

Abstract
Despite the advances in the management of patients with diabetes, diabetic nephropathy (DN) remains the most common cause of end stage renal disease (ESRD) in the US and worldwide. Inflammation and endothelial dysfunction appear to play a central role in the onset and the progression of DN. Recent evidence has emerged in the last decade to suggest uric acid is an inflammatory factor and may play a role in endothelial dysfunction. This has lead our group and others to explore the role of uric acid in the onset and progression of DN. In this review, we will highlight some of the animal and human studies that implicate uric acid in DN. Based on the evidence we review, we conclude the need for properly planned randomized controlled studies to lower uric acid levels and assess the impact of such therapy on diabetic kidney disease.
Introduction
Diabetic nephropathy (DN) is the most common cause of chronic kidney disease in the US and worldwide (1–2). Unfortunately, in the wake of the current epidemic of diabetes mellitus (DM), the prevalence of DN and ESRD are projected to rise (3). Different therapeutic strategies targeting DN have been explored such as tight glycemic control (4), tight blood pressure control (5), and various inhibitors of the renin angiotensin aldosterone system (RAAS) (6–8). While these therapies appear to slow the progression of kidney disease due to diabetes, none of them are curative. Hence there is a pressing interest to identify other potentially modifiable factors in the progression of DN.
Over the last 2 decades, an ample amount of scientific evidence has been generated and testifies to the role of cytokines in diabetic nephropathy (9–15). Specifically, hemodynamically- induced activation of transforming growth factor β-1 (TGFβ-1) appears to play a major role in mesangial expansion (16–18); in concert with the induction of ECM production (17, 19–24). Several biochemical mechanisms have been identified to explain the adverse effects of hyperglycemia on the kidney, including protein kinase C (PKC), the (mitogen activate protein) MAP kinase pathway, in addition to activation of the polyol pathway, increased accumulation of advanced glycation products, and oxidative stress (25–31). Despite the strides that we have made in understanding the factors that contribute to the evolution and the progression of diabetic kidney disease, this growing knowledge has yet to culminate in new therapeutic approaches. This is partially due to the extreme complexity of the underlying process. But also, some potent mediators of diabetic kidney disease are not viable or safe therapeutic targets. For example, as enticing as it has been to target TGFβ-1 for the treatment of diabetic nephropathy, TGFβ-1 carries out multiple vital biologic functions (32–33). Importantly, it is a primary regulator of the immune system (34–35) and mice with targeted disruption of TGFβ-1 gene die within weeks of birth due to a generalized wasting syndrome characterized by multifocal mixed inflammatory cellular response and tissue necrosis (36). This explains the apprehension towards inhibiting TGFβ-1 in humans and illustrates the need for other potentially modifiable factors in DN. One such factor that has made it onto the scene in recent years is uric acid.
Uric Acid
Uric acid (Urate) is synthesized in the liver from purine compounds provided by the diet or by the endogenous pathway of purine synthesis de novo. Some uric acid is also produced in peripheral tissues, especially the intestine and kidney. Uric acid that is produced in the liver is released into the circulation in its soluble form (monosodium urate), which is readily filtered by the glomerulus. The proximal tubular cells of the kidney reabsorb most of the uric acid resulting in a normal fractional excretion of approximately 10% (37). Uric acid accumulation beyond its solubility point (6.8 mg/dL) in water defines hyperuricemia. In general, hyperuricemia develops due to uric acid overproduction, undersecretion, or both (37). It is widely accepted that when uric acid levels are chronically elevated beyond their physiological levels, uric acid deposits in the joints and soft tissues leading to inflammatory arthritis and tophi (gout). Lowering uric acid levels is key to preventing recurrent acute gout attacks (38). Serum uric acid levels have also increased in Western populations where they have been found to predict the development of insulin resistance and diabetes (39–40). The potential causal relationship between uric acid and other conditions such as chronic kidney disease, however, remains controversial.
Some authors indicate that uric acid is a potent antioxidant, and in a few studies when uric acid was administered acutely it appeared to improve endothelial function (41–43). Other experimental evidence however suggests that uric acid may induce oxidative stress once it enters cells, and as such it may be a mediator of disease (44). Consistent with this latter observation, data by our group demonstrates that even mild hyperuricemia, induced by the administration of a uricase inhibitor, causes endothelial dysfunction (45) that resolves once uric acid levels are lowered. These findings when viewed in light of the importance of endothelial dysfunction in the progression of DN prompted us to hypothesize that uric acid plays a role in diabetic induced kidney disease.
Potential Mechanisms by which Uric Acid could Mediate DN
Uric acid has several reported effects by which it may cause DN illustrated in Figure 1, including endothelial dysfunction, increased activity of the RAAS, and induction of inflammatory cascades, in addition to profibrotic cytokine activation all of which have been demonstrated to contribute to progression of microvascular disease and thereby renal injury in DN.
Figure 1.
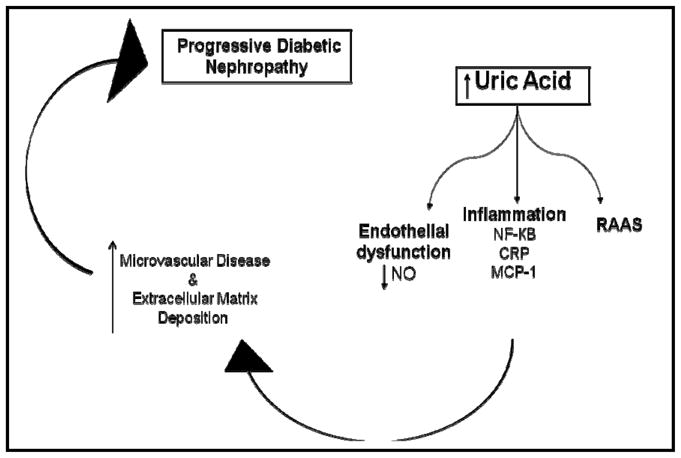
Mechanisms by which uric acid may cause diabetic nephropathy.
The effects of uric acid on the endothelium are subject of a contentious debate. On the one hand, uric acid has been shown to decrease nitric oxide (NO) production by endothelial cells in vitro (44), and it does so in association with increased CRP expression (46). Uric acid can also react with NO irreversibly leading to the formation of 6- aminouracil and may thus lead to NO depletion (47). Furthermore, hyperuricemic rats develop endothelial dysfunction (as noted by reduced urinary nitrites), and if given early, L-arginine supplementation, can prevent both the systemic and glomerular hypertension in experimental hyperuricemia (48–49). These data by our group and others suggest that uric acid leads to endothelial dysfunction. On the other hand however, some studies suggest that oxidative stress due to increased xanthine oxidase activity rather than uric acid is the major factor contributing to endothelial dysfunction. An example of such findings can be found in 2 double- blind placebo- controlled studies by George et al. (50). Patients with congestive heart failure were randomized in the first study to allopurinol or placebo and in the second study to probenecid (a uricosuric agent) or placebo. Both treatment arms in both studies had lower serum uric acid levels, but endothelial function improved only in the study where allopurinol was administered. Direct comparisons between the allopurinol and the probenecid groups were not conducted in this study, hence, it remains unclear if the favorable outcomes noted with allopurinol treatment are secondary to xanthine oxidase inhibition, lowering uric acid, or perhaps both.
In the kidney, experimental hyperuricemia causes an afferent renal arteriolopathy and tubulointerstitial fibrosis. This effect is largely mediated by activating the RAAS, as the renal injury was reversed with angiotensin converting enzyme inhibitors or angiotensin II receptor blockers but not with thiazide therapy despite all treatments lowering blood pressure (51). In this study, uric acid was shown to induce vascular smooth muscle proliferation in vitro as well, and similar to the findings in the animal kidneys, the effects of uric acid on vascular smooth muscles was reversible with the use of losartan. In addition to a direct role for uric acid in the vasculature, such data suggest uric acid effects are mediated at least partially by activation of the RAAS.
On the inflammatory front, uric acid induces the interstitial inflammation and the local expression of chemokines such as MCP-1 in the kidney (52–53), as well as COX-2 in the blood vessels (53). A direct role for uric acid in inducing inflammation is further supported by the findings that when infused into mice, uric acid increases cytokine production (TNF-α) (54). In humans with CKD, withdrawal of uric acid lowering therapy has been reported to increased urinary TGFβ-1 suggesting that hyperuricemia may contribute to the fibrotic process in patients with kidney disease (55). In addition to stimulating TGF β-1 production, hyperuricemia may activate its downstream targets. Although the transcriptional effects of TGFβ-1 are generally mediated by a group of proteins; the Smads (56), the expression of certain TGFβ-1-induced genes is mediated via the mitogen activate protein (MAP) kinase pathway (57). This pathway has also been reported to mediate uric acid effects in cell culture (58). Although the results of these studies need confirmation, such findings raise the possibility that treatment of hyperuricemia may provide a safe venue for alleviating cytokine- mediated kidney disease progression.
Uric Acid in Animal Models of Diabetic Nephropathy
Despite the wealth of evidence linking uric acid to inflammation and endothelial dysfunction, animal studies evaluating the role of uric acid in DN are sparse. This is interesting considering the critical role that endothelial dysfunction is known to play in diabetic kidney disease (59). Kosugi et al. explored the involvement of uric acid in DN in a recent study (60). In this particular study allopurinol was used to lower serum uric acid levels of diabetic (db/db) mice and to assess the downstream effects of this treatment on DN. We found that this model of type II DM develops hyperuricemia in conjunction with albuminuria, mesangial matrix expansion, and mild tubulointerstitial disease. Allopurinol significantly lowered uric acid levels, in addition to reducing albuminuria and tubulointerstitial injury. There was no effect on mesangial expansion. Allopurinol at the dose used (30 mg/kg/day) did not reduce oxidative stress in the kidney, but rather it reduced intercellular adhesion molecule 1 (ICAM-1) expression by the tubular epithelial cells. In vitro, uric acid directly induced ICAM expression in proximal tubular cells. The findings of this study strongly suggest uric acid is a mediator of inflammation and tubular injury in DN and that therapies aimed at lowering uric acid levels may be of benefit in human disease.
Other groups have also reported findings consistent with our results including evidence that uric acid inhibits proximal tubular cellular proliferation in vitro (61). This inhibitory effect of uric acid appears to be mediated by signaling pathways ultimately affecting cytoplasmic phospholipase A2 and the inflammatory transcription factor nuclear factor κ B (NF-κB).
Uric Acid as a Predictor of Human Disease
Understanding the relationship between uric acid and kidney disease in humans has been complicated by the fact that uric acid levels are elevated in patients with chronic kidney disease (CKD) due to a variety of factors including reduced GFR and diuretics use in patients with CKD (62). Even early decline in renal function is associated with an increase in serum uric acid levels (63) creating a major confounder in the interpretation of many observational studies. Another major limitation of observational studies lies with uric acid being a product of xanthine oxidase activity. Xanthine oxidase, in addition to generating uric acid, generates reactive oxidative species. Uric acid in such instances may be a marker of oxidative stress. Further complicating the debate, although many studies have shown increased uric acid levels are predictive of incident CKD and of CKD progression (64–68), some studies have not (69–71).
Notwithstanding the limitations of observational studies, several have examined uric acid as a risk factor for DN. Hovind et al. (72) at the Steno Diabetes Center explored the association between uric acid levels and micro- and macro- albuminuria in patients with type 1 DM. They enlisted 263 individuals with newly diagnosed type 1 DM between 1979 and 1984. During a median follow up of 18.1 years, 8.7% of their patients developed persistent macro-albuminuria. Although there was no association between serum uric acid levels and microalbuminuria in this study, the investigators found baseline uric acid levels were predictive of persistent macro-albuminuria; hazard ratio for the development of macro-albuminuria was 2.37 (95% C.I. 1.04–5.37) for every 100 μmol/L (equivalent to 1.7 mg/dl) increase in serum uric acid levels (P= 0.04) (Figure 2).
Figure 2.
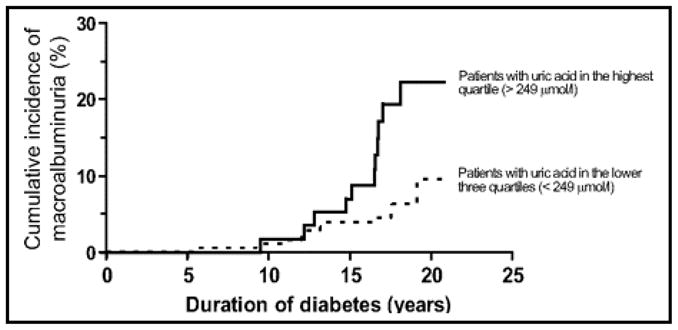
Figure from Hovind et al. (Diabetes. 2010 Oct;59(10):2695) (72). Cumulative incidence of persistent macroalbuminuria in 263 type 1 diabetic patients with onset of diabetes from 1979 to 1984. Divided by quartiles of uric acid 3 years after onset of type 1 diabetes, comparing patients with uric acid in highest quartile (> 249 umol/l) with patients with uric acid in lower 3 quartiles (< 249 umol/l); P value 0.006.
Similarly, we explored the association between uric acid and DN in participants of the Coronary Artery Calcification in Type 1 Diabetes (CACTI) Study (73). The study enrolled 1416 individuals between 19 and 56 years of age, with no known history of coronary heart disease: 652 participants with type 1 DM and 764 control participants without DM and followed them for 6 years. Our analysis revealed that baseline serum uric acid levels predict micro- or macro- albuminuria after a 6 years follow up period of 324 participants in this study; we reported that for every 1 mg/dL increase in serum uric acid levels, there was an 80% increased risk of developing DN (odds ratio 1.8; 95% C.I. 1.2–2.8; P= 0.005).
In addition to the above mentioned prospective studies linking uric acid to albuminuria, Ficociello et al. (74) investigated the impact of baseline serum uric acid levels on early decline in glomerular filtration rate (GFR) in patients with type 1 DM. They evaluated data of 355 participants in the Second Joslin Study on the Natural History of Microalbuminuria in Type 1 Diabetes. Over a follow up period of 6 years, they observed a significant association (P < 0.0002) between uric acid levels (within the normal range) and early GFR loss defined as cystatin C calculated GFR loss of > 3.3% per year.
Interventional Studies and progression of diabetic nephropathy
In contrast to the large number of observational studies examining the relation between uric acid and CKD, the number of interventional studies assessing the role of uric acid lowering therapy in slowing CKD progression is scant. One study by Siu et al. randomized 54 patients with various causes of CKD at various stages of the disease (CKD stages 2–4) to allopurinol versus control. At the end of the 12 month follow up period, a significantly larger number of participants in the control group achieved the combined end point of increased serum creatinine >40%, dialysis, or death; 16% vs 46% (P 0.015). Caution needs to be exercised in interpreting the results of this study. Although these findings suggest allopurinol therapy to lower serum uric acid levels would be of value in patients with CKD, it is important to note this was a small study and included a handful of patients with diabetic nephropathy (6 in the allopurinol group vs 7 in the control group).
More recently Goicechea et al. conducted a prospective study in 113 patients with CKD (75). Similar to the study by Siu et al, it lacked a placebo arm, but patients were randomized to either control or allopurinol and followed for 2 years. At the end of the study follow up period, the investigators found that estimated GFR decreased 3.3 ± 1.2 ml/min per 1.73 m2 in the control group as opposed to estimated GFR increased 1.3 ± 1.3 ml/min per 1.73 m2 in the allopurinol group (P = 0.018). This study also enlisted a small number of patients with diabetic kidney disease.
We found one interventional study by Momeni et al. that was a double blind randomized placebo controlled trial that evaluated the impact of lowering uric acid on DN (76). Forty patients with type 2 DM were randomized to receive either allopurinol or placebo and the patients’ proteinuria was monitored. In addition to lowering serum uric acid levels, 24 hour urinary protein excretion was reduced in the treatment group after a 4- month follow up period. This study has many limitations including the small number of patients, the short duration of follow up, and the lack of assessment of kidney function, but indicates that low dose allopurinol is safe in patients with DN and suggest that the effects of allopurinol on proteinuria are complementary to traditional DN treatment since all participants were either on an angiotensin converting enzyme inhibitor or an angiotensin receptor blocker.
Of interest is a post hoc analysis of the Reduction of Endpoints in Non-Insulin-Dependent Diabetes Mellitus With the Angiotensin II Antagonist Losartan (RENAAL) (77). Losartan in addition to lowering blood pressure and reducing proteinuria has a uric acid lowering effect. This analysis aimed to assess if some of the reported benefit of losartan on renal outcomes in RENAAL was due to the uric acid lowering effect of losartan by determining the relation between the change in serum uric acid levels in the first 6 months of the study and the composite outcome of doubling of serum creatinine or end stage renal disease. The authors reported that the renoprotective effect of losartan was attenuated from 22% to 17% after adjusting for the treatment effect on uric acid levels, supporting the hypothesis that uric acid may be a modifiable contributor to CKD progression.
Despite the promising results of these studies, in the absence of a randomized, adequately powered, placebo- controlled study, it remains unknown if treatment of asymptomatic hyperuricemia is implemental in slowing CKD progression in patients with DN.
Uric acid and mortality in patients with diabetic kidney disease
End stage renal disease is a much feared complication of CKD in general and DN specifically, yet longitudinal follow up demonstrates that patients with CKD are more likely to die from a cardiovascular event prior to requiring dialysis or kidney transplantation (78) and multiple studies have shown CKD to be an independent risk factor for cardiovascular events and mortality (79–83). Although patients with CKD have a higher prevalence of traditional (Framingham) risk factors for cardiovascular disease (84–85), the increased risk of cardiovascular disease in this patient population is only partially explained by the traditional risk factors of the general population (86). Several nontraditional risk factors may play a role in the development and progression of accelerated atherosclerosis noted in individuals with CKD.
Uric acid has been proposed to contribute to cardiovascular disease in this patient population, and a few studies have examined the correlation between hyperuricemia and cardiovascular disease in CKD (71, 87–90) with conflicted findings. Except for the study by Madero et al. that analyzed data from the Modification of Diet in Renal Disease (MDRD) study (71), most of these studies included patients with diabetes (87–90), and 2 were conducted in patients with ESRD on dialysis (87–88). These studies in patients with ESRD identified a J- shaped association between serum uric acid levels and mortality where high as well as low serum uric acid levels were poor predictors of outcomes. The negative impact of low serum uric acid levels may be related to poor nutritional status or to loss of the antioxidant qualities of uric acid in a high risk population.
The association between serum uric acid levels and coronary artery disease is not well studied in the literature with only few reports. In 1993, Rathman et al. reported a positive correlation between serum uric acid levels and coronary artery disease in women (91) in a study involving 7847 patients with type I and type II diabetes. More recently, we found that serum uric acid levels are predictive of progressive coronary calcification in patients with type I diabetes. Baseline serum UA predicted CAC progression with an odds ratio of 1.30 for each 1 SD change [0.2 mg/dL] ([95% CI 1.07–1.58], P = 0.007). This was noted only in the absence of significant CKD, but the small number of patients with CKD in this study (n=105) likely precluded the identification of such a relationship in patients with CKD.
Similar to CKD progression, the potential impact of lowering uric acid on cardiovascular disease and events in patients with diabetes is not known. In the study by Goicechea et al. (75), in addition to reducing the risk of CKD progression, allopurinol therapy seemed to be associated with reduced cardiovascular risk, although the results of this study should be interpreted with caution as it included only 113 participants. The potential for modifying cardiovascular risk in patients with CKD is furthered by a recent study of 67 patients with CKD (53 of whom completed and study) (92). Despite the small n, this was a randomized double- blinded placebo- controlled study where patients were followed for 9 months. At the end of the study, allopurinol significantly reduced left ventricular hypertrophy (P = 0.036), improved endothelial function (P = 0.009), and improved the pulse wave velocity (P = 0.015) suggesting a potential role for allopurinol in CKD. Of note, similar to many of the studies discussed above, this study had only 6 patients with diabetes. Further studies are needed to address the potential cardiovascular benefit from lowering serum uric acid levels in patients with DN.
Conclusion
DN is a complex disease. Several therapeutic options are available for the treatment of patients with DN, however the disease remains incurable. There is evidence that directly links uric acid to the progression of kidney disease in DM. While more studies are needed to further understand the role of uric acid in DN, the potential for modifying chronic kidney disease in diabetic patients by lowering serum uric acid levels is demonstrates promise and should be tested in well designed randomized controlled studies in the future. Such studies should also evaluate the impact of lowering uric acid levels on cardiovascular events and mortality, considering the high burden of cardiovascular disease and mortality in patients with DN.
Acknowledgments
This review was funded by the following grants: 1K23DK088833 – 01, HL-68607, and K23DK075630, as well by the Veteran’s Administration Medical Center at Denver, CO.
Footnotes
Conflict of Interest: Dr. Takahiko Nakagawa is listed as an inventor on therapies to lower uric acid with the goal of preventing metabolic syndrome, hypertension, and CKD progression.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Atlas of End-Stage Renal Disease. Bethesda: National Institutes of Health, NIDDK; 2005. USRDS 2005 Annual Data Report. [Google Scholar]
- 2.KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. Am J Kidney Dis. 2007 Feb;49(2 Suppl 2):S12–154. doi: 10.1053/j.ajkd.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Stenvinkel P. Chronic kidney disease: a public he priorityalth and harbinger of premature cardiovascular disease. J Intern Med. 2010 Aug 4; doi: 10.1111/j.1365-2796.2010.02269.x. [DOI] [PubMed] [Google Scholar]
- 4.Keen H. The Diabetes Control and Complications Trial (DCCT) Health Trends. 1994;26(2):41–3. [PubMed] [Google Scholar]
- 5.Schrier RW, Estacio RO, Mehler PS, Hiatt WR. Appropriate blood pressure control in hypertensive and normotensive type 2 diabetes mellitus: a summary of the ABCD trial. Nat Clin Pract Nephrol. 2007 Aug;3(8):428–38. doi: 10.1038/ncpneph0559. [DOI] [PubMed] [Google Scholar]
- 6.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993 Nov 11;329(20):1456–62. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 7.Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001 Sep 20;345(12):851–60. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 8.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001 Sep 20;345(12):861–9. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 9.Border WA, Noble NA. Evidence that TGF-beta should be a therapeutic target in diabetic nephropathy. Kidney international. 1998 Oct;54(4):1390–1. doi: 10.1046/j.1523-1755.1998.00127.x. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Hong SW, Iglesias-de la Cruz MC, Isono M, Casaretto A, Ziyadeh FN. The key role of the transforming growth factor-beta system in the pathogenesis of diabetic nephropathy. Renal failure. 2001 May-Jul;23(3–4):471–81. doi: 10.1081/jdi-100104730. [DOI] [PubMed] [Google Scholar]
- 11.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proceedings of the National Academy of Sciences of the United States of America. 2000 Jul 5;97(14):8015–20. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci U S A. 1993 Mar 1;90(5):1814–8. doi: 10.1073/pnas.90.5.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura T, Fukui M, Ebihara I, Osada S, Nagaoka I, Tomino Y, et al. mRNA expression of growth factors in glomeruli from diabetic rats. Diabetes. 1993 Mar;42(3):450–6. doi: 10.2337/diab.42.3.450. [DOI] [PubMed] [Google Scholar]
- 14.Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996 Apr;45(4):522–30. doi: 10.2337/diab.45.4.522. [DOI] [PubMed] [Google Scholar]
- 15.Sharma K, Ziyadeh FN. Renal hypertrophy is associated with upregulation of TGF-beta 1 gene expression in diabetic BB rat and NOD mouse. Am J Physiol. 1994 Dec;267(6 Pt 2):F1094–01. doi: 10.1152/ajprenal.1994.267.6.F1094. [DOI] [PubMed] [Google Scholar]
- 16.Riser BL, Cortes P, Yee J, Sharba AK, Asano K, Rodriguez-Barbero A, et al. Mechanical strain- and high glucose-induced alterations in mesangial cell collagen metabolism: role of TGF-beta. J Am Soc Nephrol. 1998 May;9(5):827–36. doi: 10.1681/ASN.V95827. [DOI] [PubMed] [Google Scholar]
- 17.Riser BL, Ladson-Wofford S, Sharba A, Cortes P, Drake K, Guerin CJ, et al. TGF-beta receptor expression and binding in rat mesangial cells: modulation by glucose and cyclic mechanical strain. Kidney international. 1999 Aug;56(2):428–39. doi: 10.1046/j.1523-1755.1999.00600.x. [DOI] [PubMed] [Google Scholar]
- 18.Ziyadeh FN, Sharma K, Ericksen M, Wolf G. Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose is mediated by autocrine activation of transforming growth factor-beta. J Clin Invest. 1994 Feb;93(2):536–42. doi: 10.1172/JCI117004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ayo SH, Radnik RA, Garoni JA, Glass WF, 2nd, Kreisberg JI. High glucose causes an increase in extracellular matrix proteins in cultured mesangial cells. Am J Pathol. 1990 Jun;136(6):1339–48. [PMC free article] [PubMed] [Google Scholar]
- 20.Haneda M, Kikkawa R, Horide N, Togawa M, Koya D, Kajiwara N, et al. Glucose enhances type IV collagen production in cultured rat glomerular mesangial cells. Diabetologia. 1991 Mar;34(3):198–200. doi: 10.1007/BF00418276. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi K, Epstein M, Loutzenhiser R, Forster H. Impaired myogenic responsiveness of the afferent arteriole in streptozotocin-induced diabetic rats: role of eicosanoid derangements. J Am Soc Nephrol. 1992 May;2(11):1578–86. doi: 10.1681/ASN.V2111578. [DOI] [PubMed] [Google Scholar]
- 22.Bidani AK, Griffin KA, Picken M, Lansky DM. Continuous telemetric blood pressure monitoring and glomerular injury in the rat remnant kidney model. Am J Physiol. 1993 Sep;265(3 Pt 2):F391–8. doi: 10.1152/ajprenal.1993.265.3.F391. [DOI] [PubMed] [Google Scholar]
- 23.Riser BL, Cortes P, Zhao X, Bernstein J, Dumler F, Narins RG. Intraglomerular pressure and mesangial stretching stimulate extracellular matrix formation in the rat. J Clin Invest. 1992 Nov;90(5):1932–43. doi: 10.1172/JCI116071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riser BL, Cortes P, Heilig C, Grondin J, Ladson-Wofford S, Patterson D, et al. Cyclic stretching force selectively up-regulates transforming growth factor-beta isoforms in cultured rat mesangial cells. Am J Pathol. 1996 Jun;148(6):1915–23. [PMC free article] [PubMed] [Google Scholar]
- 25.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998 Jun;47(6):859–66. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 26.Haneda M, Araki S, Togawa M, Sugimoto T, Isono M, Kikkawa R. Mitogen-activated protein kinase cascade is activated in glomeruli of diabetic rats and glomerular mesangial cells cultured under high glucose conditions. Diabetes. 1997 May;46(5):847–53. doi: 10.2337/diab.46.5.847. [DOI] [PubMed] [Google Scholar]
- 27.Haneda M, Kikkawa R, Arimura T, Ebata K, Togawa M, Maeda S, et al. Glucose inhibits myo-inositol uptake and reduces myo-inositol content in cultured rat glomerular mesangial cells. Metabolism: clinical and experimental. 1990 Jan;39(1):40–5. doi: 10.1016/0026-0495(90)90145-3. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki D, Miyata T, Saotome N, Horie K, Inagi R, Yasuda Y, et al. Immunohistochemical evidence for an increased oxidative stress and carbonyl modification of proteins in diabetic glomerular lesions. J Am Soc Nephrol. 1999 Apr;10(4):822–32. doi: 10.1681/ASN.V104822. [DOI] [PubMed] [Google Scholar]
- 29.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. The New England journal of medicine. 1988 May 19;318(20):1315–21. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 30.Koya D, Haneda M, Nakagawa H, Isshiki K, Sato H, Maeda S, et al. Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC beta inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. Faseb J. 2000 Mar;14(3):439–47. doi: 10.1096/fasebj.14.3.439. [DOI] [PubMed] [Google Scholar]
- 31.Derubertis FR, Craven PA. Activation of protein kinase C in glomerular cells in diabetes. Mechanisms and potential links to the pathogenesis of diabetic glomerulopathy. Diabetes. 1994 Jan;43(1):1–8. doi: 10.2337/diab.43.1.1. [DOI] [PubMed] [Google Scholar]
- 32.Barnard JA, Lyons RM, Moses HL. The cell biology of transforming growth factor beta. Biochimica et biophysica acta. 1990 Jun 1;1032(1):79–87. doi: 10.1016/0304-419x(90)90013-q. [DOI] [PubMed] [Google Scholar]
- 33.Massague J. The transforming growth factor-beta family. Annual review of cell biology. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- 34.Prud'homme GJ, Piccirillo CA. The inhibitory effects of transforming growth factor-beta-1 (TGF-beta1) in autoimmune diseases. J Autoimmun. 2000 Feb;14(1):23–42. doi: 10.1006/jaut.1999.0339. [DOI] [PubMed] [Google Scholar]
- 35.McCartney-Francis NL, Frazier-Jessen M, Wahl SM. TGF-beta: a balancing act. Int Rev Immunol. 1998;16(5–6):553–80. doi: 10.3109/08830189809043009. [DOI] [PubMed] [Google Scholar]
- 36.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992 Oct 22;359(6397):693–9. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edwards NL. The role of hyperuricemia and gout in kidney and cardiovascular disease. Cleve Clin J Med. 2008 Jul;75( Suppl 5):S13–6. doi: 10.3949/ccjm.75.suppl_5.s13. [DOI] [PubMed] [Google Scholar]
- 38.Dalbeth N, Stamp L. Allopurinol dosing in renal impairment: walking the tightrope between adequate urate lowering and adverse events. Semin Dial. 2007 Sep-Oct;20(5):391–5. doi: 10.1111/j.1525-139X.2007.00270.x. [DOI] [PubMed] [Google Scholar]
- 39.Kodama S, Saito K, Yachi Y, Asumi M, Sugawara A, Totsuka K, et al. Association between serum uric acid and development of type 2 diabetes. Diabetes care. 2009 Sep;32(9):1737–42. doi: 10.2337/dc09-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson RJ, Perez-Pozo SE, Sautin YY, Manitius J, Sanchez-Lozada LG, Feig DI, et al. Hypothesis: could excessive fructose intake and uric acid cause type 2 diabetes? Endocr Rev. 2009 Feb;30(1):96–116. doi: 10.1210/er.2008-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waring WS, McKnight JA, Webb DJ, Maxwell SR. Uric acid restores endothelial function in patients with type 1 diabetes and regular smokers. Diabetes. 2006 Nov;55(11):3127–32. doi: 10.2337/db06-0283. [DOI] [PubMed] [Google Scholar]
- 42.Scott GS, Cuzzocrea S, Genovese T, Koprowski H, Hooper DC. Uric acid protects against secondary damage after spinal cord injury. Proc Natl Acad Sci U S A. 2005 Mar 1;102(9):3483–8. doi: 10.1073/pnas.0500307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waring WS, Adwani SH, Breukels O, Webb DJ, Maxwell SR. Hyperuricaemia does not impair cardiovascular function in healthy adults. Heart. 2004 Feb;90(2):155–9. doi: 10.1136/hrt.2003.016121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zharikov S, Krotova K, Hu H, Baylis C, Johnson RJ, Block ER, et al. Uric acid decreases NO production and increases arginase activity in cultured pulmonary artery endothelial cells. Am J Physiol Cell Physiol. 2008 Nov;295(5):C1183–90. doi: 10.1152/ajpcell.00075.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, Mu W, et al. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005 May;67(5):1739–42. doi: 10.1111/j.1523-1755.2005.00273.x. [DOI] [PubMed] [Google Scholar]
- 46.Kang DH, Park SK, Lee IK, Johnson RJ. Uric acid-induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol. 2005 Dec;16(12):3553–62. doi: 10.1681/ASN.2005050572. [DOI] [PubMed] [Google Scholar]
- 47.Gersch C, Palii SP, Kim KM, Angerhofer A, Johnson RJ, Henderson GN. Inactivation of nitric oxide by uric acid. Nucleosides Nucleotides Nucleic Acids. 2008 Aug;27(8):967–78. doi: 10.1080/15257770802257952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mazzali M, Hughes J, Kim YG, Jefferson JA, Kang DH, Gordon KL, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001 Nov;38(5):1101–6. doi: 10.1161/hy1101.092839. [DOI] [PubMed] [Google Scholar]
- 49.Sanchez-Lozada LG, Tapia E, Lopez-Molina R, Nepomuceno T, Soto V, Avila-Casado C, et al. Effects of acute and chronic L-arginine treatment in experimental hyperuricemia. American journal of physiology. 2007 Apr;292(4):F1238–44. doi: 10.1152/ajprenal.00164.2006. [DOI] [PubMed] [Google Scholar]
- 50.George J, Carr E, Davies J, Belch JJ, Struthers A. High-dose allopurinol improves endothelial function by profoundly reducing vascular oxidative stress and not by lowering uric acid. Circulation. 2006 Dec 5;114(23):2508–16. doi: 10.1161/CIRCULATIONAHA.106.651117. [DOI] [PubMed] [Google Scholar]
- 51.Mazzali M, Kanellis J, Han L, Feng L, Xia YY, Chen Q, et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Renal Physiol. 2002 Jun;282(6):F991–7. doi: 10.1152/ajprenal.00283.2001. [DOI] [PubMed] [Google Scholar]
- 52.Roncal CA, Mu W, Croker B, Reungjui S, Ouyang X, Tabah-Fisch I, et al. Effect of elevated serum uric acid on cisplatin-induced acute renal failure. American journal of physiology. 2007 Jan;292(1):F116–22. doi: 10.1152/ajprenal.00160.2006. [DOI] [PubMed] [Google Scholar]
- 53.Kang DH, Nakagawa T, Feng L, Watanabe S, Han L, Mazzali M, et al. A role for uric acid in the progression of renal disease. J Am Soc Nephrol. 2002 Dec;13(12):2888–97. doi: 10.1097/01.asn.0000034910.58454.fd. [DOI] [PubMed] [Google Scholar]
- 54.Netea MG, Kullberg BJ, Blok WL, Netea RT, van der Meer JW. The role of hyperuricemia in the increased cytokine production after lipopolysaccharide challenge in neutropenic mice. Blood. 1997 Jan 15;89(2):577–82. [PubMed] [Google Scholar]
- 55.Talaat KM, el-Sheikh AR. The effect of mild hyperuricemia on urinary transforming growth factor beta and the progression of chronic kidney disease. Am J Nephrol. 2007;27(5):435–40. doi: 10.1159/000105142. [DOI] [PubMed] [Google Scholar]
- 56.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000 Dec;1(3):169–78. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 57.Choi ME. Mechanism of transforming growth factor-beta1 signaling. Kidney Int Suppl. 2000 Sep;77:S53–8. [PubMed] [Google Scholar]
- 58.Kanellis J, Watanabe S, Li JH, Kang DH, Li P, Nakagawa T, et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension. 2003 Jun;41(6):1287–93. doi: 10.1161/01.HYP.0000072820.07472.3B. [DOI] [PubMed] [Google Scholar]
- 59.Nakagawa T, Sato W, Glushakova O, Heinig M, Clarke T, Campbell-Thompson M, et al. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007 Feb;18(2):539–50. doi: 10.1681/ASN.2006050459. [DOI] [PubMed] [Google Scholar]
- 60.Kosugi T, Nakayama T, Heinig M, Zhang L, Yuzawa Y, Sanchez-Lozada LG, et al. The Effect of Lowering Uric Acid on Renal Disease in the Type 2 Diabetic db/db Mice. Am J Physiol Renal Physiol. 2009 May 20; doi: 10.1152/ajprenal.00092.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han HJ, Lim MJ, Lee YJ, Lee JH, Yang IS, Taub M. Uric acid inhibits renal proximal tubule cell proliferation via at least two signaling pathways involving PKC, MAPK, cPLA2, and NF-kappaB. Am J Physiol Renal Physiol. 2007 Jan;292(1):F373–81. doi: 10.1152/ajprenal.00104.2006. [DOI] [PubMed] [Google Scholar]
- 62.Johnson RJ, Kang DH, Feig D, Kivlighn S, Kanellis J, Watanabe S, et al. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension. 2003 Jun;41(6):1183–90. doi: 10.1161/01.HYP.0000069700.62727.C5. [DOI] [PubMed] [Google Scholar]
- 63.Rosolowsky ET, Ficociello LH, Maselli NJ, Niewczas MA, Binns AL, Roshan B, et al. High-normal serum uric acid is associated with impaired glomerular filtration rate in nonproteinuric patients with type 1 diabetes. Clin J Am Soc Nephrol. 2008 May;3(3):706–13. doi: 10.2215/CJN.04271007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hsu CY, Iribarren C, McCulloch CE, Darbinian J, Go AS. Risk factors for end-stage renal disease: 25 -year follow-up. Arch Intern Med. 2009 Feb 23;169(4):342–50. doi: 10.1001/archinternmed.2008.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Obermayr RP, Temml C, Gutjahr G, Knechtelsdorfer M, Oberbauer R, Klauser-Braun R. Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol. 2008 Dec;19(12):2407–13. doi: 10.1681/ASN.2008010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weiner DE, Tighiouart H, Elsayed EF, Griffith JL, Salem DN, Levey AS. Uric acid and incident kidney disease in the community. J Am Soc Nephrol. 2008 Jun;19(6):1204–11. doi: 10.1681/ASN.2007101075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Borges RL, Hirota AH, Quinto BM, Ribeiro AB, Zanella MT, Batista MC. Uric acid as a marker for renal dysfunction in hypertensive women on diuretic and nondiuretic therapy. J Clin Hypertens (Greenwich) 2009 May;11(5):253–9. doi: 10.1111/j.1751-7176.2009.00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park JT, Kim DK, Chang TI, Kim HW, Chang JH, Park SY, et al. Uric acid is associated with the rate of residual renal function decline in peritoneal dialysis patients. Nephrol Dial Transplant. 2009 Nov;24(11):3520–5. doi: 10.1093/ndt/gfp272. [DOI] [PubMed] [Google Scholar]
- 69.Chonchol M, Shlipak MG, Katz R, Sarnak MJ, Newman AB, Siscovick DS, et al. Relationship of uric acid with progression of kidney disease. Am J Kidney Dis. 2007 Aug;50(2):239–47. doi: 10.1053/j.ajkd.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 70.Sturm G, Kollerits B, Neyer U, Ritz E, Kronenberg F. Uric acid as a risk factor for progression of non-diabetic chronic kidney disease? The Mild to Moderate Kidney Disease (MMKD) Study. Exp Gerontol. 2008 Apr;43(4):347–52. doi: 10.1016/j.exger.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 71.Madero M, Sarnak MJ, Wang X, Greene T, Beck GJ, Kusek JW, et al. Uric acid and long-term outcomes in CKD. Am J Kidney Dis. 2009 May;53(5):796–803. doi: 10.1053/j.ajkd.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hovind P, Rossing P, Tarnow L, Johnson RJ, Parving HH. Serum uric acid as a predictor for development of diabetic nephropathy in type 1 diabetes - an inception cohort study. Diabetes. 2009 May 1; doi: 10.2337/db09-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jalal DI, Rivard CJ, Johnson RJ, Maahs DM, McFann K, Rewers M, et al. Serum uric acid levels predict the development of albuminuria over 6 years in patients with type 1 diabetes: findings from the Coronary Artery Calcification in Type 1 Diabetes study. Nephrol Dial Transplant. 2010 Jun;25(6):1865–9. doi: 10.1093/ndt/gfp740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ficociello LH, Rosolowsky ET, Niewczas MA, Maselli NJ, Weinberg JM, Aschengrau A, et al. High-normal serum uric acid increases risk of early progressive renal function loss in type 1 diabetes: results of a 6-year follow-up. Diabetes Care. 2010 Jun;33(6):1337–43. doi: 10.2337/dc10-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goicoechea M, de Vinuesa SG, Verdalles U, Ruiz-Caro C, Ampuero J, Rincon A, et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol. 2010 Aug;5(8):1388–93. doi: 10.2215/CJN.01580210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Momeni A, Shahidi S, Seirafian S, Taheri S, Kheiri S. Effect of allopurinol in decreasing proteinuria in type 2 diabetic patients. Iran J Kidney Dis. 2010 Apr;4(2):128–32. [PubMed] [Google Scholar]
- 77.Miao Y, Ottenbros SA, Laverman GD, Brenner BM, Cooper ME, Parving HH, et al. Effect of a Reduction in Uric Acid on Renal Outcomes During Losartan Treatment: A Post Hoc Analysis of the Reduction of Endpoints in Non-Insulin-Dependent Diabetes Mellitus With the Angiotensin II Antagonist Losartan Trial. Hypertension. 2011 Jul;58(1):2–7. doi: 10.1161/HYPERTENSIONAHA.111.171488. [DOI] [PubMed] [Google Scholar]
- 78.Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med. 2004 Mar 22;164(6):659–63. doi: 10.1001/archinte.164.6.659. [DOI] [PubMed] [Google Scholar]
- 79.Brantsma AH, Bakker SJ, Hillege HL, de Zeeuw D, de Jong PE, Gansevoort RT. Cardiovascular and renal outcome in subjects with K/DOQI stage 1–3 chronic kidney disease: the importance of urinary albumin excretion. Nephrol Dial Transplant. 2008 Dec;23(12):3851–8. doi: 10.1093/ndt/gfn356. [DOI] [PubMed] [Google Scholar]
- 80.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004 Sep 23;351(13):1296–305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 81.McCullough PA, Jurkovitz CT, Pergola PE, McGill JB, Brown WW, Collins AJ, et al. Independent components of chronic kidney disease as a cardiovascular risk state: results from the Kidney Early Evaluation Program (KEEP) Arch Intern Med. 2007 Jun 11;167(11):1122–9. doi: 10.1001/archinte.167.11.1122. [DOI] [PubMed] [Google Scholar]
- 82.McCullough PA, Li S, Jurkovitz CT, Stevens L, Collins AJ, Chen SC, et al. Chronic kidney disease, prevalence of premature cardiovascular disease, and relationship to short-term mortality. Am Heart J. 2008 Aug;156(2):277–83. doi: 10.1016/j.ahj.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 83.Weiner DE, Tighiouart H, Stark PC, Amin MG, MacLeod B, Griffith JL, et al. Kidney disease as a risk factor for recurrent cardiovascular disease and mortality. Am J Kidney Dis. 2004 Aug;44(2):198–206. doi: 10.1053/j.ajkd.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 84.Whaley-Connell AT, Sowers JR, Stevens LA, McFarlane SI, Shlipak MG, Norris KC, et al. CKD in the United States: Kidney Early Evaluation Program (KEEP) and National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am J Kidney Dis. 2008 Apr;51(4 Suppl 2):S13–20. doi: 10.1053/j.ajkd.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 85.Rao MV, Qiu Y, Wang C, Bakris G. Hypertension and CKD: Kidney Early Evaluation Program (KEEP) and National Health and Nutrition Examination Survey (NHANES), 1999–2004. Am J Kidney Dis. 2008 Apr;51(4 Suppl 2):S30–7. doi: 10.1053/j.ajkd.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 86.Sarnak MJ, Coronado BE, Greene T, Wang SR, Kusek JW, Beck GJ, et al. Cardiovascular disease risk factors in chronic renal insufficiency. Clin Nephrol. 2002 May;57(5):327–35. doi: 10.5414/cnp57327. [DOI] [PubMed] [Google Scholar]
- 87.Suliman ME, Johnson RJ, Garcia-Lopez E, Qureshi AR, Molinaei H, Carrero JJ, et al. J-shaped mortality relationship for uric acid in CKD. Am J Kidney Dis. 2006 Nov;48(5):761–71. doi: 10.1053/j.ajkd.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 88.Hsu SP, Pai MF, Peng YS, Chiang CK, Ho TI, Hung KY. Serum uric acid levels show a 'J-shaped' association with all-cause mortality in haemodialysis patients. Nephrol Dial Transplant. 2004 Feb;19(2):457–62. doi: 10.1093/ndt/gfg563. [DOI] [PubMed] [Google Scholar]
- 89.Weiner DE, Tighiouart H, Elsayed EF, Griffith JL, Salem DN, Levey AS, et al. The relationship between nontraditional risk factors and outcomes in individuals with stage 3 to 4 CKD. Am J Kidney Dis. 2008 Feb;51(2):212–23. doi: 10.1053/j.ajkd.2007.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Navaneethan SD, Beddhu S. Associations of serum uric acid with cardiovascular events and mortality in moderate chronic kidney disease. Nephrol Dial Transplant. 2009 Apr;24(4):1260–6. doi: 10.1093/ndt/gfn621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rathmann W, Hauner H, Dannehl K, Gries FA. Association of elevated serum uric acid with coronary heart disease in diabetes mellitus. Diabete Metab. 1993;19(1 Pt 2):159–66. [PubMed] [Google Scholar]
- 92.Kao MP, Ang DS, Gandy SJ, Nadir MA, Houston JG, Lang CC, et al. Allopurinol benefits left ventricular mass and endothelial dysfunction in chronic kidney disease. J Am Soc Nephrol. 2011 Jul;22(7):1382–9. doi: 10.1681/ASN.2010111185. [DOI] [PMC free article] [PubMed] [Google Scholar]