments are strengthened, while helix-helix interactions between segment IIS6 and IIIS6 remain unchanged. Obviously, replacements in position I781, with the more flexible19 but less hydrophobic threonine, or other less hydrophobic residues, leads to pronounced changes in helix-helix interactions, which might stabilize the open conformation as suggested previously.9
MD simulations suggest changes in segments IIS6 and IIIS6 by I781T/P
Our models, together with MD simulations, suggest complex conformational changes caused by mutation I781T (Figs. 2A, B and E and 3C and D). Similar changes were observed for I781P (data not shown) and I781T/N785G (Figs. 2D and G and 4B). Proline may, however, induce additional effects caused by the lack of the backbone hydrogen bond and steric constrains, leading to bending and destabilization of helices.20-23
The functional properties of the partial rescue mutant I781T/N785A (Fig. 6) can be explained by a helix stabilizing effect of alanine (Figs. 2C and F and 4A). I781T/N785G, or I781T/N785L, did not rescue wild-type current kinetics (Fig. 6D and E and Table 1). It is therefore unlikely that the partial rescue of I781T/N785A is based on additional hydrophobic interactions. The different effects of the single mutants (N785G shifted the activation curve to the right, N785A did not conduct barium currents and N785L induced no significant shift) suggest that other properties than flexibility and hydrophobicity in this position play a role. The elucidation of the rescue mechanism warrants further research. Molecular dynamics simulations revealed additional information, which could never be obtained from static models. The inserted threonine seems to affect the conformation of helix IIIS6 via disruption of hydrophobic helix-helix interactions between the two domains in the open channel conformation (L786 interacting with I1190, see Fig. 3B and D). As a consequence the stability of helix IIIS6 is decreased, which leads to partial unwinding at residues M1187-F1194 (Figs. 2B and E and 3C and D). Interestingly, key inactivation determinants have been identified in the lower part of segment IIIS6 (reviewed in ref. 10). We speculate that structural changes in the adjacent segment IIIS6 may contribute to simultaneous changes in channel activation and inactivation caused by point mutations in the lower part of IIS6.9 Hence, key residues affecting CaV1.2 inactivation (IFV motif10) have previously been identified in the allosterically affected region of IIIS6.
Summarizing, our MD simulations reveal significant effects of mutations in position 781 on sequentially distant residues, especially on the adjacent IIIS6 helix. The assumption that mutational effects are due solely to localized changes does not appear to be valid in this case. More complex conformational changes, involving neighboring segments, should also be considered. Our results suggest that molecular modeling combined with molecular dynamics simulations may prove effective in identifying and analyzing these more complicated situations.
Materials and Methods
Experimental procedures
Mutagenesis
The CaV1.2 α1-subunit coding sequence (GenBank™ X15539) in-frame 3′ to the coding region of a modified green fluorescent protein (GFP) was kindly donated by Dr. M. Grabner.11 For electrophysiological studies we used the plasmid lacking the GFP tag. Mutations in segment IIS6 of the CaV1.2 α1-subunit were introduced by the “gene SOEing” technique.12
The mutated fragments were cloned into a BamHI-AflII-cassette (nt 1265 and 2689, numbering according to the CaV1.2 α1-subunitcoding sequence). All constructs were checked by restriction site mapping and sequencing.
Cell culture and transient transfection
Human embryonic kidney tsA-201 cells were grown at 5% CO2 and 37°C to 80% confluence in Dulbecco’s modified Eagle’s/F-12 medium supplemented with 10% (v/v) foetal-calf serum and 100 units/ml penicillin/streptomycin. Cells were split via trypsin + EDTA and plated on 35 mm Petri dishes (Falcon) at 30–50% confluence ~16 h before transfection. Subsequently tsA-201 cells were co-transfected with cDNAs encoding wild-type or mutant CaV1.2 α1-subunits with auxiliary β1a as well as α2-δ1 subunits. The transfection of tsA-201 cells was performed using the FUGENE6 Transfection Reagent (Roche) following standard protocols.
Ionic current recordings and data acquisition
Barium currents (IBa) through voltage-gated Ca2+ channels were recorded at 22–25°C using the patch-clamp technique13 by means of an Axopatch 200A patch clamp amplifier (Axon Instruments, Foster City) 36–48 h after transfection. The extracellular bath solution contained (in mM): BaCl2 5, MgCl2 1, HEPES 10, choline-Cl 140, titrated to pH 7.4 with methanesulfonic acid. Patch pipettes with resistances of 1 to 4 MΩ were made from borosilicate glass (Clark Electromedical Instruments, UK) and filled with pipette solution containing (in mM): CsCl 145, MgCl2 3, HEPES 10, EGTA 10, titrated to pH 7.25 with CsOH. All data were digitized using a DIGIDATA 1200 interface (Axon Instruments, Foster City), smoothed by means of a four-pole Bessel filter and stored on computer hard disc. 100 ms current traces were sampled at 10 kHz and filtered at 5 kHz; for the steady state inactivation protocol, currents were sampled at 1 kHz and filtered at 0.5 kHz; and tail currents were sampled at 50 kHz and filtered at 10 kHz. Leak currents were subtracted digitally using average values of scaled leakage currents elicited by a 10 mV hyperpolarizing pulse or electronically by means of an Axopatch 200 amplifier (Axon Instruments, Foster City). Series resistance and offset voltage were routinely compensated. The pClamp software package (Version 7.0 Axon Instruments, Inc.,) was used for data acquisition and preliminary analysis. OriginLab Corporation (www.originlab.com) was employed for analysis and curve fitting.
The voltage-dependence of activation was determined from current-voltage (I–V) curves that were fitted according to the following modified Boltzmann term:
where Vrev, extrapolated reversal potential; V, membrane potential; I, peak current; Gmax, maximum membrane conductance; V0.5, act, voltage for half-maximal activation; and kact, slope factor. The time course of current activation was fitted to a mono-exponential function:
where I(t), current at time t; A, the amplitude coefficient; τ, time constant; C a constant. The voltage-dependence of IBa inactivation (inactivation curve) was measured using a multi-step protocol to account for run-down. The pulse sequence was applied every 40 seconds from a holding potential of −100 mV. Inactivation curves were drawn according to a Boltzmann equation:
where V, membrane potential; V0.5, inact, midpoint voltage; k, slope factor and Iss, fraction of non-inactivating current. Data are given as Mean ± S.E. Statistical significance was assessed with the Student’s unpaired t-test.
Confocal imaging
Membrane localization of the non conducting channel construct N785A was analyzed by means of confocal microscopy. Confocal images were obtained ~30 h after transfection and acquired with a Zeiss LSM-510 confocal laser scanning microscope, using a x63 (1.4 NA) oil immersion objective. Data illustrated are representative for 15–20 tsA-201 cells from three independent experiments. The plasma membrane was stained with 1 μM FM4-64 (amphiphilic styryl dye, Molecular Probes). Images were acquired using an argon laser (excitation, 488 nm; emission BP505-530 nm emission filter) for the GFP-tagged CaV1.2 α1-subunits and a He-Ne laser (excitation, 543 nm; emission filter, LP650 nm) for FM4-64.
Molecular dynamics simulations of the open CaV1.2 mutants
To gain information about structural effects of mutations at position 781, we generated mutants I781T and I781P9 using the WHAT IF software.14 The mutant channels were energy minimized using 10000 steps of steepest descent with the minimization tool in Gromacs.15 Mutant channels were embedded in a lipid bilayer of POPE, and simulated using the same parameters as for the wild-type Cav1.2, as described in the accompanying paper. Default protonation states, based on calculations with the software Propka 1.0.1,16 which showed default values for all residues in the region of the 781 hotspot, were used. Channels were simulated five times with different random starting velocities for 10 ns for the first run of each channel, followed by four repeating runs for 6 ns each, resulting in a total simulation time of 34 ns for each channel.
Acknowledgements
We thank Boris Zhorov for suggesting mutation N785A and Thomas Peterbauer for help with confocal microscopy, and Marion Maw for initiating this work. This project was supported by FWF grants 15914 and P19614-B11 (to Steffen Hering) and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (to H. Robert Guy).
References
- 1.Cannon SC. Pathomechanisms in channelopathies of skeletal muscle and brain. Annu Rev Neurosci. 2006;29:387–415. doi: 10.1146/annurev.neuro.29.051605.112815. [DOI] [PubMed] [Google Scholar]
- 2.Striessnig J, Hoda JC, Koschak A, Zaghetto F, Müllner C, Sinnegger Brauns MJ, Wild C, Watschinger K, Trockenbacher A, Pelster G. L-type Ca2+ channels in Ca2+ channelopathies. Biochem Biophys Res Comm. 2004;322:1341–6. doi: 10.1016/j.bbrc.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 3.Pietrobon D. Calcium channels and channelopathies of the central nervous system. Mol Neurobiol. 2002;25:31–50. doi: 10.1385/MN:25:1:031. [DOI] [PubMed] [Google Scholar]
- 4.Hans M, Luvisetto S, Williams ME, Spagnolo M, Urrutia A, Tottene A, Brust PF, Johnson EC, Harpold MM, Stauderman KA, Pietrobon D. Functional consequences of mutations in the human alpha1A calcium channel subunit linked to familial hemiplegic migraine. J Neurosci. 1999;19:1610–9. doi: 10.1523/JNEUROSCI.19-05-01610.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hemara Wahanui A, Berjukow S, Hope CI, Dearden PK, Wu SB, Wilson Wheeler J, Sharp DM, Lundon Treweek P, Clover GM, Hoda JC, Striessnig J, Marksteiner R, Hering S, Maw MA. A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation. Proc Natl Acad Sci USA. 2005;102:7553–8. doi: 10.1073/pnas.0501907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kraus RL, Sinnegger MJ, Glossmann H, Hering S, Striessnig J. Familial hemiplegic migraine mutations change alpha1A Ca2+ channel kinetics. J Biol Chem. 1998;273:5586–90. doi: 10.1074/jbc.273.10.5586. [DOI] [PubMed] [Google Scholar]
- 7.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager Flusberg H, Priori SG, Sanguinetti MC, Keating MT. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Hope CI, Sharp DM, Hemara Wahanui A, Sissingh JI, Lundon P, Mitchell EA, Maw MA, Clover GM. Clinical manifestations of a unique X-linked retinal disorder in a large New Zealand family with a novel mutation in CACNA1F, the gene responsible for CSNB2. Clin Exp Ophthalmol. 2005;33:129–36. doi: 10.1111/j.1442-9071.2005.00987.x. [DOI] [PubMed] [Google Scholar]
- 9.Hohaus A, Beyl S, Kudrnac M, Berjukow S, Timin EN, Marksteiner R, Maw MA, Hering S. Structural determinants of L-type channel activation in segment IIS6 revealed by a retinal disorder. J Biol Chem. 2005;280:38471–7. doi: 10.1074/jbc.M507013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hering S, Berjukow S, Sokolov S, Marksteiner R, Weiss RG, Kraus R, Timin EN. Molecular determinants of inactivation in voltage-gated Ca2+ channels. J Physiol. 2000;528:237–49. doi: 10.1111/j.1469-7793.2000.t01-1-00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grabner M, Dirksen RT, Suda N, Beam KG. The II-III loop of the skeletal muscle dihydro-pyridine receptor is responsible for the Bi-directional coupling with the ryanodine receptor. Proc Natl Acad Sci USA. 1998;95:1903–8. [Google Scholar]
- 12.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–8. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 13.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Eur J Physiol. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 14.Vriend G, WHAT IF. A molecular modeling and drug design program. J Mol Graph. 1990;8:52–6. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 15.van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–18. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–21. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 17.Hackos DH, Chang TH, Swartz KJ. Scanning the intracellular S6 activation gate in the shaker K+ channel. J Gen Phys. 2002;119:521–31. doi: 10.1085/jgp.20028569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armstrong CM. Voltage-gated K channels. Sci STKE. 2003:10. doi: 10.1126/stke.2003.188.re10. [DOI] [PubMed] [Google Scholar]
- 19.Deupi X, Olivella M, Govaerts C, Ballesteros JA, Campillo M, Pardo L. Ser and Thr residues modulate the conformation of pro-kinked transmembrane alpha-helices. Biophys J. 2004;86:105–15. doi: 10.1016/S0006-3495(04)74088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacArthur MW, Thornton JM. Influence of proline residues on protein conformation. J Mol Biol. 1991;218:397–412. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]
- 21.Cordes FS, Bright JN, Sansom MS. Proline-induced distortions of transmembrane helices. J Mol Biol. 2002;323:951–60. doi: 10.1016/s0022-2836(02)01006-9. [DOI] [PubMed] [Google Scholar]
- 22.Barlow DJ, Thornton JM. Helix geometry in proteins. J Mol Biol. 1988;201:601–19. doi: 10.1016/0022-2836(88)90641-9. [DOI] [PubMed] [Google Scholar]
- 23.Blaber M, Zhang XJ, Matthews BW. Structural basis of amino acid alpha helix propensity. Science. 1993;260:1637–40. doi: 10.1126/science.8503008. å . [DOI] [PubMed] [Google Scholar]