

Abstract
The functional characterization of genes expressed during mammalian retinal development remains a significant challenge. Gene targeting to generate constitutive or conditional loss of function knockouts remains cost and labor intensive, as well as time consuming. Adding to these challenges, retina expressed genes may have essential roles outside the retina leading to unintended confounds when using a knockout approach. Furthermore, the ability to ectopically express a gene in a gain of function experiment can be extremely valuable when attempting to identify a role in cell fate specification and/or terminal differentiation.
We present a method for the rapid and efficient incorporation of DNA plasmids into the neonatal mouse retina by electroporation. The application of short electrical impulses above a certain field strength results in a transient increase in plasma membrane permeability, facilitating the transfer of material across the membrane 1,2,3,4. Groundbreaking work demonstrated that electroporation could be utilized as a method of gene transfer into mammalian cells by inducing the formation of hydrophilic plasma membrane pores allowing the passage of highly charged DNA through the lipid bilayer 5. Continuous technical development has resulted in the viability of electroporation as a method for in vivo gene transfer in multiple mouse tissues including the retina, the method for which is described herein 6, 7, 8, 9, 10.
DNA solution is injected into the subretinal space so that DNA is placed between the retinal pigmented epithelium and retina of the neonatal (P0) mouse and electrical pulses are applied using a tweezer electrode. The lateral placement of the eyes in the mouse allows for the easy orientation of the tweezer electrode to the necessary negative pole-DNA-retina-positive pole alignment. Extensive incorporation and expression of transferred genes can be identified by postnatal day 2 (P2). Due to the lack of significant lateral migration of cells in the retina, electroporated and non-electroporated regions are generated. Non-electroporated regions may serve as internal histological controls where appropriate.
Retinal electroporation can be used to express a gene under a ubiquitous promoter, such as CAG, or to disrupt gene function using shRNA constructs or Cre-recombinase. More targeted expression can be achieved by designing constructs with cell specific gene promoters. Visualization of electroporated cells is achieved using bicistronic constructs expressing GFP or by co-electroporating a GFP expression construct. Furthermore, multiple constructs may be electroporated for the study of combinatorial gene effects or simultaneous gain and loss of function of different genes. Retinal electroporation may also be utilized for the analysis of genomic cis-regulatory elements by generating appropriate expression constructs and deletion mutants. Such experiments can be used to identify cis-regulatory regions sufficient or required for cell specific gene expression 11. Potential experiments are limited only by construct availability.
Protocol
1. Plasmid preparation for electroporation
The DNA concentration required for electroporation is 5μg/μl. This typically requires the desired plasmids to be amplified using a Maxi-prep (Qiagen) or equivalent method followed by a purification and concentration of the DNA to the working amount. The following steps describe the preparation of DNA to the working amount.
Aliquot 100 μg of DNA (from Maxi-prep or equivalent) and dilute the volume to 100 μL for ease of manipulation.
Add phenol; the amount of phenol should be calculated to obtain an approximately 60% DNA/volume ratio (μg DNA:total volume) i.e.: 100 μg DNA (in 100 μL) plus 67 μL phenol (100 μg:167 μL). Mix thoroughly; do not pipette up and down as this may cause excessive shearing of the DNA.
Spin the DNA for 5 minutes at 14000 RPM at room temperature.
Collect supernatant and add chloroform using the same DNA:total volume ratio in step 1.2; mix as described in step 1.2 and spin at 14000 RPM for 5 minutes at room temperature.
Collect the supernatant, add 10 % supernatant volume of 3 M sodium acetate to the DNA solution, mix gently and add 2.5 X the supernatant volume of 100% ethanol. Mix by inversion. DNA should precipitate out of solution during the mixing.
Spin the DNA at 4°C for 10 minutes at 14000 RPM.
Rinse with 350 μl of 70 % ethanol, spin at 14000 RPM for 5 minutes at 4°C.
Air dry the pellet then dissolve the DNA in 1 X PBS (Cell biology grade) to 5μg/μL. Do not overdry the sample as this will make it very difficult to dissolve into PBS.
Add Fast Green dye (10% stock) to the DNA solution, the final concentration of dye is 0.1%, to act as an injection tracer.
2. Subretinal injection of DNA
The following steps are performed with the assistance of a stereomicroscope. Once practiced the process of eye opening, incision, and injection takes less than 1.5 minutes which is not enough time for a properly anesthetized pup to recover. Using a sharp 30-gauge needle, carefully open the eye by cutting along the fused junctional epithelium (Fig. 1C). Do not apply excessive force as this may result in cutting of the underlying eye. Avoid cutting beyond the range of the eyelid junction as this will result in bleeding which can obscure the injection.
Anesthetize the newborn mice on ice for several minutes (Fig. 1A), do not bury pups in the ice as this may result in mortality. The length of time on ice is variable from pup to pup, typically 5 minutes is sufficient but mice should be carefully monitored as individuals may respond very quickly or may require longer exposure to ensure appropriate anesthesia. Conduct a paw pinch with hemostats to check for withdrawal reflex.
In order to facilitate the injection of DNA to the subretinal space the eye must first be opened. Swab the eye to be injected with a 70% isopropyl alcohol prep (Tyco healthcare) and identify the fused junctional epithelium where the two eyelids come together (Fig. 1B).
Using a sharp 30-gauge needle, carefully open the eye by cutting along the fused junctional epithelium (Fig. 1C). Do not apply excessive force as this may result in cutting of the underlying eye. Avoid cutting beyond the range of the eyelid junction as this will result in bleeding which can obscure the injection.
To facilitate penetration of the eye by the blunt ended injection needle use the tip of a 30-guage needle make a small incision in the sclera near the junction with the cornea (Fig. 1E). Do not penetrate too deeply as this may result in a puncture of the lens.
Draw 0.3 μl of DNA solution into a 33 gauge blunt ended injection needle using the syringe gradients to measure the volume individually for each injection (needle outer diameter 0.52 mm, inner diameter 0.13 mm; Exmire microsyringe; Ito corp). Insert the needle into the incision until the resistance of the opposing scleral wall is felt. Be careful to avoid penetrating the lens as the needle is passed through the vitreal chamber (Fig. 2B).
Slowly inject 0.3 μl of DNA solution into the subretinal space using the index finger or thumb to depress the syringe plunger. The experimenter must be careful to control the speed of plunger depression so that the rate at which the DNA solution is injected into the subretinal space does not outpace the rate at which the DNA solution spreads within subretinal space (Fig. 2C). The DNA solution is viscous so it is important that the needle is not pressed too tightly against the opposing scleral wall as this may result in the DNA not being injected or not spreading evenly. A successful injection will result in an even spread of DNA solution in a portion of the subretinal space. Regions of retina with and without underlying green tracer should be evident when rotating the injected animal.
3. Electroporation
Electroporation is performed using a 10 mm diameter tweezer electrode (Model #522; BTX Instruments). Soak the tweezer electrode in PBS in order to maximize electrical conductivity from the electrode to the animal and place the head of the injected pup between the electrodes with the injected eye adjacent to the positive pole electrode and the non-injected eye adjacent to the negative pole electrode (Fig. 3B).
Apply five square pulses using a pulse generator: each pulse is 80 volts and 50 milliseconds in duration with a 950 millisecond interval between pulses.
The electroporated pups must now be warmed until they recover from the ice anesthesia. This can be done by placing the pups under a warming lamp or on top of a slide warmer. If using a slide warmer ensure that an appropriate pad is placed between the metal surface and the recovering pup. Upon recovery return the electroporated pups to their mother.
4. Preparation of eyes for analysis
The time of harvesting and analysis of the electroporated retinas is performed subject to the objectives of the experiment. GFP expression can be grossly visualized 3 days after electroporation. For cryoembedding and sectioning proceed as follows.
Sacrifice electroporated animals at the desired timepoint. Disect out the electroporated eye and fix in 4% paraformaldehyde at 4°C for 50 minutes.
Carefully remove the retina from the eye by micro-dissecting away the sclera, cornea, lens, choroid and retinal pigmented epithelium. The dissected retinas may be analyzed under fluorescence to grossly determine the efficiency of electroporation. Transfer the retinas to 30% sucrose solution overnight at 4°C.
Freeze retinas into O.C.T. cryo-embedding media (Sakura Finetek USA) and store at -80°C. Retinas may now be sectioned on a cryotome and captured onto glass slides.
5. Representative Results:
Examples of P0 neonatal retinas electroporated with a pCAG-EGFP expression plasmid are presented at 3 days following electroporation (P3) and 14 days following electroporation (P14) in figure 4. The area of retinal tissue electroporated is variable from experiment to experiment depending on the extent of tissue damage incurred during the injection and evenness of the spread of the DNA solution into the subretinal space. Typically 90-100% of electroporated retinas will feature cells that have successfully incorporated and expressed the introduced plasmid. However, when factoring in tissue morphology and electroporation efficiency only 40-60 % of electroporated retinas will be suitable for comparative analysis. Rosette formation and retinal detachment at the site of needle penetration is nearly always observed and this region should not be used for analysis. Successful DNA spread during the microinjection results in efficiently electroporated tissue lateral to the site of needle penetration free from rosettes and detachment. Gliosis is also an important experimental concern and nearly always occurs at the site of needle penetration. However, as with rosette formation and retinal detachment, gliosis is typically not significant in laterally electroporated tissue. Immunohistochemical markers such as glial fibrillary acidic protein (GFAP) can be utilized to determine whether levels of reactive gliosis in analyzed regions fall within acceptable thresholds. Sectioned retinas demonstrating that the morphology of the electroporated retina remains intact and that EGFP expression labels individual electroporated cells are presented. Examples 3 days following a P0 electroporation (Fig. 4A-C) and 14 days following a P0 electroporation (Fig. 4D-F) are presented.
In P3 retinas the majority of electroporated cells are located in the neuroblastic layer (NBL) of the retina, and do not demonstrate distinct morphological characteristics of any of the differentiated neural or glial cells of the retina (Fig. 4B, C). By P14 electroporated cells can be found in the outer nuclear layer (ONL) and the inner nuclear layer (INL) of the retina. Furthermore the various labeled cells now display characteristic morphological hallmarks of differentiated neurons including interneurons of the INL, photoreceptors, and Müller glia (Fig. 4E, F).
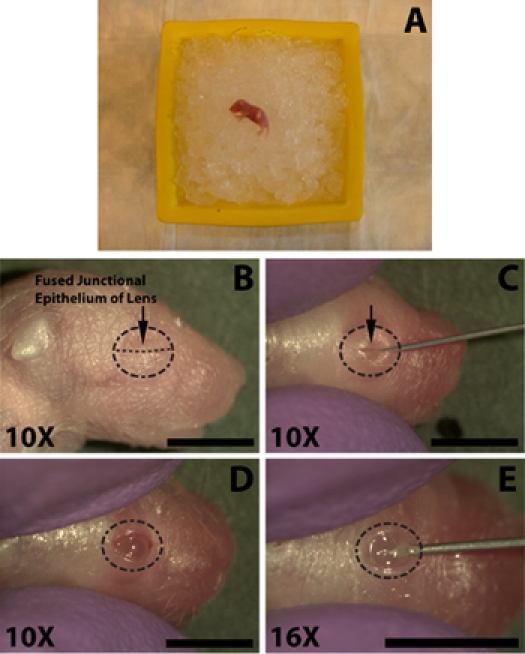 Figure 1. Anesthesia of a newborn (P0) mouse pup and opening of eye for subretinal injection of DNA solution. A) Neonatal pup placed on a bed of crushed ice for anesthetization. Eye to be injected is placed down against the ice. B) The location of the fused junctional epithelium (B, arrow) of the eyelids to be cut for eye opening. C) Cutting of the eyelids along the fused junctional epithelium (C, arrow) to expose the eye. D) The opened eye following cutting at the fused junction. E) An incision is made into the eye beneath in the sclera beneath the cornea to facilitate easy insertion of the blunt ended micro-injection syringe. Scale bars, B, C, D, E: 4mm.
Figure 1. Anesthesia of a newborn (P0) mouse pup and opening of eye for subretinal injection of DNA solution. A) Neonatal pup placed on a bed of crushed ice for anesthetization. Eye to be injected is placed down against the ice. B) The location of the fused junctional epithelium (B, arrow) of the eyelids to be cut for eye opening. C) Cutting of the eyelids along the fused junctional epithelium (C, arrow) to expose the eye. D) The opened eye following cutting at the fused junction. E) An incision is made into the eye beneath in the sclera beneath the cornea to facilitate easy insertion of the blunt ended micro-injection syringe. Scale bars, B, C, D, E: 4mm.
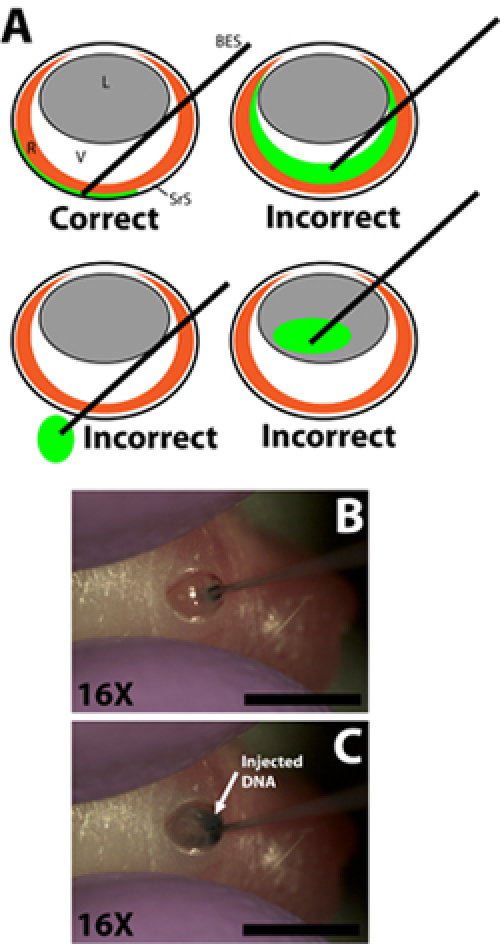 Figure 2. Insertion of the blunt ended micro-injection syringe and injection of DNA plasmid solution into the subretinal space. A) Cartoon schematic demonstrating the correct insertion of the micro injection syringe into the eye and settlement of the syringe tip into the subretinal space. Injection of the DNA solution into the vitreous chamber, passage of the syringe through the eye into the socket, or injection into the lens will result in failed experiments with few if any electroporated cells. B) Penetration of the syringe into the incision made in the scleral wall and settlement of the syringe tip into the subretinal space. C) Injection of the DNA solution into the subretinal space. Scale bars, B, C: 4mm. Abbreviations as follows: R-retina, L-lens, V-vitreous, SrS-subretinal space, BES-blunt ended syringe.
Figure 2. Insertion of the blunt ended micro-injection syringe and injection of DNA plasmid solution into the subretinal space. A) Cartoon schematic demonstrating the correct insertion of the micro injection syringe into the eye and settlement of the syringe tip into the subretinal space. Injection of the DNA solution into the vitreous chamber, passage of the syringe through the eye into the socket, or injection into the lens will result in failed experiments with few if any electroporated cells. B) Penetration of the syringe into the incision made in the scleral wall and settlement of the syringe tip into the subretinal space. C) Injection of the DNA solution into the subretinal space. Scale bars, B, C: 4mm. Abbreviations as follows: R-retina, L-lens, V-vitreous, SrS-subretinal space, BES-blunt ended syringe.
 Figure 3. Orientation of the tweezer electrode onto the mouse for electroporation. A) Cartoon schematic demonstrating the orientation of the positive and negative paddles of the tweezer electrode relative to the electroporated eye. Green represents the location of injected DNA into the subretinal space. Dashed arrows represent the electrophoretic movement of negatively charged injected DNA towards the positive electrode. DNA electrophoresis occurs from the subretinal space adjacent to the negative electrode into the retina which is oriented towards the positive electrode. B) The positive paddle of the tweezer is placed adjacent to the DNA microinjected eye and the negative paddle is placed adjacent to the non-injected eye. C) High magnification image of tweezer electrode placement on the neonatal mouse. Dashed lines represent the direction of electrophoretic movement of DNA towards the positive electrode. Scale bar, C: 5mm.
Figure 3. Orientation of the tweezer electrode onto the mouse for electroporation. A) Cartoon schematic demonstrating the orientation of the positive and negative paddles of the tweezer electrode relative to the electroporated eye. Green represents the location of injected DNA into the subretinal space. Dashed arrows represent the electrophoretic movement of negatively charged injected DNA towards the positive electrode. DNA electrophoresis occurs from the subretinal space adjacent to the negative electrode into the retina which is oriented towards the positive electrode. B) The positive paddle of the tweezer is placed adjacent to the DNA microinjected eye and the negative paddle is placed adjacent to the non-injected eye. C) High magnification image of tweezer electrode placement on the neonatal mouse. Dashed lines represent the direction of electrophoretic movement of DNA towards the positive electrode. Scale bar, C: 5mm.
 Figure 4. Neonatal mouse retinas (P0) electroporated with pCAG-EGFP and analyzed at postnatal day 3 (P3) and postnatal day 14 (P14). A-C) Confocal images of a sectioned P3 retina electroporated with pCAG-EGFP at P0. The majority of electroporated cells sit in the retinal neuroblastic layer (NBL). (D-F) Confocal images of a sectioned P14 retina electroporated with pCAG-EGFP at P0. Electroporated cells can be identified in the outer nuclear layer (ONL) and the inner nuclear layer (INL) of the retina. Scale Bars, A-F: 50 μm.
Figure 4. Neonatal mouse retinas (P0) electroporated with pCAG-EGFP and analyzed at postnatal day 3 (P3) and postnatal day 14 (P14). A-C) Confocal images of a sectioned P3 retina electroporated with pCAG-EGFP at P0. The majority of electroporated cells sit in the retinal neuroblastic layer (NBL). (D-F) Confocal images of a sectioned P14 retina electroporated with pCAG-EGFP at P0. Electroporated cells can be identified in the outer nuclear layer (ONL) and the inner nuclear layer (INL) of the retina. Scale Bars, A-F: 50 μm.
Discussion
In vivo electroporation represents a rapid and efficient method for the transformation of retinal cells with DNA expression plasmids. This method allows the experimenter to perform gain of function studies by ectopically introducing a gene of interest under the control of a ubiquitously expressed promoter or to perform loss of function studies by using shRNA constructs targeting genes of interest. Furthermore, multiple DNA plasmids can be electroporated simultaneously, allowing the experimenter to analyze multiple gene effects or to knock down one gene while introducing a second gene of interest. Electroporation of plasmids driving expression of Cre recombinase can also be utilized to eliminate gene expression in a mosaic fashion where animals containing a floxed gene of interest are available. This method is particularly valuable for the determination of intrinsic vs. extrinsic gene function. Conversely electroporation of a gene back into the retinas of conventional knockout mice allows the experimenter to perform rescue studies attempting to reverse phenotypes identified in knockouts. Mice of any background strain can be utilized for electroporation subject to the requirements of the study being performed. However, it is best, where possible, to use strains which demonstrate strong maternal behavior, i.e. Swiss Webster or CD1, in order to minimize the possibility of rejection of electroporated pups. Albino laboratory strains are easier to work with since the spread of the DNA solution during microinjection can be easily visualized. Strains with darkly pigmented eyes such as C57BL/6J are more challenging since the Fast Green tracer can not be discriminated against the pigment of the retinal pigmented epithelium.
The number of cells electroporated is extensive however the ability to electroporate progenitor populations competent to generate the earliest-born cell types is limited. Late-born cells including photoreceptors, bipolar cells, Müller glia, and amacrine cells are easily electroporated using this method. Conversely, early-born cells including retinal ganglion cells, horizontal cells, and subsets of amacrine cells are typically not electroporated using this method. A modification to this protocol in which whole embryonic retinas are dissected, electroporated in vitro and then cultured as explants can be utilized to identify genes regulating specification of early-born cell classes7.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was funded by NIH R01EY020560-01 and by a W.M. Keck Young Scholar in Medical Research Award. The authors would like to thank Joseph Bedont for his assistance during the imaging of retinal preparations and injections.
References
- Neumann E, Rosenheck K. Permeability changes induced by electric impulses in vesicular membranes. J. Membr. Biol. 1972;10:279–290. doi: 10.1007/BF01867861. [DOI] [PubMed] [Google Scholar]
- Turnbull RJ. Letter: Letter: An alternate explanation for the permeability changes induced by electrical impulses in vesicular membranes. J. Membr. Biol. 1973;14:193–196. doi: 10.1007/BF01868077. [DOI] [PubMed] [Google Scholar]
- Zimmermann U, Schulz J, Pilwat G. Transcellular ion flow in Escherichia coli B and electrical sizing of bacterias. Biophys. J. 1973;13:1005–1013. doi: 10.1016/S0006-3495(73)86041-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinosita K, Tsong TY. Voltage-induced pore formation and hemolysis of human erythrocytes. Biochim. Biophys. Acta. 1977;471:227–242. doi: 10.1016/0005-2736(77)90252-8. [DOI] [PubMed] [Google Scholar]
- Neumann E, Schaefer-Ridder M, Wang Y, Hofschneider PH. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1982;1:841–845. doi: 10.1002/j.1460-2075.1982.tb01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz M, Eberhart J, Mastick GS, Krull CE. Sparking new frontiers: using in vivo electroporation for genetic manipulations. Dev. Biol. 2001;233:13–21. doi: 10.1006/dbio.2001.0181. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. U. S. A. 2004;101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. U. S. A. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi A. Pias3-dependent SUMOylation directs rod photoreceptor development. Neuron. 2009;61:234–246. doi: 10.1016/j.neuron.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi A. The orphan nuclear hormone receptor ERRbeta controls rod photoreceptor survival. Proc. Natl. Acad. Sci. U. S. A. 2010;107:11579–11584. doi: 10.1073/pnas.1000102107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS. A Core Paired-Type and POU Homeodomain-Containing Transcription Factor Program Drives Retinal Bipolar Cell Gene Expression. J. Neurosci. 2008;28:7748–7764. doi: 10.1523/JNEUROSCI.0397-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]