

Abstract
Two electrode voltage clamp electrophysiology (TEVC) is a powerful tool to investigate the mechanism of ion transport1 for a wide variety of membrane proteins including ion channels2, ion pumps3, and transporters4. Recent developments have combined site-specific fluorophore labeling alongside TEVC to concurrently examine the conformational dynamics at specific residues and function of these proteins on the surface of single cells.
We will describe a method to study the conformational dynamics of membrane proteins by simultaneously monitoring fluorescence and current changes using voltage-clamp fluorometry. This approach can be used to examine the molecular motion of membrane proteins site-specifically following cysteine replacement and site-directed fluorophore labeling5,6. Furthermore, this method provides an approach to determine distance constraints between specific residues7,8. This is achieved by selectively attaching donor and acceptor fluorophores to two mutated cysteine residues of interest.
In brief, these experiments are performed following functional expression of the desired protein on the surface of Xenopus leavis oocytes. The large surface area of these oocytes enables facile functional measurements and a robust fluorescence signal5. It is also possible to readily change the extracellular conditions such as pH, ligand or cations/anions, which can provide further information on the mechanism of membrane proteins4. Finally, recent developments have also enabled the manipulation of select internal ions following co-expression with a second protein9.
Our protocol is described in multiple parts. First, cysteine scanning mutagenesis proceeded by fluorophore labeling is completed at residues located at the interface of the transmembrane and extracellular domains. Subsequent experiments are designed to identify residues which demonstrate large changes in fluorescence intensity (<5%)3 upon a conformational change of the protein. Second, these changes in fluorescence intensity are compared to the kinetic parameters of the membrane protein in order to correlate the conformational dynamics to the function of the protein10. This enables a rigorous biophysical analysis of the molecular motion of the target protein. Lastly, two residues of the holoenzyme can be labeled with a donor and acceptor fluorophore in order to determine distance constraints using donor photodestruction methods. It is also possible to monitor the relative movement of protein subunits following labeling with a donor and acceptor fluorophore.
Keywords: Cellular Biology, Issue 51, membrane protein, two electrode voltage-clamp, biophysics, site-specific fluorophore labeling, microscopy, conformational dynamics
Protocol
1. Protein Expression
Clone the membrane protein of interest into a vector suitable for Xenopus laevis oocyte expression such as pTLN11 or pSG01MX12. Optimized vectors contain both 5' and 3' Xenopus laevis β-Globin untranslated regions11,12, a unique restriction site, and an RNA promoter site located before the 5' UTR11. These components are required for optimal expression in oocytes, linearization of the plasmid before mRNA synthesis and mRNA synthesis, respectively.
2. Cysteine Mutation
A Kyte-Doolittle plot is employed to identify the transmembrane domains of the targeted protein using the protein's amino acid sequence (ProtScale, ExPASy)13. The data is displayed as hydrophobicity vs. sequence number. Residues that display positive values are most likely found in the transmembrane domain. Use this plot to determine the most probable residues located at the interface between the transmembrane and extracellular domains that will be mutated to cysteine. As determining the interface of the transmembrane domain and extracellular region cannot be predicted with complete certainty using the Kyte-Doolittle plot, it is important to examine a range of residues which are predicted to be at the interface. This region of the protein is selected for the predicted movement of the fluorophore.During a conformational change, the fluorophore will move between a hydrophilic and hydrophobic environment or between a more water quenching and less water quenching environment. Each of these predicted changes in environment will result in a change in fluorescence intensity.
QuickChange Site-Directed Mutagenesis Kit (Stratagene) is used to generate single extracellularly accessible cysteine constructs. In brief, mix 5 μL of 10X reaction buffer, 2 μL of 5 ng/μL plasmid DNA, 1.25 μL each of 100 ng/μL forward and reverse primer, 1 μL of dNTP (100 mM), and 39.5 μL of double-distilled water. Lastly, add 1 μL of PfuTurbo DNA polymerase (Stratagene). The primers used for this step should be designed with the cysteine mutation at an extracellular residue of interest.
Program a thermal cycler to the following specifications: 1 cycle at 95°C for 30 seconds, 12-18 cycles at 95°C for 30 seconds, 55°C for 1 minute, and 68°C for 1 minute per kb plasmid length. The annealing temperature (55°C ) is calculated directly from the melting temperature of each primer. A final step can be programmed to hold the temperature at 4°C if the reaction is performed overnight. The number of cycles depends on the mutation. Use 12 cycles for point mutations, 16 cycles for a single amino acid change, or 18 cycles for multiple amino acid insertions.
Add 1 μL of DpnI (New England Biolabs) to the reaction mixture, mix by pipetting, and centrifuge (6000 rpm) for 1 minute. Incubate for 1 hour at 37°C in water bath.
Thaw top 10 electro-competent cells (Invitrogen) on ice and incubate sterile SOC media (20 mg/ml tryptone, 5 mg/mL yeast extract, 1 mg/mL NaCl, 0.4 mg/mL KCl, 0.02 M Mg2+, 0.02 M glucose, ddH2O) at 37°C until needed. Aliquot 20 μL of cells into 1.5 mL centrifuge tubes and add 1 μL of digested DNA to the cells.
Fill the Cell-Porator (Life Technologies) with ice water, set the capacitance to 330 μF, charge rate to fast, and resistance on the Voltage-Booster to 4 kΩ.
Pipette 20 μL of the cell/DNA solution into cell-porator cuvettes. Place the cuvettes into the Cell-Porator, close the lid, connect the power cord, and turn the dial to the correct cuvette. Set the power supply to charge and hold ‘up' until the voltage reads 430. Turn the dial to arm and press trigger. If there is a popping sound, retry the electroporation.
Pipette the cells from the cuvette and transfer them to 1.5 mL of SOC media. Incubate for 1.5 hours at 37°C with shaking.
Transfer 30 μL of the SOC/cell solution to LB/ampicillin agar plates (10 mg/mL tryptone, 5 mg/mL yeast extract, 10 mg/mL NaCl, 15 mg/mL agar, ddH2O and 100 ng/μL ampicillin). Spread the cells on the plate and incubate the plate at 37°C overnight.
Harvest single colonies from the plates and inoculate 5 mL cultures containing LB/ampicillin (10 mg/mL tryptone, 5 mg/mL yeast extract, 10 mg/mL NaCl,100 ng/μL ampicillin ddH2O) media. Shake at 37°C for 16-20 hours.
Perform a DNA miniprep using the Nucleospin Plasmid kit (Macherey-Nagel). Qualitatively examine the product by agarose gel electrophoresis. Quantitiate the yield of DNA using a Nanodrop 2000c Spectrophotometer (Thermo Scientific).
Verify insertion of mutation by DNA sequencing.
3. mRNA Synthesis
To linearize the DNA, digest 3 μg of plasmid DNA in 50 L reaction volume with 5 μL appropriate buffer and 1 μL of restriction enzyme for 1 hour at 37.0°C.
Using the High-Pure PCR Product Purification Kit (Roche Applied Science), add 350 μL binding buffer to the digest vial and vortex briefly. This kit is used as it contains guanidiniumisothiocyanate which makes the product RNA grade.
Place spin columns in collection tubes and load the column with digest DNA solution. Centrifuge at 18,000g for 20 seconds and discard flow-through.
Add 500 μL of wash buffer to the column. Centrifuge 20 seconds at 18,000g and discard flow through.
Add 200 μL of wash buffer to the column. Centrifuge 30 seconds or until column is dry at 18,000g.
Place the spin column in an autoclaved 1.5 mL centrifuge tube and add 50 μL of DEPC-treated water to the column. Elute by centrifuging for 30 seconds at 18,000g.
Concentrate the eluted DNA to approximately 15 μL which results in 0.5 μg in 3 μL of solution.
Choose the correct mMessage mMachine Kit (Ambion) according to the promoter site sequence in the plasmid DNA (SP6, T7). Add 5 μL 1 NTP-cap mix, 3 μL linearized DNA from the previous step, 1 μL transcription buffer, and 1 μL of appropriate RNA polymerase. Incubate for 1.5-2 hours at 37°C.
Add 12.5 μL of LiCl from kit, 15 μL DEPC water, mix briefly (do not vortex), centrifuge at 11,000g for 10 seconds, and store at -20°C for at least 30 minutes for precipitation.
Pre-cool centrifuge to 4°C and centrifuge at least 15 minutes at full speed after precipitation.
After centrifugation a brown pellet should be visible at the bottom of the tube. Carefully and completely remove the supernatant and add 150 μL of 70% RNA grade ethanol cooled to -20°C. Centrifuge at 4°C for 5 minutes at full speed.
Remove the supernatant completely, briefly dry (1-2 min) in an Eppendorf Vacufuge Plus, and dissolve the now white pellet in 12 μL of DEPC water. Quantitiate the yield of mRNA using a Nanodrop 2000c Spectrophotometer (Thermo Scientific).
4. Oocyte Removal
This protocol has been approved by WPI's Institutional Animal Care and Use Committee.
Prior to operation, the frog should be fasted for 12 hours to prevent vomiting during surgery.
Make a solution of 0.5-3.0 g MS-222 dissolved in water. Add sodium bicarbonate until the solution has a pH 7.0-8.0. MS-222 is used as an anesthetic for the frog during the surgery. It is important to wear nitrile gloves at all times while handling MS-222 and prepare the solution under a chemical hood
Immerse the frog in the MS-222 solution until the frog has lost righting and toe pinch reflexes. To check unresponsiveness, pinch the toe of the frog.
Place the frog on the dorsal side on the absorbent side of a wet diaper pad. This avoids damage to the skin of the frog. Keep a wet paper towel close to the frog at all times, as the frog must be kept moist throughout the surgery. If the eyes of the frog are open, it is important to moisten them with saline solution. If the frog shows signs of awakening, pour the anesthetic solution onto the frog and wait until the frog becomes unresponsive. Perform a second toe pinch before continuing the procedure.
Make a small coelomic incision to either the left or right of the frog's midline. Incisions should be made on alternating sides with subsequent surgeries.
Bring the ovary to the exterior of the frog and remove the ovary. Avoid touching the ovaries to the frog's skin. If bleeding occurs, apply pressure with a sterile Q-tip until the bleeding stops.
Suture the incision using an interrupted suture pattern with 3.0-4.0 Ethicon Vicryl suture material (Johnson & Johnson). When suturing, use surgical instruments to tie one knot at a time and avoid touching the skin of the frog. It is also important to suture the coelomic and skin layers separately. This is consistent with the NIH Guidelines for egg and oocyte harvesting in Xenopus laevis (see: http://oacu.od.nih.gov/arac/XenopusOocyte_101007_Fnl.pdf).
After the surgery, the frog must not undergo operation for at least 2 months. It should also be determined if the frog is healthy enough to undergo subsequent surgeries. A maximum of 6 surgeries can be performed on each frog with the 6th surgery being terminal.
5. Oocyte Defoculation and mRNA Injection
The isolated ovarian lobe is divided with scissors into smaller parts. The clumps are transferred into Ringer's Solution + Ca2+ with collagenase (8.6 g/L NaCl, 0.3 g/L KCl, and 0.33 g/L CaCl2).
Gently shake the digest solution for 2 hrs at 18°C. If most oocytes are separated, wash oocytes with Ringer's Solution – Ca2+ (8.6 g/L NaCl and 0.3 g/L KCl). Incubate oocytes for ten minutes in Ringer's Solution – Ca2+.
Transfer oocytes to Ringer's Solution + Ca2+.
Inject oocytes with 25 ng of mRNA, in a final volume of 50 nL with the Nanoject II Auto-Nanoliter Injector (Drummond), with 3.5" Drummond capillary tubes. Note: The amount of mRNA varies according to the target protein.
After injection, incubate oocytes 3-7 days in Ringer's Solution (90 mM NaCl, 2 mM KCl, 5 mM MOPS, 2 mM CaCl2, pH 7.4) and 1 mg/mL gentamycin at 18°C in the dark8.
6. Site-specific Fluorescence Labeling
Prior to measurement, incubate oocytes for 45 min in loading buffer (110 mM NaCl, 2.5 mM sodium citrate, and 10 mM MOPS/Tris at pH 7.4) and 45 min in post-loading buffer (100 mM NaCl, 1 mM CaCl2, 5 mM BaCl2, 5 mM NiCl2, and 10 mM MOPS/Tris at pH 7.4) to increase the intracellular Na+ concentration14.
For fluorescence measurements, incubate oocytes in a post-loading buffer that contains 5 μM of the desired fluorophore [ex. tetramethylrhodamine-6-maleimide (TMRM) or fluorescein-5-maleimide (FM)] for 5-10 minutes. After fluorophore labeling, wash the oocytes exhaustively with dye-free post-loading buffer3.
7. Electrophysiology
Make microelectrodes by pulling borosilicate capillaries (1B150F-4, World Precision Instruments) using a micropipette puller (Model PC-10, Narishige). Fill the electrodes with 3 M KCl and test the resistance. The resistance should be between 0.5-1.5 MΩ15.
Equip the fluorescence microscope (Carl Zeiss, Axio Examiner Fluorescence Microscope) with a 535DF50 excitation filter, a 565 EFLP emission filter, and a 570DRLP dichroic mirror (Omega Optical) for cysteine scanning experiments.
Place the oocyte in a RC-10 chamber (Warner Instruments) on the fluorescence microscope stage and gently insert the fabricated electrodes into the oocyte. Using a Turbo Tec-05X amplifier (NPI Electronics), hold the membrane potential at a constant value and measure the ion flux across the membrane following solution exchange or by changing the membrane potential. For concurrent fluorescence measurements, use a 100 W tungsten light source to excite the donor fluorophore and a PIN-022A photodiode (United Detector Technologies) to detect changes in fluorescence intensity. Both the amplifier and photodiode are interfaced, via a Digidata 1440A data acquisition system (Axon Instruments), with a computer utilizing pCLAMP10 software (Axon Instruments) for data acquisition and recording.
For cysteine scanning, measure the change in fluorescence intensity at stationary current using voltage steps that are controlled by pCLAMP 10 software (Molecular Devices). Depending on the membrane protein of interest, the extracellular solution is designed to ensure stationary current.
Correlate changes in fluorescence intensity to functional measurements of the targeted protein.
8. Anisotropy Measurements and Determination of Distance Constraints.
Anisotropy measurements should be taken alongside distance measurements8 to calculate the range of κ2 values. Anisotropy measures the relative mobility of the fluorophore due to rotation16. Using polarized filters (Linos Photonics Inc.), measure the parallel and perpendicular light emitted by FM or TMRM following excitation with polarized light at amino acid residues of interest. The measurements are taken under solution exchange conditions8 and constant membrane potential to determine the mobility of the fluorophores.
Anisotropy is given by r = (I||-I⊥)/(I|| + 2I⊥) where I|| is the parallel light emitted and I⊥ is the perpendicular light emitted16. To calculate the error in distance measurements, κ2max = 2/3(1 + Frd + Fra + 3Frd * Fra) and κ2min = 2/3(1-(Frd + Fra)/2) where Frd = (rd/ro)0.5 and Fra = (ra/ro)0.5; ra is anisotropy of TMRM, rd is anisotropy of FM, and ro is fundamental anisotropy of each fluorophore8.
To measure distance constraints, use a holoenzyme which has two accessible extracellular cysteine residues. As the oocytes will be held at a constant membrane potential and therefore constant distance during the measurements, there does not need to be a fluorescence change as a function of the conformational state of the protein. Incubate the oocytes in post loading buffer containing 1 μM FM (acceptor fluorophore) and 4 μM TMRM (donor fluorophore) for 30 minutes on ice in the dark, or only 1 μM FM. This allows for measurements to be made for holoenzymes labeled with and without an acceptor fluorophore8. Equip the microscope with a 475DF40 excitation filter, 530DF30 emission filter, and 505DRLP dichroic mirror.
Measure the time dependence of donor bleaching in the presence and absence of the acceptor fluorophore. During the time course of these measurements, solution flow should be continuous. Otherwise, a heat induced fluorescence increase over time may be observed. The difference in photodestruction with and without the acceptor fluorophore is used to calculate the distance between the two residues. Holoenzymes containing only the donor fluorophore will experience a faster photodestruction rate than holoenzymes with an acceptor fluorophore. Holoyenzymes containing both a donor and acceptor fluorophore will exhibit slow photodestruction rates when in close proximity to one another8.
Using the Clampex feature in pClamp10, average the results of the photodestruction of donor fluorophore for a minimum of four oocyte recordings.
Once the results have been averaged, use an exponential function (monoexponential, biexponential) to obtain a curve of best fit. The curve of best fit can be used to determine the time constant for the photodestruction of donor fluorophore with and without the acceptor.
Determine the efficiency of energy transfer given by the equation E = 1-ΓDA/ΓD17, where GDA refers to the time constant for the donor/acceptor fluorophore pair and ΓD refers to the time constant for only the donor fluorophore.
Using the Förster equation17, E = 1/(1+R6/Ro6), the distance between labeled cysteine residues can be determined; E is the FRET efficiency, R is the distance between donor and acceptor fluorophore, and Ro is the 50% efficiency distance for the donor and acceptor pairing17. Ro is governed by the equation Ro = (9.7x103 JΦDn-4 κ2) where J is the normalized spectral overlap of donor emission and acceptor absorption, ΦD is the quantum yield of donor emission without an acceptor fluorophore, n is the index of refraction, and κ2 is the orientation factor for dipole-dipole interactions8.
9. Relative Movement of Protein Subunits
Use double cysteine constructs which are labeled with acceptor and donor fluorophores. For these experiments, the change in distance between the two cysteine residues will be measured. Since this is the case, the fluorescence intensity following labeling at each residue should be independent of the conformational state of the protein8. Thus, any change in fluorescence resonance energy transfer will be the result of a change of distance between the donor and acceptor fluorophores.
Equip the microscope with a 475AF40 excitation filter, 595AF60 emission filter and 505DRLP dichroic mirror. For these experiments, the donor is excited and the fluorescence of the acceptor fluorophore is measured. Use the appropriate method (voltage, ligand, solution exchange) to activate the membrane protein and simultaneously measure the change in fluorescence intensity. An increase in fluorescence intensity will indicate that the two fluorophores and thus two residues are moving closer together. A decrease in fluorescence intensity will indicate that the two fluorophores and thus two residues are moving further apart. Finally, no change in fluorescence intensity will indicate that the two fluorophores and thus two residues are at the same distance upon the change in conformation of the protein.
10. Representative Results
Labeling extracellular cysteine residues with specific fluorophores enables the investigation of the movement of membrane proteins upon conformational changes. A typical voltage clamp fluorometry trace is shown in Figure 1. The change in fluorescence intensity (lower trace), which is the movement of the fluorophore from a more hydrophilic to more hydrophobic environment or from a more water quenching to less water quenching environment, results from a conformational change in the protein upon solution exchange (upper trace).
This technique can be extended to determine distance constraints between two residues. Such experimental results are shown in Figure 2. In the presence of an acceptor flurophore (red), photobleaching occurs at a slower rate than without an acceptor fluorophore (black). The rate of photobleaching is directly related to the distance between two fluorophores according to the Förster equation17. Such results can be correlated to different conformational states of the protein as verified by two electrode voltage clamp experiments performed in tandem.
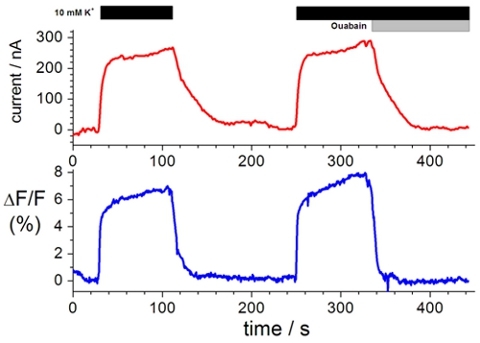 Figure 1. Representative recording of ion transport and changes in fluorescence intensity. Top, current clamp measurements in the presence of Na+ test solution and K+ test solution in the presence of 10 μM and 10 mM ouabain. Bottom, changes in florescence intensity measured in tandem with current clamp measurements3,8,19.
Figure 1. Representative recording of ion transport and changes in fluorescence intensity. Top, current clamp measurements in the presence of Na+ test solution and K+ test solution in the presence of 10 μM and 10 mM ouabain. Bottom, changes in florescence intensity measured in tandem with current clamp measurements3,8,19.
 Figure 2. Time Dependence of Photobleaching. Donor photodestruction is measured in the absence (black) and presence of acceptor fluorophore (red). Each trace is the average of 4 oocyte recordings.
Figure 2. Time Dependence of Photobleaching. Donor photodestruction is measured in the absence (black) and presence of acceptor fluorophore (red). Each trace is the average of 4 oocyte recordings.
Discussion
The experimental approach that we describe combines site-specific fluorophore labeling and two electrode voltage clamp to investigate the relationship between structure and function of membrane proteins. This technique can be used to obtain time-resolved information on the conformational dynamics of membrane proteins during ion transport. Furthermore, this approach can be tailored to work with various proteins such as ion pumps, ion channels and transporters.
In addition to investigating the conformational dynamics at a specific residue, it is also possible to use fluorescence resonance energy transfer to determine distance constraints within a holoenzyme. The determination of distances between residues as well as measuring the relative movement between subunits can help solve key questions involving gating mechanisms.
Disclosures
No conflicts of interest declared.
References
- Cole KS, Moore JW. Potassium Ion Current in the Squid Giant Axon: Dynamic Characteristic. Biophys. J. 1960;1:1–14. doi: 10.1016/s0006-3495(60)86871-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela M, Toro L, Stefani E. Novel voltage clamp to record small, fast currents from ion channels expressed in Xenopus oocytes. Biophys. J. 1992;61:78–82. doi: 10.1016/S0006-3495(92)81817-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempski RE, Friedrich T, Bamberg E. The beta subunit of the Na+/K+-ATPase follows the conformational state of the holoenzyme. J. Gen. Physiol. 2005;125:505–520. doi: 10.1085/jgp.200409186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AJ, Zhou JJ. Xenopus oocytes as an expression system for plant transporters. BBA - Biomembranes. 2000;1465:343–358. doi: 10.1016/s0005-2736(00)00148-6. [DOI] [PubMed] [Google Scholar]
- Cha A, Bezanilla F. Characterizing Voltage-Dependent Conformational Changes in the Shaker K+ Channel with Fluorescence. Neuron. 1997;19:1127–1140. doi: 10.1016/s0896-6273(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Mannuzzu LM, Moronne MM, Isacoff EY. Direct Physical Measure of Conformational Rearrangement Underlying Potassium Channel Gating. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- Koch HP, Larsson PH. Small-scale molecular motions accomplish glutamate uptake in human glutamate transporters. J. Neurosci. 2005;25:1730–1736. doi: 10.1523/JNEUROSCI.4138-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempski RE, Hartung K, Friedrich T, Bamberg E. Fluorometric Measurements of Intermolecular Distances between the α- and β-Subunits of the Na+/K+-ATPase. J. Biol. Chem. 2006;281:36338–36338. doi: 10.1074/jbc.M604788200. [DOI] [PubMed] [Google Scholar]
- Geys SA, Bamberg E, Dempski RE. Ligand-Dependent Effects on the Conformational Equilibrium of the Na+,K+-ATPase As Monitored by Voltage Clamp Fluorometry. Biophys. J. 2009;96:4561–4561. doi: 10.1016/j.bpj.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geibel S, Kaplan JH, Bamberg E, Friedrich T. Conformational Dynamics of the Na+/K+-ATPase Probed by Voltage Clamp Fluorometry. Proc. Natl. Acad. Sci. U.S.A. 2003;100:964–969. doi: 10.1073/pnas.0337336100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz L, Pusch M, Jentsch TJ. Heteromultimeric CLC chloride channels with novel properties. Proc. Natl. Acad. Sci. U.S.A. 1996;93:13362–13366. doi: 10.1073/pnas.93.23.13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mruk K, Kobertz WR. Discovery of a Novel Activator of KCNQ1-KCNE1 K+ Channel Complexes. Biophys. J. 2009;177a doi: 10.1371/journal.pone.0004236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A method for diplaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Rakowski RF. Charge movement by the Na/K pump in Xenopus oocytes. J. Gen. Physiol. 1993;101:117–144. doi: 10.1085/jgp.101.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda SP, Hegemann P. Glu 87 of Channelrhodopsin-1 Causes pH-dependent Color Tuning and Fast Photocurrent Inactivation. Photochem. Photobiol. 2009;85:564–569. doi: 10.1111/j.1751-1097.2008.00519.x. [DOI] [PubMed] [Google Scholar]
- Cha A, Bezanilla F. Structural implications of fluorescence quenching in the Shaker K+ channel. J. Gen. Physiol. 1998;112:391–391. doi: 10.1085/jgp.112.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster T. Ann Physik. 1948;2:55–75. [Google Scholar]
- Dong X, Stothard P, Forsythe IJ, Wishart DS. PlasMapper: a web server for drawing and auto-annotating plasmid maps. Nucleic Acids Res. 2004;32:W660–W664. doi: 10.1093/nar/gkh410. [DOI] [PMC free article] [PubMed] [Google Scholar]