

Abstract
Organotypic cultures of neuronal tissue were first introduced by Hogue in 1947 1,2 and have constituted a major breakthrough in the field of neuroscience. Since then, the technique was developed further and currently there are many different ways to prepare organotypic cultures. The method presented here was adapted from the one described by Stoppini et al. for the preparation of the slices and from Gogolla et al. for the staining procedure 3,4.
A unique feature of this technique is that it allows you to study different parts of the brain such as hippocampus or cerebellum in their original structure, providing a big advantage over dissociated cultures in which all the cellular organization and neuronal networks are disrupted. In the case of the cerebellum it is even more advantageous because it allows the study of Purkinje cells, extremely difficult to obtain as dissociated primary culture. This method can be used to study certain developmental features of the cerebellum in vitro, as well as for electrophysiological and pharmacological experiments in both wild type and mutant mice.
The method described here was designed to study the effect of apoptotic stimuli such as Fas ligand in the developing cerebellum, using TUNEL staining to measure apoptotic cell death. If TUNEL staining is combined with cell type specific markers, such as Calbindin for Purkinje cells, it is possible to evaluate cell death in a cell population specific manner. The Calbindin staining also serves the purpose of evaluating the quality of the cerebellar cultures.
Keywords: Neuroscience, Issue 51, Cerebellum, Organotypic, Fas, Apoptosis, Purkinje cell
Protocol
1. Organotypic Cerebellar Cultures:
To successfully generate organotypic cerebellar cultures use mice between P0-P3 or P8-P12. For our experiment we used mice in the P8-P12 range because this developmental stage was more appropriate for our study.
- Prepare the dissection and the culture medium:
- Brain dissection and slice preparation are carried out in low sodium Artificial Cerebrospinal Fluid (ACSF) containing 1mM calcium chloride, 10mM D-Glucose, 4mM potassium chloride, 5mM magnesium chloride, 26mM sodium bicarbonate, 246mM sucrose and phenol red solution (1:1000), pH 7.3. To sterilize the solution, filter it through a .22 μm filter and store at 4 °C.
- Cerebellar slices are cultured in 75% Minimum Essential Medium Eagle (MEM), 25% heat-inactivated horse serum, 25mM HEPES, 1mM glutamine, 5mg/ mL glucose, penicillin (100 U/ mL) and streptomycin (100 U/ mL). Filter the medium through a 0.22 μm filter and store at 4 °C for a maximum of two weeks.
Dissect brain in ice-cold ACSF medium previously bubbled with Carbogen (95% oxygen and 5% carbon dioxide) Note, that saturation with Carbogen will change the pH of the solution so it will shift from red to orange. The dissection should be performed in the minimum amount of time possible being careful not to damage the brain structure.
- Slice the brain in sagittal sections of 400 μm using a vibratome.
- Make a sagittal cut with a razor blade in one hemisphere, trying to cut as little as possible of the cerebellum; this will provide a flat surface for slicing. The cortex is used only for support to make the slicing easier but since time is an important issue it is more convenient to cut out the rostral half of the cortex with a razor blade.
- Use cyanoacrylate (Superglue) to fix the position of the brain on the metal vibratome plate. It is important to chill the plate on ice before adding the glue and to spread it well using the tip of the tube to form a thin adhesive layer. If there is an excess of Superglue, it will detach from the plate when it comes in contact with the ACSF and the sample will be lost.
- Once the brain is attached to the vibratome plate, add enough ACSF media to cover the brain and put ice below the plate to keep it cold.
- Cut 400 μm slices and transfer them with a plastic Pasteur pipette (cut the tip to widen it and avoid damage to the slices) to a 6-well cell culture plate containing ACSF on ice.
- Separate the cerebellum from the rest of the brain with the help of two insulin syringes (syringes with small diameter 28-gauge needles) under the dissecting microscope. Some slices will have Superglue attached to the edges: this is the moment to try to remove it as it can affect the viability of the slice. It is important to do it carefully without damaging the cerebellar structure.
Transfer the cerebellar slices to 6-well plates containing 30mm culture plate inserts with 0.4 μm pores. Add 1 mL of culture media per well, that was pre-conditioned by incubation for at least 2 hours at 37°C and 5% CO2 (in the cell culture incubator). Place the inserts on top of the media making sure there are no air bubbles trapped underneath and then place the cerebellar slices (1-3 per insert) on top of the inserts making sure they are fully extended and that no liquid is covering them. Aspirate excess of media if necessary. Incubate at 37°C and 5% CO2 changing the culture media every 2 or 3 days. It is best to replace only a portion of the medium (e.g. ½ volume) at once, since used medium contains slice-derived trophic factors that are important for cell survival.
2. Fas Treatment and Staining of Cerebellar Slices:
For the apoptotic challenge experiment, we first culture the slices for 5 days changing media on day in vitro 3 (DIV3). On DIV5 cultures are treated with 0.5 μg/ mL of the Fas agonist antibody Jo2 by adding it directly to the media in order to have a clean background of cell death since the changes of media can induce some mortality in the cultures. Half of the wells are left untreated as controls.
After 24 hours remove the media underneath the inserts by suction. To fix the slices, add 1 mL of ice cold 4% PFA beneath and 1 mL above the insert. Incubate for 10 minutes at room temperature.
After 10 minutes remove the PFA and wash briefly with cold PBS.
Next remove the PBS and add 1 mL beneath and 1 mL above the insert of 20% ice-cold methanol in PBS and incubate for 5 minutes at room temperature.
Wash again with PBS and add 1 mL beneath and 1 mL above the insert of 0.5% Triton-X-100 in PBS for permeabilization. Incubate for a minimum of 12 hours at 4°C.
After permeabilization, block with 20% BSA in PBS for a minimum of 4 hours at room temperature or overnight at 4°C. At this stage the slices can be kept in blocking solution for at least 3 days at 4°C.
In order to stain the slices carefully cut the piece of the membrane insert attached to the cerebellar slice and transfer it to 24-well plates containing PBS.
- The immunostaining is done sequentially, first with TUNEL reagent for one hour and then with Calbidin antibody overnight.
- Aspirate the PBS and add 250 μL off TUNEL reagent into each well making sure it covers the entire surface of the slice. Incubate at 37°C for one hour in the dark.
- After one hour remove the TUNEL mix and wash with PBS three times for 10 minutes.
- After the last wash, remove the PBS and add 250 μL of a 1/1000 dilution of the Calbindin antibody in PBS and incubate overnight at 4°C in the dark.
- Remove the primary antibody solution and wash with PBS three times for 10 minutes.
- Add 250 μL of secondary antibody diluted 1/500 in PBS and incubate for at least three hours at room temperature in the dark. The TUNEL kit we used is labeled with TMR red, therefore our secondary antibody for Calbindin is conjugated to Alexa-488.
- Wash three times for 10 minutes with PBS and mount the slices onto a glass microscope slide using a mounting media that contains DAPI.
- Image the slices with a confocal microscope using the 488 nm and 561 nm excitation wavelenght for Calbindin and TUNEL respectively.
3. Representative Results:
The main challenge of this protocol is to be able to generate healthy cerebellar slice cultures, especially since our final readout will be cell death. A healthy culture appears as one where the cell body layer is translucent and allows you to see the foliated structure of the cerebellum under the microscope (Figure 1). After few days in culture the slices start to thin and non-neuronal cells can be observed migrating away from the margins of the slice. The dark round cells (most likely macrophages) that cover the slices after few days in culture, will eventually decrease in number after 10-14 days in vitro (Figure 2) 5. The ultimate proof for the viability of the slices is to stain for different cellular markers, such as Calbindin for the Purkinje cells. Figure 3 shows a representative confocal microscope image of the Purkinje cell layer with several TUNEL positive nuclei.
It is important to consider that even if some slices received an apoptotic stimulus, they were only exposed for 24 hours. This allowed sufficient time to observe DNA fragmentation in the dying cells with the TUNEL staining, but a defined Purkinje cell layer was still present.
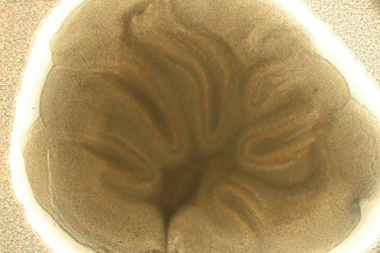 Figure 1. Healthy cerebellar slice at DIV2. This image shows the translucent cell body layer and the foliated cerebellar structure in a healthy slice.
Figure 1. Healthy cerebellar slice at DIV2. This image shows the translucent cell body layer and the foliated cerebellar structure in a healthy slice.
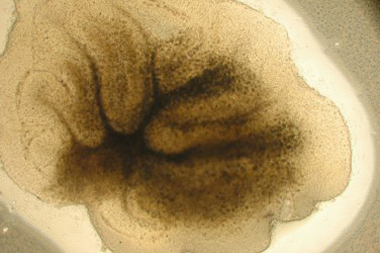 Figure 2. Healthy cerebellar slice at DIV 7. In this image we can still observe the foliated structure of the cerebellum and the appearance of dark cell bodies.
Figure 2. Healthy cerebellar slice at DIV 7. In this image we can still observe the foliated structure of the cerebellum and the appearance of dark cell bodies.
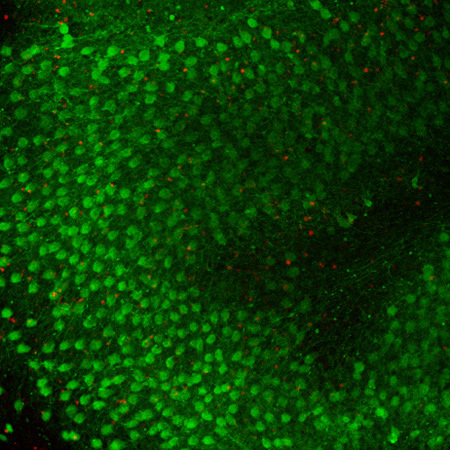 Figure 3. Representative image of a Fas-treated cerebellar slice. This confocal image shows the Purkinje cells stained with Calbindin (green) and the apoptotic cells positive for TUNEL staining (red). Purkinje cells do not appear in a single row but are clustered forming a folium-like structure.
Figure 3. Representative image of a Fas-treated cerebellar slice. This confocal image shows the Purkinje cells stained with Calbindin (green) and the apoptotic cells positive for TUNEL staining (red). Purkinje cells do not appear in a single row but are clustered forming a folium-like structure.
Discussion
This method describes one of the many possible applications of neuronal organotypic cultures and it has allowed us to study the effect of apoptotic stimuli in wild type and mutant cerebellar slices.
The most critical part of this experiment is to be able to consistently generate healthy cerebellar slice cultures. The key factors are dissection and slicing technique and the ability to perform the protocol in the least time possible, but at the same time being careful not to damage the slices. Another crucial factor is the preparation and composition of the culture media, these cultures are very sensitive, so we had to test several recipes and brands of media. Even different batches of serum or any other components of the media could affect the viability of the slices. We got the best results with the Horse Serum from Invitrogen Lot # 480116.
Once the cerebellar slices are generated one can study cerebellar development, electrophysiology, cell survival alone or in response to apoptotic stimuli, excitotoxicity or oxygen deprivation. Comparative studies using wild type and mutant cerebellar slices have been very insightful in many aspects of cerebellar function and development.
Disclosures
All animal work conducted was done in accordance with policies established by the IACUC. The Salk Institute is an AAALAC accredited facility.
Acknowledgments
This study was funded in part by the IPSEN Foundation. IMV is an American Cancer Society Professor of Molecular Biology, and holds the Irwin and Joan Jacobs Chair in Exemplary Life Sciences. This work was supported in part by grants from the NIH, Leducq Foundation, Meriaux Foundation, Ellison Medical Foundation, Ipsen/Biomeasure, Sanofi Aventis, Prostate Cancer Foundation, and the H.N. and Frances C. Berger Foundation. PAS is funded by McKnight Endowment Fund for Neuroscience and the National Institute on Drug Abuse (R01 DA019022). The authors would like to thank Mark Schmitt. Graphic art was designed by Jamie T. Simon .Sponsorship was kindly provided by Invitrogen.
References
- Hogue MJ. Human fetal ependymal cells in tissue cultures. Anat Rec. 1947;99:523–529. [PubMed] [Google Scholar]
- Hogue MJ. Review of studies of human fetal brain cells in tissue cultures. Etudes Neonatales. 1952;1:1–13. [PubMed] [Google Scholar]
- Gogolla N, Galimberti I, DePaola V, Caroni P. Staining protocol for organotypic hippocampal slice cultures. Nat Protoc. 2006;1:2452–2456. doi: 10.1038/nprot.2006.180. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Beat H, Gähwiler SMT, McKinney RA, Debanne D, Robertson RT. In: Organotypic Slice Cultures of Neural Tissue in Culturing Nerve Cells. Banker G, Goslin K, editors. Cambridge: MIT Press; 1998. pp. 461–498. [Google Scholar]