

Abstract
Purpose
To explore new strategies for effective isolation, preservation, and expansion of human corneal endothelial cells (HCECs).
Methods
Human corneal Descemet’s membrane and corneal endothelial cells were digested with collagenase A or Dispase II in supplemented hormonal epithelial medium (SHEM) for 1.5 to 16 hours. HCEC aggregates derived from collagenase A digestion were preserved in serum-free medium with low or high calcium for up to 3 weeks. Cryosections of HCEC aggregates were subjected to immunostaining with ZO-1, connexin 43, type IV collagen, laminin-5, and perlecan, and apoptosis was determined by TUNEL or cell-viability assay. For expansion, HCEC aggregates were seeded directly or after brief treatment with trypsin/EDTA in SHEM, with or without additional bovine pituitary extract (BPE), nerve growth factor (NGF), or basic fibroblast growth factor (bFGF). The resultant HCECs were immunostained with ZO-1, connexin 43, and Ki67.
Results
Digestion with collagenase A, but not Dispase, of the stripped Descemet’s membrane generated HCEC aggregates, which preserved cell–cell junctions and basement membrane components. High cell viability of HCEC aggregates was preservable in a serum-free, high-calcium, but not low-calcium, medium for at least 3 weeks. Brief treatment of HCEC aggregates with trypsin/EDTA resulted in a higher proliferation rate than without, when cultured in SHEM, and the resultant confluent monolayer of hexagonal cells retained cell–cell junctions. However, additional BPE, NGF, or bFGF did not increase cell proliferation, whereas additional BPE or bFGF disrupted cell–cell junctions.
Conclusions
Collagenase A digestion successfully harvested aggregates with viable HCECs that were preservable for at least 3 weeks in a serum-free, high-calcium medium and, with brief trypsin/EDTA treatment, expanded in the SHEM into a monolayer with hexagonal cells that exhibited characteristic cell junctions.
The corneal endothelium, a single layer of cells lining the posterior surface of the cornea and facing the anterior chamber, plays a pivotal role in regulating corneal stromal hydration and hence, transparency (for review, see Ref. 1). Unlike the corneal endothelial cells of other species, such as rabbit and bovine, human corneal endothelial cells (HCECs) are notorious for their limited proliferative capacity in vivo.2 Thus, the major means of replenishing the loss of HCECs in vivo is facilitated via cell migration and enlargement.3 Several eye diseases can cause dysfunction or a low density of HCECs, leading to sight-threatening bullous keratopathy or corneal edema (for review, see Ref. 4). At the present time, the only way to restore vision in eyes inflicted with bullous keratopathy is by transplantation of a full- or partial-thickness cadaveric donor cornea containing a healthy corneal endothelium.
Currently, there is a global shortage of donor corneas, and approximately 30% of all corneal transplantations are performed because of the aforementioned corneal endothelium diseases. Therefore, the ability to engineer the human corneal endothelium in vitro is paramount, as it may function as an alternative graft to restore vision in eyes inflicted with corneal endothelial failure. In theory, an ideal and effective engineering method should comprise three key steps: isolation of HCEC from the donor cornea, preservation of isolated HCECs for a period to allow transportation, and expansion of isolated HCECs on an appropriate in vitro environment suitable for transplantation. Furthermore, in each of these three steps, HCECs could also be influenced by the medium to which the cells are exposed.
No study has been conducted to address the aforementioned three key steps in a comprehensive manner. Regarding the method of isolating HCECs from the donor cornea, some have used the nonenzymatic method based on EDTA,5 but most have relied on enzymatic digestion using trypsin/EDTA, Dispase,6 or collagenase.7,8 However, none have systemically compared these different enzymatic methods to determine the yield and the reproducibility, nor have they considered the possibility that the digestion in a certain medium may trigger cell death. There has not been any attempt made to investigate the method of preservation of isolated HCECs. Regarding the method of expanding isolated HCECs, a significant advance has been made by Senoo et al.,9 showing that the use of EDTA to dissociate cell– cell junctions is essential for the stimulation of cellular proliferation by releasing the cell cycle block. Furthermore, they devised a unique medium supplemented with pituitary extracts and NGF to promote HCEC proliferation.5,10
In this study, we used several novel strategies to achieve effective isolation of HCECs from a small strip of Descemet’s membrane, to prolong the period of preservation of such isolated HCECs for at least 3 weeks, and to facilitate subsequent expansion of isolated HCECs into a characteristic hexagonal morphology. These accomplishments lay down a solid foundation to embark on tissue engineering of both allogeneic and autologous human corneal endothelium in the future.
Material and Methods
Dulbecco’s modified Eagle’s medium (DMEM), Ham’s/F12 medium, keratinocyte serum-free medium (KSFM) with its supplement, HEPES buffer, Hanks’ balanced salt solution (HBSS), phosphate-buffered saline (PBS), amphotericin B, gentamicin, fetal bovine serum (FBS), bovine pituitary extract (BPE), human recombinant epidermal growth factor (h-EGF), nerve growth factor (NGF), 0.25% trypsin/0.53 mM EDTA (trypsin/EDTA), and cell-viability assay reagent (Live/Dead Assay) were purchased from Invitrogen (Carlsbad, CA). Dispase II and collagenase A were obtained from Roche (Indianapolis, IN). Hydrocortisone, di-methyl sulfoxide, cholera toxin, insulin-transferrin-sodium selenite media supplement, propidium iodide, Hoechst 33342 dye, Triton X-100, bovine serum albumin (BSA), human basic fibroblast growth factor (h-bFGF), paraformaldehyde, and FITC conjugated anti-mouse IgG were purchased from Sigma-Aldrich (St. Louis, MO); mouse anti-ZO-1 antibody from BD Biosciences (Bedford, MA); mouse anti-laminin 5, type IV collagen α2 chain, perlecan, and connexin 43 antibodies from Chemicon (Temecula, CA); mouse anti-type IV collagen α1 antibody from Kamiya Biomedical (Seattle, WA); antifade mounting solution from Vector Laboratories (Burlingame, CA); mouse anti-Ki67 antibody from DakoCytomation (Carpinteria, CA); and a fluorometric TUNEL system (DeadEnd) from Promega (Madison, WI).
Isolation of HCECs
Human tissue was handled according to the Declaration of Helsinki. Eighteen corneoscleral tissues from human donor eyes were obtained from the Florida Lions Eye Bank (Miami, FL). Some of their central corneal buttons had been used for corneal transplantation. The donors’ ages were between 18 and 68 years (41.4 ± 15.8 years). All tissues were maintained at 4°C in storage medium (Optisol; Chiron Vision, Irvine, CA) for less than 10 days before study. The tissue was rinsed three times with DMEM containing 50 mg/mL gentamicin and 1.25 mg/mL amphotericin B. The central cornea was removed by a trephine of 8-mm diameter. Afterward, the Descemet’s membrane and corneal endothelial cells were stripped from the posterior surface of the peripheral corneoscleral tissue under a dissecting microscope and digested at 37°C for 1.5 to 16 hours with 2 mg/mL collagenase A in supplemented hormonal epithelial medium (SHEM), which was made of an equal volume of HEPES-buffered DMEM and Ham’s F12 supplemented with 5% FBS, 0.5% dimethyl sulfoxide, 2 ng/mL mouse EGF, 5 μg/mL insulin, 5 μg/mL transferrin, 5 ng/mL selenium, 0.5 μg/mL hydrocortisone, 1 nM cholera toxin, 50 μg/mL gentamicin, and 1.25 μg/mL amphotericin B. After digestion, HCECs formed aggregates, which were collected by centrifugation at 2000 rpm for 3 minutes to remove the digestion solution. As a control, Descemet’s membrane strips were also digested in 10 mg/mL Dispase II in SHEM and trypsin/EDTA for up to 3 hours.
Preservation of Isolated HCEC Aggregates
The resultant aggregates of HCECs were preserved in KSFM with complete supplement (storage medium 1), DMEM/F12 with KSFM supplements (storage medium 2), or DMEM/F12 with SHEM supplements without FBS (storage medium 3). All these media are serum free, one of the major differences among them is the calcium concentration, which was 0.09 mM in storage medium 1, but was 1.05 mM in storage media 2 and 3. HCEC aggregates were stored in a tissue culture incubator at 37°C for up to 3 weeks. Cell viability was determined (Live and Dead assay; Invitrogen) and also evaluated by subculturing them in SHEM.
Expansion of Isolated HCEC Aggregates
The resultant HCEC aggregates, either immediately after digestion or after a period of preservation in a storage medium, were then cultured in SHEM with or without additional growth factors such as 40 ng/mL bFGF, 0.1 mg/mL BPE, and 20 ng/mL NGF on a plastic dish under 37°C and 5% CO2. The media were changed every 2 to 3 days. Some HCEC aggregates were pretreated with trypsin/EDTA at 37°C for 10 minutes to dissociate endothelial cells before the aforementioned cultivation.
Immunostaining
HCEC aggregates were embedded in OCT and subjected to frozen sectioning. Cryosections of 4 μm were air-dried at room temperature (RT) for 30 minutes, and fixed in cold acetone for 10 minutes at −20°C. Sections used for immunostaining were rehydrated in PBS, and incubated in 0.2% Triton X-100 for 10 minutes. After three rinses with PBS for 5 minutes each and preincubation with 2% BSA to block nonspecific staining, the sections were incubated with anti-laminin 5, type IV collagen α1 and α2 chain, perlecan, ZO-1, and connexin 43 (all at 1:100) antibodies for 1 hour. After three washes with PBS for 15 minutes, the sections were incubated with a FITC-conjugated secondary antibody (goat anti-rabbit or anti-mouse IgG at 1:100) for 45 minutes. After three additional PBS washes, each for 10 minutes, they were counterstained with propidium iodide (1:1000) or Hoechst 33342 (10 μg/mL), then mounted with an antifade solution and analyzed with a fluorescence microscope. HCECs cultured in 24-well plates or chamber slides were fixed in 4% paraformaldehyde for 15 minutes at RT and stained with anti-ZO-1 and connexin 43 antibodies as just described. For immunohistochemical staining of Ki67, endogenous peroxidase activity was blocked by 0.6% hydrogen peroxide for 10 minutes. Nonspecific staining was blocked by 1% normal goat serum for 30 minutes. Cells were then incubated with anti-Ki67 antibody (1:100) for 1 hour. After three washes with PBS for 15 minutes, cells were incubated with biotinylated rabbit anti-mouse IgG (1:100) for 30 minutes, followed by incubation with ABC reagent for 30 minutes. The reaction product was developed with DAB for 5 minutes and examined by light microscope.
Cell-Viability and TUNEL Assays
Cell-viability and terminal deoxyribonucleotidyl transferase-mediated FITC-linked dUTP nick-end DNA labeling (TUNEL) assays were used to determine living and apoptotic cells, respectively. HCEC aggregates were incubated with cell-viability assay reagents for 15 minutes at RT. Live cells were distinguished by green fluorescence staining of the cell cytoplasm, and dead cells were stained with red fluorescence in the nuclei. The TUNEL assay was performed according to the manufacturer’s instructions. Briefly, cross-sections of HCEC aggregates were fixed in 4% paraformaldehyde for 20 minutes at RT and permeabilized with 1% Triton X-100. Samples were then incubated for 60 minutes at 37°C with exogenous TdT and fluorescein-conjugated dUTP, for repair of nicked 3′-hydroxyl DNA ends. Cells were treated with DNase I as the positive control, whereas negative control cells were incubated with a buffer lacking the rTdT enzyme. The apoptotic nuclei were labeled with green fluorescence.
Results
Isolation of HCECs as Cell Aggregates by Collagenase A in a Serum-Containing Medium
Under a dissecting microscope, the Descemet’s membrane was surgically stripped from the peripheral cornea of the corneoscleral ring tissue. As a result, most of the HCECs still adhered to the Descemet’s membrane, whereas some cells detached, creating regions without cells (Fig. 1A). After Dispase II digestion at 37°C for 1.5 hour in SHEM, HCECs started to aggregate but still did not detach from the Descemet’s membrane (Fig. 1B). In contrast, after the stripped Descemet’s membrane was digested in collagenase A for 1.5 hours, HCECs aggregated into considerable clusters and detached from the Descemet’s membrane (Fig. 1C), leaving an intact Descemet’s membrane behind. After 3 hours, the aggregates derived from collagenase A digestion became more compact. However, cells still did not detach from the Descemet’s membrane and started to disintegrate after Dispase II digestion (data not shown). Notably, after 16 hours of collagenase A digestion, the Descemet’s membrane was dissolved and most HCEC aggregates were compact and exhibited different sizes and shapes (Fig. 1D), whereas very few demonstrated looseness (Figs. 1D, 1E, arrows). The cell-viability assay showed that the compact aggregates were composed of viable cells exhibiting intense green fluorescence (Fig. 1F). In contrast, loosened aggregates contained dead cells (Fig. 1F). We speculated that these cells may have already been dead during storage of the donor cornea and thus were unable to form aggregates during collagenase A digestion.
Figure 1.

Isolation of HCECs as cell aggregates by collagenase A in a serum-containing medium. Descemet’s membrane was stripped from the peripheral donor cornea and most of the HCECs were adherent to the Descemet’s membrane, whereas some cells were detached in some areas (dotted lines and asterisks) (A). Dispase II digestion at 37°C for 1.5 hours, HCECs started to aggregate but still did not detach from the Descemet’s membrane (B). In contrast, collagenase A digestion for 1.5 hours resulted in frank aggregation of HCECs into clusters and complete detachment from Descemet’s membrane (C). After 16 hours’ digestion in collagenase A, most of the HCEC aggregates became compact and varied in size and shape (D) with some not forming tight aggregates (D, E, arrows). The cell-viability assay showed that the compact aggregates were composed of viable cells; however, those loosely held and disintegrated aggregates contained some dead cells (F, arrows; E is a phase-contrast micrograph of F). Bars, 100 μm.
Maintenance of Cell–Cell Junctions and Basement Membrane Components in HCEC Aggregates
To investigate whether cell–cell junctions and basement membrane components were maintained after collagenase A digestion, HCEC aggregates were embedded in OCT, prepared for cryosections, and subjected to immunostaining. The results showed that tight junction ZO-1 (Fig. 2A), gap junction connexin-43 (Fig. 2B), and such basement membrane components as type IV collagen α1 (Fig. 2C) and α2 (Fig. 2D) chains, laminin 5 (Fig. 2E), and perlecan (Fig. 2F) were all present in HCEC aggregates. Nuclear counterstaining further showed that HCECs in the aggregate were compact. The TUNEL assay confirmed that only a few apoptotic cells were present in the center of the aggregate (Figs. 2G, 2H). To investigate further whether these basement membrane components helped to maintain the viability of HCECs, collagenase-isolated aggregates were subsequently treated with Dispase II (10 mg/mL in SHEM) at 4°C for 16 hours, a treatment, as we have reported, that can remove collagen IV and laminin 5.11 The results showed that the additional Dispase II digestion did not disintegrate HCEC aggregates and that the cells within aggregates were still alive, as judged by the viability assay (data not shown). However, Dispase-treated HCEC aggregates did not readily attach to plastic in SHEM (data not shown), whereas nontreated aggregates did. These results indicate that cell–cell junctions may play a more important role in forming the aggregates and in maintaining the cell viability of HCECs than do cell–matrix interactions and that the remaining basement membrane matrix components in aggregates may play an important role in facilitating cell attachment of HCECs on plastic during subculturing.
Figure 2.

Maintenance of cell–cell junctions and basement membrane components in HCEC aggregates. Immunofluorescence staining showed ZO-1 (A), connexin-43 (B), type IV collagen α1 (C) and α2 (D) chains, laminin 5 (E), and perlecan (F) were present in HCEC aggregates. The TUNEL assay revealed only a few apoptotic cells present in the center of the aggregate (G, arrowheads; H shows the nuclear counterstaining of G). Bar, 100 μm.
Viability of HCEC Aggregates after Storage in High-Calcium, Serum-Free Medium
To investigate whether it was feasible to store HCEC aggregates for a period before future cultivation, we incubated them in different medium for 3 weeks and then subjected them to cultivation on plastic dishes in different media. When incubated in complete SHEM which containing 5% FBS, HCEC aggregates quickly attached to the plastic dish within 12 hours (data not shown). In contrast, when incubated in serum-free medium that was KSFM or DMEM/F12 based, they remained as floating aggregates (Figs. 3C, 3G). In storage medium 1, floating aggregates gradually disintegrated after 1 week (Figs. 3F, 3G). In contrast, they were organized into round spheres in storage medium 2 and 3 after 1 week (Fig. 3B) and organized into a compact round sphere for up to 3 weeks (Fig. 3C). At the end of the third week, these aggregates attached within 12 hours and spread out as an intact HCEC sheet within 4 days when seeded on plastic in SHEM (Fig. 3D). However, those preserved in storage medium 1 generated very few single cells (Fig. 3H). These results indicate that collagenase-isolated HCEC aggregates can be preserved for at least 3 weeks in a high-calcium, serum-free medium, and that such preserved aggregates still retain high HCEC viability for subsequent cultivation in a serum-containing medium.
Figure 3.

Incubation of HCEC aggregates in a high- or low-calcium, serum-free medium. HCEC aggregates harvested from collagenase A digestion (A, E) were incubated in a high-calcium, serum-free medium (A–C) or a low-calcium, serum-free medium (E–G) continuously for 1 week (B, F) and 3 weeks (C, G). At the end of the third week, aggregates were transferred to the SHEM and further cultured for 4 days. Aggregates incubated in a high-calcium, serum-free medium generated an intact HCEC monolayer (D). However, aggregates incubated in a low calcium, serum-free medium generated few scattered single cells (H). Bar, 100 μm.
Expansion of HCECs in SHEM with Higher Proliferation after a Brief Trypsin/EDTA Treatment
It has been shown that a brief EDTA treatment promotes proliferation of HCECs in organ cultures by releasing mitotic block mediated by cell–cell contacts.9 We thus wanted to determine whether the isolated HCEC aggregates would also be stimulated into higher proliferation if cell–cell contacts were also released by a brief treatment with trypsin/EDTA. To do so, HCEC aggregates harvested from one peripheral corneal rim were treated with 0.25% trypsin/0.53 mM EDTA at 37°C for 10 minutes. After the brief treatment, HCEC aggregates were dissociated into smaller clusters and single cells (Fig. 4A), after removal of digest solution by centrifuge, HCECs were suspended in SHEM and seeded into two wells of a 24-well culture plate. Most cells attached and spread out within 24 hours (Fig. 4B), and grew into patches and sheets 4 days later (Fig. 4C). After 1 week, these cells reached confluence and maintained a typical hexagonal shape (Fig. 4D). Immunostaining showed confluent cells expressed tight junction ZO-1 (Fig. 4E) and gap junction connexin-43 (Fig. 4F). These results indicated that additional brief digestion by trypsin/EDTA indeed resulted in successful expansion of HCECs into a monolayer.
Figure 4.

Expansion of HCECs from aggregates in SHEM. HCEC aggregates from a 24-year-old donor were subsequently treated with trypsin/EDTA for 10 minutes, and seeded in the SHEM. This treatment dissociated HCEC aggregates into small clusters and single cells (A). Most cells attached and spread out within 24 hours (B), and grew into patches and sheets 4 days later (C). After 1 week, the cells reached confluence and maintained a typical hexagonal shape (D). Immunostaining showed that confluent cells expressed tight junction ZO-1 (E) and gap junction connexin-43 (F). Bars, 100 μm.
To confirm further that a brief treatment with trypsin/EDTA was necessary to stimulate HCEC proliferation, we performed immunohistochemical staining for Ki67, which is expressed at all stages of the cell cycle except G0.12 As shown in Figure 5, HCEC aggregates directly seeded on plastic without trypsin/EDTA treatment also exhibited a sheetlike growth after 1 week of culturing in SHEM (Fig. 5A). Nevertheless, Ki67 positive nuclei were only occasionally observed in the periphery of the growth (Fig. 5G). In contrast, after a brief trypsin/EDTA treatment, although HCEC aggregates also resulted in a confluent cell sheet if seeded on plastic in SHEM (Fig. 5B), more cells exhibited randomly distributed Ki67-positive nuclei (Fig. 5H). This result indicated that a brief treatment of trypsin/EDTA indeed promoted cellular proliferation.
Figure 5.
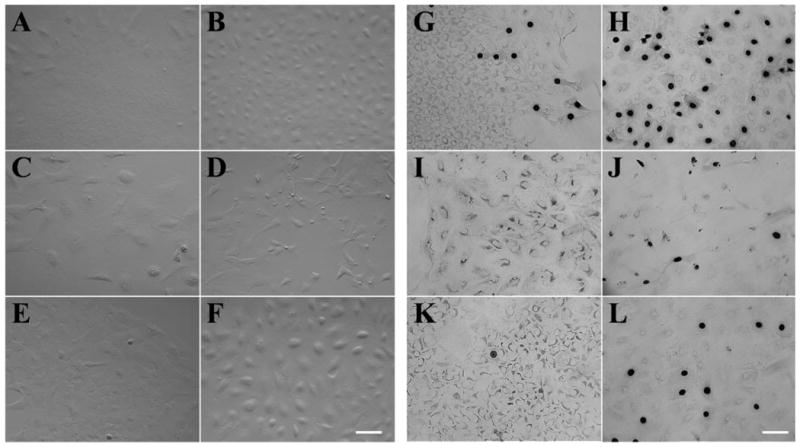
Ki67 expression of HCECs cultured under different conditions. HCEC aggregates harvested from two donors, 48 and 53 years old, without (A, C, E) or with (B, D, F) a brief trypsin/EDTA treatment were seeded on plastic in SHEM (A, B), SHEM+BPE (C, D), or SHEM+NGF (E, F) for 1 week. Immunostaining showed sporadic Ki67-positive nuclei in the periphery of HCEC sheets when aggregates were cultured in SHEM (G). In contrast, very few Ki67-positive nuclei were found in SHEM+BPE (I) and SHEM+NGF (K). Ki67-positive nuclei increased dramatically when aggregates were pretreated with trypsin/EDTA (H, J, L). However, there were more Ki67-positive nuclei in SHEM (H) than those in SHEM+BPE (J) and SHEM+NGF (L). Bar, 100 μm.
To determine whether additional supplement of growth factors in SHEM was beneficial, HCEC aggregates with or without a brief trypsin/EDTA treatment were cultured in SHEM with or without 0.1 mg/mL BPE, 20 ng/mL NGF, or 40 ng/mL bFGF for 1 week. The results showed that additional BPE stimulated more scattering of cells, resulting in the loss of an intact sheet (Fig. 5C), and if pretreated with trypsin/EDTA, this phenomenon became more prominent, and most of the cells changed to a fibroblastic shape (Fig. 5D). In contrast, additional NGF did not cause the aforementioned dramatic cell-shape change, and cells maintained an intact sheet without (Fig. 5E) or with (Fig. 5F) trypsin/EDTA treatment. Ki67 staining revealed that addition of either BPE or NGF in SHEM yield less Ki67-positive nuclei with (Figs. 5J, 5L, respectively) or without (Figs. 5I, 5K, respectively) a brief treatment of trypsin/EDTA. A similar result was obtained when bFGF was added to SHEM when compared with BPE (data not shown). These results indicated that a brief trypsin/EDTA treatment resulted in more Ki67-positive cells in all these four cultures and that cellular proliferation was not promoted by addition of any of these three growth supplements in SHEM. Addition of BPE or bFGF to SHEM resulted in a loss of a hexagonal phenotype and cell–cell junction formation, suggesting that these growth supplements may increase cell migration or differentiation instead of proliferation.
Discussion
A critical prerequisite for successful tissue engineering of the human corneal endothelium is to ensure effective isolation, preservation, and expansion from a small number of HCECs. The present study demonstrated its feasibility based on isolation by collagenase A digestion; storage in a serum-free, high-calcium medium; and expansion in SHEM after a brief treatment with trypsin/EDTA.
It has been recognized that HCECs are difficult to isolate from Descemet’s membrane because of their strong adherence to the extracellular matrix. The commonly used trypsin digestion tends to lead to cellular degeneration because a prolonged incubation time is needed to detach cells from the matrix (for review, see Ref. 13). Other groups have used Dispase digestion6 or EDTA treatment5 followed by pipetting, a method that is not only time consuming, but also may cause cell damage and decrease the yield. In our experiment, Dispase II digestion for 1.5 hour could not separate HCECs from the Descemet’s membrane, while a longer digestion resulted in cell disintegration. Engelmann et al.7 first reported the isolation of the human corneal endothelium by two-step digestion of the entire cornea with a high concentration of 5 mg/mL collagenase A for 1.5 hours followed by a low concentration of 0.4 mg/mL for up to 16 hours. Because Descemet’s membrane was not stripped, their method invariably included contaminated corneal fibroblasts, a problem that they solved by subjecting cells to a selective L-valine-free medium culture. Recently, Yokoo et al.8 used digestion with 0.2 mg/mL type IA collagenase at 37°C overnight to isolate HCECs from the stripped Descemet’s membrane. Nevertheless, they did not mention or demonstrate formation of HCEC aggregates. In our study, 2 mg/mL collagenase A digestion of the stripped Descemet’s membrane for 16 hours resulted in compact HCEC aggregates that retained high cell viability without keratocyte contamination. These aggregates were very easy to harvest by gentle centrifugation or to handle by pipetting under a dissecting microscope. They further maintained cell–cell junctions mediated by ZO-1 and connexin-43 and such basement membrane components as type IV collagen α1 and α2 chains, laminin 5, and perlecan (Fig. 2). We speculate that cell viability in HCEC aggregates was maintained mainly by cell–cell junctions, whereas basement membrane components facilitated subsequent cell adhesion to plastic.
For the first time, we demonstrated that such HCEC aggregates could become organized into a round sphere if continuously cultured in a serum-free, high-calcium medium for 3 weeks and that such preserved spheres could yield a monolayer of HCECs with a characteristic hexagonal shape (Fig. 3D). In contrast, HCEC aggregates quickly adhered to plastic within 24 hours if FBS was added during storage, or continued to degenerate when a low-calcium, serum-free medium was used (Fig. 3H). Because the formation of cell–cell junctions is calcium-dependent (for review see Ref. 14), our results further support the notion that the retention of cell–cell junctions in HCEC aggregates played an important role in achieving preservation of HCEC viability during long-term storage. Such an accomplishment is critical for solving logistic problems in transportation of donor HCECs before cell expansion and tissue engineering to be conducted at a distant site. In this experiment, endothelial sheets were generated from HCEC aggregates preserved in both storage medium 2, which contained KSFM supplements, and medium 3, which contained SHEM supplements without FBS. Further study is needed to define efficient and sufficient preservation conditions by testing different variables such as temperature and growth factors. Because the conventional corneal preservation method used by eye banks can achieve a maximum preservation period of only 14 days, after which time there is an increasing loss of HCECs (for review, see Ref. 15), it is tempting to speculate that further advances based on our new discovery are possible and will achieve better preservation of HCECs over an extended period of storage.
It is well known that HCECs have low mitotic activity in vivo, although they retain proliferative capacity.16 Investigators have successfully expanded HCECs by the use of different media supplemented with EGF,5,8 NGF,5 bFGF,6,8,17,18 or BPE5 on plastic19 or on different substrates such as laminin and chondroitin sulfate,7 fibronectin,16 type IV collagen,18 amniotic membrane,6 or a temperature-responsive culturing surface.17,18 For the first time, we showed that SHEM, which is commonly used for epithelial cell cultures, is also effective in expanding HCECs on plastic dishes. As for the culture of HCECs, it is well known that the final cell morphology depends on the cell density of the primary culture. As shown in Figures 4A and 4B, the seeding density used in our experiment was very low when HCECs from one peripheral corneal rim were seeded in two wells of a 24-well plate. However, unlike previous reports that showed that HCECs became elongated into a fibroblastic shape during expansion,16,18 we noted that HCECs maintained their shape very well during expansion in SHEM even at low seeding density (Fig. 4C). After reaching confluence, HCECs maintained a monolayer of hexagonal cells expressing cell–cell junctions mediated by ZO-1 and connexin-43 (Figs. 4E, 4F), indicating that HCECs not only can enter the cell cycle in SHEM, but also can restore their in vivo phenotype. Of note, addition of BPE (Figs. 5C, 5D) or bFGF (not shown) resulted in a significant cell shape change into a fibroblastic appearance, resembling what was reported in another medium with added BPE,16 and in another report when bFGF was added.18 Furthermore, our study showed that addition of BPE to SHEM decreased Ki67 nuclear staining (Fig. 5I. 5J), a finding different from the previous report.16 We speculated that this morphologic change caused by BPE may result from the inclusion of bFGF, a major component of BPE,20,21 which has been shown to cause mesenchymal transformation of rabbit corneal endothelial cells.22–24 Addition of bFGF to SHEM, resulting in a finding similar to that obtained with BPE, further supported this notion. Taken together, addition of BPE or bFGF to SHEM preferentially induces migration or differentiation of HCECs rather than proliferation. Furthermore, addition of NGF to SHEM also decreased HCEC proliferation, as judged by Ki67 labeling. A previous study also did not show a significant stimulatory effect of NGF on HCEC proliferation.16 Further studies are needed to determine whether other components in the SHEM can be optimized to improve expansion of HCECs.
It has been shown that ex vivo proliferation of HCECs is affected by donor ages (for review, see Ref. 1), and that HCECs from younger donors grow faster and can be passaged more times than cells from older donors.10,16,25 We have not conducted a thorough investigation of regarding donor ages. However, we did note that HCECs from a 24-year-old donor took 1 week, a shorter time, to reach confluence (Fig. 4). However, HCECs from 48- and 53-year-old donors also entered the cell cycle and expressed Ki67 (Fig. 5), indicating that our culturing method can be applied to cells from donors of a wide range of ages. Because the aforementioned procedures could expand a small stripped Descemet’s membrane into a monolayer of HCECs, we believed that it is possible to begin tissue engineering of the autologous human endothelium from a surgical biopsy sample in the future.
Acknowledgments
Supported by R01 EY06819 and R01 EY015735 (SCGT) from National Eye Institute, National Institutes of Health, Bethesda, Maryland, in part by a research grant from TissueTech, Inc., and in part by an unrestricted grant from the Ocular Surface Research and Education Foundation, Miami, FL.
Footnotes
Disclosure: W. Li, TissueTech, Inc. (F, E); A. L. Sabater, None; Y.-T. Chen, None; Y. Hayashida, TissueTech, Inc. (F, E); S.-Y. Chen, TissueTech, Inc. (F, E); H. He, TissueTech, Inc. (F, E); S.C.G. Tseng, TissueTech, Inc. (F, I, E)
References
- 1.Joyce NC. Cell cycle status in human corneal endothelium. Exp Eye Res. 2005;81:629–638. doi: 10.1016/j.exer.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 2.Laing RA, Neubauer L, Oak SS, et al. Evidence for mitosis in the adult corneal endothelium. Ophthalmology. 1984;91:1129–1134. doi: 10.1016/s0161-6420(84)34176-8. [DOI] [PubMed] [Google Scholar]
- 3.Sherrard ES. The corneal endothelium in vivo: its response to mild trauma. Exp Eye Res. 1976;22:347–357. doi: 10.1016/0014-4835(76)90227-x. [DOI] [PubMed] [Google Scholar]
- 4.Joyce NC. Proliferative capacity of the corneal endothelium. Prog Retin Eye Res. 2003;22:359–389. doi: 10.1016/s1350-9462(02)00065-4. [DOI] [PubMed] [Google Scholar]
- 5.Chen KH, Azar D, Joyce NC. Transplantation of adult human corneal endothelium ex vivo: a morphologic study. Cornea. 2001;20:731–737. doi: 10.1097/00003226-200110000-00012. [DOI] [PubMed] [Google Scholar]
- 6.Ishino Y, Sano Y, Nakamura T, et al. Amniotic membrane as a carrier for cultivated human corneal endothelial cell transplantation. Invest Ophthalmol Vis Sci. 2004;45:800–806. doi: 10.1167/iovs.03-0016. [DOI] [PubMed] [Google Scholar]
- 7.Engelmann K, Bohnke M, Friedl P. Isolation and long-term cultivation of human corneal endothelial cells. Invest Ophthalmol Vis Sci. 1988;29:1656–1662. [PubMed] [Google Scholar]
- 8.Yokoo S, Yamagami S, Yanagi Y, et al. Human corneal endothelial cell precursors isolated by sphere-forming assay. Invest Ophthalmol Vis Sci. 2005;46:1626–1631. doi: 10.1167/iovs.04-1263. [DOI] [PubMed] [Google Scholar]
- 9.Senoo T, Obara Y, Joyce NC. EDTA: a promoter of proliferation in human corneal endothelium. Invest Ophthalmol Vis Sci. 2000;41:2930–2935. [PubMed] [Google Scholar]
- 10.Zhu C, Joyce NC. Proliferative response of corneal endothelial cells from young and older donors. Invest Ophthalmol Vis Sci. 2004;45:1743–1751. doi: 10.1167/iovs.03-0814. [DOI] [PubMed] [Google Scholar]
- 11.Espana EM, Romano AC, Kawakita T, et al. Novel enzymatic isolation of an entire viable human limbal epithelial sheet. Invest Ophthalmol Vis Sci. 2003;44:4275–4281. doi: 10.1167/iovs.03-0089. [DOI] [PubMed] [Google Scholar]
- 12.Schluter C, Duchrow M, Wohlenberg C, et al. The cell proliferation-associated antigen of antibody Ki-67: a very large, ubiquitous nuclear protein with numerous repeated elements, representing a new kind of cell cycle-maintaining proteins. J Cell Biol. 1993;123:513–522. doi: 10.1083/jcb.123.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelmann K, Bednarz J, Valtink M. Prospects for endothelial transplantation. Exp Eye Res. 2004;78:573–578. doi: 10.1016/s0014-4835(03)00209-4. [DOI] [PubMed] [Google Scholar]
- 14.Brown RC, Davis TP. Calcium modulation of adherens and tight junction function: a potential mechanism for blood-brain barrier disruption after stroke. Stroke. 2002;33:1706–1711. doi: 10.1161/01.str.0000016405.06729.83. [DOI] [PubMed] [Google Scholar]
- 15.Chu W. The past twenty-five years in eye banking. Cornea. 2000;19:754–765. doi: 10.1097/00003226-200009000-00020. [DOI] [PubMed] [Google Scholar]
- 16.Joyce NC, Zhu CC. Human corneal endothelial cell proliferation: potential for use in regenerative medicine. Cornea. 2004;23:S8–S19. doi: 10.1097/01.ico.0000136666.63870.18. [DOI] [PubMed] [Google Scholar]
- 17.Hsiue GH, Lai JY, Chen KH, Hsu WM. A novel strategy for corneal endothelial reconstruction with a bioengineered cell sheet. Transplantation. 2006;81:473–476. doi: 10.1097/01.tp.0000194864.13539.2c. [DOI] [PubMed] [Google Scholar]
- 18.Sumide T, Nishida K, Yamato M, et al. Functional human corneal endothelial cell sheets harvested from temperature-responsive culture surfaces. FASEB J. 2006;20:392–394. doi: 10.1096/fj.04-3035fje. [DOI] [PubMed] [Google Scholar]
- 19.Schonthal AH, Hwang JJ, Stevenson D, Trousdale MD. Expression and activity of cell cycle-regulatory proteins in normal and transformed corneal endothelial cells. Exp Eye Res. 1999;68:531–539. doi: 10.1006/exer.1998.0634. [DOI] [PubMed] [Google Scholar]
- 20.Esch F, Baird A, Ling N, et al. Primary structure of bovine pituitary basic fibroblast growth factor (FGF) and comparison with the amino-terminal sequence of bovine brain acidic FGF. Proc Natl Acad Sci USA. 1985;82:6507–6511. doi: 10.1073/pnas.82.19.6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perantoni AO, Dove LF, Karavanova I. Basic fibroblast growth factor can mediate the early inductive events in renal development. Proc Natl Acad Sci USA. 1995;92:4696–4700. doi: 10.1073/pnas.92.10.4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HT, Kay EP. FGF-2 induced reorganization and disruption of actin cytoskeleton through PI 3-kinase, Rho, and Cdc42 in corneal endothelial cells. Mol Vis. 2003;9:624–634. [PubMed] [Google Scholar]
- 23.Ko MK, Kay EP. Regulatory role of FGF-2 on type I collagen expression during endothelial mesenchymal transformation. Invest Ophthalmol Vis Sci. 2005;46:4495–4503. doi: 10.1167/iovs.05-0818. [DOI] [PubMed] [Google Scholar]
- 24.Lee HT, Lee JG, Na M, Kay EP. FGF-2 induced by interleukin-1 beta through the action of phosphatidylinositol 3-kinase mediates endothelial mesenchymal transformation in corneal endothelial cells. J Biol Chem. 2004;279:32325–32332. doi: 10.1074/jbc.M405208200. [DOI] [PubMed] [Google Scholar]
- 25.Nayak SK, Binder PS. The growth of endothelium from human corneal rims in tissue culture. Invest Ophthalmol Vis Sci. 1984;25:1213–1216. [PubMed] [Google Scholar]