

Abstract
Transforming growth factor-βactivated kinase 1 (TAK1), a member of the MAPKKK family, was initially described to play an essential role in the TGF beta-signaling pathway, but recent evidence has emerged implicating TAK1 in the IL-1 and TNF pathways. Notably, two homologous proteins, TAB2 and TAB3, have been identified as adaptors linking TAK1 to the upstream adaptors TRAFs. However, it remains unclear whether the interaction between TAB2/TAB3 and TAK1 is necessary for its kinase activation and subsequent activation of the IKK and MAPK pathways. Here, we characterized the TAB2/TAB3-binding domain in TAK1 and further examined the requirement of this interaction for IL-1, TNF, and RANKL signaling. Through deletion mapping experiments, we demonstrated that the binding motif for TAB2/TAB2 is a non-contiguous region located within the last C-terminal 100 residues of TAK1. However, residues 479–553 of TAK1 appear to be necessary and sufficient for TAB2/TAB3 interaction. Conversely, residues 574–693 of TAB2 were shown to interact with TAK1. A green fluorescent protein (GFP) fusion protein containing the last 100 residues of TAK1 (TAK1-C100) abolished the interaction of endogenous TAB2/TAB3 with TAK1, the phosphorylation of TAK1 and prevented the activation of IKK and MAPK induced by IL-1, TNF, and RANKL. Furthermore, TAK1-C100 blocked RANKL-induced nuclear accumulation of NFATc1 and consequently osteoclast differentiation consistent with the ability of a catalytically inactive TAK1 to block RANKL-mediated signaling. Significantly, our study provides evidence that the TAB2/TAB3 interaction with TAK1 is crucial for the activation of signaling cascades mediated by IL-1, TNF, and RANKL.
INTRODUCTION
The pro-inflammatory cytokines interleukin-1 (IL-1)3 and tumor necrosis factor (TNF) elicit a critical function in the innate immune response. Following receptor activation, these cytokines induce a cascade of signaling events leading to the activation of transcription factors such as NF-κB and AP1 through upstream kinases including IκB kinase (IKK) and the mitogen-activated protein kinases (MAPKs; JNK, p38, and ERK). These events culminate in the expression and regulation of numerous pro-inflammatory genes (1,2).
Transforming growth factor-β-activated kinase 1 (TAK1), a member of the MAP3K family, was initially identified as a kinase in TGF-β signaling (3). Recent evidence has emerged indicating that TAK1 is involved in various signaling pathways, including IL-1, IL-18, TNF, and receptor activator of NF-κB ligand (RANKL) (4–7). Since TAK1 has the capacity to be activated by various cytokines, upstream adaptor molecules are critical in providing the selectivity of initiating distinct signaling events upon receptor activation.
Members of the TNF-receptor-associated factor (TRAF) family of adaptor proteins are involved in coupling activated TNF and IL-1 receptors to NF-κB and MAPKs pathways (8–11). Although TRAF proteins have no known enzymatic activity, recent evidence indicates that they act as E3s through their N-terminal really interesting new gene (RING) domain (12–14). RING-type E3s contain a series of Cys and His residues distinctly separated to constitute a novel Zn-binding domain. TRAF6 together with the heterodimeric E2 complex (Ubc13/Uev1A) facilitates the synthesis of unique Lys63 (K63)-linked poly-ubiquitin chains on itself, rather than the conventional Lys48 (K48)-linked poly-ubiquitin chains that target proteins for degradation. This unique K63-linked poly-ubiquitin chain likely provides a scaffold to recruit downstream effector molecules to activate various signaling components.
TAK1-binding protein 2 (TAB2), or its homologue TAB3, appears to be the adaptor molecule linking these TRAF molecules to activate TAK1 (15–19). Both TAB2 and TAB3 contain a highly conserved C-terminal novel zinc finger (NZF) domain, which binds preferentially to K63-linked poly-ubiquitin chains (20) (B. G. D., unpublished observations). Mutation of this Zn-finger domain abolishes the ability of TAB2 and TAB3 to bind to poly-ubiquitin chains, as well as their ability to activate TAK1 that appears to be a ubiquitin-dependent kinase of MKK and IKK (21). However, the mechanism by which the poly-ubiquitination events induce TAK1 activation has yet to be determined.
Following IL-1, TNF, or LPS stimulation, two possible TAK1 complexes exist, one which consists of TAB2-TAK1-TAB1 and another which consists of TAB3-TAK1-TAB1 that causes the activation of MAPKs and NF-κB pathways (18,21). These data suggest that TAB2 and TAB3 may play redundant roles in TAK1 activation (18). Consistent with this hypothesis, activation of NF-κB and MAPKs pathways by IL-1 and TNF is intact in TAB2-deficient mouse embryonic fibroblasts (MEFs) (22) (A. B., unpublished observations). Additionally, knockdown of both TAB2/TAB3 by siRNA results in complete loss of TAK1 activation (19).
While it appears that TAK1 activation requires poly-ubiquitination of TRAF6 and RIP1 in the IL-1 and TNF pathways, respectively, the precise role of TAB2/TAB3 in TAK1 activation has not been clearly investigated. Furthermore, the dissection of the respective roles of TAB2 and TAB3 in regulating TAK1 signaling is prevented due to the compensatory nature of these proteins. The development of a construct that could ablate the functional interaction of both TAB2/TAB3 with TAK1 could be an alternative strategy to examine the functional role of TAB2/TAB3 in regulating TAK1-dependent signaling. In this study, we demonstrated that the binding motif for TAB2/TAB3 on TAK1 is a non-contiguous region within the last C-terminal 100 residues of TAK1 (TAK1-C100), and more specifically encompassing residues 479–553 of TAK1. Furthermore, TAK1-C100 prevented endogenous interaction of TAB2 and TAK1, and blocked IL-1 and TNF signaling. Additionally, we show the requirement of TAK1 kinase activity in RANKL signaling and osteoclast differentiation. Notably, we demonstrate that TAK1-C100 abolishes RANKL-mediated activation of IKK and JNK, nuclear accumulation of NFATc1, and terminal osteoclast differentiation. Our results reveal the critical function of the TAB2/TAB3 and TAK1 interaction for eliciting TAK1-dependent signals generated by the cytokines IL-1, TNF, and RANKL.
EXPERIMENTAL PROCEDURES
Cell Lines, Reagents, and Antibodies
Human embryonic kidney 293 cells (HEK293), mouse macrophage cell line RAW264.7 (RAW), and mouse fibroblastic cell line L929 were purchased from the American Type Cell Culture Collection (Rockville, MD) and cultured as previously described (23–25). Retroviral packaging cell line GP2-293 was purchased from Clontech (Palo Alto, CA) and cultured in DMEM supplemented with 10% fetal bovine serum and antibiotics. Recombinant TNF and IL-1 were purchased from PeproTech (Rocky Hill, New Jersey). Monoclonal antibody to phospho-IκBα and rabbit polyclonal antibodies to phospho-c-Jun, phospho-p38 and phospho-TAK1 (Thr 184/187) were purchased from Cell Signaling Technology (Beverly, MA); rabbit polyclonal antibodies to JNK1, NEMO, TAB2, and monoclonal antibody to NFATc1 from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal antibody for α-tubulin was purchased from Calbiochem (San Diego, CA), and rabbit polyclonal antibody for β-actin from Cytoskeleton (Denver, CO). Goat anti-rabbit IgG-conjugated horseradish peroxidase antibody was purchased from BioRad Laboratories (Hercules, CA) and goat anti-mouse IgG-conjugated horseradish peroxidase from BD Biosciences Pharmingen (San Jose, CA). Monoclonal anti-FLAG antibody, glutathione-agarose, and tartrate resistance acid phosphatase (TRAP) kit were purchased from Sigma (St. Louis, MO). Protein A/G Agarose beads were purchased from Pierce (Rockford, IL).
Plasmids
Mouse TAK1-K63W cDNA was a generous gift from Dr. Tan (Baylor College of Medicine, Houston, TX). The cDNA encoding full-length TAK1-K63W was amplified by PCR using gene specific primers containing EcoRI (5′) and BamHI (3′) sites that was subsequently digested and cloned into p3XFLAG-CMV-14 (Sigma; St. Louis, MO). To obtain wild-type TAK1 (TAK1-WT), the K63W mutation was reversed using the QuickChange Site-Directed Mutagenesis kit (Stratagene; La Jolla, CA). The various C-terminal truncated forms of TAK1 Δ1-Δ5 were generated by PCR using gene specific primers containing EcoRI (5′) and BamHI (3′) sites that was subsequently digested and cloned into p3XFLAG-CMV-14. To generate the fragments encoding residues 479–579 (TAK1-Δ6) and 479–513 (TAK1-Δ7), we performed PCR with gene specific primers containing ScaI (5′) and BamHI (3′) sites and after digesting with the appropriate enzymes, the fragments were fused to the sequence encoding residues 1–300 of TAK1 and inserted into p3XFLAG-CMV-14. Using full-length TAK1 coding sequence, TAK1-Δ8, -Δ9, and -Δ10 constructs were generated using the QuickChange Site-Directed Mutagenesis kit. Retroviral vectors expressing FLAG-tagged TAK1-WT and TAK1-K63W were generated by PCR and inserted into a modified pMX vector (26,27) containing an IRES-GFP and a puromycin selectable marker. The GFP-TAK1-C100, GFP-TAK1 479–520, GFP-TAK1 505–536, GFP-TAK1 505–547, GFP-TAK1 505–553, GFP-TAK1 479–553 fusion protein constructs were generated by PCR using gene specific primers corresponding to residues 479–579, 479–520, 505–536, 505–547, 505–553, 479–553 of TAK1 with SalI (5′) and BamHI (3′) sites and inserted in-frame with GFP in the pLEGF-C1 retroviral vector (Clontech; Palo Alto, CA). The entire GFP-TAK1-C100 fragment was subsequently cloned into pMX-puro. The entire TAB2 cDNA was sub-cloned (EcoRI/XbaI) by PCR into a modified pCR3 mammalian expression vector (Invitrogen; Carlsbad, CA) containing two tandem FLAG sequences at the N-terminus. Human TAB3 cDNA was purchased from ATCC (Rockville, MD) and initially cloned into a pCR3-T7 vector using BamHI (5′) and NotI (3′) sites. We subcloned it from pCR3-T7-TAB3 into pGEX-KG between BamHI (5′) and a blunted SalI (3′) site. Expression vectors encoding FLAG-RANK, FLAG-TRAF6, GST-IκBα, and GST-cJun have been previously described (28,29). All constructs were verified by automated DNA sequencing.
Recombinant Proteins
Purification of all GST fusion proteins has been previously described (28,29). Recombinant RANKL has been previously described (24,25).
Isothermal Titration Calorimetry (ITC) and Data Analysis
Prior to analysis, purified GST fusion proteins consisting of TAK1 (479–579) and TAB2 (574–693) in which the GST portion was cleaved with thrombin were further purified by gel filtration (Superdex 200). Following overnight dialysis with identical buffer (50 mM NaH2PO4, pH 7.4, 1 mM tris(2-carboxyethel)phosphine hydrochloride), the protein was concentrated in Amicon Ultra-4 tubes (Millipore) with a 5-kDa molecular mass cutoff. ITC measurements were performed at 25°C using a VP-ITC machine that was connected to a computer with ORIGIN software (Microcal Inc, Northampton, MA). Prior to titration, protein samples were centrifuged at 10,000 r.p.m. at 4°C for 10 min and degassed by vacuum aspiration for ~10 min. The calorimeter cell and titration syringe were extensively rinsed with dialysis buffer (50 mM NaH2PO4, pH 7.4, 1 mM tris (2-carboxyethel)phosphine hydrochloride). The calorimetric titrations were carried out with 20–45 injections of 5–10 μl TAK1 residues 479–579 (0.5–0.8 μM), spaced 180 s apart, into the sample cell containing a solution of 1.334 ml TAB2 residues 574–693 (20–80 μM). After subtracting the heat of dilution, the association constant (KA), enthalpy change (ΔH) and the stoichiometry (N) were obtained by fitting the thermograms to a one-set binding site model using ORIGIN software. The remaining thermodynamic parameters, the dissociation constant (KD), free energy change (ΔG), and the entropy change (ΔS) were calculated from the relationships: KA−1 = KD and -RTlnKA = ΔG = ΔH - TΔS.
Transfection and Retroviral Infection
Transfection of HEK293 cells were performed using Calcium Phosphate Transfection kit (Invitrogen; Carlsbad, CA) according to the manufacturer’s instructions. For production of retroviral supernatants, GP2-293 cells were co-transfected with the indicated retroviral vector and pVSV-G (Clontech; Palo Alto, CA) using Fugene 6 transfection reagent (Boehringer Mannheim; Indianapolis, IN) according to manufacturer’s instructions. Twelve hours after transfection, the medium was replaced with fresh medium. Viral supernatants were collected after 48–72 hours and used to infect HEK293, RAW264.7, or L929 cells. After two days, selection was initiated with G418 (500 μg/ml) or puromycin (4 μg/ml). Stable pools of cells or clones were maintained in G418 (300 μg/ml) or puromycin (2 μg/ml).
Reporter Gene Assays
HEK293 cells were plated into 6-wells plates and transfected as above together with an NF-κB-driven luciferase reporter construct (0.5 μg/well). The total amount of DNA was adjusted to 2.5 μg/well with empty vector. Thirty-six hours after transfection, cells were harvested and lysates were analyzed for luciferase activity using Dual-Luciferase Reporter Assay System from Promega (Madison, WI).
Protein Extraction, Immunoprecipitation, and Western Blotting
Cells were harvested, washed two times with ice-cold phosphate-buffered saline (PBS) and then lysed in buffer containing 20 mM TRIS, pH 7.4, 250 mM NaCl, 1 mM DTT, 1 mM sodium orthovanadate, 2 mM EDTA, 1% Triton X-100, 2 mg/ml leupeptin, and 2 mg/ml aprotinin for 20 minutes on ice. Cellular debris was removed by centrifugation at 13K rpm for 15 minutes. Protein was estimated by using the BioRad Protein Assay dye. For immunoprecipitation, equivalent amounts of cell lysate were incubated with various antibodies for 2 hours at 4°C under rotation. Then 15 μl of Protein A/G agarose beads were added and incubated for an additional hour at 4°C under rotation. The immune complex was washed 3 times with lysis buffer and one time with low-salt buffer (20 mM TRIS, pH 7.4, 25 mM NaCl, 1 mM DTT). For GST pull down assays, glutathione agarose beads coated with indicated GST fusion proteins were mixed with equivalent amounts of cell lysate for 2 hours at 4°C under rotation. The protein complexes were washed 3 times with lysis buffer and one time with low-salt buffer. For immunoblotting, the immunoprecipitates, GST pulled-down protein complexes, or cell lysates were resolved on SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (BioRad; Hercules, CA). The membranes were immunoblotted with indicated antibodies followed by the appropriate secondary antibody and the visualized by using the Enhanced Chemiluminescence (ECL) Western Blotting system (Amersham; Piscataway, NJ).
In Vitro Kinase Assays
JNK and NEMO activities were analyzed using GST-Jun and GST-IκBα as substrates as previously described (23–25). Briefly, cells were plated in 6-well or 10 cm plates and the next day they were starved overnight. The following day, cells were treated with the indicated cytokine and washed three times in ice-cold PBS. Cells were lysed for 20 min on ice in lysis buffer and cellular debris was removed by centrifugation at 13k rpm for 15 minutes. Equivalent amount of proteins were immunoprecipitated with anti-JNK1 or anti-NEMO antibodies as described above. The kinase assay was performed in a 2x reaction mix containing 50 mM HEPES, pH 7.4, 20 mM MgCl2, 2 mM DTT, 2.5 μg of GST-Jun or GST-IκBα, and 10 μCi of [γ-32P] ATP at 30°C for 20 min. Reactions were stopped by addition of SDS-sample buffer and subjected to 10% SDS-PAGE. Phosphorylation of GST-Jun or GST- IκBα was detected using autoradiography. The incorporation of [γ-32P] ATP into the substrate was quantitated with a Phosphorimager using ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Osteoclast Differentiation
Stable RAW cell lines were plated in 24-well plates at 1 × 104 cells/well in triplicate, and the next day treated with 100 ng/ml with RANKL. After 5 days, cells were fixed and stained for TRAP after which pictures were taken of representative fields. Osteoclast formation was assessed by counting the number of TRAP-positive multi-nucleated cells per well. In some cases prior to fixing the cells, pictures were taken with a 10× objective lens under bright or fluorescent light.
RESULTS
Binding of TAB2 Occurs in a Non-contiguous Region Within the C-terminal Tail of TAK1
While previous studies have shown that TAB2 could interact with the C-terminus of TAK1, the precise domain in TAK1 that is required for TAB2 binding has not been mapped. To further map the region of TAK1 that is required to bind TAB2, we took two alternative approaches. In the first approach, we created a series of C-terminal deletion mutants of FLAG-tagged TAK1 (Fig. 1A) and co-transfected full-length TAK1 (TAK1-FL) or the indicated TAK1 deletion mutants with FLAG-TAB2 in HEK293 cells. The cell lysates were either immunoprecipitated with anti-TAB2 and immunoblotted with anti-FLAG or immunoblotted with anti-FLAG. TAB2 co-precipitated with only TAK1-FL, TAK1-Δ5 and TAK1-Δ6 (Fig. 1B). Analysis of these TAK1 deletion mutants indicated that TAB2 appears to interact with residues 479–547 of TAK1, since TAK1-Δ5 and TAK1-Δ6 both interacted with TAB2, but not TAK1-Δ1-Δ4 or TAK1-Δ7. In confirmation of these results, we performed GST-fusion pull down experiments with either GST or GST-TAB2 and lysates from transiently transfected HEK293 cells programmed to express various TAK1 deletion mutants. Similar to the co-immunoprecipitation experiments, only TAK1-FL, TAK1-Δ5, and TAK1-Δ6 interacted with GST-TAB2 (Fig. 1C). These results demonstrate that the region of TAK1 comprised between the residues 479–547 interacts with TAB2.
FIGURE 1. Defining the region of TAK1 that interacts with TAB2 and TAB3.

A, Schematic diagram of TAK1 constructs used in this study. The N-terminal region of TAK1 containing the kinase domain is highlighted in black. The C-terminal region is in white. The numbers for mouse TAK1 represent the N- and C-terminal residues of the respective construct. B, The region between residues 479–547 of TAK1 is required to bind TAB2. HEK293 cells were transiently transfected with empty vector (−) or co-transfected with FLAG-TAB2 in the absence or presence of the indicated FLAG-TAK1 constructs. After 36 h, cells were harvested and lysates were immunoprecipitated with anti-TAB2 and immunoblotted with anti-FLAG (top). Expression of the proteins in the cell lysates was determined by immunoblotting with anti-FLAG (bottom). C, TAB2 binds a motif between residues 479–547 within the C-terminal region of TAK1. HEK293 cells were transiently transfected with empty vector (−) or with the indicated FLAG-TAK1 constructs. After 36 h, cells were harvested and lysates were mixed with glutathione agarose beads coated with either GST (G) or GST-TAB2 (T). Bound proteins were determined by immunoblotting with anti-FLAG (top) and expression of the proteins in the lysates was determined by immunoblotting with anti-FLAG (bottom). D–E, Residues 521–545 are not essential for TAB2 interaction. HEK293 cells were transiently transfected with empty vector (−) or co-transfected with FLAG-TAB2 in the absence or presence of the indicated FLAG-TAK1 constructs and immunoprecipitation with anti-TAB2 (D) or a GST pull down (E) was performed essentially as described above. F, TAB3 binds to a similar region on TAK1. HEK293 cells were transiently transfected with empty vector (−) or with the indicated FLAG-TAK1 constructs. After 36 h, cells were harvested and lysates were mixed with glutathione agarose beads coated with either GST (G) or GST-TAB3 (T). Bound proteins were determined by immunoblotting with anti-FLAG (top) and expression of the proteins in the lysates was determined by immunoblotting with anti-FLAG (bottom).
In the alternative approach, we compared the primary amino acid sequence within the last 100 residues of TAK1 from multiple species and designed three internal TAK1 deletion mutants that lacked residues between 508–547 (Fig. 1A; Δ8–Δ10). We analyzed the ability of these TAK1 mutants to interact with co-transfected TAB2 in HEK293 cells. TAB2 co-precipitated with TAK1-FL and TAK1-Δ8, but weakly with TAK1-Δ9 and not at all with TAK1-Δ10 (Fig. 1D). Similarly, GST-TAB2 also interacted with TAK1-FL and TAK1-Δ8, but weakly with TAK1-Δ9 and not at all with TAK1-Δ10 (Fig. 1E), suggesting that residues 521–545 are not essential for the interaction but residues 509–520 are important.
TAB3, a Homologue of TAB2, Binds to the Same Region of TAK1
TAB3 has recently been described as functional homologue of TAB2 (17,18). To determine whether the TAB2-binding region in TAK1 interacts also with TAB3, we transiently transfected HEK293 cells with various TAK1 deletion mutants and examined their ability to precipitate with GST-TAB3. Similar to TAB2, GST-TAB3 interacted only with TAK1-FL, TAK1-Δ6, and TAK1-Δ8 (Fig. 1F). Notably, TAK1-Δ9 interacted weakly with GST-TAB2, but not at all with GST-TAB3 suggesting a slight variation in the binding motif.
A GFP-fusion Protein Containing the C-terminal 100 Residues of TAK1 Binds Both TAB2 and TAB3
The data presented thus far indicate that residues 509–520 of TAK1 are important for binding both TAB2 and TAB3. To determine whether this region is sufficient to interact with TAB2/TAB3, we created green fluorescent protein (GFP) fusion proteins containing residues 505–536 or 505–547 of TAK1 and examined their ability to bind TAB2; however, both of these constructs failed to interact with TAB2 and TAB3 (data not shown). Since both TAB2 and TAB3 interacted with TAK1-Δ6, which contains the C-terminal 100 residues of TAK1, we generated additional GFP-fusion proteins consisting of residues 479–579 (TAK1-C100), 479–520 (TAK1 479–520), 505–553 (TAK1 505–553), and 479–553 (TAK1 470–553) of TAK1 (Fig. 2A). The GFP-fusion protein containing TAK1-C100 and TAK1 479–553 interacted with both TAB2 and TAB3 by co-immunoprecipitation and GST-fusion pull down experiments, while GFP-fusion proteins containing TAK1 479–520 or TAK1 505–553 failed to interact with TAB2 and TAB3 (Fig. 2B–C). Collectively, these data indicate that the interaction of TAB2/TAB3 with TAK1 resides in a non-contiguous region of TAK1, and suggests that residues 509–520 of TAK1 are important for this interaction, but not sufficient for binding TAB2 and TAB3 (see Discussion).
FIGURE 2. TAK1-C100 interacts with both TAB2 and TAB3 and prevents TRAF6-mediated phosphorylation of IκBα.
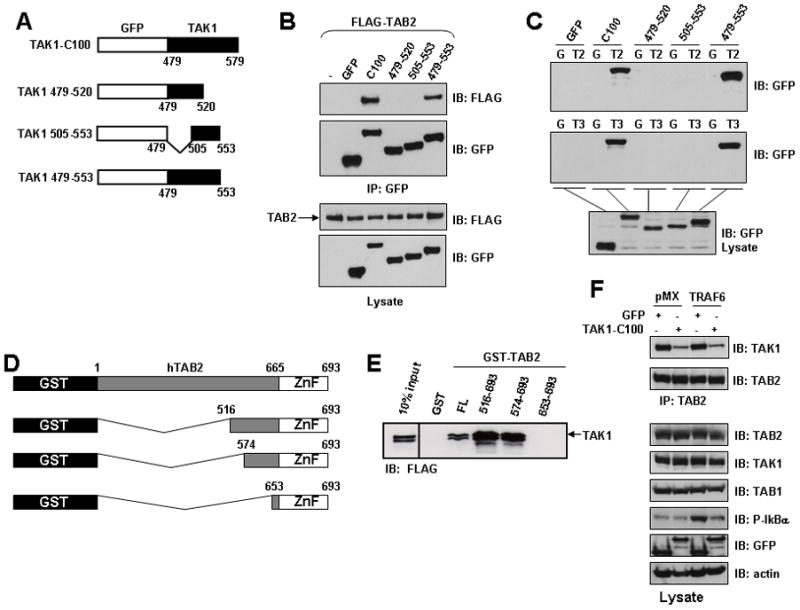
A, Schematic diagram of the GFP fusion proteins TAK1-C100, TAK1 479–520, TAK1 505–553 and TAK1 479–553. The indicated residues of TAK1 were fused to the C-terminus of GFP in the retroviral vector pLEGFP-C1. B, Co-immunoprecipitation of TAK1-C100 and 479–553 with TAB2. HEK293 cells were transiently co-transfected with the indicated constructs. After 36 h, cells were harvested and immunoprecipitated with anti-GFP and bound proteins were visualized by immunoblotting with anti-FLAG. The membrane was stripped and probed with anti-GFP. Expression of the proteins in the lysates was determined by immunoblotting with anti-GFP or anti-FLAG. C, TAK1-C100 and 479–553 bind TAB2 and TAB3. HEK293 cells were transiently transfected with the indicated fusion proteins. After 36 h, cells were harvested and lysates were mixed with glutathione agarose beads coated with either GST (G), GST-TAB2 (T2) or GST-TAB3 (T3). Bound proteins were visualized by immunoblotting with anti-GFP (top) and expression of the proteins in the lysates was determined by immunoblotting with anti-GFP (bottom). D, Schematic diagram of the GST-TAB2 fusion proteins encompassing the C-terminal domain of TAB2. The indicated GST-fusion proteins were constructed with human TAB2. ZnF, Zinc-finger domain. E, Residues 574–653 of TAB2 are sufficient to bind to TAK1. HEK293 cells were transfected with an expression plasmid encoding FLAG-tagged full-length TAK1. Equal amounts of cell lysate were incubated with GST or the different GST-TAB2 mutants immobilized on glutathione agarose beads. After incubation for 2 h at 4°C, the beads were washed, and bound proteins were separated by SDS-PAGE and the bound TAK1 was detected by immunoblotting with an anti-FLAG. Ten percent of the extract used for the pull-down assay is shown on the left. F, TAK1-C100 significantly impairs the interaction between endogenous TAB2 and TAK1 resulting in loss of TRAF6-medieated IKK activation. HEK293 cells stably expressing empty vector (pMX) or TRAF6 were subsequently infected with retrovirus expressing either GFP or TAK1-C100. After neomycin selection, cells were harvested and lysates were immunoprecipitated with anti-TAB2 and then immunoblotted with the indicated antibodies (top two panels). Expression of the indicated proteins in the lysates was visualized with their respective antibody (bottom 6 panels).
The C-terminal 100 Residues of TAK1 Binds to Residues 574–693 of TAB2
Since we mapped a small region of TAK1 that is necessary for interaction with TAB2/TAB3, we also determined the region of TAB2 that interacts with TAK1. We constructed a series of GST-fusion proteins containing the C-terminal region of TAB2 (Fig. 2D) and similar to previous experiments, we examined their ability to interact with TAK1. HEK293 cells were transfected with FLAG-tagged TAK1 and cell lysates were used in GST pull-down experiments. TAK1 interacted with TAB2 mutants containing residues 516–693 or 574–693, but not with residues containing primarily the C-terminal Zn-finger domain of TAB2 (653–693) (Fig. 2E). These data suggest that a region between residues 574–653 of TAB2 is necessary to interact with TAK1. Consistent with these observations, mutation of the Zn-finger domain (C670/673A) of TAB2 did not prevent TAK1 from interacting with TAB2 (data not shown). In confirmation of our findings, we examined quantitatively the interaction between recombinant proteins consisting of the C-termini of both TAK1 (479–579) and TAB2 (574–693) by isothermal titration calorimetry. Measurements from this experiment indicated a stiochiometric-binding ratio of 1:1 between TAK1 and TAB2 with a calculated KD of 65 μM (data not shown). Taken together, these results indicate that the C-terminal 100 residues of TAK1 are sufficient to interact with the C-terminus of TAB2.
TAK1-C100 Significantly Impairs TAB2-TAK1 Interaction and Inhibits TRAF6-induced Activation of IKK
We hypothesized that since TAB2/TAB3 both interacted with the C-terminal 100 residues of TAK1, expression of only the C-terminal tail of TAK1 may function as a dominant negative mutant and block subsequent TAK1 activation and downstream signaling. To explore this possibility, we created a retroviral vector expressing a GFP-fusion protein consisting of the C-terminal 100 residues of TAK1 (TAK1-C100) (Fig. 2A). While a somewhat smaller region was sufficient to interact with TAB2/TAB3, we opted to use TAK1-C100 to ensure correct folding and increase the ability to interact with both TAB2 and TAB3.
The activation of IKK by TRAF6 has been reported to involve the K63-linked auto-ubiquitination of TRAF6, which serves as a scaffold to recruit TAB2 by its ability to selectively bind K63-linked poly-ubiquitin chains (12,21,30,31) (B. G. D., unpublished observations). The formation of a TRAF6-TAB2-TAK1 complex may cause conformational changes in the catalytic domain of TAK1 thereby inducing its kinase activity. HEK293 cells stably expressing TRAF6 or empty vector (pMX) were infected with retrovirus expressing either GFP or TAK1-C100 and selected with neomycin for two weeks. After selection, the cells were harvested and lysates were examined for the interaction between TAB2 and TAK1 by immunoprecipitation with anti-TAB2 and immunoblotting with anti-TAK1. The interaction between TAB2-TAK1 complex was significantly impaired in cells expressing TAK1-C100 compared to the cells expressing only GFP (Fig. 2F). We observed equivalent expression of all of the exogenous proteins as well as TAB1 indicating that the loss of the TAB2-TAK1 complex was not due to downregulation of any of these proteins (Fig. 2F). Furthermore, expression of TRAF6 caused activation of IKK as judged by immunoblotting with anti-phospho IκBα, which was significantly reduced in TRAF6-expressing cells that also expressed TAK1-C100 (Fig. 2F). These data reveal the essential requirement of the TAB2-TAK1 interaction for TAK1-dependent activation of the NF-κB pathway.
TAK1-C100 Blocks NF-κB Activation by RANK, TRAF6, and TAB2
Since TAK1 lies downstream of both RANK and TAB2, we next determined whether TAK1-C100, through its ability to sequester both TAB2/TAB3 could inhibit NF-κB activation induced by RANK and TAB2. HEK293 cells were transiently co-transfected with RANK and TAB2 in combination with GFP or TAK1-C100 together with a NF-κB luciferase reporter vector. While expression of GFP had no effect, TAK1-C100 significantly inhibited RANK- and TAB2-induced NF-κB activation (Fig. 3A). Furthermore, we generated HEK293 cells stably expressing either GFP or TAK1-C100 and then examined the ability of RANK, TRAF6, and TAB2 to activate an NF-κB luciferase reporter in these cell lines. As previously observed, the RANK-, TRAF6-, and TAB2-induced NF-κB activation was significantly impaired in cells expressing TAK1-C100 compared to GFP alone (Fig. 3B). Taken together, these results demonstrate that the interaction between TAB2/TAB3 and TAK1 is critical for TAK1 activation and subsequent NF-κB activation.
FIGURE 3. TAK1-C100 blocks RANK-, TRAF6- and TAB2-induced NF-κB activation.
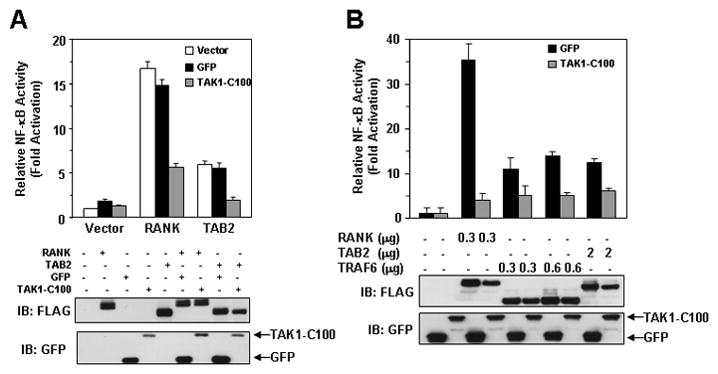
A, RANK- and TAB2-induced NF-κB activation is blocked by TAK1-C100. HEK293 cells were transiently transfected with the indicated constructs together with an NF-κB luciferase reporter vector. After 36 h, cells were harvested and luciferase activity was measured as described in “Experimental Procedures”. Expression of the indicated proteins was detected by immunoblotting the lysates with anti-FLAG (lower panel). B, RANK, TRAF6, and TAB2-induced NF-κB activation is dependent on the interaction between TAB2/3 and TAK1. HEK293 cells stably expressing GFP or TAK1-C100 were transfected with the indicated constructs together with an NF-κB luciferase reporter vector. Cells were processed as described in (A).
TAK1-C100 Inhibits Signaling by IL-1 and TNF
The essential role of TAK1 in activation of NF-κB by IL-1 and TNF has been reported in embryonic fibroblasts lacking TAK1 (32,33). To determine whether TAK1-C100 inhibits IL-1 and TNF signaling, we generated L929 (mouse fibroblast) cell lines stably expressing either GFP or TAK1-C100. Non-infected L929 cells were used as autofluorescence staining control (Fig. 4A). Next, serum-starved cells were stimulated with either IL-1 or TNF for the indicated times and cell lysates prepared. Stable expression of TAK1-C100, but not GFP, inhibited both IL-1-and TNF-induced phosphorylation of IκBα, c-Jun, and p38 (Fig. 4B), which suggests that TAK1-C100 function as a dominant negative inhibitor by blocking the interaction between TAB2/TAB3 and TAK1. To further confirm this hypothesis, we stimulated the two different cell lines with IL-1 and examined whether TAK1 was present in the TAB2 complex by immunoprecipitating with anti-TAB2 antibody. After IL-1 stimulation, L929 cells expressing GFP showed increased amounts of TAK1 co-precipitating with TAB2, but TAK1 was absent in the anti-TAB2 complex from cells expressing TAK1-C100 (Fig. 4C), although equivalent amounts of TAB2 were immunoprecipitated. Furthermore, using a phospho-specific antibody directed against the active form of TAK1 (15,34), the phosphorylation of TAK1 induced by IL-1 stimulation was also inhibited in cells expressing TAK1-C100 (Fig. 4C). These data reveal an essential role of the TAB2-TAK1 complex for both IL-1 and TNF signaling.
FIGURE 4. Interaction between TAB2 and TAK1 is required for IL-1- and TNF-induced signaling.

A, Stable expression of GFP or TAK1-C100 in L929 cells. L929 murine fibroblasts were infected with retrovirus expressing either GFP (pMX-GFP) or TAK1-C100 (pMX-GFP-TAK1-C100). After puromycin selection, pictures were taken with a 10× objective lens under bright light or fluorescence. Non-infected L929 cells were used as autofluorescence staining control. B, TAK1-C100 inhibits the phosphorylation of IκBα, c-Jun, and p38 induced by IL-1 and TNF. The indicated cell lines were plated in 10 cm dishes and serum starved overnight. The next day, the cells were stimulated with either IL-1 (left panels) or TNF (right panels) for the indicated times. The cells were harvested and lysates were subjected to SDS-PAGE and immunoblotted with the indicated antibodies. The membrane was stripped and reprobed with anti-actin. C, TAK1-C100 blocks the interaction between TAB2 and TAK1 and inhibits the phosphorylation of TAK1. The indicated cell lines were treated as in (A) and the lysates immunoprecipitated with anti-TAB2 and then immunoblotted with anti-TAK1 and anti-TAB2 (upper panels). The lysates were subjected to SDS-PAGE and immunoblotted with the indicated antibodies (bottom panels).
TAK1 is Required for RANKL-induced Osteoclast Differentiation
From the data presented above, TAK1-C100 blocked NF-κB activation induced by RANK and its essential modulator TRAF6 (Fig. 3), which suggests that TAK1 may function in the RANKL-RANK pathway. As a prelude to investigating the TAB2-TAK1 complex in the RANK pathway, we first sought to determine whether TAK1 is required for osteoclast differentiation in vitro. The mouse macrophage cell line RAW264.7 (RAW) was infected with a control retrovirus (pMX) or expressing either FLAG-tagged TAK1 (TAK1-WT) or a catalytically inactive mutant of TAK1 (TAK1-K63W) followed by selection with puromycin. After selection, cells were examined for their ability to differentiate into TRAP-positive osteoclast following RANKL treatment for 5 days. Results from this experiment revealed that only control cells or cells expressing TAK1-WT have the ability to form osteoclasts, while cells expressing TAK1-K63W failed to form osteoclasts (Fig. 5A–B). These data demonstrate that the kinase activity of TAK1 is crucial for in vitro RANKL-stimulated osteoclast differentiation.
FIGURE 5. TAK1 is required for osteoclast differentiation.
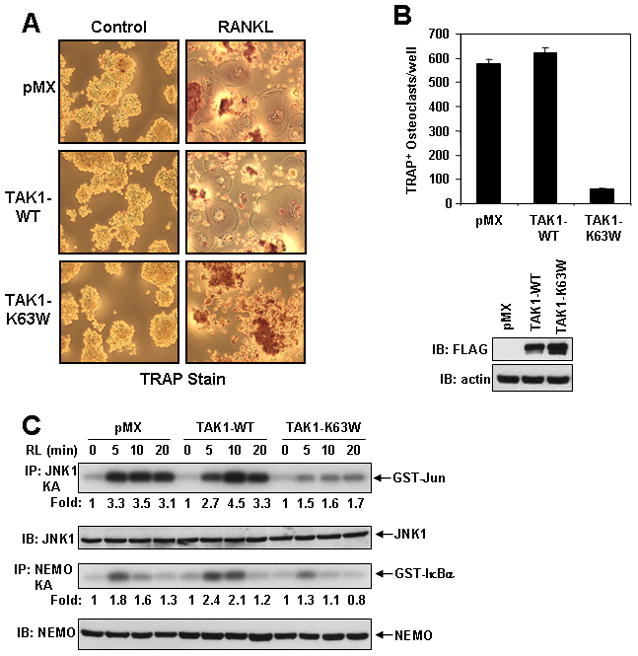
A–B, Catalytically inactive TAK1 inhibits RANKL-mediated osteoclast differentiation. RAW cells stably expressing empty vector (pMX), FLAG-TAK1 (TAK1-WT), or catalytically inactive TAK1 (TAK1-K63W) were plated under osteoclastic conditions and treated the next day with RANKL (100 ng/ml). After 5 days, cells were fixed and stained for TRAP. Pictures of representative wells were taken with a 10× objective lens (A) and the number of TRAP-positive multi-nucleated osteoclasts per well were counted and represented as the average per well (in triplicate) from two independent experiments (B). The expression of TAK1-WT and TAK1-K63W was detected by immunoblotting with anti-FLAG antibody. The membrane was stripped and reprobed with anti-actin. C, TAK1-K63W inhibits RANKL-induced JNK and IKK activation. The indicated RAW cell lines were treated with RANKL (100 ng/ml) for the indicated times and the cells harvested. Lysates were subjected to immunoprecipitation with either JNK1 or NEMO antibodies and in vitro kinase assays performed using GST-Jun or GST-IκBα respectively, as substrates. The incorporation of 32P into the substrates was quantitated with a Phosphorimager and ImageQuant software, and represented as fold activation compared to time zero. Lysates were also immunoblotted with JNK1 and NEMO antibodies.
Catalytically Inactive TAK1 Inhibits RANKL-induced IKK and JNK Activation
Loss of osteoclast differentiation by RANKL in cells expressing TAK1-K63W may reflect the absence of IKK and/or JNK activation by RANKL. To test this hypothesis, we examined the activation of IKK and JNK after RANKL stimulation in RAW cells stably expressing empty vector, TAK1-WT, or TAK1-K63W. The RANKL-mediated activation of both IKK and JNK was significantly reduced in cells expressing TAK1-K63W as compared to either cells expressing empty vector or TAK1-WT (Fig. 5C). These data demonstrate that TAK1-K63W functions as a dominant negative regulator of RANKL-mediated osteoclast differentiation most likely by blocking both IKK and JNK activation.
TAK1-C100 Inhibits RANKL-mediated Osteoclast Differentiation and Signaling
The data presented thus far have demonstrated that the interaction between TAB2 and TAK1 is essential for activation of NF-κB and MAPK induced by IL-1 and TNF or by transient expression of RANK, TAB2, and TRAF6. We next addressed whether the interaction of TAK1 with TAB2 is required for RANKL-mediated signaling and osteoclast differentiation. We generated RAW cells stably expressing GFP or TAK1-C100 and selected individual clones expressing each protein for further analysis. First, we examined RANKL-mediated osteoclast differentiation in these clones expressing either GFP or TAK1-C100. Expression of TAK1-C100, but not GFP alone, prevented RANKL-dependent osteoclast differentiation as visualized by TRAP-positive staining (Fig. 6A). As shown graphically, TAK1-C100 significantly reduced the number of TRAP-positive osteoclasts induced by RANKL (Fig. 6B). These results demonstrate that the interaction between TAB2 and TAK1 is necessary for RANKL-induced osteoclast differentiation.
FIGURE 6. RANKL-mediated osteoclast differentiation and signaling is dependent on the interaction between TAB2/3 and TAK1.

A–B, Blocking the interaction between TAB2/3 and TAK1 inhibits osteoclast differentiation. RAW clones stably expressing GFP or TAK1-C100 were plated under osteoclastic conditions and the next day treated with RANKL (100 ng/ml). After 5 days, the cells were fixed, TRAP stained, and pictures were taken using a 10× objective lens as indicated (A). The number of TRAP-positive multi-nucleated osteoclasts per well were counted and represented as the average per well (in triplicate) from two independent experiments (B). The expression of GFP and TAK1-C100 was detected by immunoblotting with anti-GFP antibody. The membrane was stripped and reprobed with anti-actin. C, TAK1-C100 inhibits RANKL-induced JNK and IKK activation. RAW cells stably expressing GFP or TAK1-C100 were treated with RANKL (100 ng/ml) for the indicated times. Cells were harvested and lysates were processed as described in Fig. 5C. D, TAK1-C100 abolishes RANKL-induced NFATc1 accumulation in the nucleus. RAW cells stably expressing GFP or TAK1-C100 were plated at 5 × 104 cells/well in 6-wells plates and the next day stimulated with RANKL (100 ng/ml) for the indicated times. Cells were harvested and nuclear extracts were subjected to SDS-PAGE and immunoblotted with anti-NFATc1 (upper panels). The membrane was stripped and reprobed with anti-tubulin (lower panels).
We next examined the activation of IKK and JNK upon RANKL stimulation in RAW cells expressing GFP or TAK1-C100. The RANKL-induced activation of both JNK and IKK was notably impaired in cells expressing TAK1-C100 as compared to cells expressing only GFP (Fig. 6C). Furthermore, the transcription factor NFATc1 plays an important role in terminal differentiation of osteoclast progenitors (35–39). We examined for expression of NFATc1 in nuclear extracts following RANKL stimulation for zero, 12, 24, and 48 hours in cells expressing either GFP or TAK1-C100. RAW cells expressing GFP exhibited nuclear accumulation of NFATc1 after RANKL stimulation, which was completely abolished in cells expressing TAK1-C100 (Fig. 6D). Taken together these results demonstrate that the interaction between TAB2/TAB3 and TAK1 is required to obtain full activation of TAK1, which in turn activates both the IKK complex and MAPKs pathways. Moreover, interfering with the interaction between TAB2 and TAK1 also results in the loss of induction of NFATc1 by RANKL.
DISCUSSION
In the IL-1 and TNF pathways, genetic ablation of TAK1 in mice has implicated this MAP3K in the activation of IKK and JNK, which results in the induction of NF-κB and AP1 transcription factors, respectively (32,33). However, IL-1 and TNF activate these pathways in the absence of either TAB1 or TAB2; the later of which is likely compensated its homologue TAB3. The functional role of TAB1 in this process remains unclear at present. Previous studies have demonstrated that TRAF6 interacts with TAB2 in an IL-1-dependent manner, resulting in the formation of a TRAF6-TAB2-TAK1 complex that is required for activation of NF-κB and JNK (15). In this model, TAB2 acts as a bridge linking TRAF6 to TAK1 and thereby mediating the activation of TAK1 in the IL-1 pathway. Similarly, since TRAF6 is essential for RANKL-mediated signaling (11,40–44), engagement of the RANK pathway may use the identical TRAF6-TAB2-TAK1 complex for its signaling leading to osteoclast differentiation. In contrast, stimulation by TNF causes the site-specific K63 ubiquitination of RIP1 in the TNFR1 complex by an unidentified E3 ligase (possibly TRAF2 or cIAP1), which serves to recruit TAB2 and NEMO resulting in the receptor recruitment of the TAK1 (through the NZF ubiquitin binding domain of TAB2/3) and IKK complexes (45). While this model demonstrates an essential requirement of TAK1 in activating these signaling cascades, the functional interaction between TAB2/TAB3 and TAK1 remains elusive due to a suitable cell system lacking both TAB2 and TAB3.
We hypothesized that disruption of the constitutive interaction between TAB2/TAB3 may interfere with TAK1 activation since previous data indicated that silencing both TAB2 and TAB3 by siRNA knockdown abolished IL-1 and TNF signaling (19). We sought an alternative strategy “to neutralize” both TAB2 and TAB3 by developing a construct that could interact with both TAB2/TAB3 and block their interaction with TAK1. To test our hypothesis, we first characterized the region of TAK1 that is necessary to interact with TAB2 and TAB3. Through deletion mapping, co-immunoprecipitation, and GST pull-down experiments, we demonstrated that residues 509–520 of TAK1 appear to be important for binding both TAB2 and TAB3, but not sufficient. However, a larger region encompassing residues 479–553 of TAK1 was sufficient for interacting with TAB2 and TAB3.
Further analysis utilizing programs to predict secondary structure (46) of the C-terminal 100 residues of TAK1 indicated an alpha-helical structure (Fig. 7). In brief, the last 100 residues of TAK1 are predicted to form four alpha helices (Helix 1, 490–505; Helix 2, 512–526; Helix 3, 536–549; Helix 4, 553–573). Since TAK1-Δ5, which terminated with residue 547, and GFP-TAK1 479–553 interacted with TAB2/TAB3, Helix 4 probably does not contribute to this interaction as summarized in Fig. 7. We also generated GFP-fusion constructs encompassing Helix 1 and part of Helix 2 (TAK1 479–520), Helix 2 (TAK1 505–536), both Helix 2 and 3 (TAK1 505–547 and 505–553), or Helix 1, 2, and 3 (TAK1 479–553), but all of these constructs except TAK1 479–553 failed to interact with TAB2/TAB3. These results suggest that the C-terminal 100 residues of TAK1, with the exception of Helix 4, forms a tertiary conformation and that eliminating any one or more of these predicted helices may interfere with the intermolecular contacts required for the proper folding of this domain. Nonetheless, the C-terminal region of TAK1 appears as a functional domain that is required for interacting with TAB2/TAB3.
FIGURE 7. Predicted secondary structure of TAK1-C100 and summary of the interaction of TAK1 mutants with TAB2 and TAB3.
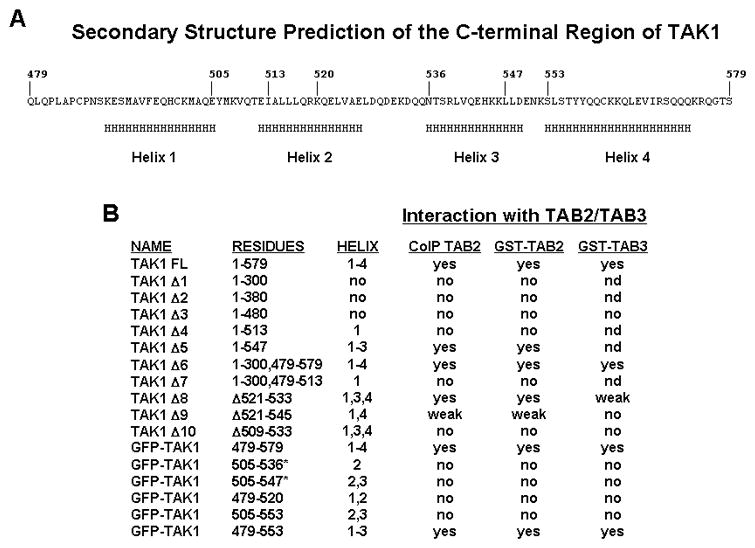
A, Schematic diagram of the C-terminal 100 residues of TAK1. A secondary structure prediction program (46) was used on the last 100 residues of TAK1 and regions of alpha-helical structure (H) were assigned based upon a confidence level of greater that 80%. B, Summary of the interaction between TAK1 and TAB2/TAB3 based upon results depicted in Figures 1 and 2. Yes, positive interaction; no, no interaction; weak, weak interaction; nd, not determined; *, data not shown.
Since TAK1-C100 was able to bind both TAB2 and TAB3, we next investigated whether this region could functionally block the interaction of endogenous TAB2/TAB3 and TAK1. Using a retroviral delivery system, we presented evidence that a GFP fusion protein consisting of TAK1-C100 was able to significantly impair the constitutive interaction of TAB2/TAB3 with TAK1. Furthermore, using L929 mouse fibroblasts stably expressing TAK1-C100 we clearly demonstrated that interfering with the TAB2/TAB3-TAK1 interaction blocked NF-κB and MAPKs signaling upon IL-1 and TNF stimulation through the inhibition of TAK1 phosphorylation. Moreover, expression of TAK1-C100 in HEK293 cells abolished the ability of RANK, TRAF6, and TAB2 to activate an NF-κB luciferase reporter. Collectively, these data support a critical role of the interaction of either TAB2 or TAB3 with TAK1 for its activation and therefore its ability to stimulate the IKK and JNK pathways in response to IL-1 and TNF stimulation.
Analogous to the IL-1 signal transduction pathway, RANKL requires engagement of the TRAF6 machinery for its signaling since TRAF6-deficiency results in complete loss of RANKL signaling and osteoclast differentiation (11,40). Notably, the ubiquitin ligase activity of TRAF6 and its auto-ubiquitination are essential for NF-κB and JNK activation in osteoclast progenitors (B. L. and B. G. D., unpublished observations). Ectopic expression of a catalytically inactive TAK1 abolished RANKL-mediated IKK and JNK activation and consequently the differentiation of these cells into TRAP-positive osteoclasts, suggesting an essential role of TAK1 in this process. In support of this hypothesis, RAW cells stably expressing TAK1-C100 showed dramatic loss of RANKL-mediated IKK and JNK activation and osteoclast differentiation. Furthermore, the nuclear accumulation of NFATc1, a critical transcription factor for osteoclast differentiation, was considerably reduced in TAK1-C100 expressing RAW cells. These results place TAK1 as a potential upstream activator of NFATc1 in the formation of multi-nucleated osteoclasts.
In summary, we have demonstrated in this study the fundamental role of TAB2/TAB3 as key adaptors between different receptor complexes and a central kinase, TAK1, involved in multiple pathways. By identifying a region of TAK1 that is necessary for interacting with TAB2/TAB3, we were able to develop a model system to explore the functional requirement of this interaction in signaling by IL-1, TNF, and RANKL. Indeed, TAB2-deficient MEFs showed normal activation of downstream signaling pathways upon IL-1 and TNF stimulation, suggesting that the TAB2 homologue TAB3 compensates for the absence of TAB2 (data not shown). Nevertheless, in the current proposed model (47), blocking TAB2-TAK1 and TAB3-TAK1 interactions would likely prevent the recruitment of TAK1 to the TRAF6 and TRAF2 complexes and consequently TAK1 activation. Further investigation will be necessary to understand the role of TAB2/TAB3 in TAK1-dependent signaling possibly by generation of conditional knockout mice deficient in both TAB2 and TAB3.
Acknowledgments
We wish to generously thank Dr. Kitamura for providing the pMX vectors; Dr. Akira and Ninomiya-Tsuji for providing the TAB2 wild type and knockout MEFs; and the structural biology groups at Memorial Sloan-Kettering Cancer Center for generous access to the Micro Calorimetry System. The DNA Sequencing Core facility is funded in part through a grant from the NCI (CA16672DAF). This work was supported in part by institutional start-up funds (B. G. D.) and NIH RO1-AI 45937 (H. W.).
Footnotes
This study was supported in part by Start-up development funds from The University of Texas MD Anderson Cancer Center to B. G. D.
The abbreviations used are: IL-1, interleukin-1; IP, immunoprecipitation; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; TNF, tumor necrosis factor; TAB1, 2, 3, TAK1 binding protein 1, 2, 3; GST, glutathione S-transferase; DTT, 1,4-Dithiothreitol; GFP, green fluorescent protein; TRAF, TNF receptor-associated factor; TGF-β, transforming growth factor β; TAK1, TGF-β-activated kinase 1; IKK, IκB kinase; RANK, receptor activator of NF-κB; MEF, mouse embryonic fibroblast; WT, wild type; HEK, human embryonic kidney; NF-κB, nuclear factorκB; ERK, extracellular signal-regulated kinase; NFAT, nuclear factor of differentiation factor; RANKL, RANK ligand; TRAP tartrate-resistant acid phosphatase; PI3K, phosphoinositide-3OH kinase; MAP3K, MAPKK kinase; NZF, novel zinc finger; LPS, lipopolysaccharide; MKK, MAP kinase kinase; RING, really interesting new gene; SDS, sodium dodecylsulfate; PBS, phosphate-buffered saline.
References
- 1.Dinarello CA. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 2.Baud V, Karin M. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 4.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. Nature. 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 5.Irie T, Muta T, Takeshige K. FEBS Lett. 2000;467:160–164. doi: 10.1016/s0014-5793(00)01146-7. [DOI] [PubMed] [Google Scholar]
- 6.Sakurai H, Miyoshi H, Toriumi W, Sugita T. J Biol Chem. 1999;274:10641–10648. doi: 10.1074/jbc.274.15.10641. [DOI] [PubMed] [Google Scholar]
- 7.Wald D, Commane M, Stark GR, Li X. Eur J Immunol. 2001;31:3747–3754. doi: 10.1002/1521-4141(200112)31:12<3747::aid-immu3747>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 8.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 9.Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa JL, Ferrick D, Hum B, Iscove N, Ohashi P, Rothe M, Goeddel DV, Mak TW. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- 10.Silverman N, Maniatis T. Genes Dev. 2001;15:2321–2342. doi: 10.1101/gad.909001. [DOI] [PubMed] [Google Scholar]
- 11.Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M, Yamamoto T, Inoue J. Genes Cells. 1999;4:353–362. doi: 10.1046/j.1365-2443.1999.00265.x. [DOI] [PubMed] [Google Scholar]
- 12.Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. J Biol Chem. 2006 doi: 10.1074/jbc.M608867200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen ZJ. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen ZJ, Bhoj V, Seth RB. Cell Death Differ. 2006 doi: 10.1038/sj.cdd.4401869. [DOI] [PubMed] [Google Scholar]
- 15.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, Ninomiya-Tsuji J, Matsumoto K. Mol Cell. 2000;5:649–658. doi: 10.1016/s1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 16.Kishida S, Sanjo H, Akira S, Matsumoto K, Ninomiya-Tsuji J. Genes Cells. 2005;10:447–454. doi: 10.1111/j.1365-2443.2005.00852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin G, Klika A, Callahan M, Faga B, Danzig J, Jiang Z, Li X, Stark GR, Harrington J, Sherf B. Proc Natl Acad Sci U S A. 2004;101:2028–2033. doi: 10.1073/pnas.0307314101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung PC, Nebreda AR, Cohen P. Biochem J. 2004;378:27–34. doi: 10.1042/BJ20031794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishitani T, Takaesu G, Ninomiya-Tsuji J, Shibuya H, Gaynor RB, Matsumoto K. Embo J. 2003;22:6277–6288. doi: 10.1093/emboj/cdg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ. Mol Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 21.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 22.Sanjo H, Takeda K, Tsujimura T, Ninomiya-Tsuji J, Matsumoto K, Akira S. Mol Cell Biol. 2003;23:1231–1238. doi: 10.1128/MCB.23.4.1231-1238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darnay BG, Ni J, Moore PA, Aggarwal BB. J Biol Chem. 1999;274:7724–7731. doi: 10.1074/jbc.274.12.7724. [DOI] [PubMed] [Google Scholar]
- 24.Polek TC, Talpaz M, Darnay BG, Spivak-Kroizman T. J Biol Chem. 2003;278:32317–32323. doi: 10.1074/jbc.M302518200. [DOI] [PubMed] [Google Scholar]
- 25.Ye H, Arron JR, Lamothe B, Cirilli M, Kobayashi T, Shevde NK, Segal D, Dzivenu OK, Vologodskaia M, Yim M, Du K, Singh S, Pike JW, Darnay BG, Choi Y, Wu H. Nature. 2002;418:443–447. doi: 10.1038/nature00888. [DOI] [PubMed] [Google Scholar]
- 26.Kitamura T. Int J Hematol. 1998;67:351–359. doi: 10.1016/s0925-5710(98)00025-5. [DOI] [PubMed] [Google Scholar]
- 27.Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, Kumagai H. Exp Hematol. 2003;31:1007–1014. [PubMed] [Google Scholar]
- 28.Darnay BG, Aggarwal BB. Ann Rheum Dis. 1999;58(Suppl 1):I2–I13. doi: 10.1136/ard.58.2008.i2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darnay BG, Haridas V, Ni J, Moore PA, Aggarwal BB. J Biol Chem. 1998;273:20551–20555. doi: 10.1074/jbc.273.32.20551. [DOI] [PubMed] [Google Scholar]
- 30.Sun L, Chen ZJ. Curr Opin Cell Biol. 2004;16:119–126. doi: 10.1016/j.ceb.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 32.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Nat Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 33.Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee KY, Bussey C, Steckel M, Tanaka N, Yamada G, Akira S, Matsumoto K, Ghosh S. Genes Dev. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakurai H, Miyoshi H, Mizukami J, Sugita T. FEBS Lett. 2000;474:141–145. doi: 10.1016/s0014-5793(00)01588-x. [DOI] [PubMed] [Google Scholar]
- 35.Ishida N, Hayashi K, Hoshijima M, Ogawa T, Koga S, Miyatake Y, Kumegawa M, Kimura T, Takeya T. J Biol Chem. 2002;277:41147–41156. doi: 10.1074/jbc.M205063200. [DOI] [PubMed] [Google Scholar]
- 36.Hirotani H, Tuohy NA, Woo JT, Stern PH, Clipstone NA. J Biol Chem. 2004;279:13984–13992. doi: 10.1074/jbc.M213067200. [DOI] [PubMed] [Google Scholar]
- 37.Gohda J, Akiyama T, Koga T, Takayanagi H, Tanaka S, Inoue J. Embo J. 2005;24:790–799. doi: 10.1038/sj.emboj.7600564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang H, Ryu J, Ha J, Chang EJ, Kim HJ, Kim HM, Kitamura T, Lee ZH, Kim HH. Cell Death Differ. 2006 doi: 10.1038/sj.cdd.4401882. [DOI] [PubMed] [Google Scholar]
- 39.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 40.Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, Inoue J. Embo J. 2001;20:1271–1280. doi: 10.1093/emboj/20.6.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizukami J, Takaesu G, Akatsuka H, Sakurai H, Ninomiya-Tsuji J, Matsumoto K, Sakurai N. Mol Cell Biol. 2002;22:992–1000. doi: 10.1128/MCB.22.4.992-1000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, Yokochi T, Oda H, Nakamura K, Ida N, Wagner EF, Taniguchi T. Nature. 2002;416:744–749. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- 43.Wu H, Arron JR. Bioessays. 2003;25:1096–1105. doi: 10.1002/bies.10352. [DOI] [PubMed] [Google Scholar]
- 44.Takayanagi H. J Mol Med. 2005;83:170–179. doi: 10.1007/s00109-004-0612-6. [DOI] [PubMed] [Google Scholar]
- 45.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 46.Rost B, Sander C. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 47.Israel A. Trends Immunol. 2006;27:395–397. doi: 10.1016/j.it.2006.07.003. [DOI] [PubMed] [Google Scholar]