

Abstract
Stimulation of synaptic NMDA receptors (NMDARs) induces neuroprotection, while extrasynaptic NMDARs promote excitotoxic cell death. Neuronal expression of cyclooxygenase-2 (COX-2) is enhanced by synaptic NMDARs, and although this enzyme mediates neuronal functions, COX-2 is also regarded as a key modulator of neuroinflammation and is thought to exacerbate excitotoxicity via overproduction of prostaglandins. This raises an apparent paradox: synaptic NMDARs are pro-survival yet are essential for robust neuronal COX-2 expression. We hypothesized that stimulation of extrasynaptic NMDARs converts COX-2 signaling from a physiological to a potentially pathological process. We combined HPLC-electrospray ionization-tandem MS-based mediator lipidomics and unbiased image analysis in mouse dissociated and organotypic cortical cultures to uncover that synaptic and extrasynaptic NMDARs differentially modulate neuronal COX-2 expression and activity. We show that synaptic NMDARs enhance neuronal COX-2 expression, while sustained synaptic stimulation limits COX-2 activity by suppressing cellular levels of the primary COX-2 substrate, arachidonic acid (AA). In contrast, extrasynaptic NMDARs suppress COX-2 expression while activating phospholipase A2, which enhances AA levels by hydrolysis of membrane phospholipids. Thus, sequential activation of synaptic then extrasynaptic NMDARs maximizes COX-2-dependent prostaglandin synthesis. We also show that excitotoxic events only drive induction of COX-2 expression through abnormal synaptic network excitability. Finally, we show that nonenzymatic lipid peroxidation of arachidonic and other polyunsaturated fatty acids is a function of network activity history. A new paradigm emerges from our results suggesting that pathological COX-2 signaling associated with models of stroke, epilepsy, and neurodegeneration requires specific spatiotemporal NMDAR stimulation.
Introduction
NMDA-type glutamate neurotransmitter receptors (NMDARs) conduct Ca2+ and Na+ ions through membranes and mediate synaptic plasticity. However, under pathological circumstances, stimulation of NMDARs initiates deregulation of intracellular Ca2+ homeostasis and excitotoxic cell death. The dual role of NMDARs in function and pathology is explained by receptor location, i.e., synaptic or extrasynaptic. Synaptic NMDARs are thought to initiate survival signaling, while extrasynaptic NMDARs are linked to Ca2+ deregulation and cell death. Many examples of opposing effects of synaptic and extrasynaptic NMDARs on neuronal signaling pathways and survival outcomes have been reported, and some key mediators of these pathways have recently been reviewed (Hardingham and Bading, 2010).
Phospholipases A2 (PLA2s) cleave the polyunsaturated fatty acids (PUFAs) arachidonic acid (AA; 20:4,n−6) and docosahexaenoic acid (DHA; 22:6, n-3) from the sn-2 position of membrane phospholipids by ester hydrolysis (Bazan, 2003). NMDARs enhance AA release from phospholipids through Ca2+-dependent activation of PLA2s (Dumuis et al., 1988; Stella et al., 1995; Taylor et al., 2008). Cyclooxygenase-2 (COX-2) is a heme-containing enzyme that catalyzes the rate-limiting step to prostaglandin (PG) synthesis by adding molecular oxygen to free, unesterified AA (Smith et al., 2000). PGs engage bioactivity through activation of G-protein-coupled receptors and are required for certain forms of synaptic plasticity (Kaufmann et al., 1996; Chen et al., 2002; Chen and Bazan, 2005; Sang et al., 2005; Akaneya and Tsumoto, 2006; Savonenko et al., 2009; Yang et al., 2009; Koch et al., 2010; Le et al., 2010) and neurovascular coupling (Niwa et al., 2000; Gordon et al., 2008); however, overproduction of PGs by COX-2 appears to mediate neuronal injury, aberrant plasticity, and inflammatory/immune responses in ischemia-reperfusion damage (Nogawa et al., 1997; Nakayama et al., 1998; Iadecola et al., 2001; Kawano et al., 2006), neurodegenerations (Hunot et al., 2004; Liang et al., 2005; Block et al., 2007; Kotilinek et al., 2008; Keene et al., 2010) and epilepsy (Yamagata et al., 1993; Tu and Bazan, 2003; Lee et al., 2007; Koch et al., 2010). Given that synaptic NMDARs induce neuroprotection and that COX-2 has been linked to a variety of brain injuries, the observation that synaptic NMDARs increase COX-2 expression (Yamagata et al., 1993) is an apparent paradox.
The role of extrasynaptic NMDARs in COX-2 expression and activity has never been investigated. We hypothesized that stimulation of extrasynaptic NMDARs results in overproduction of neuronal PGs by COX-2, thereby converting physiological PG signaling from synaptic NMDARs to a potentially pathological process. We combined HPLC-electrospray ionization (ESI)-tandem MS (MS/MS)-based mediator lipidomics and unbiased image analysis in mouse dissociated and organotypic cortical cultures to show that synaptic NMDARs drive COX-2 expression with relatively low PG formation, while subsequent extrasynaptic NMDAR activation induces remarkably enhanced COX-2 enzymatic activity via increased AA release. We find that synaptic activity suppresses cellular AA levels and nonenzymatic lipid (auto)peroxidation, while extrasynaptic NMDAR stimulation specifically couples to PLA2 activation, AA release, and peroxidation. In addition, we show that glutamate transporter blockade initiates excitotoxicity followed by aberrant network activity and enhanced COX-2 expression, without remarkably increased COX-2 activity, highlighting the importance of spatiotemporal patterning in NMDAR-dependent COX-2 signaling. This is the first process reported to require such spatiotemporal NMDAR stimulation and may represent a key determinant of physiological versus pathological COX-2 signaling.
Materials and Methods
Dissociated cortical cultures.
Cortices from C57BL/6 mouse pups (postnatal day 0) of either sex were dissected and minced in ice-cold dissection medium containing (in mm): 73.6 Na2SO4, 27.0 K2SO4, 12.6 MgCl2, 0.23 CaCl2, 1.4 HEPES, 18 glucose, 1.0 sodium kynurenate, and 0.001% phenol red. Tissue was transferred to digestion medium (dissection medium plus 3.7 mm l-cysteine, 10 U/ml papain from papaya latex, and 0.75 KU/ml DNase I from bovine pancreas) and incubated at 37°C for 40 min. The tissue was rinsed twice with dissection medium and twice with growth medium (Neurobasal A plus B27 supplement (Invitrogen), 1 mm l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, and 1% rat serum). The tissue was then triturated in growth medium plus 0.75 KU/ml DNase I using a series of 9 inch Pasteur pipettes flame polished to successively smaller bore sizes. The resulting suspensions of dissociated cells were pooled and diluted to the desired density using Opti-MEM I (Invitrogen) plus 20 mm glucose. Diluted cell suspensions were seeded on polystyrene tissue culture plates coated with 0.8–1.3 μg/cm2 laminin from Engelbreth-Holm-Swarm murine sarcoma basement membrane and 5.0–8.3 μg/cm2 poly-d-lysine hydrobromide (molecular weight 30,000–70,000). After 3 h the seeding medium was replaced with fresh growth medium. Cultures were treated with 4.8 μm cytosine arabinoside on day in vitro (DIV) 4. Cultures were fed by adding 1 volume of fresh growth medium on DIV 4 and replacing 50% of the resulting medium volume on DIV 8. Experiments were conducted on DIV 9–11. Dissociated cultures were ∼80% neuronal as assessed by NeuN immunofluorescence (IF).
Organotypic coronal slice cultures.
Brains from 7-d-old C57BL/6 mice of either sex were removed and embedded in 4% low melt agarose in dissection medium [HBSS (Invitrogen) plus 36.5 mm glucose]. Coronal slices (350 μm) were cut using a vibratome, and slices obtained from approximately the level of first appearance of the striatum to the caudal third of hippocampus were transferred to PTFE membrane cell culture inserts (Millipore) in 30 mm dishes containing 1.1 ml of preequilibrated growth medium. Growth medium consisted of 50% Basal Medium Eagle/25% Earle's Balanced Salt Solution (Invitrogen)/25% horse serum plus 36.5 mm glucose and antibiotics as above. Cultures were fed by replacing half of the growth medium every 2 d. Experiments were conducted on DIV 7–8. All cultures [dissociated and organotypic coronal slice cultures (OTCs)] were maintained in humidified 5% CO2/95% air and 37°C.
Stimulations.
On DIV 9, dissociated cultures were switched from growth medium to defined stimulation medium (TM0) plus the indicated drugs. TM0 is composed of 90% salt-glucose-glycine (SGG) solution and 10% MEM [(+) Earle's salts, (−) glutamine; Invitrogen] plus antibiotics as above. SGG is composed of the following (in mm): 114 NaCl, 26.1 NaHCO3, 5.3 KCl, 1 MgCl2, 2 CaCl2, 1 HEPES, 1 glycine, 30 glucose, 0.5 sodium pyruvate, 0.001% phenol red. Network disinhibition was elicited for 24 h with the GABAA receptor antagonist bicuculline (50 μm) and the broad spectrum K+-channel blocker 4-aminopyridine (250 μm) (hereafter referred to as bic/4-AP). When used, NMDAR antagonists were added at the same time as bic/4-AP or TBOA (dl-threo-β-benzyloxyaspartic acid). MK-801 was used at 10 μm. Unless indicated, NMDA challenge was made in the presence of (μm) 1 TTX, 40 CNQX, and 5 nifedipine, all added 5 min before NMDA. Unless indicated, NMDA was used at 100 μm. Aspirin (1 mm), NS-398 (10 μm), and pyrrphenone (1 μm) were added 1 h before NMDA challenge. TBOA was used at 30 μm. OTCs were switched to TM0 ± bic/4-AP on DIV 7, stimulated for 24 h, and then challenged with 100 μm NMDA for 1 h.
Immunofluorescence microscopy and morphonuclear analysis imaging method.
Cells were fixed in 3.7% neutral buffered formalin, permeabilized for 20 min in 0.1% Triton X-100 plus 20 μm Hoechst 33258 in PBS, and blocked with 10% goat serum in PBS plus 1% BSA for 2 h at room temperature. Cells were incubated with primary antibodies in PBS plus 1% BSA for 1 h at room temperature. Primary antibodies were rabbit anti-COX-2 (Cayman) or rabbit anti-GAD65/67 (Millipore). Excess primary antibody was removed with three washes in PBS plus 1% BSA, and cells were incubated with secondary antibodies for 1 h. Secondary antibody was goat anti-rabbit Alexa 488 conjugate (Invitrogen). One 3 × 3 tile mosaic representing a total area of 1.921 mm2 was acquired from the center of each well using a Zeiss 510 Meta laser confocal microscope and LSM 510 Meta software. Hoechst and Alexa 488 signals were acquired on separate channels at 8-bit depth. Images were imported into NIH image analysis software ImageJ and batch processed using custom macros. An Otsu autothreshold was applied to each image of Hoechst-stained nuclei, and the area of each detected object was recorded. Objects with areas <10 μm2 were excluded from analysis (Fig. 1o). To estimate percentage non-pyknotic nuclei, we chose a size cutoff value above which objects were assumed to be non-pyknotic. The size cutoff for pyknosis was chosen based on the shapes of nuclear size distributions from populations of cells subjected to increasing concentrations of NMDA challenge (Fig. 1k). We estimated the number of nuclei in overlapping bunches by dividing the area of the bunch by the modal nuclear size from a sparsely seeded population of control cells (Fig. 1o), and the quotient of this operation was tallied with the non-pyknotic nuclei, as bunching tended to correlate with decreased injury and pyknosis. Objects with areas >3 times the modal nuclear size were assumed to constitute an overlapping bunch. Figure 1j shows colorized examples of how individual objects were tallied.
Figure 1.

MAIM applied to disinhibited and NMDA-challenged dissociated cortical cultures. a–j, Representative images from a MAIM applied to dissociated cortical cultures challenged with bath NMDA with and without a 24 h bic/4-AP pretreatment. Bath NMDA challenge was 1 h and the cells were fixed and stained with Hoechst 33258 after 8 h recovery. The boxed area in a is expanded in b. Scale bar, 40 μm. Application of an Otsu autothreshold to each field of Hoechst-stained nuclei defined discrete areas of Hoechst positivity. In j, the red, green, and blue objects were counted as (respectively): 1 pyknotic nucleus, 1 non-pyknotic nucleus, 3 non-pyknotic nuclei. k, MAIM was used to quantify nuclear size distributions following NMDA challenge in spontaneously active (top) and disinhibited (bottom) cultures. l, Data from the top of k are expressed as cumulative percentage. m, n, Summary of data from k expressed as standard box plots (m) and average percentage non-pyknotic nuclei (n) with individual values from each replicate (squares). The nuclear size cutoff for defining pyknotic vs non-pyknotic nuclei is represented by the dashed lines in k and q. *p < 0.0005, n = 4, two-sample t test. o, Top and middle, Nuclear size distributions of the control group from (k) before and after exclusion of very small (<10 μm2) objects. Bottom, Untreated cells plated at a significantly lower (see p; *p = 0.003, n = 4, two-sample t test) density have a narrow nuclear size distribution with a prominent mode at 77 μm2. q, Individual values of nuclear size in spontaneously active cultures challenged with 40 μm NMDA, plotted by replicate.
To quantify COX-2 expression, an Otsu autothreshold was applied to each image from the group with highest expression (e.g., bic/4-AP or TBOA for 24 h), and the average threshold value obtained by this procedure was then applied to all COX-2 IF images from all treatment groups. A lower limit for size exclusion was applied, and all remaining objects were counted as COX-2 “positive” neurons (see Fig. 3n, Method A). To insure that the thresholding process did not introduce bias into our analysis of COX-2 IF, we applied a second method (see Fig. 3o, Method B) in which the COX-2 channel was not thresholded. Instead, we used the boundaries of Hoechst-positive areas from a morphonuclear analysis imaging method (MAIM) (as in Fig. 1d,g,j) to define regions of interest within the COX-2 channel. This approach returned a value for COX-2 IF that was associated with each cell regardless of the associated COX-2 IF signal intensity. To measure GAD65/67 expression, we applied an Intermodes autothreshold to each field and selected a lower size limit as indicated. GAD65/67 IF signal tended to be more diffuse than the COX-2 signal, which was primarily limited to soma and proximal dendrites as determined by double-staining with anti-MAP2. The Intermodes autothreshold was used to assess GAD65/67 signal because compared with Otsu and other threshold algorithms, it returned clearly discrete areas that were more reflective of somatic expression, which facilitated counting.
Figure 3.

Synaptic and extrasynaptic NMDARs differentially modulate neuronal COX-2 expression. a–l, Representative micrographs from simultaneous MAIM and unbiased image analysis of COX-2 IF applied in cortical cultures subjected to differing patterns of synaptic and extrasynaptic NMDAR stimulation over 24 h. Scale bar, 40 μm. m, Network disinhibition increased COX-2 IF that always colocalized with neuronal marker MAP2. Scale bar, 15 μm. n, o, Neuronal COX-2 expression quantified using two independent image analysis approaches. #p = 0.001, n = 5, ANOVA with Fisher's least significant difference (LSD) test. p, Simultaneous quantification of cell death using MAIM. **p < 0.0005, n = 5, two-sample t test. q, The number of nuclei per unit area remained invariant across all treatments. r, Size distribution histograms of COX-2-positive objects identified by Otsu thresholding. The dashed line indicates the lower limit for size exclusion. s, Western blots for COX-2 expression using actin as a loading control. *p = 0.035, ANOVA.
HPLC-ESI-MS/MS mediator lipidomics.
Lipidomics experiments were conducted using dissociated cortical cultures grown on 6-well culture plates (∼1 million cells/well; 2 ml of stimulation medium/well; 3 wells pooled/sample) or OTCs. OTC coronal slices from different anatomical regions were equally represented in all treatments to minimize variation due to biomass input. Samples were collected after 1 h of NMDA challenge or as indicated and 1250 pg of each internal standard was added. Cells from dissociated cultures and overlaid stimulation medium were sampled and processed separately. Stimulation medium was acidified and applied directly to preequilibrated BondElut C18 solid phase extraction (SPE) columns (Varian). Cells were scraped into 75% methanol plus 0.75 g/L BHT, sonicated, stored at −80°C for ≥1 h, and then accelerated at 4000 RCF for 20 min. Supernatants were diluted to <15% methanol (MeOH), acidified and applied to preequilibrated SPE columns. Columns were sequentially rinsed with 15% MeOH, water, and hexane. Target compounds were eluted with methyl formate, and eluate was dried under a UHP nitrogen stream. Extracts were resuspended in 25 μl initial conditions mobile phase. Extracts (20 μl injection volume) were loaded on C18 or chiral analytical columns (Fig. 2) connected to a TSQ Quantum Ultra triple quadrupole mass spectrometer (Thermo Scientific) equipped with a HESI-II electrospray ionization probe (operated in negative ion mode), and Accela autosampler, UV/PDA detector, and HPLC pump. The instrument was operated in full scan mode as indicated or multiple reaction monitoring mode (Table 1). Target compounds were quantified by the method of surrogate internal standards in XCalibur software.
Figure 2.

HPLC conditions and analytical columns used for HPLC-ESI-MS/MS-based mediator lipidomic analysis. HPLC Method 1 was used in conjunction with MRM operation for all quantitative assessment of target compounds. HPLC Method 2 was used to confirm the chirality of aspirin-triggered compounds. PGD2 and PGE2 are not well resolved by HPLC Method 1 since their mass spectra are closely related. HPLC Method 3 was used to resolve PGD2 and PGE2 for qualitative analysis. Analytical columns were maintained at 27°C.
Table 1.
MRM Strategy
| Compound | Parent m/z | Product m/z | Interpretation | CE | Average RT | RT SD | |
|---|---|---|---|---|---|---|---|
| Di-/trihydroxy-PUFA, PGs | PGD/E2 | 351 | 271 | M − 2H2O − CO2 | 18 | 5.23 | 0.06 |
| PGD2-d4 | 355 | 275 | M − 2H2O − CO2 | 20 | 5.27 | 0.04 | |
| PGF2α | 353 | 309 | M − CO2 | 20 | 5.63 | 0.10 | |
| TXB2 | 369 | 169 | Double ring cleavage | 20 | 5.96 | 0.04 | |
| RvD1 | 375 | 141 | 7Mc − H | 19 | 6.07 | 0.05 | |
| LXA4 | 351 | 115 | 5Mc − H | 18 | 6.19 | 0.06 | |
| PGI2 | 351 | 215 | M − 2H2O | 21 | 6.20 | 0.07 | |
| NPD1 | 359 | 206 | 10Cm − H | 18 | 13.35 | 0.08 | |
| Monohydroxy-PUFA | 20-HDHA | 343 | 241 | 20Cc + H − CO2 | 15 | 24.06 | 0.05 |
| 15-HETE | 319 | 219 | 15Cc + H | 15 | 24.07 | 0.05 | |
| 16-HDHA | 343 | 233 | 16Cc + H | 15 | 24.34 | 0.04 | |
| 17-HDHA | 343 | 201 | 17Cc + H − CO2 | 16 | 24.53 | 0.03 | |
| 11-HETE | 319 | 167 | 11Cc + H | 18 | 24.54 | 0.03 | |
| 13-HDHA | 343 | 193 | 13Cc + H | 15 | 24.74 | 0.05 | |
| 10-HDHA | 343 | 153 | 10Cc + H | 16 | 25.01 | 0.04 | |
| 8-HETE | 319 | 155 | 8Mc − H | 17 | 25.01 | 0.03 | |
| 12-HETE | 319 | 179 | 12Cc + H | 16 | 25.02 | 0.05 | |
| 14-HDHA | 343 | 205 | 14Cc + H | 14 | 25.03 | 0.04 | |
| 11-HDHA | 343 | 149 | 11Mc − H − CO2 | 15 | 25.56 | 0.05 | |
| 9-HETE | 319 | 151 | 9Mm | 16 | 25.59 | 0.05 | |
| 7-HDHA | 343 | 141 | 7Mc − H | 14 | 25.70 | 0.04 | |
| 5-(S)-HETE-d8 | 327 | 309 | M − H2O | 14 | 26.07 | 0.04 | |
| 5-HETE | 319 | 115 | 5Mc − H | 16 | 26.34 | 0.06 | |
| 8-HDHA | 343 | 189 | 8Mm | 15 | 26.37 | 0.03 | |
| 4-HDHA | 343 | 101 | 4Mc − H | 16 | 27.84 | 0.05 | |
| Parent PUFA | DHA-d5 | 332 | 288 | M − CO2 | 15 | 35.33 | 0.02 |
| DHA | 327 | 283 | M − CO2 | 13 | 35.37 | 0.01 | |
| AA | 303 | 259 | M − CO2 | 15 | 35.46 | 0.02 |
Single-/multiple-reaction monitoring (S/MRM) collision-induced dissociation transitions used in conjunction with HPLC Method 1 to quantify target compounds. The underlined compounds were used as surrogate internal standards for the corresponding PUFA classes (parent PUFA, monohydroxy-PUFA, and di-/trihydroxy-PUFA plus PGs). Transitions were picked from the four or five most abundant daughter ions produced by optimization of collision energy and tube lens offset. For mono-, di-, and trihydroxy-PUFA, only alpha cleavage or alpha cleavage plus peripheral-cut transitions were used. For compounds with similar parent mass and RT, transitions were picked to ensure minimal overlap of product ion spectra. CE, Collision (induced dissociation) energy (eV); RT, retention time in minutes.
Calcium imaging.
Cells were loaded with 11 μm Fluo-3 AM in TM0 (minus phenol red, plus 0.1% Pluronic F-127) for 20 min at 37°C. Excess dye was removed with two washes in phenol red free TM0, and imaging was conducted at room temperature on a Zeiss 510 Meta laser confocal microscope. Fluorescence intensities are reported as raw 8-bit gray values.
Results
Synaptic, not extrasynaptic, NMDARs induce neuronal COX-2 expression
Physiological synaptic activity in vivo enhances neuronal COX-2 expression in an NMDAR-dependent manner (Yamagata et al., 1993). An inference that clearly follows is that synaptic NMDARs mediate increased neuronal COX-2 expression. On the other hand, enhanced neuronal COX-2 expression under conditions that simultaneously stimulate both synaptic and extrasynaptic NMDARs mediates brain injury. For instance, cerebral ischemia increases neuronal COX-2 expression in the penumbra, and selective COX-2 inhibitor NS-398 reduces infarct volume (Nogawa et al., 1997). NMDA microinjection in neocortex enhances COX-2 expression and prostaglandin synthesis and produces lesions that are attenuated by NS-398 (Iadecola et al., 2001; Manabe et al., 2004), prostaglandin receptor antagonists (Kawano et al., 2006), and prostaglandin receptor (Kawano et al., 2006) or COX-2 (Iadecola et al., 2001; Manabe et al., 2004) deficiency. Bath NMDA and bath glutamate treatments are reported to induce neuronal COX-2 activity and expression in dissociated cultures (Hewett et al., 2000; McCullough et al., 2004; Tian et al., 2008). Oxygen-glucose deprivation in cortical cultures increases neuronal COX-2 expression (Gendron et al., 2004; Kim et al., 2007). Stimulation of extrasynaptic NMDARs specifically couples to cell death, and enhanced COX-2 expression or activity exacerbates excitotoxicity, depending on which prostaglandin receptor subtypes are involved (McCullough et al., 2004). Therefore, extrasynaptic NMDARs may regulate COX-2 expression or activity. To clarify the roles of synaptic versus extrasynaptic NMDARs in neuronal COX-2 expression, we applied unbiased COX-2 IF image analysis and simultaneous assessment of pyknosis using MAIM in dissociated cortical cultures. Enhanced synaptic activity was elicited by combination treatment with bic/4-AP. These conditions can result in a form of synaptic disinhibition characterized by network-wide (Arnold et al., 2005), synchronized (Arnold et al., 2005), and sustained (Bengtson et al., 2008; Tauskela et al., 2008) action potential firing and global calcium transients comparable to those induced by low dose bath NMDA or glutamate (Hardingham et al., 2002; Léveillé et al., 2008; Wittmann et al., 2009). Pretreatment of cultures with bic/4-AP produced remarkable resistance to a subsequent excitotoxic challenge with bath NMDA (Fig. 1n). This result suggests that bic/4-AP induced network disinhibition in our system similar to that observed in previous studies (Papadia et al., 2005, 2008; Léveillé et al., 2008, 2010).
We found that 24 h of network disinhibition, which selectively stimulates synaptic NMDARs (Hardingham et al., 2002; Léveillé et al., 2008; Hardingham and Bading, 2010), produced marked enhancement of neuronal COX-2 IF (Fig. 3) that was sensitive to the competitive NMDAR antagonist APV. In contrast, 24 h of bath NMDA at a dose (20 μm) sufficient to produce moderate pyknosis failed to enhance COX-2 expression. To rule out the possibility that the differences in the amount of Ca2+ influx resulting from synaptic activity versus bath NMDA treatment could account for the differential effects on COX-2 expression, we compared Ca2+ fluorescence signals generated by bic/4-AP treatment and 20 μm NMDA and found very similar profiles (Fig. 4). Since network disinhibition selectively stimulates synaptic NMDARs, while bath NMDA stimulates both synaptic and extrasynaptic NMDARs, these results indicate that synaptic, not extrasynaptic, NMDARs induce neuronal COX-2 expression.
Figure 4.

Synaptic activity and bath NMDA elicit similar Ca2+ profiles. a, The oscillatory component of the Ca2+ transient produced by bic/4-AP was terminated by subsequent application of NMDA. b, 20 μm NMDA alone elicited a calcium response that was similar in magnitude to that induced by bic/4-AP.
The Ptgs2 gene, which encodes COX-2, contains a cAMP response element (CRE) essential to COX-2 induction in neuronal and non-neuronal systems (Inoue et al., 1994; Smith et al., 2000; Lee et al., 2007). Synaptic NMDAR stimulation enhances CRE-dependent transcriptional events, whereas extrasynaptic NMDARs actively oppose CRE-dependent transcription (Hardingham et al., 2002; Hardingham and Bading, 2010). Additionally, whole-genome expression profiling of a very similar culture system showed that network disinhibition, but not bath glutamate, strongly induces Ptgs2 mRNA (Zhang et al., 2007). Therefore, these studies support a model in which synaptic NMDARs engage COX-2 induction, while extrasynaptic NMDARs prevent COX-2 induction via inhibition of CRE-dependent transcription or through a general failure of transcription or translation associated with excitotoxicity.
Sequential synaptic and extrasynaptic NMDAR stimulation defines the extent of COX-2 activity
Since synaptic, not extrasynaptic, NMDARs enhance COX-2 expression, we sought to determine how these receptor populations regulate COX-2 activity. We subjected dissociated cortical cultures to network disinhibition and measured PG levels associated with maximal COX-2 expression. Surprisingly, we found only small increases of prostaglandin D2 (PGD2), prostaglandin E2 (PGE2), and prostaglandin F2α (PGF2α) (Fig. 5a). Blockade of synaptic NMDARs with the noncompetitive NMDAR antagonist MK-801 prevented disinhibition-induced PG synthesis. When spontaneously active cultures were challenged with bath NMDA, which stimulates both synaptic and extrasynaptic NMDARs, PG synthesis was not enhanced (Figs. 5b, 6b). In contrast, NMDA challenge of previously disinhibited cultures produced up to 80-fold higher PG synthesis (Figs. 5b, 6b). PG synthesis was blocked by aspirin and by the COX-2 selective inhibitor NS-398, as expected. These observations demonstrate that maximal COX-2-dependent PG synthesis during excitotoxic events requires a history of enhanced network activity and synaptic NMDAR stimulation.
Figure 5.

Sequence of synaptic and extrasynaptic NMDAR stimulation controls COX-2 activity. a, b, Prostaglandin levels in cells and stimulation medium of dissociated cortical cultures treated for 24 h with bic/4AP (with or without MK-801) preceding a 1 h challenge with bath NMDA. Aspirin (ASA) and NS-398 were added 1 h before NMDA challenge. PGD2 and PGE2 are quantified together because they are not well resolved by standard HPLC conditions. *p = 0.036, **p = 0.046, n = 4, two-sample t test. #p = 0.001 vs all other NMDA-treated groups, n = 4, ANOVA with Fisher's LSD test. $p = 0.007, $$p = 0.006, $$$p = 0.001, n = 4, two-sample t test. c–f, HPLC retention times (c, d) and full mass spectra (e, f) of synaptic NMDAR-dependent, NS-398-sensitive compounds matched those of prostaglandin standards. Strigent HPLC conditions (Fig. 2, Method 3) were used to separate PGD2 and PGE2 for qualitative analysis.
Figure 6.

Prostaglandins from synaptically active cultures are increased by as little as 20 μm NMDA bath NMDA. a, AA and PG levels were measured in cells and stimulation medium of dissociated cortical cultures treated for 24 h with bic/4AP preceding a 1 h challenge with bath NMDA. *p = 0.007, **p = 0.005, ***p = 0.002, n = 4, two-sample t test vs control. b, PG responses from cultures challenged with low dose (30 μm) NMDA showed an absolute dependence on prior bic/4-AP treatment. p-value as indicated, two-sample t test vs NMDA-treated control. c, exogenous PGE2 exacerbated NMDA toxicity in cultures with a history of spontaneous activity. p-values as indicated, n = 5, two-sample t test.
To establish the threshold NMDA dose for eliciting enhanced PG responses from previously disinhibited cultures, we applied low dose bath NMDA and found that as little as 20 μm NMDA induced multifold increases in PG synthesis with concomitantly increased AA release (Fig. 6a), suggesting that increased substrate availability is critical to maximize PG synthesis. A slightly higher dose of NMDA (30 μm) produced multifold increases in PG synthesis that were strictly dependent on a history of enhanced synaptic activity (Fig. 6b).
To determine whether enhanced PG synthesis has any impact on NMDA toxicity in previously disinhibited cultures, we applied NS-398 either 24 or 1 h before NMDA challenge. Surprisingly, NS-398 had no significant impact on nuclear pyknosis outcome (data not shown); however, addition of exogenous PGE2 to spontaneously active cultures produced a significant increase in NMDA-induced pyknosis (Fig. 6c).
To define how the unesterified PUFA pool size is regulated under conditions that promote maximal prostaglandin synthesis, we quantified accumulation of unesterified AA and docosahexaenoic acid (DHA; 22,n−3) in the cells and medium of NMDA-challenged cultures, with or without a 24 h bic/4-AP pretreatment. Critically, network disinhibition alone decreased basal levels of unesterified PUFAs, particularly in the cellular compartment (Fig. 7a), and there was a corresponding decrease in PUFA peroxidation products in the stimulation medium (Fig. 7e,f). This process was not dependent on synaptic NMDARs, as MK-801 failed to unblock PUFA accumulation (data not shown). Therefore, synaptic activity decreases basal free PUFA levels, but synaptic NMDARs are neutral with respect to unesterified PUFA accumulation. In contrast, bath NMDA challenge rapidly enhanced the unesterified PUFA pool size (Figs. 6a, 7a), so extrasynaptic NMDARs must stimulate PUFA release, probably via PLA2 activation (Fig. 7b) (Dumuis et al., 1988; Stella et al., 1995; Taylor et al., 2008).
Figure 7.

Synaptic activity modulates compartment-specific unesterified PUFA sorting and basal lipid autoperoxidation. a, PUFA pool sizes in the cells and stimulation medium of dissociated cortical cultures during network disinhibition and excitotoxicity. DHA, 22:6,n−3. AA, 20:4,n−6. *p-values as indicated, n = 4, two-sample t test. #p = 0.001 vs all treatments, n = 4, ANOVA with Fisher's LSD test. n.s., not significant. b, Pyrrophenone is a selective group IVA cPLA2 inhibitor. NMDA-induced PUFA release was partially dependent on group IVA cPLA2. p-value as indicated, n = 4, two-sample t test. c, d, Simultaneous resolution of 10 unique monohydroxy-DHA (c) and six unique monohydroxy-HETE (d) regioisomer standards by HPLC-ESI-MS/MS. e, f, Activity-dependent downregulation of basal lipid peroxidation. *p-values as indicated, n = 4, two-sample t test.
Interestingly, NMDA challenge of previously disinhibited cultures revealed compartment-specific unesterified PUFA sorting: NMDA-induced accumulation of PUFAs in the stimulation medium was prominently reduced by high activity history, while accumulation in the cells was not (Fig. 7a). This indicates that the increased COX-2 load induced by synaptic NMDAR stimulation would still have ready access to unesterified AA substrate upon subsequent extrasynaptic NMDAR stimulation because COX-2 is located in membranes (Smith et al., 2000) into which unesterified PUFAs efficiently partition due to their hydrophobicity (Marszalek and Lodish, 2005).
While synaptic NMDAR-mediated currents are reduced by prolonged network disinhibition (Watt et al., 2000), isolated extrasynaptic NMDAR-mediated currents and Ca2+ signals are not affected (Bengtson et al., 2008). Additionally, bic/4-AP-induced Ca2+ loads are approximately equal to those induced by bath glutamate or NMDA, particularly at lower NMDA doses (∼20 μm) (Fig. 4) (Hardingham et al., 2002; Léveillé et al., 2008; Wittmann et al., 2009). These studies suggest that the extrasynaptic, rather than the synaptic, NMDAR-mediated component of bath NMDA treatment is more likely to account for enhanced PG synthesis during excitotoxic challenge of cultures with a high activity history via specific coupling to PLA2-dependent PUFA release (Fig. 8).
Figure 8.

Maximal prostaglandin synthesis during excitotoxicity is a function of network activity history. Spontaneous synaptic activity (condition [0]) stimulates synaptic NMDARs, while glutamate reuptake prevents stimulation of extrasynaptic NMDARs. Basal COX-2 expression is associated with prostaglandin (PG) levels near the lower limits of detection and quantification. Treatment with bic/4-AP (50 and 250 μm, respectively; condition [1]) increases synaptic activity and induces synaptic NMDAR-dependent neuroprotection (e.g., against staurosporine-induced apoptosis; vide infra). Under these conditions synaptic NMDAR-dependent COX-2 expression is markedly enhanced, while PG synthesis is only modestly elevated due to activity-dependent decreases in substrate availability. Challenge of spontaneously active cultures with bath NMDA (condition [2]) results in excitotoxic cell death and elevated PUFA levels, but COX-2 expression upon addition of NMDA is minimal; therefore, prostaglandin levels remain low. Robust synaptic NMDAR-dependent increases in COX-2 expression are probably blocked by stimulation of extrasynaptic NMDARs. When network disinhibition precedes an excitotoxic challenge (conditions [1] then [2]), PG synthesis is greatly enhanced because COX-2 load and substrate availability are both high.
Glutamate transporter blockade causes excitotoxicity, abnormal network excitability, and COX-2 induction
In our hands, 20 μm NMDA produced Ca2+ loads of magnitude similar to that of bic/4-AP (Fig. 4), and when applied for extended time periods (e.g., 24 h), produced moderate nuclear pyknosis; however, unlike bic/4-AP, this manipulation failed to enhance COX-2 expression (Fig. 3). On the other hand, there are numerous reports of COX-2 induction by bath application of NMDA. To explain this apparent discrepancy, we tested the hypothesis that excitotoxic insults must elicit enhanced synaptic activity to efficiently induce COX-2 expression. Perturbation of glutamate transporter function is associated with stroke, epilepsy, and neurodegenerative diseases (Beart and O'Shea, 2007). We used the glutamate transporter blocker TBOA to investigate how failure of glutamate transporter function impacts network activity and COX-2 function. TBOA treatment of dissociated cortical cultures caused a mild increase in pyknosis after only 3 h (Fig. 9a). This was a result of excitotoxicity caused by extrasynaptic spillover of synaptically released glutamate since blockade of spontaneous action potential firing with TTX reversed the effect of TBOA on acute pyknosis. Over the course of 60 h, there was a gradual increase in pyknosis in the control group (Fig. 9a,b). TTX accelerated the rate of pyknotic change by decreasing synaptic NMDAR signaling (Papadia et al., 2005; Wittmann et al., 2009). Surprisingly, TBOA partially prevented the TTX acceleration of pyknosis. Although TBOA alone caused a rapid (but mild) excitotoxicity, there was no further increase in pyknosis with time. By 60 h, pyknosis in the TBOA-only group was not different from pyknosis in the control group. TBOA also caused a time-dependent acidification of the stimulation medium that was prevented by TTX (Fig. 9c). These results demonstrate that TBOA causes mild excitotoxicity, followed by increased network activity.
Figure 9.
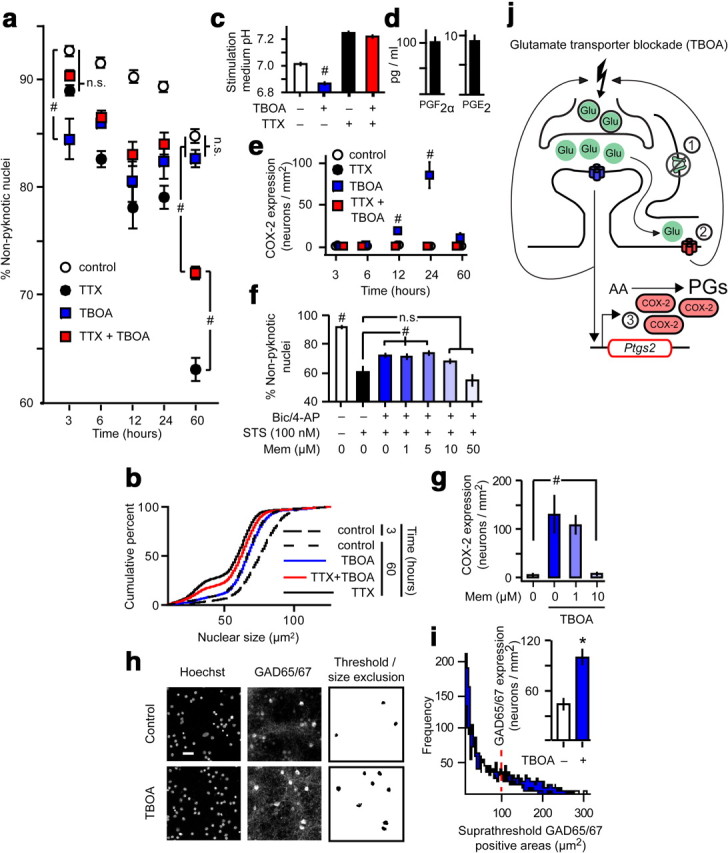
Glutamate transporter blockade induces excitotoxicity, abnormal network excitability, and COX-2 expression. a, Cell death quantified by MAIM in dissociated cultures treated with TTX and/or TBOA. #p = 0.001 as indicated vs time-matched treatments, n = 5, ANOVA with Fisher's LSD test. n.s., not significant. b, Selected nuclear size distributions from MAIM in a. c, Stimulation medium pH measured after 60 h of the indicated treatments. #p = 0.001 vs all treatments, n = 6, ANOVA with Fisher's LSD test. d, Prostaglandin levels in the medium of TBOA-treated dissociated cortical cultures. e, Simultaneous IF image analysis of COX-2 expression in cultures from a. #p = 0.001 vs all time-matched treatments, n = 5, ANOVA with Fisher's LSD test. f, Cell death quantified by MAIM after 24 h chemical induction of apoptosis with staurosporine (STS). Network disinhibition with or without memantine (Mem) proceeded for 16 h before STS treatment. #p = 0.001 as indicated or vs all treatments, n = 6, ANOVA with Fisher's LSD test. g, IF image analysis of COX-2 expression after 24 h of the indicated treatments. #p = 0.005 vs all treatments, n = 6, ANOVA with Fisher's LSD test. h, Representative micrographs used for GAD65/67 IF image analysis. Scale bar, 40 μm. i, Size distribution histograms of GAD65/67-positive areas identified by Intermodes thresholding. The dashed line indicates the lower limit for size exclusion. Inset, GAD65/67 expression quantified after 60 h TBOA treatment. *p = 0.001, n = 6, two-sample t test. j, Inhibition of glutamate transporter function with TBOA (1) increases the ambient glutamate concentration, resulting in stimulation of both synaptic and extrasynaptic NMDARs. Evidence for stimulation of extrasynaptic NMDARs (2) is observed as a rapid, mild, TTX-sensitive increase in nuclear pyknosis. Synaptic NMDAR stimulation in the context of enhanced action potential firing is required for increased COX-2 expression (3). The exact mechanism by which TBOA increases synaptic activity is unknown, but the process probably involves an NMDAR-dependent increase in neuronal excitability, and it may be terminated by compensatory GABAergic tone.
To determine whether glutamate transporter blockade controls COX-2 function, we measured COX-2 expression and PG levels in TBOA-treated cells. We found COX-2 induction comparable to that observed during bic/4-AP network disinhibition. TBOA-induced COX-2 expression peaked at 24 h and returned to baseline by 60 h (Fig. 9e). However, PG levels were not remarkably increased after 24 h TBOA (Fig. 9d) compared with basal and network disinhibition-induced levels (Figs. 4, 5). Thus, when excitotoxicity precedes enhanced network activity, prostaglandin formation is not remarkably enhanced. This highlights the importance of sequential synaptic then extrasynaptic NMDAR stimulation to maximal PG synthesis by neuronal COX-2.
The dual excitotoxic and protective effects of TBOA should be mediated by extrasynaptic and synaptic NMDARs respectively. We therefore investigated the effects of the extrasynaptic NMDAR-selective antagonist memantine (Papadia et al., 2008; Okamoto et al., 2009; Xia et al., 2010). We tested various concentrations of memantine against activity-dependent neuroprotection from chemical induction of apoptosis triggered by the protein kinase inhibitor staurosporine (Hardingham et al., 2002; Papadia et al., 2005; Léveillé et al., 2010). Activity-dependent neuroprotection by network disinhibition was only disrupted at memantine concentrations greater than or equal to 10 μm (Fig. 9f). Therefore, memantine doses up to 10 μm do not interfere with synaptic NMDAR signaling. Interestingly, 10 μm, but not 1 μm, memantine prevented TBOA-induced COX-2 expression (Fig. 9g). These results are consistent with the observation that synaptic, not extrasynaptic, NMDARs control COX-2 expression and suggest that TBOA-induced network activity occurs through an NMDAR-dependent process.
Because TBOA appeared to selectively kill a small subpopulation of cells with concomitant enhancement of synaptic activity, we hypothesized that TBOA might produce a form of network disinhibition by ablation of interneurons. On the contrary, after 60 h of TBOA treatment, GAD65/67 expression was increased, suggesting that interneurons are not ablated by TBOA (Fig. 9h,i). This fails to explain the mechanism of TBOA-induced network activity, but it does suggest a compensatory means by which TBOA-induced network activity may be terminated.
Previous studies suggest potential mechanisms for TBOA-induced elevation of synaptic activity (Fig. 9j). Low dose NMDA increases synaptic activity, and this effect has been proposed to result from NMDAR-dependent depolarization and increased likelihood of action potential firing, presynaptic facilitation, or LTP-like potentiation of non-NMDA ionotropic glutamate receptor-mediated currents (Soriano et al., 2006). Additionally, oxygen-glucose deprivation, which causes both glutamate transporter failure (Rossi et al., 2000) and increased COX-2 expression (Gendron et al., 2004; Kim et al., 2007), leads to enhanced network activity (Wahl et al., 2009).
Network activity history controls arachidonic acid and docosahexaenoic acid peroxidation in dissociated and organotypic cultures
In addition to prostaglandin synthesis, COX-2 catalyzes stereo- and regioselective lipoxygenation of PUFA to monohydroxy-PUFA [e.g., 13-monohydroxy-DHA (13-HDHA) from DHA and 11-monohydroxy-eicosatetraenoic acid (11-HETE) from AA]. Acetylation of the COX-2 active site by aspirin results in synthesis of (R)-enantiomers that possess intrinsic anti-inflammatory bioactivity and can be converted to more potent anti-inflammatory mediators (e.g., resolvins, lipoxins, and electrophilic oxo-derivatives) via subsequent enzymatic and transcellular processing (Serhan et al., 2000, 2002, 2008; Bazan, 2003, 2006; Marcheselli et al., 2003; Groeger et al., 2010). In contrast, the abundance of monohydroxy-PUFA not produced by mammalian enzymes (e.g., 8-HDHA and 9-HETE) reflects nonenzymatic lipid (auto)peroxidation. We resolved 10 mono-HDHA and 6 mono-HETE regioisomers and their parent PUFA using HPLC-ESI-MS/MS in dissociated cortical cultures subjected to network disinhibition and NMDA excitotoxicity to establish whether synaptically activated neuronal COX-2 mediates synthesis of aspirin-triggered mediators. We found that network disinhibition attenuated NMDA-induced nonenzymatic lipid peroxidation while enhancing monohydroxy-PUFA from COX-2 (Fig. 10a,b). Aspirin switched COX-2-dependent lipoxygenation of DHA and AA to preferential 17-(R)-HDHA and 15-(R)-HETE synthesis (Fig. 10a–f). These results indicate that modulation of the COX-2 active site with aspirin-like compounds (Kalgutkar et al., 1998; Carlson, 2003) to favor synthesis of (R)-monohydroxy-PUFA may be a therapeutic target in diseases characterized by abnormal network activity and neuroinflammation. Moreover, synaptic activity prevents both NMDA-induced (Fig. 10a,b) and basal (Fig. 7e,f) nonenzymatic lipid peroxidation, which is associated with cell damage.
Figure 10.

Activity history determines unesterified PUFA accumulation and peroxidation in dissociated and organotypic cultures: modulation by aspirin. a, b, Monohydroxy-PUFA levels in stimulation medium of dissociated cortical cultures following NMDA challenge with or without bic/4-AP pretreatment. #p = 0.001 vs all treatments, n = 4, ANOVA with Fisher's LSD test. n.d., not detected. c, d, Following network disinhibition of organotypic slice or dissociated cortical cultures, NMDA excitotoxicity in the presence of aspirin (ASA) triggered synthesis of nearly enantiopure (R) compounds from AA and DHA. e, f, Full mass spectra of aspirin-triggered compounds (red and blue) matched those of 15-(R)-HETE and 17-(R)-HDHA standards. g, A history of synaptic activity markedly reduced cortical opacity associated with NMDA toxicity in organotypic slice cultures. Scale bar, 300 μm. h, PGD/E2 levels in disinhibited and NMDA-challenged organotypic cultures. *p = 0.012, n = 4, two-sample t test. i, The UV spectrum of aspirin-triggered 17(R)-HDHA from disinhibited, NMDA-challenged organotypic cultures (blue) matched 17(R)-HDHA standard (black), but not ASA-free controls (gray).
To determine whether the activity history of the synaptic network is a critical determinant of excitotoxicity-induced COX-2 signaling in vivo, we subjected OTCs, which maintain closer parity to in vivo cortical architecture, to network disinhibition and NMDA challenge (Fig. 10g–i). Prostaglandin synthesis in OTCs was strongly elevated by NMDA treatment, but only in the context of elevated activity history. This result indicates that sequential synaptic then extrasynaptic NMDAR stimulation is likely to control the extent of prostaglandin formation from in vivo neuronal networks.
Discussion
We have illuminated the significance of synaptic and extrasynaptic NMDARs in neuronal COX-2 function by showing a new mechanism that amplifies PG synthesis. The temporal pattern of synaptic and extrasynaptic NMDAR stimulation is necessary for maximizing COX-2 load, substrate availability, and PG synthesis. We also demonstrate that sustained increases in synaptic activity limit the size of the unesterified PUFA pool, along with basal and excitotoxicity-induced PUFA autoperoxidation, through a synaptic NMDAR-independent mechanism. In contrast, bath NMDA rapidly increases the PUFA pool size and PUFA autoperoxidation, implying a specific role for extrasynaptic NMDARs in PUFA release from membrane phospholipids. This mechanism likely occurs via specific coupling of extrasynaptic NMDARs to cytosolic phospholipases A2 (Sanchez-Mejia and Mucke, 2010), which are stimulated by calcium to translocate to membranes.
The significance of amplified PG synthesis during excitotoxic challenge of neurons with a high activity history remains uncertain. We have suggested that sufficient stimulation of extrasynaptic NMDARs may convert COX-2 signaling from a physiological to a pathological process. We found that exogenous PGE2 can exacerbate NMDA toxicity in neurons with a history of basal synaptic activity levels; whereas blockade of PG formation in neurons with high activity history had no effect. To the extent that enhanced PG formation in previously disinhibited neurons may exacerbate excitotoxicity, these influences could be masked by the neuroprotective shield associated with synaptic activity. On the other hand, many studies have suggested that selective activation of some PG receptor subtypes (McCullough et al., 2004; Wu et al., 2007) may be a valid therapeutic strategy under certain circumstances. Therefore, enhanced PG synthesis in synaptically primed neurons may actually be a defensive response to injury. This is a complex area of investigation, and the distinct roles of the various PGs and their receptors are likely context-specific vis-à-vis pathology and cell type. No matter how these issues are ultimately settled, an understanding of this novel neuronal PG amplification mechanism will help inform debate and future investigations in this area.
We also showed that excitotoxic events can lead to abnormal network activity and enhanced COX-2 expression, explaining how this immediate-early gene enzyme can be induced by NMDARs outside the context of physiological synaptic activity. As we have demonstrated, NMDAR stimulation alone is not enough. For instance, bath NMDA (Soriano et al., 2006) treatment or oxygen-glucose deprivation (Wahl et al., 2009) of dissociated neuronal cultures should only lead to COX-2 induction if network activity is enhanced. The extracellular glutamate (or NMDA) concentration must be high enough to initiate abnormal excitability, but not so high as to cause chronic membrane depolarization. Aberrant network excitability plays an important role in neurodegenerative diseases (Palop et al., 2006, 2007) in which neuroinflammation (Block et al., 2007) and excitotoxicity (Hardingham and Bading, 2010) are prominent features.
Under physiological circumstances, why would extrasynaptic NMDARs selectively couple to maximal PG synthesis via COX-2 substrate (AA) release? Physiological stimulation of extrasynaptic NMDA receptors may occur under certain conditions. For example, during spatiotemporally dense synaptic activity, glutamate transport mechanisms may be insufficient to completely prevent spillover (Okubo et al., 2010; Okubo and Iino, 2011). Extrasynaptic NMDARs could serve as sensors of acutely increased metabolic demand. If the extrasynaptic NMDARs really are preferentially coupled to a substrate release mechanism (such as a PLA2), then the resulting release of AA and rapid “bump” in PG synthesis could signal to arteriolar vasodilation, matching blood flow to metabolic demand.
Important questions remain. Electrophysiological approaches are needed to confirm the patterns of synaptic and extrasynaptic NMDAR stimulation proposed to mediate the observations described here, and these experiments are currently underway. In addition to COX-2 load and substrate availability, are there additional mechanisms that amplify PG signaling during sequential stimulation of synaptic then extrasynaptic NMDARs? Neuronal nitric oxide synthase (nNOS) mediates NMDA neurotoxicity by S-nitrosylation of COX-2 (Tian et al., 2008), and membrane-associated guanylate kinases (MAGUKs) may couple extrasynaptic NMDARs to excessive nitric oxide production (Hardingham and Bading, 2010). Therefore, COX-2 nitrosylation by nNOS may be an important event in amplifying COX-2 signaling distal to the extrasynaptic NMDAR. Targeting of mechanisms that couple sequential synaptic then extrasynaptic NMDAR stimulation may lead to novel anti-inflammatory/neuroprotective strategies.
Footnotes
This work was supported by grants from the National Institutes of Health, National Institute of Neurological Disorders and Stroke (R01 NS046741), National Center for Research Resources (P20 RR016816), and the National Center for Complementary and Alternative Medicine (RC2 AT005909) (N.G.B.). D.T.S. is a recipient of Ruth L. Kirschstein National Research Service Awards for Individual Predoctoral MD/PhD and Other Dual Doctoral Degree Fellows (F30) Grant NS063630 from the National Institute of Neurological Disorders and Stroke. We thank Dr. Lennart Mucke for reviewing the data presented here and making constructive criticisms. We thank Dr. Giles Hardingham for providing tissue culture protocols and assistance with development of image analysis techniques. We thank Dr. Jorgelina Calandria for her help with the Western blot.
References
- Akaneya Y, Tsumoto T. Bidirectional trafficking of prostaglandin E2 receptors involved in long-term potentiation in visual cortex. J Neurosci. 2006;26:10209–10221. doi: 10.1523/JNEUROSCI.3028-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold FJ, Hofmann F, Bengtson CP, Wittmann M, Vanhoutte P, Bading H. Microelectrode array recordings of cultured hippocampal networks reveal a simple model for transcription and protein synthesis-dependent plasticity. J Physiol. 2005;564:3–19. doi: 10.1113/jphysiol.2004.077446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG. Synaptic lipid signaling: significance of polyunsaturated fatty acids and platelet-activating factor. J Lipid Res. 2003;44:2221–2233. doi: 10.1194/jlr.R300013-JLR200. [DOI] [PubMed] [Google Scholar]
- Bazan NG. Cell survival matters: docosahexaenoic acid signaling, neuroprotection and photoreceptors. Trends Neurosci. 2006;29:263–271. doi: 10.1016/j.tins.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Beart PM, O'Shea RD. Transporters for l-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 2007;150:5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson CP, Dick O, Bading H. A quantitative method to assess extrasynaptic NMDA receptor function in the protective effect of synaptic activity against neurotoxicity. BMC Neurosci. 2008;9:11. doi: 10.1186/1471-2202-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Carlson NG. Neuroprotection of cultured cortical neurons mediated by the cyclooxygenase-2 inhibitor APHS can be reversed by a prostanoid. J Neurosci Res. 2003;71:79–88. doi: 10.1002/jnr.10465. [DOI] [PubMed] [Google Scholar]
- Chen C, Bazan NG. Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;93:929–941. doi: 10.1152/jn.00696.2004. [DOI] [PubMed] [Google Scholar]
- Chen C, Magee JC, Bazan NG. Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J Neurophysiol. 2002;87:2851–2857. doi: 10.1152/jn.2002.87.6.2851. [DOI] [PubMed] [Google Scholar]
- Dumuis A, Sebben M, Haynes L, Pin JP, Bockaert J. NMDA receptors activate the arachidonic acid cascade system in striatal neurons. Nature. 1988;336:68–70. doi: 10.1038/336068a0. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Brunette E, Mealing GA, Nguyen A, Tauskela JS, Morley P. Opposing effects of cyclooxygenase-2 selective inhibitors on oxygen-glucose deprivation-induced neurotoxicity. Eur J Pharmacol. 2004;493:45–55. doi: 10.1016/j.ejphar.2004.04.026. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeger AL, Cipollina C, Cole MP, Woodcock SR, Bonacci G, Rudolph TK, Rudolph V, Freeman BA, Schopfer FJ. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat Chem Biol. 2010;6:433–441. doi: 10.1038/nchembio.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Uliasz TF, Vidwans AS, Hewett JA. Cyclooxygenase-2 contributes to N-methyl-d-aspartate-mediated neuronal cell death in primary cortical cell culture. J Pharmacol Exp Ther. 2000;293:417–425. [PubMed] [Google Scholar]
- Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S, Rakic P, Flavell RA. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:665–670. doi: 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, Morham S, Ross ME. Reduced susceptibility to ischemic brain injury and N-methylaspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci U S A. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Nanayama T, Hara S, Yokoyama C, Tanabe T. The cyclic AMP response element plays an essential role in the expression of the human prostaglandin-endoperoxide synthase 2 gene in differentiated U937 monocytic cells. FEBS Lett. 1994;350:51–54. doi: 10.1016/0014-5793(94)00731-4. [DOI] [PubMed] [Google Scholar]
- Kalgutkar AS, Crews BC, Rowlinson SW, Garner C, Seibert K, Marnett LJ. Aspirin-like molecules that covalently inactivate cyclooxygenase-2. Science. 1998;280:1268–1270. doi: 10.1126/science.280.5367.1268. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci U S A. 1996;93:2317–2321. doi: 10.1073/pnas.93.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Keene CD, Chang RC, Lopez-Yglesias AH, Shalloway BR, Sokal I, Li X, Reed PJ, Keene LM, Montine KS, Breyer RM, Rockhill JK, Montine TJ. Suppressed accumulation of cerebral amyloid beta peptides in aged transgenic Alzheimer's disease mice by transplantation with wild-type or prostaglandin E-2 receptor subtype 2-null bone marrow. Am J Pathol. 2010;177:346–354. doi: 10.2353/ajpath.2010.090840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Raval AP, Defazio RA, Perez-Pinzon MA. Ischemic preconditioning via epsilon protein kinase C activation requires cyclooxygenase-2 activation in vitro. Neuroscience. 2007;145:931–941. doi: 10.1016/j.neuroscience.2006.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch H, Huh SE, Elsen FP, Carroll MS, Hodge RD, Bedogni F, Turner MS, Hevner RF, Ramirez JM. Prostaglandin E2-induced synaptic plasticity in neocortical networks of organotypic slice cultures. J Neurosci. 2010;30:11678–11687. doi: 10.1523/JNEUROSCI.4665-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotilinek LA, Westerman MA, Wang Q, Panizzon K, Lim GP, Simonyi A, Lesne S, Falinska A, Younkin LH, Younkin SG, Rowan M, Cleary J, Wallis RA, Sun GY, Cole G, Frautschy S, Anwyl R, Ashe KH. Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain. 2008;131:651–664. doi: 10.1093/brain/awn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TD, Shirai Y, Okamoto T, Tatsukawa T, Nagao S, Shimizu T, Ito M. Lipid signaling in cytosolic phospholipase A2alpha-cyclooxygenase-2 cascade mediates cerebellar long-term depression and motor learning. Proc Natl Acad Sci U S A. 2010;107:3198–3203. doi: 10.1073/pnas.0915020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Dziema H, Lee KH, Choi YS, Obrietan K. CRE-mediated transcription and COX-2 expression in the pilocarpine model of status epilepticus. Neurobiol Dis. 2007;25:80–91. doi: 10.1016/j.nbd.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léveillé F, El GF, Gouix E, Lecocq M, Lobner D, Nicole O, Buisson A. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J. 2008;22:4258–4271. doi: 10.1096/fj.08-107268. [DOI] [PubMed] [Google Scholar]
- Léveillé F, Papadia S, Fricker M, Bell KF, Soriano FX, Martel MA, Puddifoot C, Habel M, Wyllie DJ, Ikonomidou C, Tolkovsky AM, Hardingham GE. Suppression of the intrinsic apoptosis pathway by synaptic activity. J Neurosci. 2010;30:2623–2635. doi: 10.1523/JNEUROSCI.5115-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer's disease. J Neurosci. 2005;25:10180–10187. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe Y, Anrather J, Kawano T, Niwa K, Zhou P, Ross ME, Iadecola C. Prostanoids, not reactive oxygen species, mediate COX-2-dependent neurotoxicity. Ann Neurol. 2004;55:668–675. doi: 10.1002/ana.20078. [DOI] [PubMed] [Google Scholar]
- Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, Hardy M, Gimenez JM, Chiang N, Serhan CN, Bazan NG. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem. 2003;278:43807–43817. doi: 10.1074/jbc.M305841200. [DOI] [PubMed] [Google Scholar]
- Marszalek JR, Lodish HF. Docosahexaenoic acid, fatty acid-interacting proteins, and neuronal function: breastmilk and fish are good for you. Annu Rev Cell Dev Biol. 2005;21:633–657. doi: 10.1146/annurev.cellbio.21.122303.120624. [DOI] [PubMed] [Google Scholar]
- McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose ME, Stetler RA, Isakson PC, Chen J, Graham SH. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc Natl Acad Sci U S A. 1998;95:10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa K, Araki E, Morham SG, Ross ME, Iadecola C. Cyclooxygenase-2 contributes to functional hyperemia in whisker-barrel cortex. J Neurosci. 2000;20:763–770. doi: 10.1523/JNEUROSCI.20-02-00763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, Zhang D, Vincent Chen HS, Tong G, Hayden MR, Lipton SA. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo Y, Iino M. Visualization of glutamate as a volume transmitter. J Physiol. 2011;589:481–488. doi: 10.1113/jphysiol.2010.199539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo Y, Sekiya H, Namiki S, Sakamoto H, Iinuma S, Yamasaki M, Watanabe M, Hirose K, Iino M. Imaging extrasynaptic glutamate dynamics in the brain. Proc Natl Acad Sci U S A. 2010;107:6526–6531. doi: 10.1073/pnas.0913154107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–773. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J Neurosci. 2005;25:4279–4287. doi: 10.1523/JNEUROSCI.5019-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Léveillé F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- Sanchez-Mejia RO, Mucke L. Phospholipase A2 and arachidonic acid in Alzheimer's disease. Biochim Biophys Acta. 2010;1801:784–790. doi: 10.1016/j.bbalip.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang N, Zhang J, Marcheselli V, Bazan NG, Chen C. Postsynaptically synthesized prostaglandin E2 (PGE2) modulates hippocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J Neurosci. 2005;25:9858–9870. doi: 10.1523/JNEUROSCI.2392-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savonenko A, Munoz P, Melnikova T, Wang Q, Liang X, Breyer RM, Montine TJ, Kirkwood A, Andreasson K. Impaired cognition, sensorimotor gating, and hippocampal long-term depression in mice lacking the prostaglandin E2 EP2 receptor. Exp Neurol. 2009;217:63–73. doi: 10.1016/j.expneurol.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. 2000;192:1197–1204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 2006;26:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Pellerin L, Magistretti PJ. Modulation of the glutamate-evoked release of arachidonic acid from mouse cortical neurons: involvement of a pH-sensitive membrane phospholipase A2. J Neurosci. 1995;15:3307–3317. doi: 10.1523/JNEUROSCI.15-05-03307.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauskela JS, Fang H, Hewitt M, Brunette E, Ahuja T, Thivierge JP, Comas T, Mealing GA. Elevated synaptic activity preconditions neurons against an in vitro model of ischemia. J Biol Chem. 2008;283:34667–34676. doi: 10.1074/jbc.M805624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AL, Bonventre JV, Uliasz TF, Hewett JA, Hewett SJ. Cytosolic phospholipase A2 alpha inhibition prevents neuronal NMDA receptor-stimulated arachidonic acid mobilization and prostaglandin production but not subsequent cell death. J Neurochem. 2008;106:1828–1840. doi: 10.1111/j.1471-4159.2008.05527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Kim SF, Hester L, Snyder SH. S-nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. Proc Natl Acad Sci U S A. 2008;105:10537–10540. doi: 10.1073/pnas.0804852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu B, Bazan NG. Hippocampal kindling epileptogenesis upregulates neuronal cyclooxygenase-2 expression in neocortex. Exp Neurol. 2003;179:167–175. doi: 10.1016/s0014-4886(02)00019-5. [DOI] [PubMed] [Google Scholar]
- Wahl AS, Buchthal B, Rode F, Bomholt SF, Freitag HE, Hardingham GE, Rønn LC, Bading H. Hypoxic/ischemic conditions induce expression of the putative pro-death gene Clca1 via activation of extrasynaptic N-methyl-d-aspartate receptors. Neuroscience. 2009;158:344–352. doi: 10.1016/j.neuroscience.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, Turrigiano GG. Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron. 2000;26:659–670. doi: 10.1016/s0896-6273(00)81202-7. [DOI] [PubMed] [Google Scholar]
- Wittmann M, Queisser G, Eder A, Wiegert JS, Bengtson CP, Hellwig A, Wittum G, Bading H. Synaptic activity induces dramatic changes in the geometry of the cell nucleus: interplay between nuclear structure, histone H3 phosphorylation, and nuclear calcium signaling. J Neurosci. 2009;29:14687–14700. doi: 10.1523/JNEUROSCI.1160-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Wang Q, Liang X, Andreasson K. Divergent effects of prostaglandin receptor signaling on neuronal survival. Neurosci Lett. 2007;421:253–258. doi: 10.1016/j.neulet.2007.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- Yang H, Zhang J, Breyer RM, Chen C. Altered hippocampal long-term synaptic plasticity in mice deficient in the PGE2 EP2 receptor. J Neurochem. 2009;108:295–304. doi: 10.1111/j.1471-4159.2008.05766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Steijaert MN, Lau D, Schütz G, Delucinge-Vivier C, Descombes P, Bading H. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron. 2007;53:549–562. doi: 10.1016/j.neuron.2007.01.025. [DOI] [PubMed] [Google Scholar]