

Abstract
Transcription factors within cellular gene networks control the spatiotemporal pattern and levels of expression of their target genes by binding to cis-regulatory elements (CREs), short (˜300-600 bp) stretches of genomic DNA which can lie upstream, downstream, or within the introns of the genes they control. CREs (i.e., enhancers/promoters) typically consist of multiple clustered binding sites for both transcriptional activators and repressors1-3. They serve as logical integrators of transcriptional input giving a unitary output in the form of spatiotemporally precise and quantitatively exact promoter activity. Most studies of mammalian cis-regulation to date have relied on mouse transgenesis as a means of assaying the enhancer function of CREs4-5. This technique is time-consuming, costly and, on account of insertion site effects, largely non-quantitative. On the other hand, quantitative assays for mammalian CRE function have been developed in tissue culture systems (e.g., dual luciferase assays), but the in vivo relevance of these results is often uncertain.
Electroporation offers an excellent alternative to traditional mouse transgenesis in that it permits both spatiotemporal and quantitative assessment of cis-regulatory activity in living mammalian tissue. This technique has been particularly useful in the analysis of cis-regulation in the central nervous system, especially in the cerebral cortex and the retina6-8. While mouse retinal electroporation, both in vivo and ex vivo, has been developed and extensively described by Matsuda and Cepko6-7,9, we have recently developed a simple approach to quantify the activity of photoreceptor-specific CREs in electroporated mouse retinas10. Given that the amount of DNA that is introduced into the retina by electroporation can vary from experiment to experiment, it is necessary to include a co-electroporated 'loading control' in all experiments. In this respect, the technique is very similar to the dual luciferase assay used to quantify promoter activity in cultured cells.
When assaying photoreceptor cis-regulatory activity, electroporation is usually performed in newborn mice (postnatal day 0, P0) which is the time of peak rod production11-12. Once retinal cell types become post-mitotic, electroporation is much less efficient. Given the high rate of rod birth in newborn mice and the fact that rods constitute more than 70% of the cells in the adult mouse retina, the majority of cells that are electroporated at P0 are rods. For this reason, rod photoreceptors are the easiest retinal cell type to study via electroporation. The technique we describe here is primarily useful for quantifying the activity of photoreceptor CREs.
Protocol
1. Construction of the electroporation chamber
Order a microslide, BTX Model 453 with a 3.2mm gap (Harvard Apparatus #45-0105) (Fig. 2A). The metal rails should be completely sealed to the bottom of the slide.
Use a Dremel tool to cut a handle off a plastic microcentrifuge tube rack. Cut the handle into 5 small rectangular pieces each with the following dimensions: length 0.8cm, height 0.6cm, width 0.3cm (Fig. 2B). These plastic pieces are reusable spacers that will be used to mold individual wells in the microslide chamber.
Insert the plastic spacers between the metal rails of the microslide at even intervals. The spacers should fit snugly (Fig. 2C).
Cut the tip off a P200 pipette tip, fit the tip to a 3ml syringe, and fill the syringe with 100% silicone rubber aquarium sealant. Fill the gaps between the plastic spacers with sealant. Be sure to fill the gaps from the bottom up so that no bubbles form between the sealant plug and the base of the microslide.
Place a metal rod atop the plastic spacers and fasten it with binder clips, so that the spacers are held in place while the sealant dries (Fig. 2D-E). Let the sealant dry overnight.
Remove the binder clips, metal rod, and plastic spacers (Fig. 2F). Use a scalpel blade to clean the sealant off the top of the metal rails. Use a dissecting microscope to examine the microslide and ensure that no bubbles are present at the bottom of the silicone dams. Remove any sealant film that may have formed in the wells such that bare metal is exposed inside the wells. Fill one well with water and make sure that the water doesn’t leak into the adjacent well(s). Repeat for all wells.
The finished microslide fits in the plastic dish with the metal poles adjacent to the window in the side of the dish (Fig. 2G); the electrodes will eventually be attached to the metal poles.
2. DNA preparation
Add plasmid DNA to a 1.5mL microcentrifuge tube on ice and bring the volume up to 150μl with distilled water. Multiple plasmid species may be combined for co-electroporation (e.g., an experimental DsRed construct and a control GFP construct). When calculating the amount of DNA to add to the tube, keep in mind that the final volume of the DNA aliquot will be 60μl. Typically, each construct is used at a final concentration of 0.5μg/μl.
Precipitate the DNA by adding 15μl 3M sodium acetate (pH 5.2) and 450μl 100% ethanol. Invert or tap the tube several times to mix.
Spin down the DNA at 4°C, 13,200 rpm, for 30 minutes. Wash the pellet with 70% ethanol, then spin it down again at 4°C, 13,200 rpm, for 15 minutes. Air-dry the pellet until semi-translucent, about 7 minutes, then resuspend in 54μl sterile water. Add 6μl sterile 10X PBS (pH 7.4) and mix.
3. Eye collection
Sterilize the electroporation chamber and all instruments with 70% ethanol. While eye collection and dissection need not be performed in a tissue culture hood, disinfect your gloves and benchtop with ethanol and attempt to maintain sterile conditions throughout the procedure.
Prepare Petri dishes with dissection medium (1:1 ratio of DMEM:F12, 100U/ml penicillin, 100μg/ml streptomycin, 0.29mg/ml L-glutamine, and 5μg/ml insulin): two 35mm dishes each with 3ml medium and one 60mm dish with 6ml medium. This step should be performed in a tissue culture hood.
Disinfect the head and neck of a newborn (postnatal day 0) mouse pup with 70% ethanol. Quickly decapitate with scissors and transfer the head to a sterile 100mm dish.
Cut away the scalp with small scissors to expose the eyes. Use curved forceps to gently scoop the eye out of the orbit, and place the eye in a 35mm dish containing dissection medium. It may be helpful to remove the eyes under a dissecting microscope at low power.
Repeat steps 3.3 and 3.4 until all eyes have been collected. Keep eyes in dissection medium at room temperature while dissecting. You will need 3-4 eyes per DNA aliquot.
4. Retinal dissection
Use 70% ethanol to disinfect both a razor blade and the wrapper of a sterile plastic transfer pipette. Cut the tip off the pipette with the blade so that it can suck up a whole eye. Store the pipette in the plastic wrapper when not in use.
Transfer one eye from the 35mm dish to the 60mm dish. Under the dissecting microscope at high power, use fine forceps to remove any tissue, such as extraocular muscles and fat, from the surface of the eye. Then, remove the optic nerve by pinching it off at the base.
In order to isolate the retina, poke a small hole in the sclera at the limbus. Insert one prong from both pairs of forceps into the hole (tangential to the retinal surface) and gently tear open the sclera/RPE. In albino mice, the sclera and RPE appear shiny relative to the retinal tissue, which is a homogeneous matte gray color. In pigmented mice, the RPE is black. Leave the lens in place.
Use the transfer pipette to move the dissected retina into the other 35mm dish with medium.
Repeat steps 4.2 to 4.4 until all eyes have been dissected.
Store the retinas in a 37°C tissue culture incubator until you are ready to electroporate.
5. Preparation for electroporation
Prepare 35mm dishes of medium. For each DNA aliquot to be electroporated, you will need one dish of dissection medium and one dish of culture medium (dissection medium plus 10% FBS). Label the dishes appropriately.
Use a P200 pipette and sterile 1X PBS to wash out the chambers in the electroporation dish. Each chamber holds a volume of 60-100μl. Wash out each chamber three times.
Fill the chambers with the DNA aliquots. Any unused chambers should be filled with 60μl 1X PBS. Connect the electrodes to the electroporation dish.
Use the following settings on the electroporator: mode, LV; voltage, 30V; pulse length, 50 msec; number of pulses, 5; interval, 950 msec; polarity, unipolar.
6. Electroporation
Use fine forceps to grasp the retinas by the lens and transfer them into the electroporation chambers. Each chamber holds up to 3-4 mouse retinas (Fig. 3).
Use forceps to line up the retinas such that the lens leans against the metal bar attached to the positive electrode. Clean the forceps with a Kimwipe after each transfer to avoid carry-over of DNA from one chamber to the next.
Once all retinas are aligned, press “Start” on the electroporator. Tiny bubbles should form on the metal bar attached to the negative electrode.
Disconnect the electrodes and turn off the electroporator.
Use forceps to gently move the retinas away from the chamber walls.
Use a sterile transfer pipette to transfer the retinas from the chambers into the 35mm dishes containing dissection medium.
Wash out each chamber three times with sterile 1X PBS, then rinse with sterile water. Spray dish with 70% ethanol.
7. Placing retinas on filters for culture
Use a transfer pipette to transfer the retinas into the 35mm dishes containing culture medium.
Label the wells of a sterile 6-well culture plate and fill each well with 3ml culture medium.
Use sterile forceps to place round Whatman Nuclepore filters, shiny side up, atop the medium in each well.
Under a dissecting microscope, use a sterile transfer pipette to transfer the retinas onto the filter, lens-side-down. If the retina lands lens-side-up, pick it up with the pipette and attempt to place it again. Do not place more than 4 retinas on one filter, and make sure that the droplets of medium surrounding each retina remain separate from the other droplets. Note that we culture the retina with the lens intact, although other protocols call for the removal of the lens prior to this step9.
Place the culture plate in a 37°C tissue culture incubator (5% CO2) and grow for the desired amount of time, typically eight days. In our experience, changing the medium is unnecessary during the eight day culture period, although a medium change may be required for longer culture periods.
8. Harvesting and flatmounting fluorescent retinal explants
Replace the culture medium in each well with 4% paraformaldehyde / 1X PBS. If the retinas remain stuck to the filters, use forceps to flip the filters over and gently peel the retinas off the filter. Incubate in paraformaldehyde for 30 min. at room temperature. Protect the retinas from light to avoid bleaching the fluorescence.
Rinse the retinas twice for 10 minutes in 1X PBS.
Use a disposable pipette to transfer the retinas onto a glass slide in a small drop of PBS. Under a fluorescent dissecting microscope, use forceps to flip the retinas so that they are electroporated-side-up (i.e., lens-side-down).
Place glass “feet” made from crushed coverslips (glass shards about 1-2 mm in diameter) at the corners of the slide; these feet prevent flattening of the retina. Place an intact glass coverslip over the slide so that it covers the retinas and rests on the feet. If necessary, use a pipette to add more PBS between the slide and the coverslip.
9. Imaging and quantification of fluorescence in flatmount
Use a fluorescent compound microscope equipped with a monochrome camera to image the flatmounted retina at low power (4X objective) in the red and green channels. All retinas must be imaged with the same exposure time for a given fluorescent channel to enable comparison of fluorescence intensities. Make sure that the pixels are not saturated in any image, or else accurate quantification will be impossible. Export images in grayscale TIFF format.
Open the image set for one retina (i.e.., red channel and green channel) in ImageJ software (http://rsbweb.nih.gov/ij/). For the sake of this tutorial, the green fluorescent channel (GFP protein) is the control construct that is constant across all retinas in the experiment. The red fluorescent channel (DsRed protein) is the experimental construct that varies for each set of retinas. The images should be in grayscale.
In ImageJ, select the control green image and specify a circle of interest with diameter 100 units (Analyze/Tools/ROI manager/More/Specify). Duplicate this circle (ROI manager/Add) to create eight circles total. Move circles 1-5 to select five regions that are uniformly electroporated, avoiding the outer edges of the retina and the region overlying the lens (Fig. 4A). Also, select three regions (circles 6-8) outside the retina/lens to measure background fluorescence. Select the red image, un-check the “Show all” box in ROI manager, and re-check the box. All eight circles should appear on the red image. De-select all circle coordinates in ROI manager.
With the red image selected, record the mean pixel value for all circles of interest (ROI manager/Measure); measurements 1-8 should appear, where 1-5 are the red retinal measurements and 6-8 are the red background measurements. Select the green image and record the mean pixel value; measurements 9-16 should appear, where 9-13 are the green retinal measurements and 14-16 are the green background measurements. Copy the measurement data into Excel for analysis (Fig. 5).
Average the three background measurements in both the red and the green channels. Subtract the red background average from each of the five retinal measurements in the red channel; repeat for the green channel. For each retinal region-of-interest, divide the background-subtracted red measurement by the background-subtracted green measurement in order to normalize the experimental red level to the control green level (Fig. 5).
Determine the average and standard deviation of all normalized measurements for a given DsRed construct (e.g., 5 measurements per retina times 3 separate retinas). In order to quantitatively compare the results of electroporations carried out on different days, always include a “standard” DsRed/GFP precipitation in each electroporation set. Relative expression values across experiments can be compared by normalizing to the expression level of this “standard.”
10. Representative Results:
A good electroporation results in expression of the DNA construct(s) across 1/4 to 1/3 of the retinal surface (Fig. 4A). Since rod photoreceptors in particular are efficiently transduced, this technique is ideal for quantifying photoreceptor-specific promoter activity (Fig. 4B). We previously used this approach to quantify a range of promoter variants from the rod-specific Rho and Gnat1 loci10. We found that it is possible to quantify promoter activity over a nearly 300-fold range.
Figure 5 is a sample dataset from a single electroporated retina. In this particular example, the experimental construct pNrl(1.1kb)-DsRed was measured in the red channel and the control construct pNrl(3.2kb)-GFP was measured in the green channel. A complete dataset for the pNrl(1.1kb)-DsRed construct would consist of 6-9 retinas measured in this manner, and standard deviation would be calculated based on all “DsRed normalized to GFP” values. If we were comparing the expression level of pNrl(1.1kb)-DsRed to, for example, pNrl(0.8kb)-DsRed, then both constructs would need to be electroporated with the same GFP control (e.g., pNrl(3.2kb)-GFP) and imaged at the same exposure times. It is possible to pool data collected on different days if a standard DsRed/GFP electroporation is performed on each day (e.g., pNrl(3.2kb)-dsRed + pNrl(3.2kb)-GFP). For each experimental construct, the normalized DsRed level would subsequently be normalized to the normalized DsRed level of the “standard” (pNrl(3.2kb)-dsRed).
The technique we describe here is primarily useful for quantifying the activity of photoreceptor CREs10,13. Cell-type specific cis-regulatory activity can also be quantified in rarer retinal cell types such as bipolar cells14, but this usually requires that the areas of interest to be quantified be selected in vertical cross-sections rather than in flatmount preparations. The same is true of CREs which drive expression in multiple cell types such as photoreceptors and bipolar cells. The experimental procedures are otherwise similar.
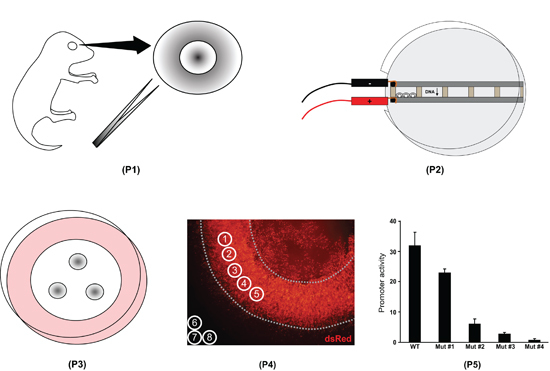 Figure 1. Overview of the retinal explant electroporation procedure. First, whole eyes are isolated from postnatal day 0 mouse pups and the retinas are dissected (P1). Second, the retinas are placed in chambers filled with DNA and electroporated (P2). Third, the retinas are placed on filters and cultured for eight days (P3). Fourth, the retinal explants are fixed, mounted on slides, and imaged. Fluorescence intensity is measured with ImageJ software (P4). Fifth, the ImageJ data are processed in a spreadsheet program to quantify the difference in activity of various promoters (P5).
Figure 1. Overview of the retinal explant electroporation procedure. First, whole eyes are isolated from postnatal day 0 mouse pups and the retinas are dissected (P1). Second, the retinas are placed in chambers filled with DNA and electroporated (P2). Third, the retinas are placed on filters and cultured for eight days (P3). Fourth, the retinal explants are fixed, mounted on slides, and imaged. Fluorescence intensity is measured with ImageJ software (P4). Fifth, the ImageJ data are processed in a spreadsheet program to quantify the difference in activity of various promoters (P5).
 Figure 2. Construction of the electroporation dish. A) Unmodified microslide chamber from Harvard Apparatus, BTX model 453 (catalog #45-0105). B) A Dremel tool is used to cut the handle off a plastic tube rack. The handle is cut into rectangular spacers with the following dimensions: length 0.8cm, height 0.6cm, width 0.3cm. C) The plastic spacers are fitted into the microslide chamber at equal intervals. Aquarium sealant is injected into the gaps between the spacers (not shown). D) A metal bar is placed over the spacers. E) The bar and spacers are clamped onto the slide with binder clips to hold everything in place as the sealant dries overnight. F) The spacers are removed and the wells are tested to ensure that they are watertight. G) The finished slide fits into the plastic dish with the metal bars adjacent to the window in the side of the dish.
Figure 2. Construction of the electroporation dish. A) Unmodified microslide chamber from Harvard Apparatus, BTX model 453 (catalog #45-0105). B) A Dremel tool is used to cut the handle off a plastic tube rack. The handle is cut into rectangular spacers with the following dimensions: length 0.8cm, height 0.6cm, width 0.3cm. C) The plastic spacers are fitted into the microslide chamber at equal intervals. Aquarium sealant is injected into the gaps between the spacers (not shown). D) A metal bar is placed over the spacers. E) The bar and spacers are clamped onto the slide with binder clips to hold everything in place as the sealant dries overnight. F) The spacers are removed and the wells are tested to ensure that they are watertight. G) The finished slide fits into the plastic dish with the metal bars adjacent to the window in the side of the dish.
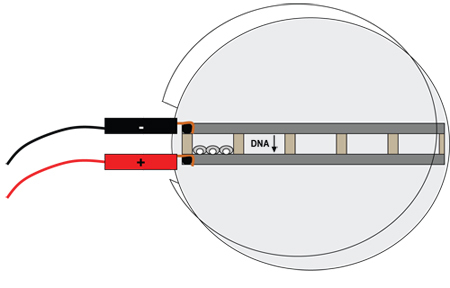 Figure 3. Diagram of the electroporation dish with retinas. The chambers are filled with DNA solutions (up to five different solutions at a time). Retinas are placed in the chambers and oriented so that the lens is leaning against the metal bar connected to the positive electrode; three or four retinas will fit in each of the five chambers. The electrical current will cause the negatively-charged DNA molecules to move into the retinal cells.
Figure 3. Diagram of the electroporation dish with retinas. The chambers are filled with DNA solutions (up to five different solutions at a time). Retinas are placed in the chambers and oriented so that the lens is leaning against the metal bar connected to the positive electrode; three or four retinas will fit in each of the five chambers. The electrical current will cause the negatively-charged DNA molecules to move into the retinal cells.
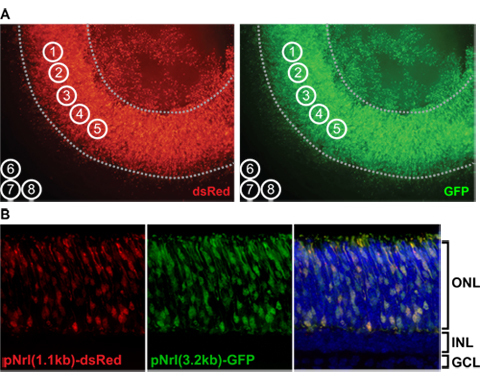 Figure 4. A) ImageJ measurement of retinal fluorescence levels in flatmount. Grayscale flatmount images in the DsRed (experimental) and GFP (control) channels are opened in ImageJ software; note that these images have been colored for illustrative purposes only. Five measurement circles (1 through 5) are placed over uniformly electroporated regions, avoiding the edges and lens (dotted lines). Three measurement circles (6 through 8) are placed outside the retina to determine background fluorescence levels. B) Cross-sectional images of an electroporated retinal explant at high power. The explant was fixed at postnatal day 8, cryoprotected in 30% sucrose/1X PBS overnight at 4°C, embedded in OCT, and cryo-sectioned at 12μm. The fluorescent constructs pNrl(1.1kb)-DsRed and pNrl(3.2kb)-GFP are expressed in photoreceptor cells in the outer nuclear layer (ONL). INL, inner nuclear layer; GCL, ganglion cell layer.
Figure 4. A) ImageJ measurement of retinal fluorescence levels in flatmount. Grayscale flatmount images in the DsRed (experimental) and GFP (control) channels are opened in ImageJ software; note that these images have been colored for illustrative purposes only. Five measurement circles (1 through 5) are placed over uniformly electroporated regions, avoiding the edges and lens (dotted lines). Three measurement circles (6 through 8) are placed outside the retina to determine background fluorescence levels. B) Cross-sectional images of an electroporated retinal explant at high power. The explant was fixed at postnatal day 8, cryoprotected in 30% sucrose/1X PBS overnight at 4°C, embedded in OCT, and cryo-sectioned at 12μm. The fluorescent constructs pNrl(1.1kb)-DsRed and pNrl(3.2kb)-GFP are expressed in photoreceptor cells in the outer nuclear layer (ONL). INL, inner nuclear layer; GCL, ganglion cell layer.
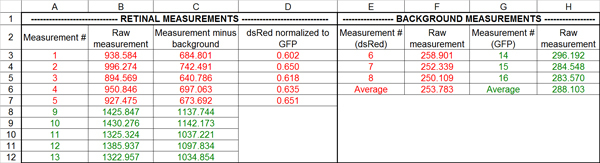 Figure 5. Processing of fluorescence data using Excel. In step 1, the mean pixel value for each measurement circle is copied into the spreadsheet (cells B3-B12, F3-F5, H3-H5). Measurements #1-5 are the DsRed retinal values and #6-8 are the DsRed background values; measurements #9-13 are the GFP retinal values and #14-16 are the GFP background values. Note that measurement #1 and #9 correspond to the same measurement circle, as do measurements #2 and #10, and so on. In step 2, the average background value for the DsRed and GFP channels is calculated (cells F6, H6). In step 3, the average background is subtracted from every retinal measurement (cells C3-C12). In step 4, each background-subtracted DsRed measurement is normalized to its corresponding GFP measurement (cells D3-D7).
Figure 5. Processing of fluorescence data using Excel. In step 1, the mean pixel value for each measurement circle is copied into the spreadsheet (cells B3-B12, F3-F5, H3-H5). Measurements #1-5 are the DsRed retinal values and #6-8 are the DsRed background values; measurements #9-13 are the GFP retinal values and #14-16 are the GFP background values. Note that measurement #1 and #9 correspond to the same measurement circle, as do measurements #2 and #10, and so on. In step 2, the average background value for the DsRed and GFP channels is calculated (cells F6, H6). In step 3, the average background is subtracted from every retinal measurement (cells C3-C12). In step 4, each background-subtracted DsRed measurement is normalized to its corresponding GFP measurement (cells D3-D7).
Discussion
Explant electroporation is a simple means of quantifying cis-regulatory activity in the developing mouse retina. Compared to cis-regulatory analysis via mouse transgenesis, electroporation is much cheaper, requiring only newborn mouse pups, DNA, dissecting instruments, and electroporation/tissue culture equipment. It is also far less time consuming: one experiment requires only a few hours of preparation time, a culture period of about eight days, and a few hours at the end of the experiment for imaging and data analysis. Explant electroporation is also superior to cell culture-based cis-regulatory analysis since actual retinal tissue is utilized. The retina develops quite normally in explant culture—it forms three distinct cellular layers (outer nuclear layer, inner nuclear layer, and ganglion cell layer), although photoreceptors fail to elaborate outer segments.
A further advantage is that explant electroporation is highly reproducible. The same constructs electroporated into different retinas, even on different days, typically results in the same relative expression levels. Furthermore, since the electroporated plasmids are thought to be maintained episomally in the nucleus and are not incorporated into chromosomes, they do not appear to be subject to the same integration site effects that bedevil cis-regulatory analysis performed in transgenic mice.
Explant electroporation does have several limitations. First, only cells that are still in the cell cycle can be efficiently transduced by electroporation15. At P0, rods and other later-born retinal cell types (bipolar cells, amacrine cells, M ller glia) are the main cell populations targeted by this method. Electroporation of cone photoreceptors by P0 electroporation has been reported16 but the efficiency appears to be low. A second limitation is that explant culture beyond two weeks results in progressive malformation of the retina and is therefore not recommended. If promoter quantification is required at late timepoints, however, an in vivo electroporation9 may be performed, followed by retinal dissection at the desired timepoint, flat-mounting of the dissected retina, and quantification as described in section 9. A third limitation is that this assay is only moderately high-throughput. Unlike cell culture-based assays that can test hundreds of constructs in a single experiment, the technique described in this protocol requires a minimum of one whole mouse retina per construct. Thus, only a couple dozen constructs can reasonably be electroporated in a day.
One additional caveat with respect to the quantification of promoter activity using the present approach is that there is a potential for 'bleed-through' of DsRed fluorescence into the GFP channel, particularly when assaying very strong promoters. The reason for this is that the emission spectrum of DsRed partially overlaps that of GFP. To circumvent this issue, optimized emission filters should be used that minimize the spectral overlap between DsRed and GFP. When such optimized filter sets are not available, another potential solution would be to use a blue-shifted fluorescent protein (e.g., BFP or CFP) in lieu of GFP.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors would like to thank Karen Lawrence for her help with the section describing construction of the electroporation chamber.
References
- Carroll SB, Grenier JK, Weatherbee SD. From DNA to diversity : molecular genetics and the evolution of animal design. 2nd edn. Blackwell Pub; 2005. [Google Scholar]
- Davidson EH. Genomic regulatory systems : development and evolution. Academic Press; 2001. [Google Scholar]
- Ptashne M, Gann A. Genes & signals. Cold Spring Harbor Laboratory Press; 2002. [Google Scholar]
- Blow MJ. ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet. 2010;42:806–810. doi: 10.1038/ng.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A. 2004;101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoTurco J, Manent JB, Sidiqi F. New and improved tools for in utero electroporation studies of developing cerebral cortex. Cereb Cortex. 2009;19:120–125. doi: 10.1093/cercor/bhp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Analysis of gene function in the retina. Methods Mol Biol. 2008;423:259–278. doi: 10.1007/978-1-59745-194-9_19. [DOI] [PubMed] [Google Scholar]
- Lee J, Myers CA, Williams N, Abdelaziz M, Corbo JC. Quantitative fine-tuning of photoreceptor cis-regulatory elements through affinity modulation of transcription factor binding sites. Gene Ther. 2010;17:1390–1399. doi: 10.1038/gt.2010.77. [DOI] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. II. Autoradiographic analysis of cell generation using tritiated thymidine. J Comp Neurol. 1979;188:263–272. doi: 10.1002/cne.901880205. [DOI] [PubMed] [Google Scholar]
- Young RW. Cell differentiation in the retina of the mouse. Anat Rec. 1985;212:199–205. doi: 10.1002/ar.1092120215. [DOI] [PubMed] [Google Scholar]
- Corbo JC. CRX ChIP-seq reveals the cis-regulatory architecture of mouse photoreceptors. Genome Res. 2010;20:1512–1525. doi: 10.1101/gr.109405.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Matsuda T, Cepko CL. A core paired-type and POU homeodomain-containing transcription factor program drives retinal bipolar cell gene expression. J Neurosci. 2008;28:7748–7764. doi: 10.1523/JNEUROSCI.0397-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karra D, Dahm R. Transfection techniques for neuronal cells. J Neurosci. 2010;30:6171–6177. doi: 10.1523/JNEUROSCI.0183-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi A, Peng GH, Chen S, Blackshaw S. Pias3-dependent SUMOylation controls mammalian cone photoreceptor differentiation. Nat Neurosci. 2010;13:1059–1065. doi: 10.1038/nn.2618. [DOI] [PMC free article] [PubMed] [Google Scholar]