

SUMMARY
Dark- and light-adaptation of retinal neurons allows our vision to operate over an enormous light intensity range. Here we report a novel mechanism which controls the light sensitivity and operational range of rod-driven bipolar cells that mediate dim-light vision. Our data indicate that the light responses of these cells are enhanced by sustained chloride currents via GABAC receptor channels. This sensitizing GABAergic input is controlled by dopamine D1 receptors, with horizontal cells serving as a plausible source of GABA release. Our findings expand the role of dopamine in vision from its well-established function of suppressing rod-driven signals in bright light to enhancing the same signals under dim illumination. They further reveal a novel role for GABA in sensitizing the circuitry for dim-light vision, thereby complementing GABA’s traditional role in providing dynamic feedforward and feedback inhibition in the retina.
INTRODUCTION
During the day/night cycle, our visual system faces the challenge of operating over a light intensity range that covers more than 9 orders of magnitude (Rodieck, 1998). To meet this challenge the retina undergoes dark- and light-adaptation at all levels of processing, including the various stages of rod-driven circuitry which mediate dim light vision (Dunn et al., 2006; Shapley and Enroth-Cugell, 1984). The types of retinal neurons participating in the primary rod circuit and addressed in this study are illustrated in Figure 1A. Rod photoreceptors provide glutamatergic input to a single class of rod bipolar cells that depolarize upon light stimulation (depolarizing “ON” bipolar cells, DBCs), a response triggered by cessation of glutamate release from rod synapses. Axon terminals of rod DBCs are located in the inner retina where they form synapses with AII-amacrine cells. The signals are further processed by cone ON-bipolar and retinal ganglion cells and transmitted to the brain via the optic nerve.
Figure 1. Reduced sensitivity and operational range of rod-driven DBCs in D1R−/− mice and localization of D1R in the retina.

(A) Cartoon illustrating the cell types discussed in this study. See text for details. (B) Representative ERG recordings from WT and D1R−/− mice under dark- and light-adapted conditions. Light intensity for flash and background light is given in units of photoexcited rhodopsin molecules per rod (R*/rod) and photoexcited rhodopsin molecules per rod per second (R*/rod/s), respectively. (C) Sensitivities of rod-driven ERG b-waves were determined for five dopamine receptor knockout mice, normalized to the dark sensitivity of WT mice (S/Sdark, WT) and plotted as a function of background light intensity (mean ± SEM). Here and in figures below see Table S1 for the summary of fitting parameters. (D) D1R immunostaining in WT and D1R−/− retinal cross-sections (green). Nuclei are stained with Hoechst (blue). (E) WT retinal cross-section stained for D1R and calbindin, which labels horizontal cells in the OPL/INL and a class of amacrine cells in the INL/IPL. (F) Confocal tangential z-sections of a retina flat-mount co-stained for D1R and the rod DBC marker, PKCα. Positions of individual z-sections relative to DBC morphology are illustrated in cartoons on the right from each panel. Abbreviations: ONL – outer nuclear layer; OPL – outer plexiform layer; INL – inner nuclear layer; IPL – inner plexiform layer; GCL – ganglion cell layer. Scale bars: 25 μm.
The strength and duration of light signals traveling through the rod-driven circuit is shaped by two classes of retinal interneurons (Wassle, 2004). Amacrine cells regulate the synaptic output of rod DBCs by GABAergic and glycinergic inputs, providing both lateral and temporal inhibitory feedback (Chavez et al., 2010; Eggers and Lukasiewicz, 2006; Tachibana and Kaneko, 1987). Horizontal cell axon terminals provide lateral feedback inhibition directly onto rods (Babai and Thoreson, 2009) and potentially feedforward inhibition onto bipolar cell dendrites (Yang and Wu, 1991). However, the precise mechanisms by which horizontal cells communicate with other neurons remain controversial (Kamermans and Spekreijse, 1999). It also remains unknown whether horizontal cells play a direct role in setting the light sensitivity of the rod-driven circuitry.
Dopamine, another major neurotransmitter in the retina, is produced by a single class of amacrine cells (Figure 1A) and has been long known to modulate retinal circuitry to favor cone-driven pathways during the daytime (Witkovsky, 2004). The goal of this study was to investigate whether dopamine is involved in controlling the light sensitivity and adaptation of rod-driven DBCs. We now demonstrate that dopamine is also critical for sensitizing rod-driven DBC responses in the dark and under dim light. This sensitizing effect of dopamine is mediated only by D1-type dopamine receptors (D1R), with horizontal cells serving as a plausible dopamine target. We further demonstrate that this D1R-dependent mechanism is conveyed through a GABAergic input via GABAC receptors (GABACR) expressed in rod-driven DBCs. Taken together, these observations reveal entirely novel roles of dopamine and GABA in the retina circuitry. They expand the role of dopamine from a messenger of bright light adaptation to a facilitator of dim-light vision and expand the role of GABA from a strictly inhibitory transmitter to a sensitizer of the rod-driven circuit.
RESULTS
The role of dopamine D1 receptor in setting light sensitivity of rod-driven DBCs
To elucidate whether dopamine can regulate rod-driven circuitry at the level of DBCs, we examined their function in knockout mouse lines each lacking one of the five mammalian dopamine receptors (D1R−/−, D2R−/−, D3R−/−, D4R−/− and D5R−/−). We used the non-invasive technique of electroretinography (ERG), which characterizes the DBC light-responses in vivo without perturbing any neuronal connections and surrounding neurotransmitter levels, or altering intra- and extracellular ion concentrations (Robson and Frishman, 1998).
A typical dark-adapted ERG evoked by a dim flash consists mainly of a positive signal, the “b-wave”, which reflects the cumulative depolarization of rod DBCs (Robson and Frishman, 1998; Robson et al., 2004). We found that the ERG b-wave amplitude of D1R−/− mice was smaller than of WT controls, particularly in the presence of adapting background illumination (Figure 1B). The corresponding response sensitivities, determined for each level of background light as a ratio between the maximal b-wave amplitude and half-saturating flash intensity, normalized to the WT dark-adapted values, are plotted in Figure 1C. This analysis demonstrates that absence of D1R expression reduces the rod DBC “operational range”, the range of background light intensities over which a detectable ERG response can be evoked (see Supplemental Experimental Procedures for explanation of how cone-driven contributions were excluded from this analysis). Similar results were obtained upon pharmacological blockade of D1R in wild type (WT) mice (Figure S1). We also showed that the retinal morphology in D1R−/− mice was normal, ruling out a role of anatomical abnormality as the cause of the ERG phenotype (Figure S2). This phenotype was strictly specific for D1R−/− mice and was not observed in mice lacking the other dopamine receptors, D2R, D3R, D4R and D5R (Figure 1C).
Immunostaining of WT retinas, using D1R−/− retinas as controls, demonstrated that D1R is expressed in both the inner and outer plexiform layers (Figures 1D and 1E; see also Veruki and Wassle, 1996). Although, D1R expression is observed in a subset of cone bipolar cells (e.g. Veruki and Wassle, 1996), we did not detect D1R signals in rod DBCs when we systematically examined individual confocal z-sections through the entire DBC length in retinal flat-mounts co-stained for D1R and a rod DBC marker, PKCα (Figure 1F). This indicates that any dopaminergic regulation of rod DBC responses is mediated by another neurotransmitter’s input from other retinal neurons.
The effect of dopamine on rod-driven DBCs is conveyed via a GABAergic input
Because light-responses of rod DBCs are regulated by GABAergic inputs from amacrine and potentially horizontal cells (McCall et al., 2002; Suzuki et al., 1990; Tachibana and Kaneko, 1987; Yang and Wu, 1991) (Figure 1A), we hypothesized that the phenotype of D1R−/− mice may originate from an alteration of one of these mechanisms. GABA acts on rod DBCs through two types of GABA receptors, GABAAR and GABACR, both chloride channels (e.g. Chavez et al., 2010; Lukasiewicz and Shields, 1998; McCall et al., 2002). Therefore, we examined b-wave responses in mice where individual GABA receptor expression was eliminated or pharmacologically blocked. We first analyzed GABACR knockout (GABACR−/−) mice (McCall et al., 2002) and found that they display a phenotype strikingly similar to that of D1R−/− mice (Figures 2A and 2B). GABACR−/− and D1R−/− mice had both a substantial reduction in dark-sensitivity (~40% and ~55%, respectively) and compression of the operational range. In contrast, blocking GABAAR-mediated input pharmacologically (there is no knockout available that removes all GABAARs from DBCs) did not affect either dark-sensitivity or operational range of rod-driven b-waves (Figure 2B). These data reveal that GABACRs regulate the light-sensitivity of rod-driven DBCs and raise the possibility that the effect of the D1R knockout may be explained by an altered GABACR-mediated input onto rod-driven DBCs. We also note that these GABACR-mediated effects on rod DBCs cannot be explained by altered rod photoreceptor synaptic output since rods do not express GABACRs (Enz et al., 1995).
Figure 2. The sensitivity and operational range of rod-driven DBC responses are regulated by GABACR.

(A) ERG recordings from WT and GABACR−/− mice under dark- and light-adapted conditions. (B) ERG b-wave sensitivity plots for the following mice and conditions: WT, GABACR−/−, WT injected with the GABAAR antagonist SR-95531, WT injected with GABACR antagonist TPMPA, D1R−/− injected with TPMPA. (C) ERG recordings from dark- or light-adapted WT and D1R−/− mice with and without GABA injections. (D) WT and D1R−/− ERG b-wave sensitivity plots in the absence or presence of injected GABA.
In reciprocal experiments, we measured ERG responses after intraocular injections of GABA (Figures 2C and 2D). As has been reported previously, GABA increased b-wave amplitudes (Naarendorp and Sieving, 1991; Robson et al., 2004). Despite this amplitude increase, GABA did not affect the sensitivity or operational range of b-wave responses in WT mice (Figure 2D). Although GABA injections into D1R−/− mice also increased b-wave amplitudes, in this case both the light-sensitivity and operational range of b-waves were restored to those observed in WT animals. (These phenomena were phenocopied in WT mice after pharmacological block of D1R; Figure S1). These data show that the lack of D1R-mediated signaling can be completely masked by exogenous GABA, consistent with a role for D1R in modulating a GABAergic input onto rod-driven DBCs.
Interestingly, intraocular injections of glycine, which normally provides lateral feedback onto rod DBC axons via chloride currents through glycine receptor channels, reduced the b-wave sensitivity functions in both WT and D1R−/− mice, rather than restoring the loss of sensitivity and operational range in the D1R knockout, as occurred in case of GABA injections into the eyes of D1R−/− mice (Figure S3). This suggests that the GABAC-dependent mechanism revealed in this study is specific and implies a difference in ionic microenvironments surrounding GABACR and glycine receptors.
Our hypothesis that D1R mediates a GABAergic input onto rod-driven DBCs predicts that pharmacological blockade of GABACRs should not further reduce b-wave sensitivity in D1R−/− mice. Indeed, inactivation of GABACRs in WT mice with the specific antagonist, TPMPA, phenocopied the effect of the GABACR knockout, but added little additional effect to the b-wave phenotype of D1R−/− animals, except under dark-adapted conditions at which the absence of D1R may not have completely eliminated effects of GABACR on the bipolar cells (Figure 2B). Taken together, these data indicate the presence of a sensitizing dopamine-dependent GABACR-mediated input onto rod-driven DBCs, a mechanism responsible for increasing DBC light-sensitivity and extending their operational range.
Sustained GABACR-mediated input onto rod DBCs hyperpolarizes their resting membrane potential and decreases their input resistance
To gain further insight into how GABACRs could sensitize rod DBCs we first analyzed maximal amplitudes of dark-adapted rod-driven ERG b-waves (Rmax,dark, Figure 3), which would be proportional to the extent of DBC depolarization upon a saturating light response (Robson et al., 2004). The Rmax,dark in D1R−/− and GABACR−/− mice were reduced by ~25% and 50%, respectively, suggesting that sustained dopamine/GABACR-mediated chloride currents in WT retina normally extend the voltage range between the resting potential and maximal light-evoked depolarization. The role of the GABACR in defining this range was further confirmed by GABA injections, which increased Rmax,dark ~2-fold in WT and D1R−/− mice, but caused no increase in GABACR−/− animals. This suggests that normally the GABACR-dependent current is not saturated and can be further activated by increases in GABA beyond its tonic physiological level.
Figure 3. Normalized maximal dark-adapted amplitudes of rod-driven b-waves differ across mice with and without GABACR-meditated input.
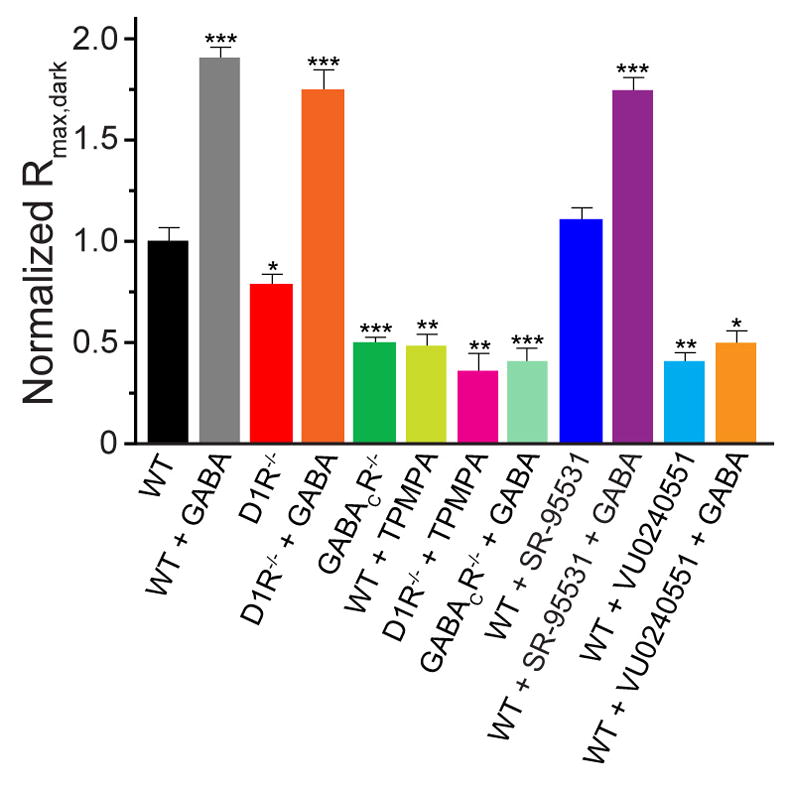
Paired t-tests between WT and each other animal/condition yielded p-values <0.025 (*), <0.005 (**) and <0.001 (***).
The concept that a tonic GABACR-mediated chloride current makes a major contribution to setting the voltage range of rod DBC ERG responses presumes that the chloride equilibrium potential in the resting state is negative to the resting potential. This chloride influx would hyperpolarize a rod DBC in a manner comparable (see Figure 4A) to the potassium outflux traditionally considered to fulfill this function (e.g. Tessier-Lavigne et al., 1988). The equivalent circuit in Figure 4B illustrates that the electrochemical gradients of chloride and potassium are completely interchangeable and either can hyperpolarize the rod DBCs and provide the electrical driving force for the light-induced cation influx that occurs at their dendrites.
Figure 4. Rod-driven DBC responses are sensitized by GABACR-dependent chloride influx.

(A) Cartoon illustrating the role of KCC2 in chloride extrusion from rod DBCs and the roles of chloride and potassium fluxes in tonic rod DBC hyperpolarization. (B) Equivalent circuit illustrating exchangeability and additivity of chloride and potassium conductances in creating the driving force for light-induced cation influx resulting in DBC depolarization. Under these conditions, the resting potential is defined as Vm = (gK·EK + gCl·ECl)/(gK + gCl), where gK and gCl are potassium and chloride conductances, and EK and ECl are potassium and chloride reversal potentials, respectively. (C) Retinal cross-section from a WT mouse immunostained for KCC2 and the rod DBC marker, PKCα (see Figure 1 for abbreviations of retina layers). (D) Confocal tangential z-section through bipolar cell somata of a flat-mounted retina co-stained for KCC2 and PKCα. Position of the z-section relative to DBC morphology is shown in the cartoon on the left. Scale bars in (C) and (D): 10 μm. (E) ERG b-wave sensitivity plots for WT mice with and without injections of the KCC2 antagonist VU0240551, or with co-injection of VU0240551 and GABA. Ethanol (95%), the vehicle for VU0240551, was injected in WT eyes as a control; the presence of ethanol shifted the sensitivity plot for WT mice, but the relative reduction in sensitivity in GABACR−/− vs. WT and in KCC2 blockade vs. WT+ethanol was similar.
The chloride equilibrium in rod-driven DBCs is maintained by the K+/Cl− co-transporter KCC2 which extrudes KCl from these cells (Figure 4A). KCC2 is expressed throughout all major rod DBC compartments (Vu et al., 2000; Zhang et al., 2007). Our own co-immunostaining of KCC2 with the rod DBC-specific marker, PKCα, revealed the most abundant KCC2 staining in rod DBC axons and cell bodies (Figure 4C), consistent with (Vu et al., 2000; Zhang et al., 2007). The latter is particularly well-seen in retina flat-mounts in which rod DBC cell bodies are well-distinguished from those of other bipolar cells, also positive for KCC2 (Figure 4D). Although an early report suggested that DBC dendrites also express the NKCC1 transporter which would accumulate chloride inside rod DBCs (Vardi et al., 2000; see also Li et al., 2008), the authors subsequently attributed this result to cross-reactivity of the “T4” antibody and showed that NKCC1 found in the outer plexiform layer of the retina is expressed in horizontal cells (Zhang et al., 2007). The lack of NKCC1 in rod DBCs is also consistent with the demonstration that the chloride reversal potentials are the same at the dendritic and axonal ends of rod DBCs in retinal slices (Satoh et al., 2001), which argues against chloride transport in opposite directions at these locations.
Based on this evidence, we predicted that inactivation of KCC2 in WT mice should shift the chloride equilibrium potential to a more positive value and reduce the amplitude of DBC light-response and the ERG b-wave. Indeed, intraocular injections of a KCC2 blocker reduced dark-sensitivity, operational range and Rmax,dark of WT rod-driven b-waves (Figures 3 and 4E), closely resembling reductions seen in GABACR−/− mice. Furthermore, the effects of the KCC2 blockade were not restored by exogenous GABA, which is consistent with a disrupted chloride gradient.
Another prerequisite for our hypothesis is the existence of a tonic hyperpolarizing GABA-mediated current. GABACRs are particularly well-suited for this function due to lack of GABA-dependent desensitization (Amin and Weiss, 1994). Tonic GABACR-mediated currents have been observed in goldfish bipolar cell terminals (Hull et al., 2006; Jones and Palmer, 2009), but have not been observed previously in mammalian rod DBCs. To directly document this current, we conducted patch clamp recordings from rod DBCs in retinal slices. We supplemented our slices with a low concentration of exogenous GABA (5 μM) in the bath to replace GABA lost by perfusion, following the experimental paradigm reviewed in (Glykys and Mody, 2007). These experiments revealed the presence of a tonic GABAergic current of ~7 pA in WT rod DBCs that was antagonized by TPMPA (Figures 5A and 5B). This current was absent in GABACR−/− rod DBCs. We next tested whether this GABACR-mediated sustained current could change the rod DBC resting membrane potential. When we measured the resting membrane potential in current clamp with zero holding current, TPMPA depolarized the resting potential of WT rod DBCs, but had no effect on GABACR−/− rod DBCs (Figure 5C).
Figure 5. WT rod DBCs have a GABACR-mediated tonic current that hyperpolarizes their resting potential and decreases their input resistance.

(A) Representative currents in WT and GABACR−/− rod DBCs in the presence of 5 μM GABA evoked by bath application of 50 μM TPMPA. (B) The average change in tonic current caused by TPMPA in WT (n=7) and GABACR−/− rod DBCs (n=6) differed significantly (p=0.001). (C) −/− rod DBCs (n=5; TPMPA depolarizes the resting potential (Vrest) of WT (n=8) but not GABACR p =0.03). (D) The linear range of current-voltage relationships of representative WT and GABACR−/− rod DBCs in the presence and absence of 50 μM TPMPA. A change in slope was observed in 6 out of 8 WT cells and in none of the 5 GABACR−/− rod DBCs. (E) Input resistances of WT rod DBCs were increased in the presence of TPMPA, but were not changed in GABACR−/− rod DBCs. The input resistances calculated from the slopes of current-voltage curves were: 0.8 ± 0.2 GΩ and 1.1 ± 0.08 GΩ (mean ± SEM, p = 0.03) for WT rod DBCs in the absence and presence of TPMPA, respectively; 1.26 GΩ ± 0.4 and 1.20 ± 0.07 GΩ for GABACR−/− rod DBCs in the absence and presence of TPMPA, respectively. The input resistance of WT rod DBCs in the presence of TPMPA was similar to GABACR−/− rod DBCs in the presence or absence of TPMPA.
This tonic GABACR-mediated conductance is expected to change the input resistance of WT rod DBCs and reduce the degree of depolarization caused by light-dependent synaptic inputs. To test this idea, we determined the input resistance in WT rod DBCs by measuring voltage changes in response to current injection in the presence or absence of TPMPA. As predicted, blocking GABACRs in WT rod DBCs increased the slope of the current-voltage (I-V) plot and, consequently, the rod DBC input resistance (Figures 5D and 5E). In contrast, TPMPA did not significantly alter the input resistance of GABACR−/− rod DBCs. This result is consistent with current clamp recordings by (Euler and Masland, 2000) who found that a blockade of GABAergic inputs reduces the dynamic range of intact rod DBCs in retinal slices.
The steeper I–V relation observed in the absence of the GABAergic input should reduce the dynamic range of the ERG b-wave light responses in D1R−/− and GABACR−/− mice. Indeed, the rod-driven b-wave stimulus-response curves, both in the dark and at each background light intensity, obtained from both D1R−/− and GABACR−/− mice displayed a systematic ~2-fold decrease in their dynamic range, defined as the range of intensities covering between 5% to 95% of the maximal response (Figure 6), which served as a reason for decreased overall operational range as illustrated in Figures 1C, 2B and 2D.
Figure 6. Knockout or blockade of D1R and/or GABACR reduces the dynamic range of ERG b-wave responses.
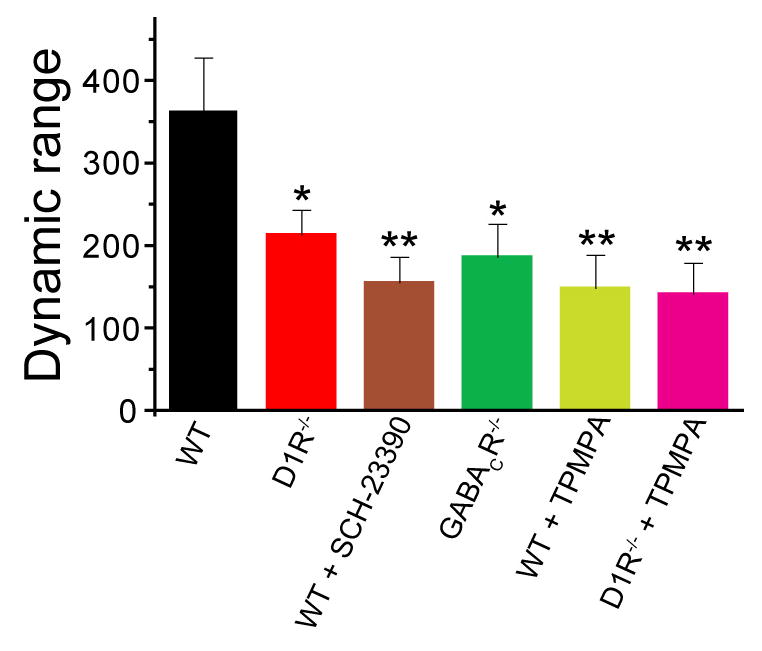
The response dynamic range for each animal/condition (i.e. the range of intensities covering between 5% to 95% of the maximal b-wave response) was calculated as 102.56/n (Thibos and Werblin, 1978), where n is the Hill coefficient for the rod-driven b-wave component calculated from fitting b-wave stimulus-response curves to Equation 1 (see Supplementary Experimental Procedures). The data were averaged from all recording sets obtained in the dark and in the presence of background light. Paired t-tests between WT and each other animal/condition yielded p-values of <0.05 (*) and <0.01 (**).
Altogether, our results argue that GABACRs mediate a tonic, sensitizing chloride current that hyperpolarizes WT rod DBCs and decreases their input resistance, thereby extending the amplitude and operational range of their depolarizing light responses.
Potential cellular sites of the dopamine-dependent GABA release
In the final set of experiments we aimed to identify the cellular source of the dopamine-dependent GABAergic input onto rod DBCs. Electrophysiological studies have described the most prevalent GABACR-mediated chloride currents in rod DBC axon terminals (e.g. (Eggers and Lukasiewicz, 2006). However, their dendrites also display a distinct GABACR-mediated chloride conductance, documented in ferret (Shields et al., 2000), which is consistent with specific GABACR immunostaining of rod DBC dendrites and its absence in GABACR−/− rod DBCs (McCall et al., 2002). Figure 7 shows that short GABA puffs evoked GABACR-mediated chloride currents in both the axon and dendritic terminals of the same WT rod DBCs in the mouse. Complete suppression of GABA-dependent currents could only be achieved by blocking both GABAA and GABAC receptors. Interestingly, the relative contributions of GABAAR- and GABACR-dependent currents were similar for dendrites and axon terminals (Figures 7C and 7D). The latter finding is consistent with results obtained for rat (Euler and Wassle, 1998) and mouse rod DBC axon terminals (McCall et al., 2002). Therefore, both axons and dendrites could be considered as potential sites of sustained GABAergic inputs.
Figure 7. GABAergic currents are mediated by GABAA and GABAC receptors in both dendrites and axon terminals of rod DBCs.

(A) and (B) Representative current responses to brief puffs of GABA delivered focally onto the axon terminals in the IPL (A) or dendrites in the OPL (B) of the same rod DBC in the presence or absence of the GABAAR antagonist bicuculline (500 μM) or a mixture of bicuculline with the GABACR antagonist TPMPA (50 μM). (C) and (D) Averaged normalized charge transfer (Q/Qmax ) of rod DBCs in response to GABA puffs in the IPL and OPL and in the presence of bicuculline alone or bicuculline +TPMPA. In both cases, bicuculline reduced responses to 80% of the control in the IPL and 66% in the OPL (n=6). TPMPA + bicuculline essentially eliminated responses in both IPL and OPL (7% vs 12% in IPL vs. OPL; n=6). An ANOVA compared the reduction in these responses from control. Both OPL and IPL responses were significantly different in the presence of bicuculline as well as bicuculline + TPMPA (p < 0.001). There was no difference when similar conditions were compared between IPL and OPL.
Furthermore, both axons and dendrites of rod DBCs are located postsynaptically to cells displaying strong immunostaining for D1R and GABA (amacrine and horizontal cells, respectively; Figure 1D and Figure S4). The expression pattern of KCC2 both on rod DBC axons and somas immediately adjacent to the relatively short dendrites (Figures 4C and 4D) predicts an efficient chloride extrusion over the whole length of the rod DBC and therefore does not favor either amacrine or horizontal cells as a major source of the GABAergic input.
We therefore searched for evidence that GABA release from either amacrine or horizontal cells could be regulated via D1R by analyzing the patterns of GABA immunostaining of both cell types in WT and D1R−/− mice. The most intriguing result was obtained for horizontal cells. In WT mice GABA staining of these cells increased by ~3.5-fold upon illumination (Figure 8A). This increase was observed in both horizontal cell bodies and axons forming synapses with rod DBC dendrites. The latter was identified using neurofilament staining as an axonal marker (Figure 8B). Remarkably, this light-dependency of GABA immunostaining was completely abolished in horizontal cells of D1R−/− mice, in which the amount of GABA remained at a constant high level (Figures 8A and 8C). A similar examination of GABA immunostaining in amacrine cells did not reveal any systematic light-dependent changes in either animal type (Figure S5). These results make horizontal cells a potential site for the D1R-dependent mechanism revealed in our study.
Figure 8. Horizontal cells serve as a putative site of D1R-regulated GABA release.

(A) GABA immunostaining of horizontal cells in single tangential confocal sections of WT and D1R−/− retinas. The position of optical sections is illustrated on the cartoon above. Animals were light-conditioned as indicated in the panel. (B) GABA co-immunostaining with neurofilaments in a single tangential confocal section through the horizontal cell layer in WT retina. (C) Quantification of GABA immunostaining in horizontal cells from mice subjected to different levels of background illumination (mean ± SEM; 3 to 6 retinas were analyzed for each condition; p-values for the difference between animal types at the same condition are shown on the top of each pair).
DISCUSSION
Our results demonstrate that the sensitivity and operational range of rod-driven vision are increased by dopamine-dependent GABAergic inputs onto rod DBCs. These findings expand the function of dopamine in the retina from its traditional role of establishing cone-vision dominance in daytime to acting as an enhancer of rod-driven circuitry. A previous study in zebrafish indicated that dopamine is required for the transmission of rod signals downstream from DBCs (Li and Dowling, 2000). We now show that dopamine increases the sensitivity of rod-driven responses at the level of DBCs. Together, these results indicate that dopamine regulates the entire primary rod pathway of mammalian vision. The fact that the desensitizing effect of the D1R knockout is observed in both dark-adapted mice and mice exposed to dim background light is consistent with significant levels of total dopamine (Nir et al., 2000) and dopamine release (Mills et al., 2007) even in dark-adapted retinas.
Mechanistic hypothesis of rod DBC sensitization by GABA
We propose that a dopamine-dependent GABAergic input causes two interrelated effects. First, a tonic GABA input hyperpolarizes rod DBCs and increases the driving force for cations entering the cell during the depolarizing light response. Second, a sustained chloride current caused by this GABAergic input broadens the dynamic range of rod DBC light responses because it imposes a mild shunting inhibition on the depolarizing light response. Ultimately, both aspects of the sustained GABACR-mediated input sensitize rod-driven vision by making rod DBC responses larger and operating over a broader light intensity range.
This role of chloride in providing a driving force on cations during depolarizing light-responses complements the more traditional role of potassium in fulfilling this function and, electrically, the contributions of these two ions are interchangeable and additive (Figure 4B). However, the regulation of these two currents is different: while the potassium conductance is likely to be defined by the channel and transporter activities confined to rod DBCs, our results suggest that the chloride conductance is further regulated by a dopaminergic input to a GABA-releasing cell. Moreover, our data suggest that rod DBCs take a two-fold advantage from maintaining large chloride gradients. The well-established role of this gradient is to enable strong, stimulus-dependent, transient GABAergic feedback inhibition from amacrine cells (Chavez et al., 2010; Tachibana and Kaneko, 1987), which adjusts the amplitude and kinetics of rod DBC light- or electrically-evoked responses (Eggers and Lukasiewicz, 2006; Roska et al., 2000). We now argue that the same chloride gradient also sensitizes their light-responses via small sustained currents. Interestingly, the same chloride channel, GABACR, is used in both cases (though GABAAR is used for the dynamic feedback as well), which requires the transient GABACR-dependent current mediating the dynamic feedback to be significantly larger than the sustained current. This is entirely consistent with observations by us and others (Naarendorp and Sieving, 1991; Robson et al., 2004) that increasing extracellular GABA by intraocular injections increases rod DBC light-response amplitudes, indicating that GABA is bound only to a fraction of GABACRs in the dark.
Do horizontal or amacrine cells serve as the primary site of the dopamine-dependent GABA release?
Another point raised in our study relates to the cellular origin of the dopamine-dependent GABA release. The light-dependency of GABA staining in horizontal cells abolished in D1R−/− mice makes these cells a potential candidate. Horizontal cells have long been known to contain GABA (Deniz et al., 2011; Guo et al., 2010; Schwartz, 1987; Vardi et al., 1994; Wassle and Chun, 1989); Figure S4), but the role of GABA release from horizontal cells, at least for the rod circuit, remains poorly understood. For instance, the recently reported inhibitory feedback from these cells onto rod terminals does not appear to rely on GABA (Babai and Thoreson, 2009). Horizontal cells display the strongest D1R immunostaining in the mouse retina (Figure 1E) and express D1R in close proximity to the processes of dopaminergic amacrine cells (Figure S4). The hyperpolarizing light-responses of horizontal cells are also known to be regulated by dopamine via D1-type receptors (Hankins and Ikeda, 1994; Knapp et al., 1990; Mangel and Dowling, 1985; Yang et al., 1988). Furthermore, depolarization of horizontal cells favors GABA release in isolated cells (Schwartz, 1987) and dopamine, acting via D1R, shifts the membrane potential of horizontal cells to more depolarized values (Hankins and Ikeda, 1994).
Combined with the observation that dendrites of rod DBCs have robust GABACR-mediated currents, these properties of horizontal cells allow the following interpretation of our GABA immunostaining data. We suggest that horizontal cells in D1R−/− mice release less GABA than horizontal cells in WT mice under all illumination conditions used in our study. This idea relies on the assumptions that the intensity of GABA immunostaining in horizontal cells measures the retention of GABA, thereby serving as a reciprocal measure of the amount of GABA released, and that GABA synthesis is not grossly affected by light. Accordingly, the sensitivity of rod-driven ERG b-waves in D1R−/− mice remained lower than in WT mice even at the highest tested background level of 800 R*/rod/s. Interestingly, horizontal cell coupling via gap junctions is also controlled by a D1R-dependent mechanism in the same light intensity range as analyzed in our study (e.g. Weiler et al., 2000), which raises a possibility that the two phenomena are inter-dependent. Testing these ideas and elucidating mechanistic details of the dopamine-dependent GABA release is the goal of future studies.
We should stress, however, that an alternative model in which GABA release originates primarily from amacrine cells remains plausible as well, particularly because sustained GABACR-mediated currents have been observed in axon terminals of mixed rod/cone DBCs in goldfish retina (Hull et al., 2006; Jones and Palmer, 2009). Another argument in favor of this possibility is the observation by Euler and Masland (2000) that an application of GABA receptor blockers in retinal slices decreased the dynamic range of intact rod DBC light responses, but not DBCs which with severed axon terminals. In principle, it is also conceivable that GABA is released at both locations. A critical future approach to identify relative dendritic and axonal contributions to sustained chloride currents would be to generate conditional knockout mice lacking D1R from specific retina neuron types.
Dopamine-dependent modulation of GABAergic outputs, particularly via D1R, is a critical mechanism in the physiology and pathology of multiple brain functions (Carlsson et al., 2001; Greengard, 2001). The present study extends this modulatory interaction to the retina, where it plays a crucial role in dim-light vision via sustained sensitization of rod bipolar cells.
EXPERIMENTAL PROCEDURES
Details on the animal strains used, electrophysiological recordings and immunohistochemical procedures are available in the Supplemental Information.
Electroretinography
ERGs were recorded essentially as described (Herrmann et al., 2010). b-wave sensitivities were determined as the ratio between the maximal rod-driven response amplitude and half-saturating flash intensity (parameters derived from fits of b-wave stimulus response curves with a hyperbolic function). In all figures, b-wave sensitivities were normalized to the sensitivity of dark-adapted WT mice and fitted by the Weber-Fechner equation; fitting parameters are summarized in Table S1.
Whole cell rod DBC recordings in retina slices
Preparation of retina slices, whole-cell voltage and current clamp recordings, bathing and pipette solutions are described in (McCall et al., 2002) and (Eggers et al., 2007). Rod DBCs were identified by their characteristic morphology after filling them with lucifer yellow (0.05%) or sulfarhodamine (0.01 %) included in the recording electrode. To record tonic GABAergic currents, cells were voltage-clamped at 0 mV in the presence of 5 μM GABA. The resting potential was determined in the current clamp mode with zero holding current. Input resistance was also measured in current clamp, from voltage changes due to current injections. To record transient GABA-evoked currents in dendrites and axon terminals of rod DBCs, cells were voltage clamped to the reversal potential for cations (0 mV) and currents were evoked by 30 ms GABA puffs (100–300 μM) onto dendrites or their axon terminals.
Immunohistochemistry
Immunostaining was performed essentially as described (Herrmann et al., 2010). To analyze light-dependent GABA immunostaining in retinal neurons, dark-adapted mice were exposed for 5 min to background illumination of varying intensities. Mice were sacrificed, retinas fixed and stained with a mixture of anti-GABA and anti-calbindin antibodies for 3 days. Following incubation with secondary antibodies for visualization of GABA and calbindin immunostaining in different color channels, flat-mounted retinas were analyzed by confocal microscopy. To quantify the light-dependent dynamics of GABA staining in horizontal cells, we first selected a single optical section representing the outer plexiform layer and displaying the most intense calbindin immunostaining. We next measured the intensity of GABA immunostaining co-localizing with calbindin staining in the same section.
Supplementary Material
Acknowledgments
We thank M.E. Burns for critically reading an earlier version of the manuscript. This work was supported by the NIH grants EY10336 (VYA), MH073853 (MC), EY06671 (LJF), EY014701 (MAMc), EY5722 (to Duke University) and RPB (MAMc).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amin J, Weiss DS. Homomeric rho 1 GABA channels: activation properties and domains. Receptors Channels. 1994;2:227–236. [PubMed] [Google Scholar]
- Babai N, Thoreson WB. Horizontal cell feedback regulates calcium currents and intracellular calcium levels in rod photoreceptors of salamander and mouse retina. J Physiol. 2009;587:2353–2364. doi: 10.1113/jphysiol.2009.169656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML. Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol. 2001;41:237–260. doi: 10.1146/annurev.pharmtox.41.1.237. [DOI] [PubMed] [Google Scholar]
- Chavez AE, Grimes WN, Diamond JS. Mechanisms underlying lateral GABAergic feedback onto rod bipolar cells in rat retina. J Neurosci. 2010;30:2330–2339. doi: 10.1523/JNEUROSCI.5574-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz S, Wersinger E, Schwab Y, Mura C, Erdelyi F, Szabo G, Rendon A, Sahel JA, Picaud S, Roux MJ. Mammalian retinal horizontal cells are unconventional GABAergic neurons. J Neurochem. 2011;116:350–362. doi: 10.1111/j.1471-4159.2010.07114.x. [DOI] [PubMed] [Google Scholar]
- Dunn FA, Doan T, Sampath AP, Rieke F. Controlling the gain of rod-mediated signals in the Mammalian retina. J Neurosci. 2006;26:3959–3970. doi: 10.1523/JNEUROSCI.5148-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers ED, Lukasiewicz PD. GABA(A), GABA(C) and glycine receptor-mediated inhibition differentially affects light-evoked signalling from mouse retinal rod bipolar cells. J Physiol. 2006;572:215–225. doi: 10.1113/jphysiol.2005.103648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers ED, McCall MA, Lukasiewicz PD. Presynaptic inhibition differentially shapes transmission in distinct circuits in the mouse retina. J Physiol. 2007;582:569–582. doi: 10.1113/jphysiol.2007.131763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enz R, Brandstatter JH, Hartveit E, Wassle H, Bormann J. Expression of GABA receptor rho 1 and rho 2 subunits in the retina and brain of the rat. Eur J Neurosci. 1995;7:1495–1501. doi: 10.1111/j.1460-9568.1995.tb01144.x. [DOI] [PubMed] [Google Scholar]
- Euler T, Masland RH. Light-evoked responses of bipolar cells in a mammalian retina. J Neurophysiol. 2000;83:1817–1829. doi: 10.1152/jn.2000.83.4.1817. [DOI] [PubMed] [Google Scholar]
- Euler T, Wassle H. Different contributions of GABAA and GABAC receptors to rod and cone bipolar cells in a rat retinal slice preparation. J Neurophysiol. 1998;79:1384–1395. doi: 10.1152/jn.1998.79.3.1384. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I. Activation of GABAA receptors: views from outside the synaptic cleft. Neuron. 2007;56:763–770. doi: 10.1016/j.neuron.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294:1024–1030. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- Guo C, Hirano AA, Stella SL, Jr, Bitzer M, Brecha NC. Guinea pig horizontal cells express GABA, the GABA-synthesizing enzyme GAD 65, and the GABA vesicular transporter. J Comp Neurol. 2010;518:1647–1669. doi: 10.1002/cne.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankins M, Ikeda H. Early abnormalities of retinal dopamine pathways in rats with hereditary retinal dystrophy. Doc Ophthalmol. 1994;86:325–334. doi: 10.1007/BF01203555. [DOI] [PubMed] [Google Scholar]
- Herrmann R, Lobanova ES, Hammond T, Kessler C, Burns ME, Frishman LJ, Arshavsky VY. Phosducin regulates transmission at the photoreceptor-to-ON-bipolar cell synapse. J Neurosci. 2010;30:3239–3253. doi: 10.1523/JNEUROSCI.4775-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Li GL, von Gersdorff H. GABA transporters regulate a standing GABAC receptor-mediated current at a retinal presynaptic terminal. J Neurosci. 2006;26:6979–6984. doi: 10.1523/JNEUROSCI.1386-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Palmer MJ. Activation of the tonic GABAC receptor current in retinal bipolar cell terminals by nonvesicular GABA release. J Neurophysiol. 2009;102:691–699. doi: 10.1152/jn.00285.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamermans M, Spekreijse H. The feedback pathway from horizontal cells to cones. A mini review with a look ahead. Vision Res. 1999;39:2449–2468. doi: 10.1016/s0042-6989(99)00043-7. [DOI] [PubMed] [Google Scholar]
- Knapp AG, Schmidt KF, Dowling JE. Dopamine modulates the kinetics of ion channels gated by excitatory amino acids in retinal horizontal cells. Proc Natl Acad Sci U S A. 1990;87:767–771. doi: 10.1073/pnas.87.2.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, McKernan K, Shen W. Spatial and temporal distribution patterns of Na-K-2Cl cotransporter in adult and developing mouse retinas. Vis Neurosci. 2008;25:109–123. doi: 10.1017/S0952523808080164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Dowling JE. Effects of dopamine depletion on visual sensitivity of zebrafish. J Neurosci. 2000;20:1893–1903. doi: 10.1523/JNEUROSCI.20-05-01893.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasiewicz PD, Shields CR. A diversity of GABA receptors in the retina. Semin Cell Dev Biol. 1998;9:293–299. doi: 10.1006/scdb.1998.0238. [DOI] [PubMed] [Google Scholar]
- Mangel SC, Dowling JE. Responsiveness and receptive field size of carp horizontal cells are reduced by prolonged darkness and dopamine. Science. 1985;229:1107–1109. doi: 10.1126/science.4035351. [DOI] [PubMed] [Google Scholar]
- McCall MA, Lukasiewicz PD, Gregg RG, Peachey NS. Elimination of the rho1 subunit abolishes GABA(C) receptor expression and alters visual processing in the mouse retina. J Neurosci. 2002;22:4163–4174. doi: 10.1523/JNEUROSCI.22-10-04163.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills SL, Xia XB, Hoshi H, Firth SI, Rice ME, Frishman LJ, Marshak DW. Dopaminergic modulation of tracer coupling in a ganglion-amacrine cell network. Vis Neurosci. 2007;24:593–608. doi: 10.1017/S0952523807070575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naarendorp F, Sieving PA. The scotopic threshold response of the cat ERG is suppressed selectively by GABA and glycine. Vision Res. 1991;31:1–15. doi: 10.1016/0042-6989(91)90068-g. [DOI] [PubMed] [Google Scholar]
- Nir I, Haque R, Iuvone PM. Diurnal metabolism of dopamine in the mouse retina. Brain Res. 2000;870:118–125. doi: 10.1016/s0006-8993(00)02409-4. [DOI] [PubMed] [Google Scholar]
- Robson JG, Frishman LJ. Dissecting the dark-adapted electroretinogram. Doc Ophthalmol. 1998;95:187–215. doi: 10.1023/a:1001891904176. [DOI] [PubMed] [Google Scholar]
- Robson JG, Maeda H, Saszik SM, Frishman LJ. In vivo studies of signaling in rod pathways of the mouse using the electroretinogram. Vision Res. 2004;44:3253–3268. doi: 10.1016/j.visres.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Rodieck RW. The First Steps in Seeing. Sinauer Associates; 1998. [Google Scholar]
- Roska B, Nemeth E, Orzo L, Werblin FS. Three levels of lateral inhibition: A space-time study of the retina of the tiger salamander. J Neurosci. 2000;20:1941–1951. doi: 10.1523/JNEUROSCI.20-05-01941.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Kaneda M, Kaneko A. Intracellular chloride concentration is higher in rod bipolar cells than in cone bipolar cells of the mouse retina. Neurosci Lett. 2001;310:161–164. doi: 10.1016/s0304-3940(01)02120-6. [DOI] [PubMed] [Google Scholar]
- Schwartz EA. Depolarization without calcium can release gamma-aminobutyric acid from a retinal neuron. Science. 1987;238:350–355. doi: 10.1126/science.2443977. [DOI] [PubMed] [Google Scholar]
- Shapley RM, Enroth-Cugell C. Visual adaptation and retinal gain controls. Prog Retin Res. 1984;3:263–346. [Google Scholar]
- Shields CR, Tran MN, Wong RO, Lukasiewicz PD. Distinct ionotropic GABA receptors mediate presynaptic and postsynaptic inhibition in retinal bipolar cells. J Neurosci. 2000;20:2673–2682. doi: 10.1523/JNEUROSCI.20-07-02673.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Tachibana M, Kaneko A. Effects of glycine and GABA on isolated bipolar cells of the mouse retina. J Physiol. 1990;421:645–662. doi: 10.1113/jphysiol.1990.sp017967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Kaneko A. gamma-Aminobutyric acid exerts a local inhibitory action on the axon terminal of bipolar cells: evidence for negative feedback from amacrine cells. Proc Natl Acad Sci U S A. 1987;84:3501–3505. doi: 10.1073/pnas.84.10.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Attwell D, Mobbs P, Wilson M. Membrane currents in retinal bipolar cells of the axolotl. J Gen Physiol. 1988;91:49–72. doi: 10.1085/jgp.91.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibos LN, Werblin FS. The response properties of the steady antagonistic surround in the mudpuppy retina. J Physiol. 1978;278:79–99. doi: 10.1113/jphysiol.1978.sp012294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardi N, Kaufman DL, Sterling P. Horizontal cells in cat and monkey retina express different isoforms of glutamic acid decarboxylase. Vis Neurosci. 1994;11:135–142. doi: 10.1017/s0952523800011172. [DOI] [PubMed] [Google Scholar]
- Vardi N, Zhang LL, Payne JA, Sterling P. Evidence that different cation chloride cotransporters in retinal neurons allow opposite responses to GABA. J Neurosci. 2000;20:7657–7663. doi: 10.1523/JNEUROSCI.20-20-07657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veruki ML, Wassle H. Immunohistochemical localization of dopamine D1 receptors in rat retina. Eur J Neurosci. 1996;8:2286–2297. doi: 10.1111/j.1460-9568.1996.tb01192.x. [DOI] [PubMed] [Google Scholar]
- Vu TQ, Payne JA, Copenhagen DR. Localization and developmental expression patterns of the neuronal K-Cl cotransporter (KCC2) in the rat retina. J Neurosci. 2000;20:1414–1423. doi: 10.1523/JNEUROSCI.20-04-01414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassle H. Parallel processing in the mammalian retina. Nat Rev Neurosci. 2004;5:747–757. doi: 10.1038/nrn1497. [DOI] [PubMed] [Google Scholar]
- Wassle H, Chun MH. GABA-like immunoreactivity in the cat retina: light microscopy. J Comp Neurol. 1989;279:43–54. doi: 10.1002/cne.902790105. [DOI] [PubMed] [Google Scholar]
- Weiler R, Pottek M, He S, Vaney DI. Modulation of coupling between retinal horizontal cells by retinoic acid and endogenous dopamine. Brain Res Brain Res Rev. 2000;32:121–129. doi: 10.1016/s0165-0173(99)00071-5. [DOI] [PubMed] [Google Scholar]
- Witkovsky P. Dopamine and retinal function. Doc Ophthalmol. 2004;108:17–40. doi: 10.1023/b:doop.0000019487.88486.0a. [DOI] [PubMed] [Google Scholar]
- Yang XL, Tornqvist K, Dowling JE. Modulation of cone horizontal cell activity in the teleost fish retina. II. Role of interplexiform cells and dopamine in regulating light responsiveness. J Neurosci. 1988;8:2269–2278. doi: 10.1523/JNEUROSCI.08-07-02269.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XL, Wu SM. Feedforward lateral inhibition in retinal bipolar cells: input-output relation of the horizontal cell-depolarizing bipolar cell synapse. Proc Natl Acad Sci U S A. 1991;88:3310–3313. doi: 10.1073/pnas.88.8.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LL, Delpire E, Vardi N. NKCC1 does not accumulate chloride in developing retinal neurons. J Neurophysiol. 2007;98:266–277. doi: 10.1152/jn.00288.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.