

Abstract
Previously, we showed that the mouse LIM-domain only 4 (Lmo4) gene, which encodes a protein containing two zinc-finger LIM domains that interact with various DNA-binding transcription factors, attenuates behavioral sensitivity to repeated cocaine administration. Here we show that transcription of anaplastic lymphoma kinase (Alk) is repressed by LMO4 in the striatum and that Alk promotes the development of cocaine sensitization and conditioned place preference, a measure of cocaine reward. Since LMO4 is known to interact with estrogen receptor α (ERα) at the promoters of target genes, we investigated whether Alk expression might be controlled by a similar mechanism. We found that LMO4 and ERα are associated with the Alk promoter by chromatin immunoprecipitation and that Alk is an estrogen-responsive gene in the striatum. Moreover, we show that ERα knock-out mice exhibit enhanced cocaine sensitization and conditioned place preference and an increase in Alk expression in the nucleus accumbens. These data define a novel regulatory network involved in behavioral responses to cocaine. Interestingly, sex differences in several behavioral responses to cocaine in humans and rodents have been described, and estrogen is thought to mediate some of these differences. Our data suggest that estrogen regulation of Alk may be one mechanism responsible for sexually dimorphic responses to cocaine.
Introduction
Chronic exposure to cocaine leads to long-lasting cellular, molecular, and structural adaptations in brain reward systems and concurrent behavioral changes that are thought to lead to cocaine addiction (Thomas et al., 2008). One measure of behavioral plasticity to cocaine is psychomotor sensitization. Sensitization is induced by repeated, intermittent exposure to cocaine and is observed as a progressive increase in locomotor and stereotypic activity (Robinson and Berridge, 2003). The nucleus accumbens (Acb) is a critical region for the development of cocaine sensitization (Vanderschuren and Kalivas, 2000). We showed previously that the transcriptional regulator Lim domain only 4 (Lmo4) functions in the Acb to attenuate cocaine sensitization (Lasek et al., 2010).
The LMO proteins act to modulate transcription through their ability to interact with DNA binding transcription factors (Rétaux and Bachy, 2002). In a previous study, we undertook a search for potential transcriptional targets of Drosophila LMO using gene expression microarrays. We found that levels of dAlk, which encodes the Drosophila homolog of anaplastic lymphoma kinase, are altered in dLMO mutants (Lasek et al., 2011). Here we show that mouse Alk expression increases in striatal neurons upon downregulation of Lmo4 by RNA interference (RNAi), implicating Alk as a negatively regulated target of LMO4. Treatment of mice with RNAi targeting Alk in the Acb or with an orally available ALK small-molecule inhibitor resulted in attenuated development of cocaine sensitization, indicating that regulation of Alk expression by LMO4 is functionally relevant for this behavior. We also found a role for Alk in cocaine conditioned place preference (CPP), which measures the rewarding properties of cocaine and is hypothesized to be model for drug-seeking behavior (Bardo and Bevins, 2000).
LMO4 is a known interacting partner of estrogen receptor α (ERα), and negatively regulates transcriptional activation by ERα (Singh et al., 2005). This led us to hypothesize that an interaction between LMO4 and ERα in the Acb might be important for the regulation of Alk expression. We demonstrate that Alk levels in the Acb increase upon estradiol treatment in mice, suggesting that Alk is a novel estrogen-regulated gene in the nervous system. Using chromatin immunoprecipitation (ChIP), we show that LMO4 and ERα are associated with the Alk promoter, implying a direct regulation of Alk by a transcriptional complex including LMO4 and ERα. Finally, knock-out of the Esr1 gene (encoding ERα) in mice resulted in enhanced cocaine sensitization, a trend toward increased cocaine CPP, and higher levels of ALK protein in the Acb. Together, these data identify a novel molecular mechanism that regulates cocaine reward and behavioral plasticity.
Materials and Methods
Animals.
Mice used for experiments were male C57BL/6J aged 8–12 weeks, unless otherwise indicated. All mice were group housed until they underwent stereotactic surgery, after which they were singly housed, throughout subsequent testing. ERα knockout (Esr1KO) mice were purchased from Jackson Laboratory (B6.129P2-Esr1tm1Ksk/J), where they have been maintained on a C57BL/6J background for 10 generations, and were bred in-house for behavioral experiments. Food and water were provided at all times and animals were on a 12 h light–dark cycle. All animal protocols were approved by the Ernest Gallo Clinic and Research Center Institutional Animal Care and Use Committee.
Chemicals and antibodies.
17β-Estradiol and β-estradiol 3-benzoate was purchased from Sigma. TAE684 was a generous gift from Novartis. The GFP monoclonal antibody (3E6) was purchased from Invitrogen. LMO4 (C-15) and ERα (MC-20 and HC-20) antibodies were purchased from Santa Cruz Biotechnology. The ALK antibody (ab650) was purchased from Abcam. Fluorescent and biotin-conjugated secondary antibodies were purchased from Jackson ImmunoResearch. HRP-conjugated secondary antibodies for Western blotting were purchased from GE Healthcare.
Design and cloning of short hairpin RNAs.
Small interfering RNAs (siRNAs, 19 nt) were designed against the Alk mRNA (GenBank NM_007439) using the siDesign Center at Thermo Fisher Scientific (http://www.dharmacon.com/DesignCenter/DesignCenterPage.aspx). These 19 nt target sequences were then incorporated into DNA oligonucleotides encoding short hairpin RNAs (shRNAs) and cloned into the lentiviral vector pLL3.7 as described previously (Lasek et al., 2007). The 19 nt targeting sequences for the shRNAs are as follows (5′ to 3′): shAlk.1, GCAACGCAGCAUCAAACAA; shAlk.2, GCAACGCAGCAUCAAACAA; shAlk.3, GGUUCUGGCUGCAGAUAGU.
RNAi in cell culture.
Primary mouse striatum was dissected from P1 C57BL/6J mouse pups and treated as described previously (McGough et al., 2004). Cells were plated at 6 × 105 per well on poly-d-lysine-coated 12-well dishes and transfected with 100 nm siRNA using Dharmafect3 transfection reagent (Thermo Fisher) at 4 d in vitro (DIV). RNA was isolated from cells at 11 DIV using the Qiagen RNeasy Plus kit. The 19 nt targeting sequence for siLmo4.3 is 5′-GUGUUUCACAUGUUCUACC and for siScr is 5′-GCGCUUAGCUGUAGGAUUC. These siRNAs were based on previously described shRNAs (Lasek et al., 2010). Neuro-2a cells (ATCC) were grown in DMEM plus 10% fetal bovine serum and 10% CO2. Cells were seeded into six-well dishes and transfected with pLL3.7 plasmids containing shRNA sequences using Lipofectamine 2000 and Opti-MEM media (Invitrogen) according to the manufacturer's instructions. Forty-eight hours after transfection, media was removed, 0.5 ml of Trizol reagent (Invitrogen) added to each well, and total RNA isolated according to the Trizol instructions. Total RNA was treated with RNase-free DNase (Promega) to remove genomic DNA contamination.
Quantitative RT-PCR.
RNA isolation, cDNA synthesis and quantitative RT-PCR (qPCR) were performed as described previously (Lasek et al., 2010). Probes and primers for Taqman gene expression assays were predesigned by Applied Biosystems with the following assay IDs: mouse Alk, Mm00431757_m1; mouse Esr1, Mm00433149_m1; mouse Lmo4, Mm00495373_m1. Mouse Gapdh or Actb probes and primers were used as controls for the PCRs.
Chromatin immunoprecipitation.
Striatum from eight male mice were rapidly dissected, pooled, cross-linked, and homogenized as described previously (Tsankova et al., 2004). Following homogenization, the EZ-ChIP kit (Millipore) was used according to the manufacturer's instructions. For ERα ChIP, NB41A3 cells (ATCC) were maintained in F-12K media containing 15% horse serum and 2.5% fetal bovine serum (FBS) at 37°C with 5% CO2. For ChIP experiments, 5 × 106 cells were seeded into two 10 cm dishes. One day later, cells were rinsed with warm PBS and media was changed to phenol-red free DMEM containing 10% charcoal-stripped FBS. The next day, cells were treated with 100 nm β-estradiol for 45 min. Formaldehyde was added to a final concentration of 1% to crosslink chromatin, and all subsequent steps were followed using the EZ-ChIP kit. DNA was analyzed using the Applied Biosystems 7900 system and SYBR green master mix. Sequences for primers to the mouse Alk promoter are as follows: −963F, TGGAAACCTGTGCAGACACC; −963R, CAGACCTGTCAGCCACTTGGT; −274F, GATTCTGTCCAGCGGTTTTCTC; −274R, CTTGGTTCCTCGGAGTTCAC; +158F, GCAAGACAGTGACCGACTCG; +158R, CTGCAACTTTCCAGGCTGTG; exon 29F, AAAAAGCAATCAGTGCAAGGC; exon 29R, CATGCACGAAAACTTGGCG. Experiments were repeated three times with consistent results each time.
Lentivirus production.
Lentivirus was produced in 293FT cells (Invitrogen) as described previously (Lasek et al., 2007).
Stereotactic surgeries.
Male 8- to 12-week-old C57BL/6J mice weighing 23–28 g were infused with lentivirus in the Acb as described previously (Lasek et al., 2010). Mice were allowed to recover for at least 14 d before commencing behavioral experiments.
Cocaine sensitization and CPP.
Locomotor activity measurements were performed in Plexiglas locomotor activity chambers (Med Associates). To test locomotor sensitization to cocaine, on the first two days, mice were injected with saline (10 ml/kg, i.p.) and placed into the activity chambers for 15 min to habituate them to the testing procedures. On days 3–7, drug-induced locomotor activity was measured by treating with cocaine HCl (15 mg/kg, Sigma). Mice were then immediately placed into the activity chambers and horizontal distance traveled (in centimeters) was recorded for 15 min. Cocaine sensitization was also measured 10 d later (day 17), by injecting mice with 15 mg/kg cocaine.
For CPP, mice were conditioned with cocaine in two-chambered conditioned place preference boxes, consisting of modified locomotor activity boxes (Med Associates) fitted with Plexiglas floor inserts with different textures (holes vs grooves) and wall patterns (horizontal vs vertical stripes) on each side. Mice were placed initially in the box on day 1 (without treatment) and allowed ad libitum access for 20 min. to measure their initial side preference. Mice were then assigned to the initially nonpreferred chamber for cocaine treatment. On days 2 and 4, mice were treated with 10 mg/kg cocaine (i.p.) and confined to one chamber for 15 min. On days 3 and 5, mice were treated with saline intraperitoneally and confined to the opposite chamber. On day 6, mice were placed in the boxes and allowed ad libitum access to both chambers for 20 min. Preference score was calculated as the difference (after conditioning minus before conditioning) in minutes spent in the cocaine-paired chamber.
TAE684 treatments.
Mice were treated daily with 10 mg/kg NVP-TAE684 (TAE684) in 10% 1-methyl-2-pyrrolidinone/90% PEG-300 solution by oral gavage. Treatment was administered 6 h before each cocaine or saline treatment to reach maximal plasma concentrations of TAE684 (Galkin et al., 2007) for 7 d during the cocaine sensitization experiment. During the 10 d withdrawal period following cocaine sensitization, and on day 17 of the sensitization test, mice were not treated with TAE684. For the cocaine CPP experiment, mice were treated by oral gavage with 10 mg/kg TAE684 or vehicle 5 h before each conditioning session. Mice were not treated with TAE684 on the preconditioning or postconditioning test day.
Estradiol treatments.
Mice were injected subcutaneously in the flank with 5 μg of β-estradiol 3-benzoate suspended in sesame oil at 0.1 mg/ml.
Immunohistochemistry.
Mice were quickly killed as described above and were perfused intracardially with 0.9% NaCl for 5 min, followed by 4% PFA in PBS for 10 min. The brain was removed and processed as described previously (Kharazia et al., 2003). Images were acquired using a Zeiss confocal microscope and visualized using LSM software.
Western blots.
Mouse Acb samples were rapidly dissected and snap frozen in liquid nitrogen. Samples were then thawed on ice and homogenized in 100 μl of RIPA buffer (10 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 1% SDS) containing freshly added Complete mini protease inhibitor tablet (Roche). After clarifying lysates by centrifugation, a BCA protein assay (Thermo Fisher) was performed on samples. Samples were diluted into 4× LDS loading buffer containing β-mercaptoethanol, and subjected to electrophoresis and Western blotting using the NuPAGE Novex Tris-acetate mini gels (Invitrogen) and the GE Healthcare ECL Plus Western blotting detection system according to the manufacturer's instructions. For ALK blots, blocking and antibody incubation solutions contained 5% bovine serum albumen. Blots were quantified using ImageJ software.
Statistical analysis.
Biochemical data were analyzed by Student's t test or ANOVA. Cocaine sensitization experiments were analyzed by two-way repeated-measures ANOVA with post hoc comparisons as appropriate and CPP preference scores were analyzed by t tests.
Results
Alk expression is repressed by LMO4 in the striatum
We previously found that Lmo4 expression in the Acb attenuates the development of cocaine sensitization (Lasek et al., 2010). One known function of LMO4 is to negatively regulate expression of target genes by interacting with various DNA-bound transcription factors (Sum et al., 2002; Singh et al., 2005; Lu et al., 2006b; Xu et al., 2007). We hypothesized that expression of genes controlled by LMO4 would contribute to the development of cocaine sensitization. In fruit flies, dLmo regulates the acute behavioral response to cocaine (Tsai et al., 2004) and represses the expression of anaplastic lymphoma kinase (dAlk) (Lasek et al., 2011). These data suggest that a similar regulatory mechanism might operate in mammals. To determine whether mouse Alk expression is controlled by LMO4, we transfected primary mouse striatal neurons at 4 DIV with control siRNA (siScr) or siRNA targeting Lmo4 (siLmo4.3). At 11 DIV, RNA was isolated from the transfected cells and analyzed for Lmo4 and Alk expression by qPCR. In cells transfected with siLmo4.3, we observed a 70% reduction in Lmo4 expression (Fig. 1A, p < 0.001). In these same samples, Alk expression was modestly but significantly increased by 15% (Fig. 1B, p = 0.01), suggesting that Lmo4 normally functions to repress the expression of Alk. We also examined Alk expression in the Acb of adult mice infected with a lentivirus expressing shRNA targeting Lmo4 (shLmo4.3), or a control shRNA that is predicted to not target any transcript in the mouse genome (shScr). Alk expression in shLmo4.3-infected tissue showed a trend toward enhanced expression when compared to shScr-infected tissue (2.9-fold difference, p = 0.18, data not shown). Together, these data suggest that Alk is negatively regulated by LMO4 in the striatum.
Figure 1.
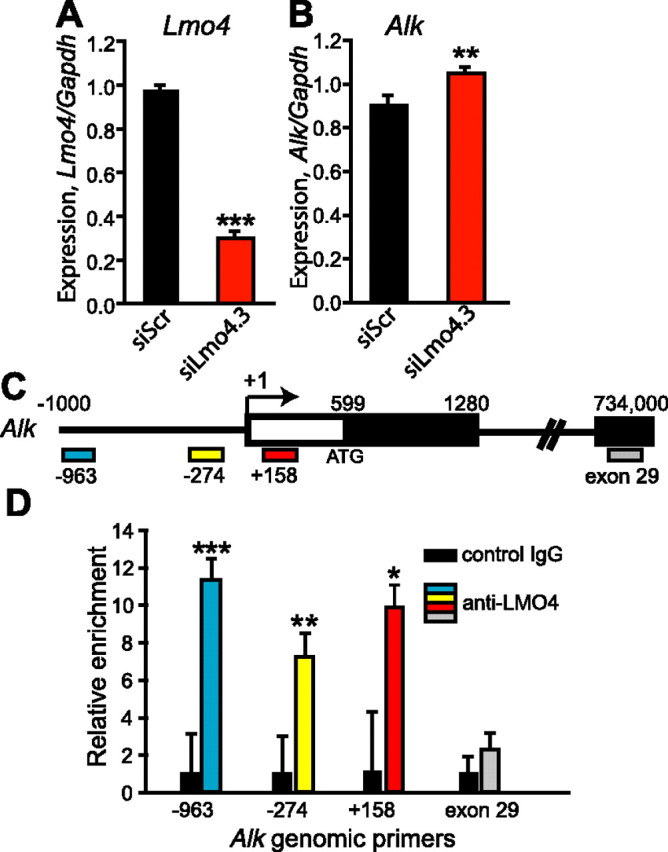
Alk expression in the striatum is repressed by LMO4. A, B, RNA was isolated from primary mouse striatal neurons transfected with siRNA targeting Lmo4 (siLmo4.3) or a control siRNA (siScr) and analyzed by qPCR for expression of Lmo4 (A) and Alk (B). Levels of each gene were normalized to Gapdh expression to control for RNA input. Data are expressed as mean ± SEM, n = 6. C, Schematic of the Alk promoter region. Colored boxes indicate the four sets of primers used for qPCR. The positions of nucleotides from the transcription start site are indicated with numbers, the first exon of the Alk gene is boxed, and shading shows the beginning of the open reading frame. The arrow indicates the direction of transcription. D, ChIP was performed using either an LMO4 (C-15) antibody or nonimmune IgG. Following ChIP, the relative amount of Alk promoter was analyzed by qPCR with each of the genomic primer sets. Black bars, Nonimmune IgG ChIP; colored bars: anti-LMO4 ChIP. Error bars are the SD of triplicate amplification reactions. ***p < 0.001, **p = 0.01, *p < 0.05.
We next examined whether LMO4 associates with the Alk promoter in striatum of adult mice using ChIP with LMO4 antibodies. Although LMO4 does not bind to DNA directly, it associates with promoters in large multiprotein complexes containing DNA binding factors. ChIP with an LMO4 antibody led to an enrichment of ∼10-fold of Alk promoter DNA as compared to control ChIP with nonimmune IgG (Fig. 1C,D). This enrichment was not observed with sequences located at the 3′ end of the Alk gene (exon 29), indicating specific binding of LMO4 to the Alk promoter. Thus, LMO4 appears to associate with the Alk promoter in striatum.
Alk promotes cocaine sensitization and reward
Since Alk expression is negatively regulated by LMO4 in the striatum, and loss of Lmo4 in Acb enhances cocaine sensitization, we hypothesized that, if this regulatory interaction was of functional significance, Alk downregulation in Acb should attenuate cocaine sensitization. To test this, we generated lentiviral vectors expressing shRNAs targeting Alk and found that all three constructs effectively reduced levels of the endogenous Alk transcript in Neuro-2a cells (Fig. 2A). Lentivirus expressing shAlk.2 or shScr was injected bilaterally into the Acb of mice. Two weeks after injection, infected Acb tissue was dissected and RNA was isolated and tested for Alk expression by qPCR. We found a 30% reduction in Alk expression in shAlk.2-infected compared to shScr-infected Acb (Fig. 2B, p = 0.016), indicating that the shAlk.2 construct was effective in reducing Alk expression in vivo. GFP expression from lentivirus was visible in the Acb after the completion of the cocaine sensitization experiment (see below), confirming sustained expression from the lentiviral vector throughout the testing period (Fig. 2C).
Figure 2.
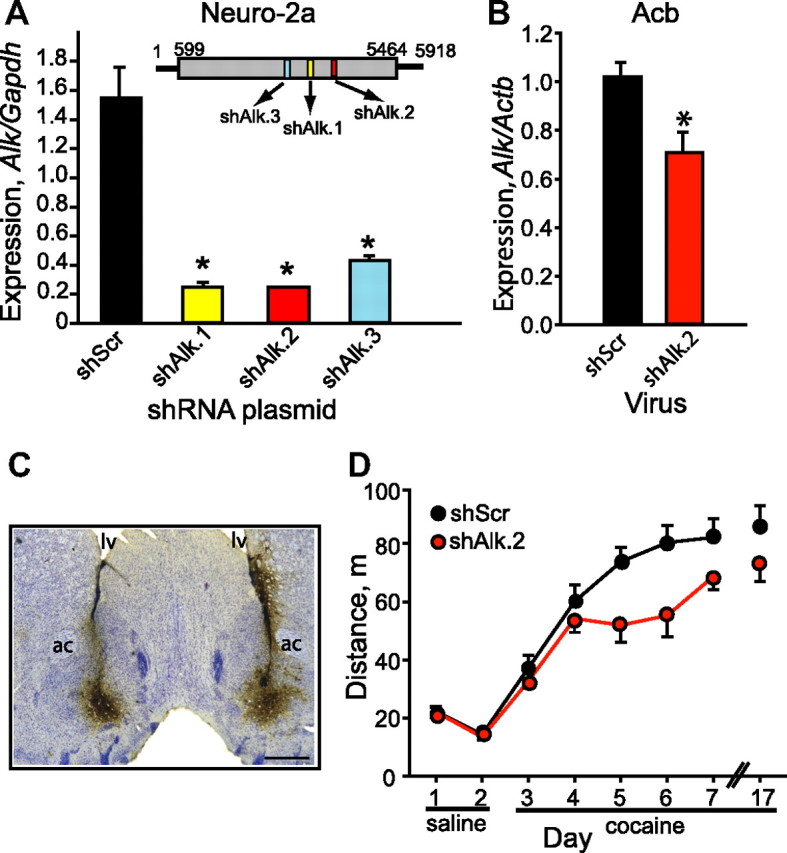
Alk in the Acb promotes cocaine sensitization. A, shRNAs targeting Alk are efficacious in cell culture. The inset in the graph diagrams the Alk transcript with the ORF shaded gray and the position of the shRNAs as colored boxes. Graph shows qPCR of Alk cDNA from Neuro-2a cells transfected with plasmids expressing one of three Alk shRNAs (shAlk.1–3) or a scrambled control shRNA (shScr). Data are presented as mean ± SD from triplicate transfections. *p < 0.001. B, qPCR indicating reduced Alk expression in the Acb of mice infected with shAlk.2 virus, *p < 0.05. C, Representative coronal section from a mouse infected with shAlk.2 lentivirus in the Acb. Section was stained with anti-GFP antibody (brown) and counterstained with Nissl (blue). ac, Anterior commissure; lv, lateral ventricle. Scale bar, 500 μm. D, Knockdown of Alk by RNAi in Acb results in attenuated cocaine sensitization. Mice were injected intraperitoneally with saline on days 1 and 2, and then injected with 15 mg/kg cocaine on days 3–7 and 17. Shown is distance traveled over a 15 min period immediately following injection. Error bars indicate SEM.
We next injected mice bilaterally in the Acb with shScr or shAlk.2 lentivirus and tested cocaine sensitization 2 weeks after surgery. Cocaine sensitization was significantly attenuated in mice expressing shAlk.2 compared to shScr control mice (treatment: F(1,25) = 6.537, p = 0.017; day: F(5,125) = 25.525, p < 0.001; treatment × day: F(5,125) = 1.717, p = 0.135, Fig. 2D), especially during the period of sensitization development. We observed a similar phenotype with a second lentiviral construct targeting Alk in the Acb (shAlk.1, data not shown). To confirm the results obtained with shRNA-mediated downregulation of Alk, we tested a small-molecule inhibitor of ALK's tyrosine kinase activity, TAE684 (Galkin et al., 2007). Mice were treated daily (days 1–7) during the cocaine sensitization experiment with 10 mg/kg TAE684 or vehicle by oral gavage 6 h before behavioral testing. Consistent with shRNA knockdown of Alk in the Acb, treatment with the ALK inhibitor resulted in delayed cocaine sensitization, particularly during the second to fourth days of cocaine injection (Fig. 3A). Data for cocaine treatment on days that mice received TAE684 (days 3–7) was analyzed by two-way repeated-measures ANOVA. There was a trend toward an effect of treatment and a treatment by day interaction (treatment: F(1,35) = 2.461, p = 0.126; day: F(4,140) = 13.196, p < 0.001; treatment × day: F(4,140) = 2.001, p = 0.098). However, a focused ANOVA on days 4–6 showed a significant effect of treatment (treatment: F(1,35) = 4.660, p = 0.038; day: F(2,70) = 0.617, p = 0.543; treatment × day: F(2,70) = 0.00257, p = 0.997).
Figure 3.

The ALK inhibitor TAE684 reduces cocaine sensitization and CPP. A, Delayed cocaine sensitization in mice treated with TAE684. Mice were administered TAE684 daily 6 h before saline or cocaine treatments and tested for sensitization as described in Figure 2D. B, Reduced cocaine CPP in mice treated with TAE684. Mice were treated daily 5 h before saline and cocaine conditioning sessions. On the test day, mice were not treated with TAE684. Shown is the preference score in minutes, *p < 0.05, n = 13. Error bars indicate SEM.
We next tested whether ALK activity might be important for the development of cocaine CPP, a measure of cocaine reward. We treated mice with TAE684 5 h before each 10 mg/kg cocaine and saline conditioning session. One day after the completion of the final conditioning session, mice were tested for CPP in the absence of both cocaine and TAE684. Mice that had been previously treated with TAE684 displayed a significantly attenuated preference for the cocaine-paired chamber compared to vehicle-treated mice (Fig. 3B). No differences in total distance traveled on test day were found between the two groups (data not shown).
Since ALK affects the development of cocaine sensitization and CPP, we asked whether Alk expression in the Acb is altered by cocaine. Alk transcript levels were analyzed in mice treated for 1, 2, or 5 d with single daily injections of 15 mg/kg cocaine or saline as a control. Acb tissue was dissected 2 and 24 h after the final injection and analyzed for Alk levels by qPCR. Alk transcript levels in the Acb were unchanged with cocaine treatment (data not shown). These results indicate that Alk expression is not affected by cocaine. However, our data indicate that ALK functions normally in the Acb of adult mice to promote the development of cocaine sensitization and that ALK activity is important for cocaine reward.
ERα is expressed in Acb and colocalizes with LMO4
Our data indicate that LMO4 regulates Alk expression through an interaction with the Alk promoter. However, LMO4 does not bind to DNA directly, but instead modulates transcription through various DNA-binding transcription factors, including ERα (Sum et al., 2002; Singh et al., 2005; Lu et al., 2006b; Xu et al., 2007). To determine whether an interaction between LMO4 and estrogen receptors in the Acb might be possible, we examined the expression of ERα in the Acb. We found that Esr1 mRNA (encoding ERα) is expressed in Acb, consistent with ERα antibody staining in mouse brain (data not shown) (Mitra et al., 2003). To visualize ERα protein expression in Acb, we immunostained Acb-containing brain sections using antibodies to ERα and LMO4. Nuclear ERα staining was visible in scattered cells throughout the Acb (Fig. 4), colocalizing in some cells with nuclear LMO4. We observed a similar pattern of ERα localization in the Acb of both male and female mice (A.W.L. and V.K., unpublished results). LMO4 was more widely expressed in Acb than ERα. Thus, ERα is expressed in Acb and is present in a subset of cells that also express LMO4.
Figure 4.
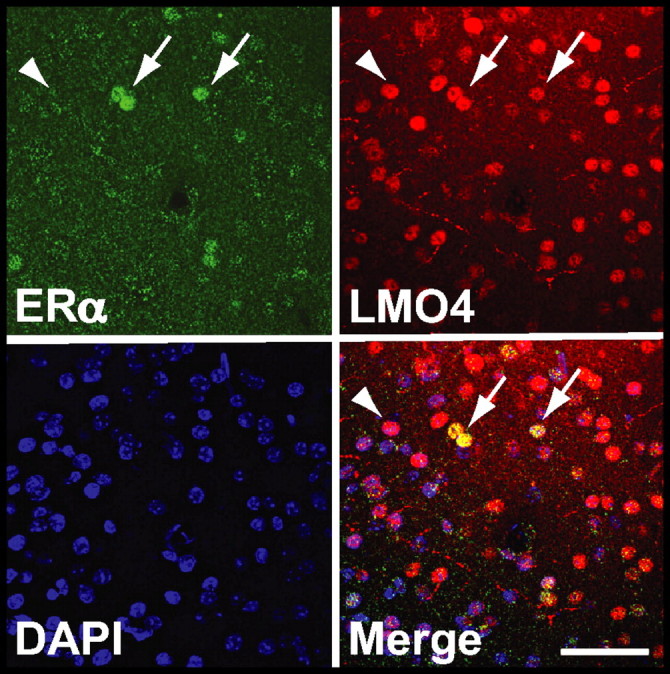
Colocalization of LMO4 and ERα in mouse Acb. A coronal section from mouse Acb was immunostained with antibodies to ERα (green) and LMO4 (red). ERα and LMO4 are colocalized in some Acb neurons (arrows). The yellow staining in the merged panel illustrates localization of ERα in a subset of LMO4-positive neurons. Many LMO4-positive neurons do not appear to express ERα (arrowhead). Nuclear (DAPI) staining is in blue. Scale bar, 50 μm.
Alk is an estrogen-regulated gene
Since LMO4 and ERα are known to directly interact and control transcription through estrogen response elements (EREs) (Singh et al., 2005), we asked whether Alk expression may be regulated by ERα. We first analyzed the Alk promoter for predicted transcription factor binding sites using the transcription element search system (TESS) (Schug, 2008). Indeed, the Alk promoter region between −2 kb and the transcription start site contains three predicted ERα half binding sites. To test whether these potential ERα binding sites are functional, we treated mice with a single injection of either 5 μg of β-estradiol 3-benzoate (EB) or sesame oil vehicle. Two hours later, brains were rapidly dissected, RNA isolated, and cDNA analyzed by qPCR for Alk expression. Alk levels were increased by 46% in Acb (p = 0.018) and 42% in caudate–putamen (CPu, p = 0.040) upon EB treatment, while remaining unchanged in frontal cortex and hippocampus (Fig. 5A). Alk levels remained elevated in the Acb for 16 h after a single EB injection (data not shown). Similarly, Alk levels in ovariectomized females treated with EB increased by 38% in the CPu (p = 0.016, data not shown). Neither expression of Lmo4 nor Esr1 changed with EB treatment in the four brain regions analyzed (data not shown). These data suggest that Alk is an estrogen-responsive gene in specific brain regions, particularly the striatum.
Figure 5.

Alk expression is regulated by β-estradiol and ERα. A, Alk levels increase in striatum with β-estradiol treatment. Mice were injected once with 5 μg of β-estradiol 3-benzoate (EB), brains were dissected 2 h later, and RNA was isolated from individual brain regions. Alk mRNA levels were measured by qPCR and normalized to β-actin (Actb) expression. B, ERα is enriched on the Alk promoter with estrogen treatment. Cross-linked chromatin was immunoprecipitated from untreated (white bars) or β-estradiol (E2)-treated (100 nm, black bars) NB41A3 cells using nonimmune IgG (control) or ERα antibodies. Shown is a representative experiment. C, ALK protein levels are increased in the Acb of Esr1KO mice. Acb from wild-type (+/+) and homozygous (−/−) Esr1KO mice was dissected and probed for ALK expression using antibodies to ALK by Western blotting. Blots were stripped and reprobed for β-actin as a loading control. Results were quantified using ImageJ analysis. *p < 0.05. Error bars indicate SEM.
To establish whether Alk is directly regulated by ERα, we performed ChIP on the endogenous Alk promoter using antibodies specific to ERα. Since ERα is sparsely expressed in mouse Acb (Fig. 4), we used a mouse neuroblastoma cell line, NB41A3, which expresses ERα and responds to β-estradiol when transfected with a synthetic reporter construct containing estrogen response elements (A.W.L. and U.H., unpublished results). Cross-linked chromatin from NB41A3 cells was immunoprecipitated with two independent ERα antibodies. Treatment of NB41A3 cells for 45 min with 100 nm β-estradiol led to a fivefold and threefold enrichment of ERα bound to the Alk promoter using the ERα MC-20 and HC-20 antibodies, respectively, compared to ChIP with nonimmune IgG (Fig. 5B). Together, these results indicate that ERα is enriched on the Alk promoter when cells are stimulated with β-estradiol, and suggest that Alk is a bona fide estrogen-regulated gene in neurons. Since Alk expression appears to be regulated by ERα, we predicted that Alk levels would be altered in Esr1KO mice. We examined ALK protein levels in the Acb of Esr1KO mice by Western blotting and found, to our surprise, that ALK protein levels were increased in the Acb of Esr1KO mice (p = 0.047, Fig. 5C), suggesting that ERα negatively regulates ALK expression (see Discussion).
ERα knock-out mice display enhanced cocaine sensitization and CPP
Estrogen is known to contribute to altered behavioral responses to cocaine. In particular, β-estradiol enhances cocaine sensitization and CPP in female rats (Sircar and Kim, 1999; Hu and Becker, 2003; Russo et al., 2003; Segarra et al., 2010). Since mice with reduced levels of Lmo4 or Alk display enhanced and reduced cocaine sensitization, respectively, and Alk expression is increased in Esr1KO mice, we hypothesized that mice lacking ERα might show enhanced cocaine sensitization. Indeed, both male and female Esr1KO mice displayed significantly enhanced development of sensitization when compared to wild-type littermates (genotype: F(2,66) = 1.45, p = 0.242; day: F(5,330) = 39.39, p < 0.001; genotype × day interaction: F(5,330) = 2.03, p = 0.030, Fig. 6A). We also tested Esr1KO mice for cocaine CPP; both male and female Esr1KO mice showed a trend toward increased cocaine CPP compared to wild-type littermates (Fig. 6B, p = 0.090), with a slight decrease in total distance traveled on test day (11% decrease, p = 0.041, data not shown). We suggest that ERα modulates cocaine sensitization and possibly CPP in mice through regulation of Alk expression in the Acb.
Figure 6.

ERα regulates the development of cocaine sensitization and CPP. A, Wild-type (+/+, closed circles), heterozygous (+/−, open circles), and homozygous (−/−, triangles) Esr1KO mice were subjected to the cocaine sensitization protocol. Since two-way ANOVA indicated no difference between males and females, data from both sexes was combined. Esr1KO mice show increased cocaine sensitization with a significant genotype × day interaction. *p < 0.05 by post hoc SNK comparisons. All data are presented as mean ± SEM. B, Trend toward enhanced cocaine CPP in Esr1KO (−/−) mice compared to wild-type (+/+) littermates. There was no significant difference between male and female mice by two-way ANOVA, so data from both sexes was combined, n = 16–18.
Discussion
Here we show that Alk is a novel transcriptional target of LMO4 and ERα that normally promotes the development of cocaine sensitization and reward. Alk expression is repressed by LMO4 and induced by β-estradiol in the striatum. Moreover, ERα associates with the Alk promoter in neuroblastoma cells, suggesting that the induction of Alk expression by β-estradiol is mediated by ERα. Yet, expression of Alk is increased in Esr1KO mice, a result that seems initially to contradict the enhanced Alk expression observed upon β-estradiol treatment (see below). We propose a model for the regulation of Alk expression by LMO4 and ERα in the Acb and its effect on behavioral responses to cocaine (Fig. 7A). Estrogen is predicted to activate transcription through ERα, resulting in increased ALK in the Acb. Higher levels of ALK in the Acb would enhance cocaine sensitization, since downregulation of Alk by RNAi in the Acb or treatment with an ALK inhibitor decreases cocaine sensitization. In this model, reducing LMO4 would also lead to increased ALK in the Acb and enhanced cocaine sensitization. The behavioral effect is consistent with what we have observed with Lmo4 gene-trap mice and mice with reduced Lmo4 expression in the Acb (Lasek et al., 2010). Similarly, we predict that increasing ALK either by increasing estrogen levels or decreasing LMO4 levels would increase cocaine reward, since treatment with the ALK inhibitor TAE684 decreases cocaine CPP.
Figure 7.

Model for regulation of Alk transcription by estrogen and LMO4. A, Estrogen (through ERα) and LMO4 have opposing effects on Alk levels in Acb and behavioral responses to cocaine. B, Model illustrating the regulation of Alk transcription by LMO4 and ERα. Under conditions of low estrogen or high LMO4, a repressive complex occupies the Alk promoter, resulting in low levels of Alk expression. Conversely, in the presence of estrogen, or if LMO4 levels decrease, repression of the Alk promoter is relieved by recruitment of a coactivator complex to the Alk promoter and Alk levels increase. We hypothesize that LMO4 recruits a histone deacetylase to the complex, since LMO4 is known to interact with HDAC2 in complex with ERα in breast cancer cells, and that ERα interacts with another DNA binding partner, such as SP1, CREB, or the AP1 complex among others (white oval).
LMO4 and ERα are known to interact directly at the promoters of estrogen-responsive genes in breast cancer cells (Singh et al., 2005). However, this is the first demonstration to our knowledge that regulation of estrogen-responsive genes in the nervous system may involve LMO4 and ERα. In our model, ERα and LMO4 physically interact to oppose each other's function in modulating transcription (Fig. 7B). Loss of LMO4 would alleviate repression of the Alk promoter, leading to increased Alk levels. Addition of estrogen would produce the same outcome, through hormone binding to ERα and potential recruitment of transcriptional coactivators to the Alk promoter. In breast cancer cells, LMO4 is found in a complex with the histone deacetylase HDAC2 (Singh et al., 2005; Wang et al., 2007). We therefore speculate that LMO4 may recruit a similar corepressor complex to the Alk promoter in striatal neurons. Surprisingly, we found that ALK protein levels are increased in the Acb of male Esr1KO mice. These data suggest that, in Acb, ERα is bound to the Alk promoter in a repressive complex in the absence of estrogen and that removal of ERα would destabilize this complex, resulting in derepression of the Alk promoter. This outcome is similar to what is observed upon downregulation of LMO4. Since an increase in ALK levels/activity is predicted to cause enhanced cocaine sensitization and reward, this hypothesis is supported by the increase in cocaine sensitization and CPP that we observed in Esr1KO mice. Based on our model, increased Alk expression and cocaine sensitization would be expected in both male and female Esr1KO mice, an effect that is independent of estrogen levels. However, we cannot formally rule out the possibility that compensatory changes in Esr1KO mice are responsible for the observed effects on Alk expression and behavioral responses to cocaine. Further investigation is required to stringently test this model.
We found that LMO4 is more widely expressed in the striatum than ERα. Our model (Fig. 7B) predicts that ERα is required to recruit LMO4 to the Alk promoter. The implication of this prediction is that Alk is not regulated throughout the entire striatum by LMO4 and estrogen, but rather in a subset of cells that express both LMO4 and ERα. Indeed, the neurons that express ERα may represent a distinct subtype in the striatum (e.g., neurons expressing dopamine D1 vs D2 receptors) that play a specific role in regulating cocaine behaviors. Determining whether this is the case is a very interesting area for future studies. In addition, LMO4 is known to bind to many different transcription factors and likely regulates other genes in neurons independently of ERα. Although the interaction between LMO4 and ERα only occurs in a subset of total cells in the striatum to affect Alk expression, this regulation still appears to be relevant for altering behavioral responses to cocaine.
Another notable point is that Alk expression is increased in male mice treated with β-estradiol. Although males have low circulating estrogen levels, testosterone is converted locally in the brain to estrogen by aromatase (Simpson and Davis, 2001), which is important for male sexual and aggressive behaviors (Wu and Shah, 2011). Thus, it is possible that Alk expression in the brain of males may be regulated by local estrogen synthesis. Females have high circulating estrogen that is produced by the ovaries. Interestingly, estrogen is known to augment several behavioral responses to cocaine in female rats, including cocaine sensitization, CPP, and intracranial self-stimulation, another measure of reward (Sircar and Kim, 1999; Hu and Becker, 2003; Russo et al., 2003; Galankin et al., 2010; Segarra et al., 2010). The molecular mechanisms responsible for this effect of estrogen on cocaine behaviors are currently unknown. We propose that the ability of estrogen to regulate transcription of specific genes, such as Alk, in the Acb is a potential mechanism by which estrogen enhances cocaine sensitization and reward. Future studies will examine more directly whether regulation of Alk expression by estrogen is responsible for enhanced cocaine sensitization and reward in females.
ALK is a receptor tyrosine kinase in the insulin receptor family. The mechanism by which ALK affects behavioral responses to cocaine is currently unknown. We found that cocaine does not alter Alk expression, yet it may affect ALK phosphorylation and signaling activity. Studies in invertebrates have provided the most information about ALK function in the nervous system. The Caenorhabditis elegans Alk homolog is hypothesized to regulate synapse differentiation (Liao et al., 2004). In Drosophila, Alk regulates retinal axon targeting and the expression of guidance molecules involved in synapse formation in target neurons (Bazigou et al., 2007). In the rat cell line PC12 and the human neuroblastoma cell line SK-N-SH, activation of ALK tyrosine kinase activity leads to neuronal differentiation and neurite outgrowth through activation of ERK (Souttou et al., 2001; Motegi et al., 2004). Thus, ALK could regulate neurite growth and targeting and synapse differentiation as well as their plasticity through ERK signaling in the adult animal during cocaine sensitization and reward. ERK signaling is clearly important for cocaine sensitization and CPP (Lu et al., 2006a). In mammals, the number of dendritic spines increases in the Acb after cocaine sensitization (Robinson and Kolb, 1999), so one may speculate that ALK plays a role in this process. We analyzed dendritic spines in Acb medium spiny neurons expressing either the shAlk.2 or the control shScr virus after repeated cocaine treatment, and did not observe any effect of reducing Alk on spine numbers (A.W.L., F.G., and U.H., unpublished results). However, ALK tyrosine kinase activity is important for cocaine sensitization and reward, since the ALK kinase inhibitor TAE684 attenuates sensitization and CPP. Future studies will address the mechanisms through which cocaine regulates ALK activity and how this, in turn, affects ALK's intracellular signaling mechanisms and, ultimately, behavioral responses to cocaine.
Footnotes
The authors declare no conflict of interest.
This work was supported by funds provided to U.H. by the State of California for medical research on alcohol and substance abuse through the University of California at San Francisco, by the Sandler Family Foundation, and by the NIH (DA14809). We are grateful to members of Nirao Shah's laboratory (Melody Wu for information on estradiol treatments and Scott Juntti for helpful discussions). We thank Chris Kliethermes for assistance with estradiol injections and brain dissections and Yun Zhang for virus production. We also thank Novartis for the generous gift of TAE684.
References
- Bardo MT, Bevins RA. Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology (Berl) 2000;153:31–43. doi: 10.1007/s002130000569. [DOI] [PubMed] [Google Scholar]
- Bazigou E, Apitz H, Johansson J, Lorén CE, Hirst EM, Chen PL, Palmer RH, Salecker I. Anterograde Jelly belly and Alk receptor tyrosine kinase signaling mediates retinal axon targeting in Drosophila. Cell. 2007;128:961–975. doi: 10.1016/j.cell.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Galankin T, Shekunova E, Zvartau E. Estradiol lowers intracranial self-stimulation thresholds and enhances cocaine facilitation of intracranial self-stimulation in rats. Horm Behav. 2010;58:827–834. doi: 10.1016/j.yhbeh.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, Xia G, Steensma R, Chopiuk G, Jiang J, Wan Y, Ding P, Liu Y, Sun F, Schultz PG, Gray NS, Warmuth M. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A. 2007;104:270–275. doi: 10.1073/pnas.0609412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Becker JB. Effects of sex and estrogen on behavioral sensitization to cocaine in rats. J Neurosci. 2003;23:693–699. doi: 10.1523/JNEUROSCI.23-02-00693.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharazia VN, Jacobs KM, Prince DA. Light microscopic study of GluR1 and calbindin expression in interneurons of neocortical microgyral malformations. Neuroscience. 2003;120:207–218. doi: 10.1016/s0306-4522(03)00282-3. [DOI] [PubMed] [Google Scholar]
- Lasek AW, Janak PH, He L, Whistler JL, Heberlein U. Downregulation of mu opioid receptor by RNA interference in the ventral tegmental area reduces ethanol consumption in mice. Genes Brain Behav. 2007;6:728–735. doi: 10.1111/j.1601-183X.2007.00303.x. [DOI] [PubMed] [Google Scholar]
- Lasek AW, Kapfhamer D, Kharazia V, Gesch J, Giorgetti F, Heberlein U. Lmo4 in the nucleus accumbens regulates cocaine sensitivity. Genes Brain Behav. 2010;9:817–824. doi: 10.1111/j.1601-183X.2010.00620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek AW, Lim J, Kliethermes CL, Berger KH, Joslyn G, Brush G, Xue L, Robertson M, Moore MS, Vranizan K, Morris SW, Schuckit MA, White RL, Heberlein U. An evolutionary conserved role for anaplastic lymphoma kinase in behavioral responses to ethanol. PLoS One. 2011;6:e22636. doi: 10.1371/journal.pone.0022636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao EH, Hung W, Abrams B, Zhen M. An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature. 2004;430:345–350. doi: 10.1038/nature02647. [DOI] [PubMed] [Google Scholar]
- Lu L, Koya E, Zhai H, Hope BT, Shaham Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006a;29:695–703. doi: 10.1016/j.tins.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Lu Z, Lam KS, Wang N, Xu X, Cortes M, Andersen B. LMO4 can interact with Smad proteins and modulate transforming growth factor-beta signaling in epithelial cells. Oncogene. 2006b;25:2920–2930. doi: 10.1038/sj.onc.1209318. [DOI] [PubMed] [Google Scholar]
- McGough NN, He DY, Logrip ML, Jeanblanc J, Phamluong K, Luong K, Kharazia V, Janak PH, Ron D. RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. J Neurosci. 2004;24:10542–10552. doi: 10.1523/JNEUROSCI.3714-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SW, Hoskin E, Yudkovitz J, Pear L, Wilkinson HA, Hayashi S, Pfaff DW, Ogawa S, Rohrer SP, Schaeffer JM, McEwen BS, Alves SE. Immunolocalization of estrogen receptor beta in the mouse brain: comparison with estrogen receptor alpha. Endocrinology. 2003;144:2055–2067. doi: 10.1210/en.2002-221069. [DOI] [PubMed] [Google Scholar]
- Motegi A, Fujimoto J, Kotani M, Sakuraba H, Yamamoto T. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J Cell Sci. 2004;117:3319–3329. doi: 10.1242/jcs.01183. [DOI] [PubMed] [Google Scholar]
- Rétaux S, Bachy I. A short history of LIM domains (1993–2002): from protein interaction to degradation. Mol Neurobiol. 2002;26:269–281. doi: 10.1385/MN:26:2-3:269. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Addiction. Annu Rev Psychol. 2003;54:25–53. doi: 10.1146/annurev.psych.54.101601.145237. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci. 1999;11:1598–1604. doi: 10.1046/j.1460-9568.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Festa ED, Fabian SJ, Gazi FM, Kraish M, Jenab S, Quinones-Jenab V. Gonadal hormones differentially modulate cocaine-induced conditioned place preference in male and female rats. Neuroscience. 2003;120:523–533. doi: 10.1016/s0306-4522(03)00317-8. [DOI] [PubMed] [Google Scholar]
- Schug J. Using TESS to predict transcription factor binding sites in DNA sequence. Curr Protoc Bioinformatics Chapter. 2008;2 doi: 10.1002/0471250953.bi0206s21. Unit 2.6. [DOI] [PubMed] [Google Scholar]
- Segarra AC, Agosto-Rivera JL, Febo M, Lugo-Escobar N, Menéndez-Delmestre R, Puig-Ramos A, Torres-Diaz YM. Estradiol: a key biological substrate mediating the response to cocaine in female rats. Horm Behav. 2010;58:33–43. doi: 10.1016/j.yhbeh.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson ER, Davis SR. Minireview: aromatase and the regulation of estrogen biosynthesis—some new perspectives. Endocrinology. 2001;142:4589–4594. doi: 10.1210/endo.142.11.8547. [DOI] [PubMed] [Google Scholar]
- Singh RR, Barnes CJ, Talukder AH, Fuqua SA, Kumar R. Negative regulation of estrogen receptor alpha transactivation functions by LIM domain only 4 protein. Cancer Res. 2005;65:10594–10601. doi: 10.1158/0008-5472.CAN-05-2268. [DOI] [PubMed] [Google Scholar]
- Sircar R, Kim D. Female gonadal hormones differentially modulate cocaine-induced behavioral sensitization in Fischer, Lewis, and Sprague-Dawley rats. J Pharmacol Exp Ther. 1999;289:54–65. [PubMed] [Google Scholar]
- Souttou B, Carvalho NB, Raulais D, Vigny M. Activation of anaplastic lymphoma kinase receptor tyrosine kinase induces neuronal differentiation through the mitogen-activated protein kinase pathway. J Biol Chem. 2001;276:9526–9531. doi: 10.1074/jbc.M007333200. [DOI] [PubMed] [Google Scholar]
- Sum EY, Peng B, Yu X, Chen J, Byrne J, Lindeman GJ, Visvader JE. The LIM domain protein LMO4 interacts with the cofactor CtIP and the tumor suppressor BRCA1 and inhibits BRCA1 activity. J Biol Chem. 2002;277:7849–7856. doi: 10.1074/jbc.M110603200. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–342. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LT, Bainton RJ, Blau J, Heberlein U. Lmo mutants reveal a novel role for circadian pacemaker neurons in cocaine-induced behaviors. PLoS Biol. 2004;2:e408. doi: 10.1371/journal.pbio.0020408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Kumar A, Nestler EJ. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci. 2004;24:5603–5610. doi: 10.1523/JNEUROSCI.0589-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (Berl) 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- Wang N, Lin KK, Lu Z, Lam KS, Newton R, Xu X, Yu Z, Gill GN, Andersen B. The LIM-only factor LMO4 regulates expression of the BMP7 gene through an HDAC2-dependent mechanism, and controls cell proliferation and apoptosis of mammary epithelial cells. Oncogene. 2007;26:6431–6441. doi: 10.1038/sj.onc.1210465. [DOI] [PubMed] [Google Scholar]
- Wu MV, Shah NM. Control of masculinization of the brain and behavior. Curr Opin Neurobiol. 2011;21:116–123. doi: 10.1016/j.conb.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Mannik J, Kudryavtseva E, Lin KK, Flanagan LA, Spencer J, Soto A, Wang N, Lu Z, Yu Z, Monuki ES, Andersen B. Co-factors of LIM domains (Clims/Ldb/Nli) regulate corneal homeostasis and maintenance of hair follicle stem cells. Dev Biol. 2007;312:484–500. doi: 10.1016/j.ydbio.2007.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]